
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
3D-ordered carbon materials by melt-shear organization for tailor-made hybrid core–shell polymer particle architectures†
S.
Vowinkel
a,
C. G.
Schäfer
a,
G.
Cherkashinin
b,
C.
Fasel
b,
F.
Roth
b,
N.
Liu
c,
C.
Dietz
c,
E.
Ionescu
b and
M.
Gallei
*a
aErnst-Berl-Institute for Chemical Engineering and Macromolecular Science, Technische Universität Darmstadt, Alarich-Weiss-Str. 4, D-64287 Darmstadt, Germany. E-mail: m.gallei@mc.tu-darmstadt.de
bMaterials Science Department, Technische Universität Darmstadt, Jovanka-Bontschits-Str. 2, D-64287 Darmstadt, Germany
cCenter of Smart Interfaces and Department of Materials Science, Technische Universität Darmstadt, Alarich-Weiss-Str. 16, D 64287 Darmstadt, Germany
First published on 10th November 2015
Abstract
The melt-shear organization technique for tailor-made polystyrene-co-polyacrylonitrile (PSAN) shell and silica core particles is investigated yielding easy-scalable carbonaceous porous films after etching and appropriate thermal treatment. Monodisperse silica core particles are surface modified and transduced to a seeded emulsion polymerization for the preparation of processable well-defined core–shell PSAN particles. Melt-shear organization for particle alignment into a colloidal crystal structure is applied prior to the thermally induced crosslinking of the PSAN shell material, followed by etching and carbonization of the porous polymeric opal film. It is shown that polymer processing and applied thermal treatment protocols are crucial and capable of maintaining the pristine particle order in the free-standing carbonaceous films. The obtained films reveal hexagonally aligned pores as part of a conductive carbonaceous matrix. Conductivity and adjustable porosity are evidenced by conductive atomic force microscopy (C-AFM) and scanning electron microscopy (SEM) measurements, respectively. The herein developed melt-shear organization technique for a novel polymer-based carbonaceous particle precursor material is shown to be a potential platform for the preparation of scalable conductive materials. The route described here will be feasible for the preparation of doped and tailor-made conductive materials with a wide range of applications in the fields of electrodes, batteries, as well as sensing and photonic band gap materials.
1. Introduction
The preparation of ordered porous materials has attracted significant attention due to tremendous potential for various applications, e.g. separation technologies, sensors, catalysis or photonic structures.1–8 Within this scientific field, control over hierarchical structures and location of chemical functionalities can be achieved by using convenient and efficient synthetic concepts for the synthesis and processing of well-defined polymer-templated precursors featuring 0D, 1D, 2D and 3D nano- and microstructures. Different templating strategies have been applied comprising of so-called hard and soft templating routes for controlling the shape and the size of the final materials after the removal of the template structure.9–19 Silica-based particles as hard templates have been used for nearly 15 years, but also in very recent studies.20–24 Especially Ozin and coworkers reported on tremendous efforts in this field of colloidal crystal templating.25,26 Compared to these hard templating methods, polymer-grafted particles have been shown to be excellent candidates for the preparation of well-defined and ordered nanocomposite materials.27–30 Carbon-based (nano)materials have attracted enormous attention due to their exciting properties and widespread potential in the fields of lithium- and sodium-ion batteries, catalysis, water purification, optoelectronic materials, and hydrogen storage.31–37 However, many approaches towards industrial scale porous carbon materials are emerging with issues as typically fragile structures featuring defects are obtained during required multi-step procedures.38 As a result, material properties are strongly and often negatively affected which therefore limit their technological potential. Compared to previously described investigations, we report here on a novel route for the preparation of carbonaceous materials based on a single-source precursor material avoiding additional infiltration steps after particle alignment. As a general promising technique for the preparation of ordered porous materials, we recently reported on the preparation of polymer-based inverse opal films for hybrid core–shell particles by using the so-called melt-shear organization technique.39 This technique has not been reported for the preparation of carbonaceous films and by this powerful technique easy-scalable and free-standing films with diameters larger than 10 cm can be obtained. In general, the basic prerequisite for the preparation of highly ordered and free-standing films using the melt-shear organization technique is the synthesis of tailor-made hard core soft shell particles featuring uniform diameters capable of undergoing colloidal crystallization. The advantages of the melt-shear technique can be summarized as follows: colloidal crystal films can be easily obtained by heating the soft polymer shell material above the glass transition temperature, Tg, while the hard core material remains unaffected, i.e. particle ordering can be carried out without the presence of solvents and dispersion media. Upon compression of the coagulated core–shell particle mass in a warm press, particles start to merge into a highly ordered colloidal crystal structure (Fig. 1).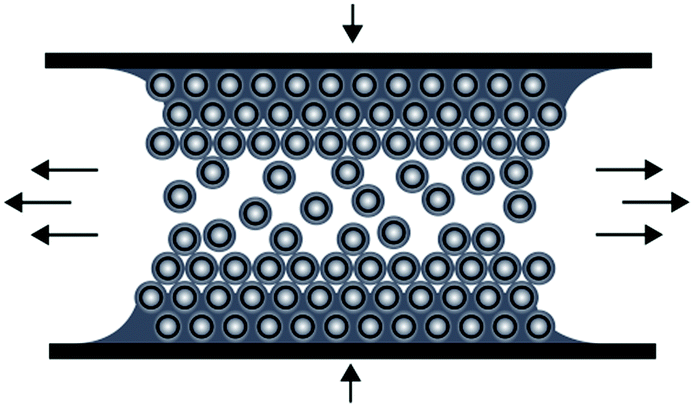 | ||
| Fig. 1 Scheme for the melt-shear organization technique for hard core soft shell particles applying a moderate pressure and temperature to a coagulated mass of tailor-made particles. | ||
The resulting polymer films have been used for a manifold of applications, e.g. as optical sensing materials due to their highly ordered domains in the range of visible light caused by (switchable) Bragg diffraction.40–46 Compared to other techniques for polymeric particle ordering, this method is extremely fast and yields ordered films in one single step. Moreover, precursor materials can be introduced into this polymer processing step avoiding additional subsequent infiltration steps. Until recently, this technique was established for pure polymer core–shell particle systems only, but it could be advantageously applied for thermo-responsive soft shell polymers having silica core particles.39 In the present study, the melt-shear organization technique for monodisperse particles consisting of polyacrylonitrile (PAN) as a copolymer with polystyrene (PS) as a shell material is investigated for the preparation of easy-scalable porous carbonaceous films. In general, PAN was reported as a suitable carbon precursor material.47–54 Tailor-made core–shell particles are prepared by Stöber synthesis protocols prior to efficient surface functionalization using bifunctional monomers followed by seeded emulsion polymerization of styrene (S) and the acrylonitrile (AN) monomers as a meltable shell material. The hybrid polymer core–shell composition is adjusted in order to improve film stability and melt shear organization processing conditions. After a moderate thermal treatment of the highly ordered opalescent films and subsequent etching of the silica core materials, carbonization is carried out by thermal treatment. It is shown that by tailoring the core–shell architecture, processing steps and applied temperature protocols, the final carbonaceous material can be tuned with respect to porosity, order and entire electrically conductive properties. The convenient scalability and the possibility of tailoring the properties by adding additional precursor materials to the carbonaceous or polymer precursor films are expected to provide a powerful platform for the generation of porous functional materials in the fields of electrodes, batteries, photonic and sensing applications.
