
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A rhodium-catalyzed transannulation of N-(per)fluoroalkyl-1,2,3-triazoles under microwave conditions – a general route to N-(per)fluoroalkyl-substituted five-membered heterocycles†
Vladimir
Motornov‡
 ab,
Athanasios
Markos‡
ac and
Petr
Beier
*a
ab,
Athanasios
Markos‡
ac and
Petr
Beier
*a
aThe Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nám. 2, CZ-166 10 Prague 6, Czech Republic. E-mail: beier@uochb.cas.cz
bHigher Chemical College, D. I. Mendeleev University of Chemical Technology of Russia, Miusskaya sq. 9, 125047 Moscow, Russian Federation
cDepartment of Organic Chemistry, Faculty of Science, Charles University, Hlavova 2030/8, CZ-128 43 Prague 2, Czech Republic
First published on 14th March 2018
Abstract
A rhodium-catalyzed transannulation via ring-opening of N-(per)fluoroalkyl-substituted 1,2,3-triazoles followed by cycloaddition with different nitriles, enol ethers, isocyanates and silyl ketene acetals under microwave heating provided a highly efficient route to previously unreported N-(per)fluoroalkyl-substituted imidazoles, pyrroles, imidazolones and pyrrolones, respectively. These reactions were found to be applicable to the synthesis of a variety of 5-membered heterocycles bearing different (per)fluoroalkyl substituents as well as both electron-donating and electron-withdrawing groups attached to the heterocyclic core.
Substituted imidazoles and pyrroles are widely used as different biologically active compounds, agrochemicals and pharmaceuticals including anticancer, antimicrobial, fungicidal and antiviral drugs.1–3 Fluorinated and in particular trifluoromethyl-containing compounds also have a broad spectrum of applications ranging from drug candidates to novel materials.4–9 However, N-CF3 and N-RF (RF = perfluoroalkyl) motifs are relatively rare10–13 and imidazoles or pyrroles with perfluoroalkyl groups attached to the nitrogen atom are almost unknown.14,15 Some azoles were trifluoromethylated in an electrophilic way but generally a mixture of regioisomers in low or moderate yields was obtained.16 Recently, we have reported the synthesis of azidoperfluoroalkanes (RFN3) by the reaction of the precursors of fluorinated carbanions with an electrophilic azide source which allowed access to N-(per)fluoroalkyl-1,2,3-triazoles by Cu(I)-catalyzed azide–alkyne cycloaddition.17,18
N-Sulfonyl-substituted 1,2,3-triazoles are known to undergo a rhodium-catalyzed ring-opening and nitrogen elimination to form a rhodium iminocarbene and subsequent insertion of the carbene or cycloaddition with different 2π-components.19–21 This transannulation reaction is a highly efficient, atom-economical method for the synthesis of five-membered heterocycles, such as imidazoles,22 pyrroles,23–25 imidazolones,26 pyrrolones,27 and others.28,29 Fused pyridotriazoles, which exist in equilibrium with the diazoform, also undergo transannulation.30,31 Transannulation was reported to be limited to N-sulfonyl triazoles with one exception being N-(1,2,4-triazolyl)-substituted triazoles which undergo transannulation with nitriles (Scheme 1).32 Both the abovementioned groups exhibit a strong –M effect, which facilitates triazole ring opening. We have proposed that the rhodium-catalyzed transannulation of N-(per)fluoroalkyl-1,2,3-triazoles might lead to a range of structurally diverse five-membered nitrogen heterocycles with (per)fluoroalkyl groups attached to the nitrogen atom and herein we report our findings in this direction (Scheme 1).
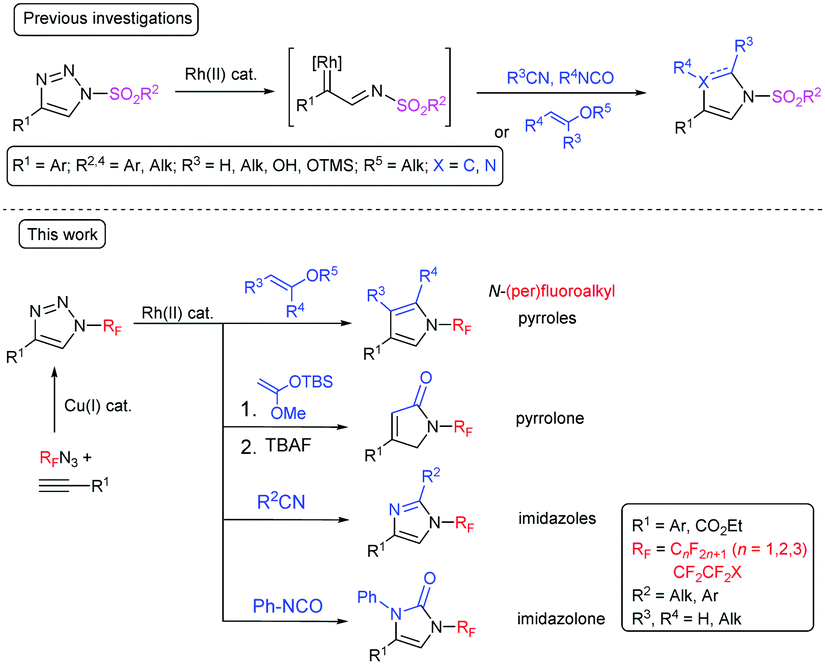 | ||
| Scheme 1 Transannulation reactions with 1,2,3-triazoles. | ||
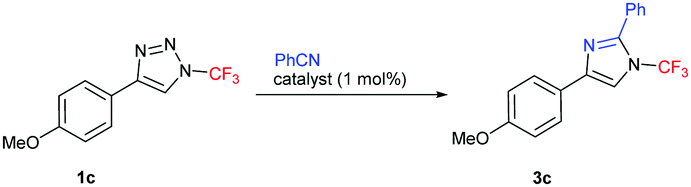
We initiated our studies using the reaction between 4-methoxyphenyl-substituted 1-trifluoromethyl-1,2,3-triazole 1c and benzonitrile with a rhodium(II) acetate dimer as a catalyst. While conventional heating of the mixture in 1,2-dichloroethane (DCE) gave poor conversion to product 3c (Table 1, entry 1), full conversion of 1c and moderate isolated yield of 3c were achieved by microwave heating (entry 2). Use of a two-fold excess of benzonitrile led to an increase in the yield to 51% (entry 3). Reaction temperature and time optimization revealed that 140 °C and 20 min were optimal. Finally, switching to Rh2(Oct)4 and using chloroform as the solvent afforded the optimized 72% isolated yield of 3c (entry 7). Importantly, the reaction did not take place without the rhodium catalyst or with CuTC (entry 8).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | PhCNb | Solvent, temp., time | Yieldc (%) |
| a Reaction conditions: 1c (0.10 mmol), PhCN (0.13–0.20 mmol), [RhII] (1 mol%), solvent (2 mL). b equiv. c Conversion of 1c was determined by 19F NMR, in brackets isolated yields of 3c. | ||||
| 1 | Rh2(OAc)4 | 1.3 | DCE, 80 °C, 17 h | 3 |
| 2 | Rh2(OAc)4 | 1.3 | DCE, 150 °C MW, 1 h | 100 (40) |
| 3 | Rh2(OAc)4 | 2 | DCE, 150 °C MW, 1 h | 100 (51) |
| 4 | Rh2(OAc)4 | 2 | DCE, 100 °C MW, 20 min | 40 (24) |