2. Results and discussion
3D-ordered carbonaceous materials were prepared by subsequent synthesis and processing steps comprising tailor-made preparation of silica core poly(styrene-co-acrylonitrile) particles (silica@PSAN) featuring appropriate core–shell composition followed by directed particle assembly in order to produce a hybrid colloidal crystal (CC) film by using the melt-shear organization technique. Thus obtained films were thermally crosslinked (xlink) at a moderate temperature via the reactions of the acrylonitrile moieties. After silica removal by etching with hydrofluoric acid (HF) and thermal treatment at higher temperatures, the conversion to 3D-ordered carbonaceous materials was achieved. The overall process is schematically depicted in Fig. 2.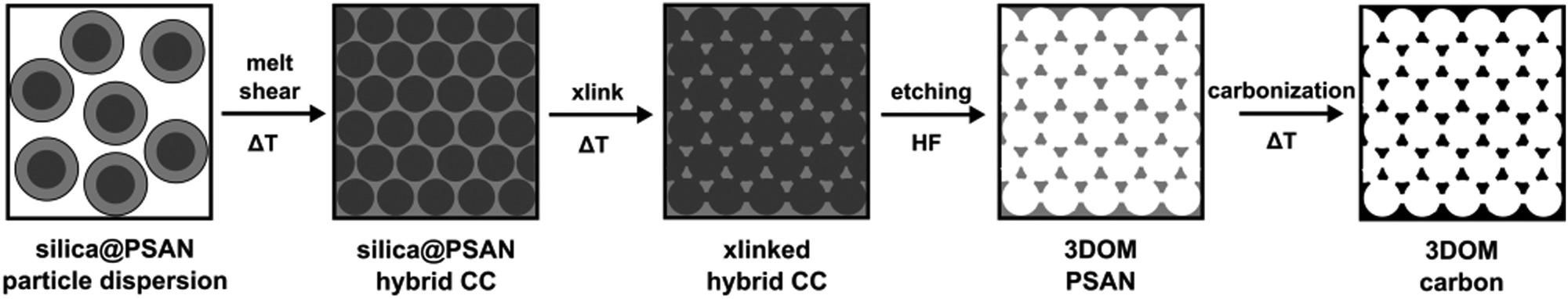 | ||
| Fig. 2 Overview scheme for the fabrication of 3D-ordered porous carbonaceous materials within this study. | ||
The following chapter is divided into different sections with the focus on introducing the typical methodology for the generation of 3D-ordered carbon-based materials. Firstly, the basic synthesis strategy for novel tailor-made silica@PSAN core–shell particles is discussed, followed by introducing the melt-shear organization towards ordered and mechanically stable hybrid silica@PSAN colloidal crystal films, which are subsequently converted into carbon materials. After extensive characterization of the obtained materials during and after the individual processing step, the evolution of the carbonization process and the associated properties of the final 3D-ordered porous carbonaceous materials are investigated in more detail.
2.1. Synthesis and characterization of silica@PSAN core–shell particles
Well-defined silica@poly(styrene-co-acrylonitrile) (PSAN) core–shell particles (Fig. 3a) were obtained via a stepwise synthesis. For this purpose, silica core particles featuring a diameter of 169 ± 4 nm as determined using TEM measurements were prepared by using tetraethoxysilane (TEOS) in ethanol through a slightly modified Stöber process as described by van Blaaderen et al.55 A crucial step for transducing silica particles into emulsion polymerization is the surface functionalization with 3-methacryloxypropyl-trimethoxysilane (MEMO), which acts as a grafting anchor for the polymer shell growth. The MEMO functionalization was evidenced by thermogravimetric analysis (TGA, Fig. S1 of the ESI†) and found to be 1.12 wt% for the silica particles. MEMO functionalized particles were capable of transferring into water without agglomeration or precipitation of the pristine particles by adding sodium dodecylsulfate (SDS) as a stabilizing agent. An emulsion polymerization of acrylonitrile and styrene (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 by weight) was carried out as described in the Experimental section, yielding silica@PSAN hybrid particles.
:30 by weight) was carried out as described in the Experimental section, yielding silica@PSAN hybrid particles.