| 5 | Rh2(OAc)4 | 2 | DCE, 140 °C MW, 20 min | 100 (44) |
| 6 | Rh2(OAc)4 | 2 | CHCl3, 140 °C MW, 20 min | 100 (59) |
| 7 | Rh2(Oct)4 | 2 | CHCl3, 140 °C MW, 20 min | 100 (72) |
| 8 | None or CuTC | 2 | CHCl3, 140 °C MW, 20 min | 0 |
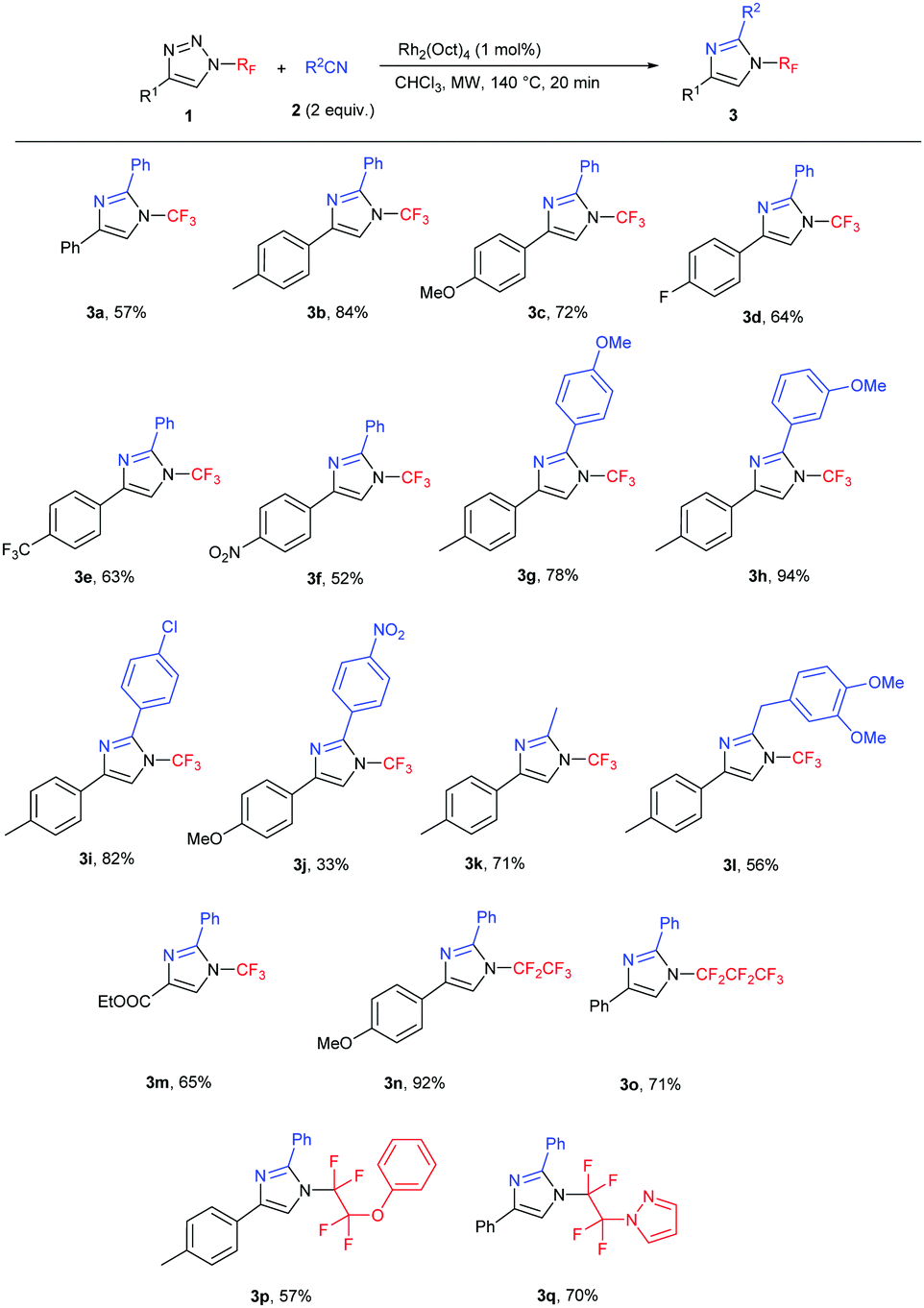
Subsequently, the scope of the reaction was studied under the optimized conditions (Table 2). We tested the transannulation reaction with nitriles on variously substituted aromatic N-perfluoroalkyl-triazoles (Scheme 3). Electron-rich substrates such as 4-methoxy- and 4-methyl-substituted triazoles (1b, 1c) were found to be more reactive and produced higher yields than electron-poor substrates (such as 1f). It is worth noting that this reaction is applicable for the synthesis of the ethoxycarbonyl-substituted aliphatic triazole 3m as well, presumably due to the ability of the CO2Et group to stabilize the forming rhodium carbenoid intermediate similarly to the aryl group despite the reported lower stability of electron-poor carbenoids.19
| a Isolated yields. |
|---|
|
We found the reaction to be rather insensitive to the electronic nature of the nitrile. The yields were excellent in the case of electron-rich, neutral and mildly electron-poor aromatic nitriles and aliphatic nitriles. Only highly electron-poor 4-nitrobenzonitrile reacted much slower under the conditions, thus providing the product in moderate yield.
Longer carbon chain N-perfluoroalkyl triazoles participated in the transannulation reaction with equal efficiency to their CF3 analogues and high yields of pentafluoroethyl- and perfluoropropyl-substituted imidazoles (3n and 3o, respectively) were obtained. Moreover, tetrafluoroethylene-containing substrates with phenoxy or pyrazolyl functional groups were tolerated, providing a route to multifunctionalized N-fluoroalkylated imidazoles 3p and 3q.
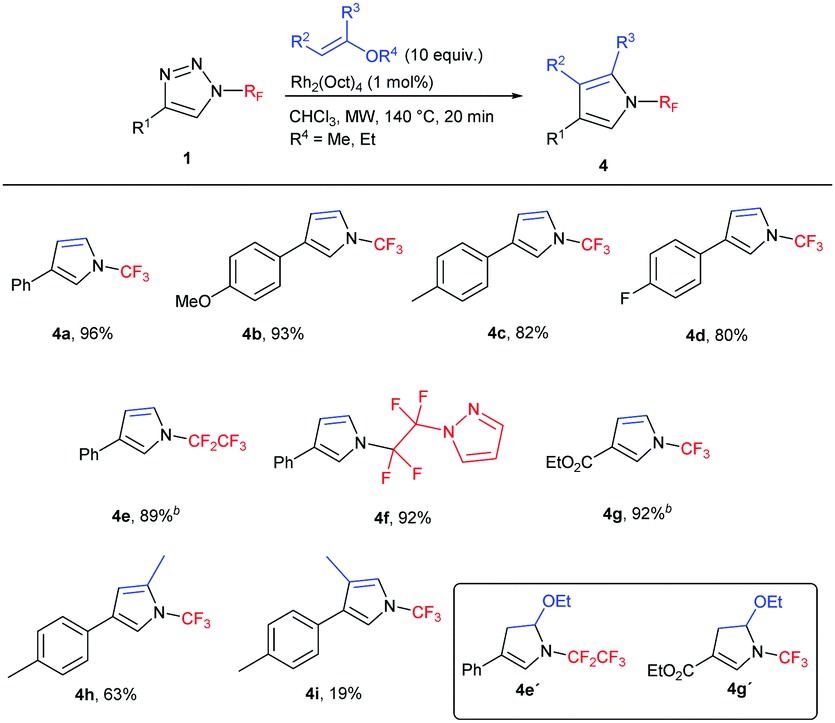
N-(Per)fluoroalkyl pyrroles are previously unreported and attractive targets, which might be accessible by the transannulation of triazoles 1 with vinyl ethers. First, the reaction with ethyl vinyl ether was tested in order to form 4,5-unsubstituted N-(per)fluoroalkyl pyrroles. Indeed, the transannulation in the presence of the Rh2(Oct)4 catalyst took place with concomitant ethanol elimination under the reaction conditions to afford pyrroles 4 in high yields (Table 3). In the case of 4e, a mixture of 4e and 4e′ (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2) formed; in the case of 4g, only compound 4g′ was observed. In both cases, the treatment of the crude reaction mixture with TsOH resulted in the formation of the desired pyrroles 4e and 4g, respectively. 2,4-Disubstituted pyrrole 4h was successfully prepared by the reaction with 2-methoxypropene. The only limitation of the methodology was observed in the preparation of 3,4-disubstituted pyrrole in the reaction with 1-ethoxypropene, where 4i was isolated in low yield.