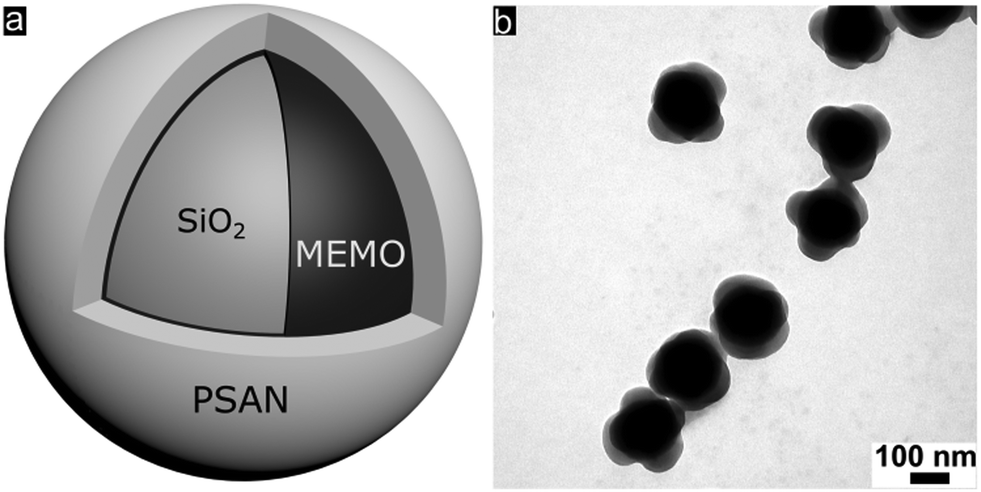 | ||
| Fig. 3 (a) Scheme of the final particle architecture of hybrid silica@PSAN core–shell particles prepared by the stepwise synthesis of silica particles via the Stöber process and subsequent functionalization with MEMO, followed by emulsion polymerization of styrene and acrylonitrile. (b) TEM image of silica@PSAN core–shell particles. | ||
In order to achieve processable materials for melt-shear organization, the particle synthesis was carried out in the presence of 1-dodecanthiol, which served both as a chain transfer agent during radical polymerization as well as a plasticizer for the coagulated particle mass. The controlled build-up of the silica@PSAN core–shell particle architecture was verified by transmission electron microscopy (TEM). In Fig. 3b, a TEM image of the final particles featuring a PSAN–shell is given. TEM images of the final PSAN-grafted particles clearly reveal the formation of a brighter shell due to less contrast of polymers compared to silica core particles during TEM measurements.
In addition to the TEM investigations, the average hydrodynamic diameter and size distributions of the particles after each step of the applied synthetic protocol were determined using dynamic light scattering (DLS). From Fig. S2 (ESI†) it can be concluded that the step of emulsion polymerization induced successively increasing hydrodynamic diameter, thus indicating the successful stepwise synthesis of silica–core PSAN–shell particles. The average diameters of the core–shell particles were determined by using TEM and DLS measurements after each synthetic step and these obtained values are in good agreement with expectations. As a result, the polymerization of styrene and acrylonitrile is evidenced to proceed quantitatively. The content of PSAN as a shell material was determined to be 60.5 vol%. It is worth mentioning that this amount was found to be the optimum for the PSAN volume fraction as a lower content of PSAN did not lead to carbonaceous films with pore interconnections. On the other side, a higher content of PSAN larger than 60.5 vol% was not suitable for the melt-shear organization technique and leads to a loss of particle order. The average diameter of the particles obtained by TEM (dTEM) and hydrodynamic diameters (dDLS) obtained by DLS measurements are compiled in Table S1 (ESI†). Noteworthily, the inhomogeneous and crumpled shell of the silica@PSAN particles can be attributed to the presence of nanocrystals during the SAN copolymerization as reported by Landfester and Antonietti.56
It can be concluded that well-defined silica@PSAN particles featuring core–shell architecture were obtained via the applied synthetic protocol. The melt-shear organization technique as well as the thermal crosslinking protocols for these novel hybrid silica@PSAN core–shell particles is discussed in the ensuing section.
2.2. Preparation and characterization of silica@PSAN films by melt-shear organization
Hybrid film disks were prepared starting from the coagulated mass of silica@PSAN particles consisting of rigid silica cores and surface-anchored PSAN shells. A full description of the process, which combines coagulation, extrusion and melt-shear organization, can be found in the Experimental section. Melt-shearing of the core–shell particles formed opal disks with an average diameter of 12 cm and a thickness of around 200 μm (Fig. 4a). The strong blue reflection color due to Bragg’s law already indicated the high order of the entire material. | ||
| Fig. 4 (a) Photograph of a silica@PSAN-based film disk (12 cm diameter) showing the strong blue reflection color. (b) TEM image of an ultrathin cross-section of the prepared film showing the highly ordered the top layers and transition to disordered deeper lying polycrystalline layers. (c) SEM image of the surface of silica@PSAN film after cross-linking at 240 °C showing the hexagonally arranged (111) plane of the silica spheres. (d) SEM image of a cross-section of silica@PSAN film after crosslinking at 240 °C showing the silica spheres embedded in the crosslinked PSAN matrix. | ||
During uniaxial compression with a radial flow profile for the particles at a temperature of 180 °C, the PSAN shells merge into a continuous molten matrix and the silica cores self-assemble in the flowing melt into hexagonally packed particle layers. Using this technique, PSAN-based particles were continuously deposited along the plates of the press, forming crystalline (111) layers resulting from a precise fcc arrangement of the hard silica cores embedded in the PSAN matrix.
Hence, (111) planes of the fcc lattice were parallelly aligned to the prepared film surfaces. This could be evidenced by ultrathin cross-sections of the corresponding film samples using TEM measurements (Fig. 4b). Well-ordered silica cores (dark) embedded in the PSAN matrix (brighter) are clearly visible. The main obstacle for the subsequent conversion into 3D-ordered carbon materials of PSAN-based materials is that softening and flowing of the silica@PSAN particles inside the film must be prevented both during etching of the silica cores and pyrolysis of the PSAN matrix. Therefore, the formation of a mechanically stable colloidal crystal film was achieved after melt-shear organization by subsequent crosslinking reaction of the PSAN matrix via thermal treatment at 240 °C under ambient air.57,58 During the thermal treatment, acrylonitrile moieties undergo inter- and intramolecular crosslinking reactions in the presence of oxygen, forming conjugated unsaturated carbon-rich polymers and hence a continuous crosslinked polymer phase is formed.47–51
In order to prove that thermal treatment did not affect the crystalline order of the PSAN-based particles in the opal films, ultrathin-sections of the samples were prepared revealing exactly the same results of particle order as given in Fig. 4b. As can be concluded from SEM images of the film topography (Fig. 4c), hexagonally arranged silica cores are still clearly visible. The spherical silica cores embedded in the polymer matrix can also be evidenced by SEM images of a corresponding cross-section (Fig. 4d).
As can be finally concluded from SEM and TEM characterizations, the melt-shear organization of herein investigated novel hybrid silica@PSAN core–shell particles led to regularly ordered colloidal crystal films, while a subsequent crosslinking reaction provided inherently stable films without the loss of the colloidal crystal order. All obtained films prepared by the melt-shear organization technique featured Bragg reflection colors evidencing the homogeneously distributed domains. In the following, the conversion of the produced colloidal crystal films into 3D-ordered carbonaceous materials will be investigated.