:1.2) formed; in the case of 4g, only compound 4g′ was observed. In both cases, the treatment of the crude reaction mixture with TsOH resulted in the formation of the desired pyrroles 4e and 4g, respectively. 2,4-Disubstituted pyrrole 4h was successfully prepared by the reaction with 2-methoxypropene. The only limitation of the methodology was observed in the preparation of 3,4-disubstituted pyrrole in the reaction with 1-ethoxypropene, where 4i was isolated in low yield.
A comparison of the reactivity of 1a with ethyl vinyl ether and vinyl acetate revealed that while in the case of ethyl vinyl ether ethanol elimination from intermediate 4a′ proceeded smoothly under the reaction conditions, whereas in the case of vinyl acetate intermediate 4a′′ preferentially undergoes hydrolysis and a mixture of the desired 4a and 5a was obtained (Scheme 2).
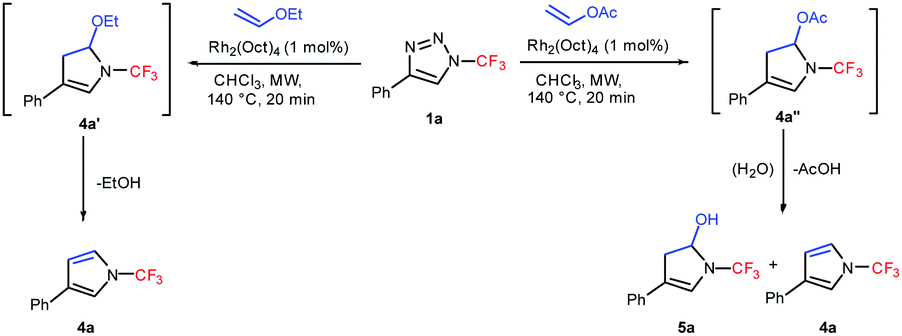 | ||
| Scheme 2 A comparison of the reactivity of 1a with ethyl vinyl ether and vinyl acetate. | ||
After having developed an efficient strategy for the preparation of unknown N-(per)fluoroalkylated imidazoles and pyrroles, we set to investigate the application of the methodology for the preparation of other unreported classes of compounds – N-perfluoroalkyl imidazolones and pyrrolones. Gratifyingly, N-trifluoromethyl-substituted imidazolone 6 was prepared by transannulation of 1b with phenylisocyanate (Scheme 3). For the preparation of N-trifluoromethyl pyrrolone 7, one-pot two-step synthesis with silyl ketene acetal, followed by the treatment with TBAF, was used (Scheme 4).
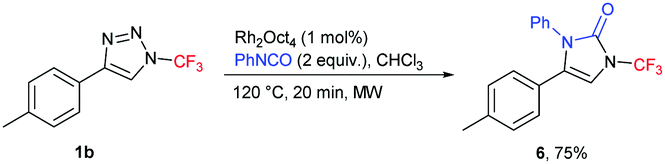 | ||
| Scheme 3 Synthesis of N-trifluoromethyl imidazolone 6. | ||
 | ||
| Scheme 4 Synthesis of N-trifluoromethyl pyrrolone 7. | ||
In order to compare the reactivities of previously described N-tosyl and our N-perfluoroalkyl triazoles in the Rh-catalyzed transannulation reaction, a competitive experiment was performed between equimolar amounts of 1d and N-tosyl triazole 8. From the composition of the reaction mixture analyzed by 19F NMR, it is clear that 1d reacted somewhat slower than 8, however, the reactivity difference is small (Scheme 5).
 | ||
| Scheme 5 Transannulation competition experiment revealing comparative reactivity of N-trifluoromethyl and N-tosyl triazoles with benzonitrile. | ||
A plausible mechanism for the transannulation process mirrors the one proposed for N-Ts triazoles. Triazole 1 upon treatment with the RhII catalyst at elevated temperature is transformed into the key rhodium iminocarbene intermediate A (Scheme 6). Formal [3+2] cycloadditions with 2π components provide zwitterionic species B–E (the actual mechanism might be more complex and can involve the formation of ylides and metallacyclobutenes or in the cases of pyrroles and pyrrolones the formation of cyclopropanes followed by the Cloke rearrangement).20,22,24 The subsequent elimination of the rhodium catalyst (and optional group deprotection or elimination) affords the final products.
 | ||
| Scheme 6 Proposed simplified mechanism for the transannulation of N-perfluoroalkyl-1,2,3-triazoles. | ||
The stability of N-perfluoroalkyl imidazoles and pyrroles under acidic or basic conditions at ambient temperature (18 h reaction time) was also investigated. Imidazole 3g was treated with 0.5 M NaOH in CD3OD. Proton–deuterium exchange (H/D 81:19) of the aromatic hydrogen was observed, suggesting its considerable C–H acidity. When the solution of 3g was treated with 1.2 M sulfuric acid in CD3OD, a shift of the aromatic hydrogen signal in 1H NMR spectrum from 7.90 ppm to 8.45 ppm was observed. Treatment of 4b with 0.5 M NaOH or 1.2 M sulfuric acid in CD3OD led to no significant change in the NMR spectra. In all the cases, no decomposition or hydrolysis of 3g or 4b was observed by NMR, showing good stability of these N-trifluoromethylated nitrogen heterocycles in acidic and basic media.
In conclusion, we have developed a highly efficient method for the synthesis of a broad range of previously unreported N-fluoroalkyl-substituted five-membered heterocycles based on a microwave heating-assisted rhodium-catalyzed transannulation of N-fluoroalkyl-substituted triazoles. This way, a variety of structurally diverse N-fluoroalkyl pyrroles, pyrrolones, imidazoles and imidazolones have been prepared in a straightforward manner. These novel classes of fluorinated compounds should be attractive for discovery programs. It was found that transannulations are not only limited to N-tosyl triazoles but also work with N-CF3 triazoles and proceed with benzonitrile with a comparable reactivity. N-Trifluoromethyl imidazoles and pyrroles are hydrolytically stable in both acidic and basic media.
This work was financially supported by the Czech Academy of Sciences (Research Plan RVO: 61388963).
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. D. Luca, Curr. Med. Chem., 2006, 13, 1–23 Search PubMed.
- M. Boiani and M. Gonzalez, Mini-Rev. Med. Chem., 2005, 5, 409–424 CrossRef CAS PubMed.
- J. T. Gupton, Heterocyclic AntitumorAntibiotics. Topics in Heterocyclic Chemistry, Springer, Berlin, Heidelberg, 2006, vol. 2 Search PubMed.
- P. V. Reddy, Organofluorine Compounds in Bilogy and Medicine, Elsevier, Amsterdam, Netherlands, 2015 Search PubMed.
- I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Wiley-Blackwell, New York, 2009 Search PubMed.
- P. Kirsch, Modern Fluoroorganic Chemistry, Wiley-VCH, Weinheim, 2nd edn, 2013 Search PubMed.