2.3. Preparation of 3D-ordered carbon materials
As described above, PSAN is an excellent precursor for carbon-based materials. In order to obtain a 3D-ordered carbonaceous material following the melt-shear organization strategy, the silica cores from the crosslinked and stabilized silica@PSAN sample (Fig. 4c and d) were removed by etching with HF. Silica is well-known to be easily removed by etching with hydrofluoric acid (HF) using hard templating methodologies. From SEM topography (Fig. 5a) and cross-section (Fig. 5b) it is evidenced that etching of the melt-sheared particle films did not affect the pristine particle, and respectively the pore order. | ||
| Fig. 5 SEM images of a crosslinked and stabilized PSAN colloidal crystal film after silica removal by etching with HF. (a) Surface topography (scale bar corresponds to 2 μm) and (b) cross-section (scale bar corresponds to 1 μm). | ||
In order to consider the decomposition behaviour of the pre-oxidized and stabilized PSAN matrix material after removal of silica cores, thermogravimetric analysis (TGA) was conducted.
As can be seen from TGA in Fig. 6, the decomposition of the stabilized PSAN started at about 300 °C and the loss of the majority of the material occurred at 400 °C (∼70 wt%), while a minor weight loss could be observed above 1000 °C (approximately 5 wt%). The residual weight reached a constant value at a temperature over about 1200 °C. In general, PS decomposes completely at temperatures of about 400 °C and no carbonaceous material remains, while PAN can be completely carbonized by further heat treatment to 1500 °C. These results coincided with SEM images of the 3D-ordered carbon, in which no significant shrinkage of the structure was observed above 400 °C. This supports the suggestion that PS decomposed and PAN remained to undergo conversion to carbon. In the following section carbonization for different samples at different temperatures was investigated with respect to the overall porous structure.
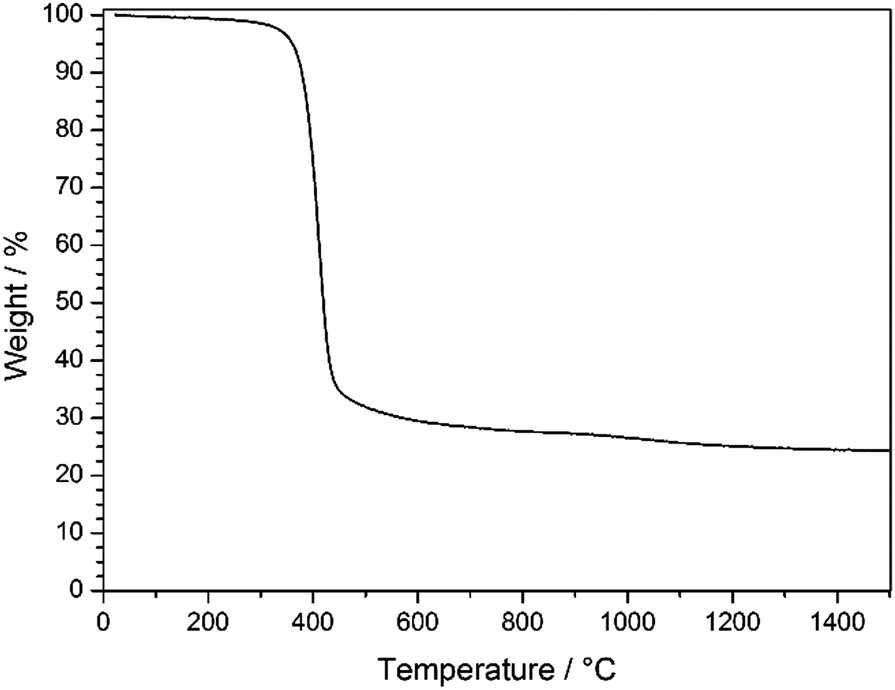 | ||
| Fig. 6 TGA of stabilized PSAN colloidal crystal film after etching with HF under argon in the temperature range of 30–1500 °C with a heating rate of 10 K min−1. | ||
TGA measurements (Fig. 6) revealed that an additional mass loss at approximately 1100 °C could be observed. Before this conversion being elucidated by using XPS and Raman spectroscopy, the change of the pore structure was investigated by using SEM.
Direct comparison of the porous material which was thermally treated at 240 °C followed by etching with HF (Fig. 7a), porous films treated at 400 °C (Fig. 7b) and films treated at 1500 °C (Fig. 7c) under an Ar atmosphere was investigated with respect to their topography by using SEM. Direct comparison of the compiled SEM images in Fig. 7 featuring the same magnification leads to the conclusion that pore evolution was accompanied by a shrinkage of the porous structure while maintaining the order.
 | ||
| Fig. 7 SEM images of the surface of (a) stabilized PSAN colloidal crystal film after thermal treatment at 240 °C and etching with HF, (b) after carbonization at 400 °C and (c) after graphitization at 1500 °C, showing the surface-parallel hexagonally arranged (111) plane of air voids in the matrix material. | ||
Therefore, from the image of the stabilized and etched sample in Fig. 7a it can be concluded that an inverted 3D-ordered structure could remarkably reproduce the pristine fcc lattice of the initial colloidal crystal structure (Fig. 4c). The sample revealed homogenously distributed domains featuring interconnected close-packed spherical voids with average diameters of 170 ± 20 nm as determined by SEM measurements. Additionally, the former contact points between adjacent silica spheres appeared as pores between the spherical voids. SEM analysis of the 3D-ordered structure after carbonization at 400 °C (Fig. 7b) demonstrated continuous and homogenous phase shrinkage due to PSAN carbonization. However, the walls of the pores appeared to be uniform and the colloidal crystalline structure remained intact. The overall average diameter of the resulting spherical pores in the as-prepared 3D-ordered carbon was determined to be 60 ± 10 nm as estimated from SEM images (Fig. 7b), which was significantly smaller than diameters of the silica spheres. This again indicates a significant shrinkage of the carbon precursors during PSAN decomposition in the carbonization stage but without losing the overall pore order. SEM investigations on the final 3D-ordered carbon after thermal treatment at 1500 °C under an Ar atmosphere (Fig. 7c) again reveal arrays of pores. Remarkably, the pristine porous precursor structure featuring a highly ordered structure was still observed, proving again the high potential of the applied processing protocols for the melt-shear organized particles and subsequent treatment. The SEM image (Fig. 7c) additionally indicates that no further shrinkage of the 3D-ordered structure took place during graphitization and the average diameters of the air voids remain at 60 ± 10 nm.
In Fig. 7c, some pore connections of the sample can be observed, which may be caused by internal tensions during the graphitization step. Nevertheless, the overall morphology of the material remained fully intact.
As can be concluded from SEM investigations compiled in Fig. 7, particle design followed by melt-shearing and appropriate thermal treatment protocols was shown to produce periodically 3D-ordered carbon structures, even though a large amount of shrinkage was observed during carbonization and graphitization of the PSAN matrix polymer. Centimeter-sized 3D-ordered carbon monoliths with regular arrays of interconnected macropores could be obtained.
2.4. Studies on the chemical structure evolution during carbonization
In order to gain more insights into the structure formation and chemical composition of the 3D-ordered precursor and final carbonaceous materials, X-ray photoelectron spectroscopy (XPS) as well as Raman spectroscopy was carried out.XPS was used to follow the carbonization process and to determine the chemical composition, which provided relevant information regarding the binding energies of carbon and nitrogen of the synthesized materials at different thermal treatment steps. XPS measurements were performed for the original PSAN sample after etching with HF and after crosslinking under ambient air at 240 °C as well as on carbonized (at 400 °C) and graphitized samples (1500 °C). The evolution of the N1s photoelectron spectra as a function of thermal treatment is shown in Fig. 8. The corresponding XPS survey spectra, as well as the C1s and O1s photoelectron spectra are given in the ESI† (Fig. S3, S5 and S6, respectively). Exemplarily, the N1s photoemission spectrum (Fig. 8) of the sample to which SEM images in Fig. 7 corresponds is narrow and can be well fitted by using one component with Ebin = 399.3 eV assigned to pyrrolic nitrogen (Fig. 8, red curve).59 The shape of the N1s photoemission spectrum evolved upon thermal treatment. The N1s photoelectron spectrum of the sample treated at 240 °C is broader and can be fitted by two components with Ebin = 399.1 eV and Ebin = 400.4 eV assigned to pyrrole and pyridone groups, respectively.59,60 Higher temperature promotes the formation of additional chemical species like pyridine with Ebin = 397.8 eV, as evidenced from the N1s photoelectron spectrum of the porous sample thermally treated at 400 °C (Fig. 7b).9 The complete disappearance of the nitrogen functional groups takes place at temperatures higher than 1500 °C (see ESI† Fig. S4 for the binding energy region of N1s photoemission).
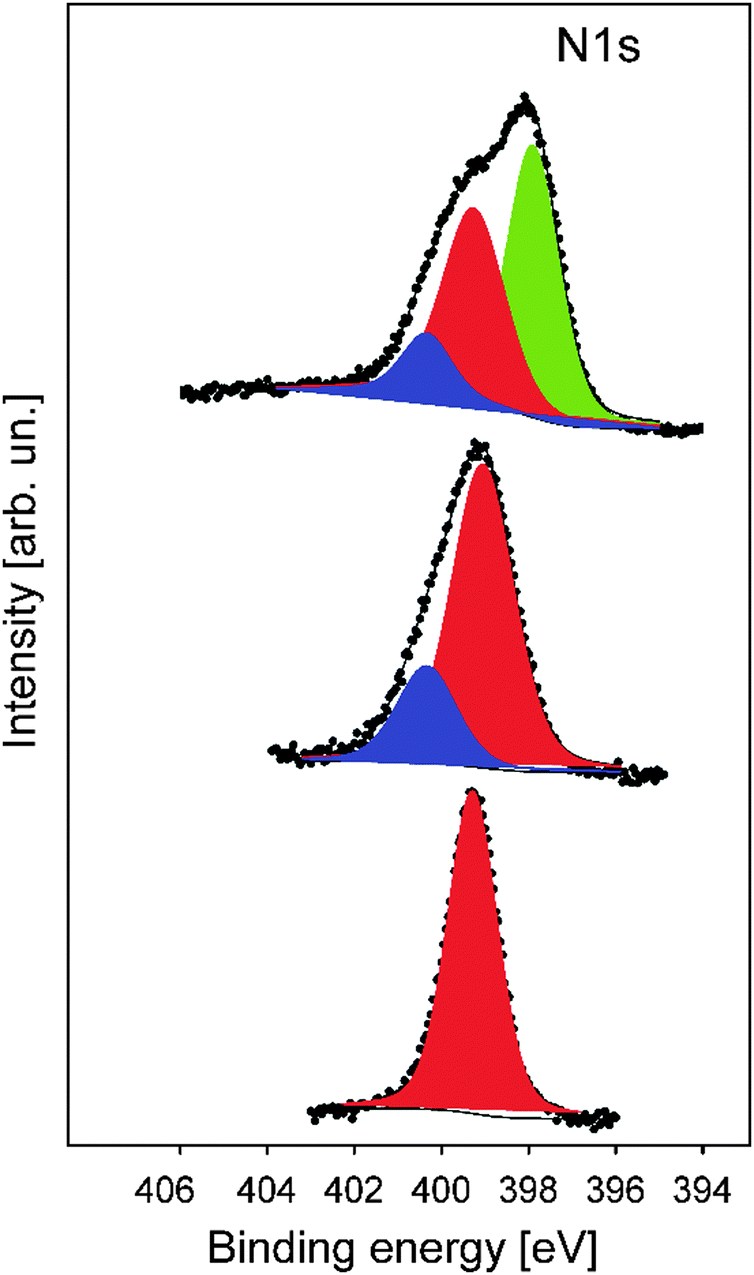 | ||
| Fig. 8 The evolution of the N1s photoelectron spectrum as a function of thermal treatment of the precursor materials: original melt-sheared particle film without thermal treatment (bottom), particle film after thermal treatment at 240 °C and etching (middle), and particle films after thermal treatment at 400 °C (top). The peak areas of pyrolic-, pyridonic- and pyridinic nitrogen are shown by red, blue and green, respectively. | ||
The shape of the C1s photoelectron spectra (Fig. S5, ESI†) of the functional carbon materials heated in the temperature range of 1500 °C evidences the presence of different carbon functional groups which can be ascribed to C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–H bonds with the binding energies of Ebin = 284.4–284.7 eV.61–63 The CN double bond with Ebin = 286 eV and some amount of CO groups with Ebin = 287.2 eV argue the incorporation of nitrogen and oxygen atoms. The increase of temperature leads to the further evolution of the aromatic rings and the appearance of carboxyl groups (COOH) with Ebin = 288.4–289.9 eV.59 The conversion of aromatic rings to a graphite-like structure is completed at T ≥ 1200 °C. The C1s photoelectron spectrum of the porous carbonaceous sample thermally treated at 1500 °C with Ebin = 284.5 eV was significantly narrower compared to the spectra of the functional carbon materials treated at low temperatures.
C and C–H bonds with the binding energies of Ebin = 284.4–284.7 eV.61–63 The CN double bond with Ebin = 286 eV and some amount of CO groups with Ebin = 287.2 eV argue the incorporation of nitrogen and oxygen atoms. The increase of temperature leads to the further evolution of the aromatic rings and the appearance of carboxyl groups (COOH) with Ebin = 288.4–289.9 eV.59 The conversion of aromatic rings to a graphite-like structure is completed at T ≥ 1200 °C. The C1s photoelectron spectrum of the porous carbonaceous sample thermally treated at 1500 °C with Ebin = 284.5 eV was significantly narrower compared to the spectra of the functional carbon materials treated at low temperatures.
The pronounced asymmetry of the C1s photoelectron line to higher binding energies is inherent for graphite-like structures.61 In summary, XPS measurements clearly revealed the conversion of the pristine PSAN shell/matrix polymer into a crosslinked and therefore stabilized film. Finally, by following the evolution of N1s photoemission spectra, the signal for the N content significantly diminished evidencing full conversion of PSAN into unsaturated carbon materials.
Raman spectroscopy was additionally used to characterize the nature of the final graphitized 3D-ordered carbonaceous material. Raman spectroscopy is a powerful (and non-destructive) technique which allows for studying lattice dynamics and vibrational spectroscopy of carbon-based materials e.g., amorphous carbons,64–66 carbon nanotubes,67,68 graphenes,69,70 graphites,70,71 diamond like carbon materials.72,73 Raman spectra of carbon-based materials are very sensitive to their nano/microstructure and consequently provide valuable information about their ordering, hybridization, defect state, etc. The first order Raman spectrum of sp2 carbon exhibits a band of E2g symmetry which relates to bond stretching of sp2 carbon pairs contained in rings or chains. This band is called the G band and appears at around 1575–1595 cm−1. Disordered or nanostructured carbon-based materials, which might contain also some amount of sp3 hybridization, exhibit additional bands in their first order Raman spectrum, such as a band of A1g symmetry which relates to breathing modes of sp2 carbon atoms within rings (the so-called D band; its position depends on the laser wavelength; ca. 1350 cm−1 at 514.5 nm), a band related to C–C sp3 vibrations (ca. 1150–1200 cm−1; A1g symmetry, T band), a D′′ band (ca. 1500 cm−1, related to amorphous carbon), as well as a D′ band (E2g symmetry, ca. 1620 cm−1, disordered graphitic lattice related to surface graphene layers). There have been numerous spectral indicators being proposed in order to characterize and describe the nature, hybridization, crystallinity/degree of ordering in carbonaceous materials.74 The structure of poly-/nanocrystalline graphites or disordered carbons are usually characterized by the average in-plane length (La, also called the lateral cluster size), the average height (so called Lc) and the average interplanar distance (i.e. in the 002 direction).
Additionally, the presence of tortuosity in disordered carbons (i.e., curvature of the graphene planes) as well as the type and density of various defects has been taken into account when describing their structure. Raman spectroscopy is able to estimate the La values for carbonaceous materials from the first-order modes.75,76 For values smaller than 2 nm, the equation proposed by Ferarri and Robertson was shown to be valid, i.e. La2 × 0.0055 Å−2 = I(D)/I(G).75 An average continuous graphene length (Leq.) parameter has been defined, which consider both La and the so-called tortuosity ratio (Rtor, being defined as the ratio between the number of phonons generated at the K point by the second-order Raman process, and the number of phonons generated at the C point in the Brillouin zone by the first-order Raman process, i.e. the ratio between the 2D band and the G band) and might be considered as an estimate of the equivalent phonon mean free path in disordered carbons.74 The Raman spectra of the graphitized 3D-ordered carbonaceous sample thermally treated at 1200 °C prepared within the present study are shown in Fig. 9. The spectral indicators of the Raman spectrum mentioned above (La, Rtor, and Leq) are compiled in Table 1.
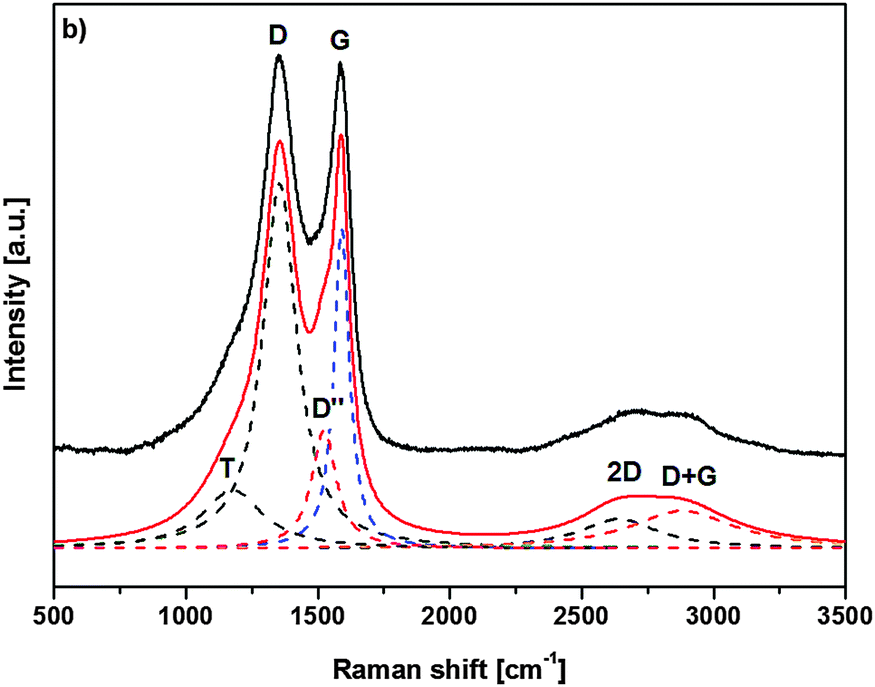 | ||
| Fig. 9 Raman spectrum of porous carbonaceous films after graphitization at 1500 °C. | ||
| Raman mode | Raman shift ω [cm−1] | FWHMa Δω [cm−1] | Peak areab [a.u.] | Peak heightb [a.u.] |
|---|---|---|---|---|
| a FWHM – full width at half maximum. b Normalized to the area/height of the D band. | ||||
| T (A1g) | 1176.5 | 275.2 | 28.1 | 16.0 |
| D (A1g) | 1354.6 | 157.0 | 100 | 100 |
| D′′ | 1522.8 | 112.3 | 23.2 | 32.5 |
| G (E2g) | 1589.9 | 70.3 | 39.2 | 87.5 |
| 2D | 2639.2 | 357.9 | 18.3 | 8.0 |
| D + G | 2881.2 | 475.9 | 31.1 | 10.3 |
| Raman indicator | |
|---|---|
| L a [nm] | 1.48 |
| R tortuosity | 0.48 |
| L eq. [nm] | 2.12 |
As can be concluded, the XPS and Raman measurements show a continuous degradation of nitrile groups during the stabilization and carbonization steps indicating cyclization and graphitization of the final 3D-ordered carbon material.
2.5. 3D-ordered carbon conductivity characterization using conductive atomic force microscopy
In order to gain insights into the conductivity of the final carbonaceous sample after melt-shearing of PSAN-based hybrid particles followed by etching and thermal treatment protocols as described above, conductive atomic force microscopy (C-AFM) measurements were carried out. Fig. 10 shows the current maps (a and b) of the final carbonaceous film, which was prepared by melt-shearing of silica@PSAN particles followed by etching and thermal treatment at 1500 °C under an Ar atmosphere, at sample potentials Vsp = +1.0 V (a) and Vsp = −1.0 V (b) in combination with a grounded tip, the corresponding topography image (c), an example of a locally measured current versus time and voltage versus time graph (d), as well as the current–voltage (I–V) spectroscopy curves (e) measured at two different sample spots marked by A (black curve) and B (red curve) in Fig. 10c. The current maps and the spectroscopy curves prove a high local conductivity of the carbon films. A clear contrast is visible within the current maps that inverts upon the change of the sign of the sample potential (cf.Fig. 10a and b). The I–V curves exhibit a strong increase/decrease in the current when increasing/decreasing the sample potential continued by saturation regimes (Vsp > 0.8 V and Vsp < −1.8 V for the red curve and Vsp > 1.2 V and Vsp < −1.8 V for the black curve) because the cantilever holder with its current amplification electronics is at the detection limit of 20 nA. Note the voltage offset of approx. −180 mV due to a device inherent electronic shift. The difference in the slope at the two sample locations originate from the contrast visible in the current maps at the sample voltages of Vsp = ±1.0 V. The sample surface roughness leads to variations in the contact area between the conductive tip and the sample causing different current flows. The contact technique used for C-AFM is restricted in the lateral resolution, hence the highly ordered colloidal crystal film structure is hardly visible in the topography image Fig. 10c. A surface root-mean-square roughness of approximately 7 nm irrespective of the applied tip-sample voltage was observed on 1 × 1 μm2 areas (see ESI,† Fig. S7 and S8). In summary, C-AFM measurements of the 3D-ordered porous carbonaceous material prove a high local conductivity at the sample surface.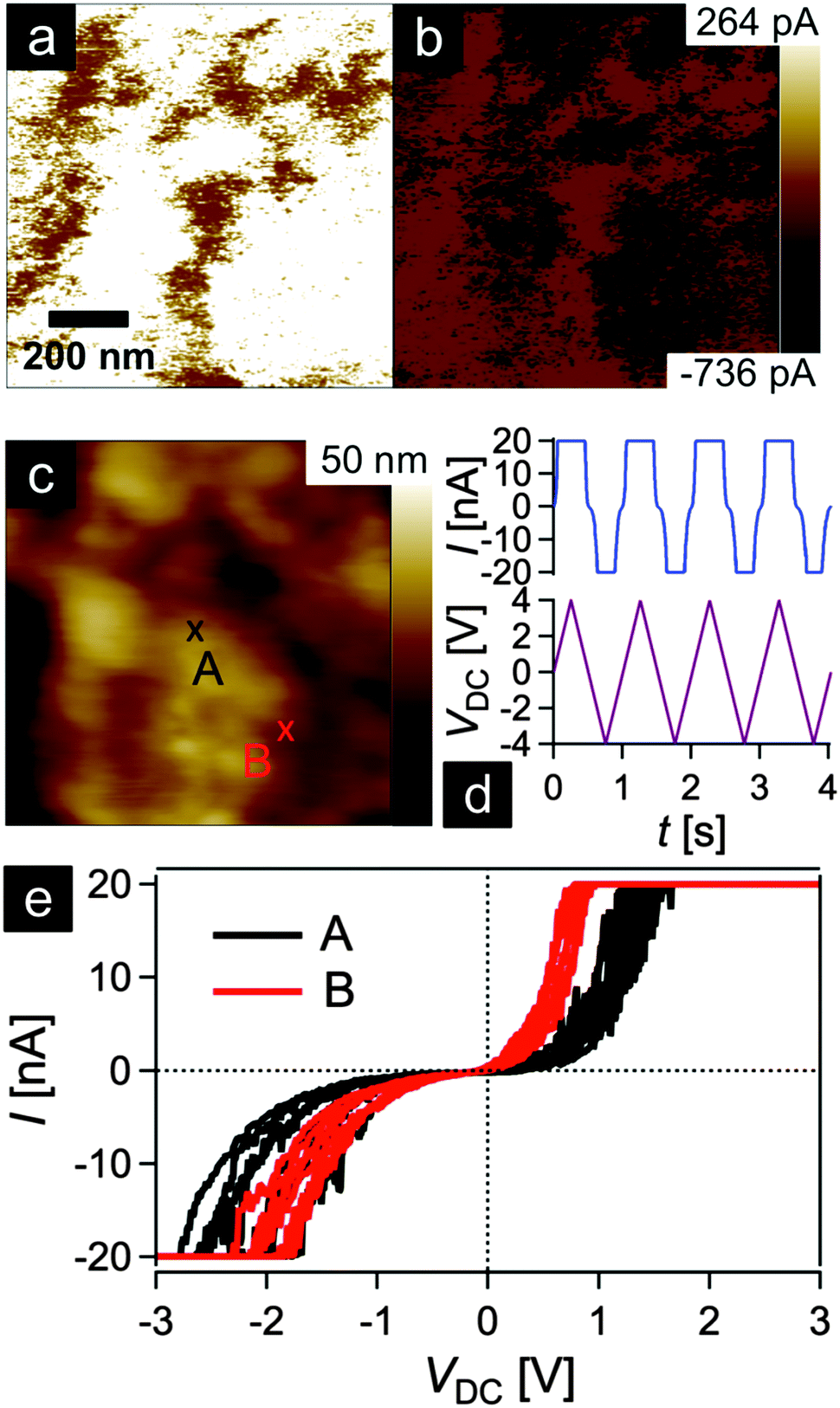 | ||
| Fig. 10 Conductive AFM experiment performed on a melt-shear organized particle film thermally treated at 1500 °C. (a and b) Current maps applying +1 V (a) and −1 V (b) to the sample surface, respectively, and the corresponding topography image (c). (d) Current measured during four triangular voltage cycles at position A marked in (c). (e) Current–voltage spectroscopy curves conducted at two different sample positions marked by A (black curve) and B (red curve) in the topography image of (c). Each curve consists of four consecutive cycles as shown by the current versus time and voltage versus time graphs (d). | ||
3. Experimental section
Materials
All chemicals and solvents were purchased from VWR, Fisher Scientific, Sigma-Aldrich and Alfa Aesar and used as received if not otherwise mentioned. Styrene (S) was obtained from BASF SE, acrylonitrile (AN) from Acros Organics. Prior to use in emulsion polymerization, the inhibitors were removed from the monomers. Therefore, S and AN were purified by passing through a column filled with basic alumina.Instrumentation
Transmission electron microscopy (TEM) measurements were performed on a Zeiss EM10 with an operating voltage of 60 kV. For investigation of the single particles, diluted dispersions were drop-casted on carbon-coated copper grids (Plano GmbH, Germany) followed by drying at room temperature. For the preparation of ultrathin sections of the colloidal films, pieces of the films were embedded in an epoxy resin (UHU endfest) and cut into slices of approximately 50 nm by using an ultramicrotome Ultracut UTC (Leica) equipped with a diamond knife. A mixture of dimethyl sulfoxide and water (50:50 by volume) was used as a floating liquid. TEM images were recorded using a slow-scan CCD camera TRS (Tröndle). Dynamic light scattering (DLS) measurements of the particles were performed on a Nanophox photon cross-correlation spectrometer (Sympatec). The experiments on diluted dispersions of the particles after each step of the particle synthesis were carried out at an angle of 90° at 25 °C. TGA was measured using a Mettler Toledo TGA1 at a heating rate of 10 K min−1. Micro-Raman spectra (10 scans, each scan lasting 3 s) were recorded using a Horiba HR800 micro-Raman spectrometer (Horiba Jobin Yvon, Bensheim, Germany) equipped with an Ar laser (wavelength 514.5 nm). The excitation line has its own interference filter (to filter out the plasma emission) and a Raman notch filter (for laser light rejection). The measurements were performed by using a grating of 1800 g mm−1 and a confocal microscope (magnification 100×, NA 0.5) with a 100 μm aperture, giving a resolution of 2–4 μm. The laser power (20 mW) was attenuated by using neutral density (ND) filters; thus, the power on the sample was in the range of 2 mW to 20 μW. SEM/EDS measurements were performed on a Philips XL30 FEG at an operating voltage of 20 kV. The samples were coated with 2 nm Cr and 14 nm Au using a Quorum Q300T D Sputtercoater. TGA was measured using a STA429 (Netzsch Gerätebau GmbH, Selb/Bavaria). The experiments were carried out in TG/DTA mode with an Argon flow of 75 mL min−1 at a heating rate of 10 K min−1. X-ray photoelectron spectroscopy (XPS) studies were carried out by using a PHI VersaProbe 5000 spectrometer equipped with the monochromatic Al-Kα radiation (hν = 1486.6 eV). The diameter of the X-ray beam spot was 200 μm. High resolution photoelectron spectra were collected at a pass energy of Epas = 23.5 eV and an electron escape angle of θ = 45°. The binding energies were calibrated with respect to the Ag3d5/2 photoelectron line and the Fermi level of a sputtered Ag foil. For the XPS measurements, the corresponding melt-sheared core–shell particles were pressed in a separate indium foils or fixed to a PHI sample holder by using a double sided tape (3 M). In the last case, the sample has no electrical contact with the spectrometer and, therefore, a dual beam charge neutralization system was used to compensate a positive charge on the surface of the sample. The measured photoelectron spectra were referred to the binding energy of the C1s photoelectron emission of the C–C bond (Ebin = 284.5 eV). The background of the XPS spectra was subtracted by using a Shirley-type function. The C1s and N1s photoelectron spectra were decomposed and fitted to different chemical states of carbon–nitrogen functional groups. The peak positions and areas were obtained by a weighed least-square fitting of model curves (70% Gaussian, 30% Lorentzian) to the experimental data. Conductive atomic force microscopy (C-AFM) measurements were performed using the ORCA system of a MFP-3D microscope from Asylum Research (Santa Barbara, USA) and PPP-EFM conductive cantilevers (nominal force constant k = 2.8 Nm−1) from Nanosensors (Neuchâtel, Switzerland) on the melt-shear organized carbon films. Tip-sample voltages in the range of −10 V to +10 V were applied while measuring the respective current through the C-AFM tip scanning in the contact mode. Tip-sample surface forces during scanning were approximately 160 nN and the tip velocity was set to 1.5 μms−1. The sensitivity of the transimpedance amplifier was 2 nAV−1. For the locally conducted current-voltage curves, a triangular-shaped voltage between −3 V and +3 V (6 V peak-to-peak) with a periodicity of 1 s in four cycles was applied to the surface at two different spots (cf.Fig. 10d).
Synthesis and functionalization of silica particles
The silica core