- J. Wang, M. Sanchez-Rosello, J. L. Acena, C. Del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Acena, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- A. F. Baxter, K. O. Christe and R. Haiges, Angew. Chem., Int. Ed., 2015, 54, 14535–14538 CrossRef CAS PubMed.
- W. J. Middleton, J. Org. Chem., 1984, 49, 4541–4543 CrossRef CAS.
- J. Yu, J. H. Lin and J. C. Xiao, Angew. Chem., Int. Ed., 2017, 56, 16669–16673 CrossRef CAS PubMed.
- T. Scattolin, K. Deckers and F. Schoenebeck, Angew. Chem., Int. Ed., 2017, 56, 221–224 CrossRef CAS PubMed.
- T. M. Sokolenko, K. I. Petko and L. M. Yagupolskii, Chem. Heterocycl. Compd., 2009, 45, 430–435 CrossRef CAS.
- G. Bissky, G.-V. Röschenthaler, E. Lork, J. Barten, M. Médebielle, V. Staninets and A. A. Kolomeitsev, J. Fluorine Chem., 2001, 109, 173–181 CrossRef CAS.
- K. Niedermann, N. Fruh, R. Senn, B. Czarniecki, R. Verel and A. Togni, Angew. Chem., Int. Ed., 2012, 51, 6511–6515 CrossRef CAS PubMed.
- Z. E. Blastik, S. Voltrova, V. Matousek, B. Jurasek, D. W. Manley, B. Klepetarova and P. Beier, Angew. Chem., Int. Ed., 2017, 56, 346–349 CrossRef CAS PubMed.
- S. Voltrova, M. Muselli, J. Filgas, V. Matousek, B. Klepetarova and P. Beier, Org. Biomol. Chem., 2017, 15, 4962–4965 CAS.
- H. M. L. Davies and A. M. Walji, Rhodium(II)-Stabilized Carbenoids Containing Both Donor and Acceptor Substituents, in Modern Rhodium-Catalyzed Organic Reactions, ed. P. A. Evans, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005 Search PubMed.
- B. Chattopadhyay and V. Gevorgyan, Angew. Chem., Int. Ed., 2012, 51, 862–872 CrossRef CAS PubMed.
- H. M. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151–5162 RSC.
- T. Horneff, S. Chuprakov, N. Chernyak, V. Gevorgyan and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 14972–14974 CrossRef CAS PubMed.
- B. Chattopadhyay and V. Gevorgyan, Org. Lett., 2011, 13, 3746–3749 CrossRef CAS PubMed.
- S. Rajasekar and P. Anbarasan, J. Org. Chem., 2014, 79, 8428–8434 CrossRef CAS PubMed.
- J. Feng, Y. Wang, Q. Li, R. Jiang and Y. Tang, Tetrahedron Lett., 2014, 55, 6455–6458 CrossRef CAS.
- S. Chuprakov, S. W. Kwok and V. V. Fokin, J. Am. Chem. Soc., 2013, 135, 4652–4655 CrossRef CAS PubMed.
- R. Q. Ran, J. He, S. D. Xiu, K. B. Wang and C. Y. Li, Org. Lett., 2014, 16, 3704–3707 CrossRef CAS PubMed.
- T. Miura, Y. Funakoshi and M. Murakami, J. Am. Chem. Soc., 2014, 136, 2272–2275 CrossRef CAS PubMed.
- J. E. Spangler and H. M. Davies, J. Am. Chem. Soc., 2013, 135, 6802–6805 CrossRef CAS PubMed.
- S. Chuprakov, F. W. Hwang and V. Gevorgyan, Angew. Chem., Int. Ed., 2007, 46, 4757–4759 CrossRef CAS PubMed.
- V. Helan, A. V. Gulevich and V. Gevorgyan, Chem. Sci., 2015, 6, 1928–1931 RSC.
- M. Zibinsky and V. V. Fokin, Org. Lett., 2011, 13, 4870–4872 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc01446a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |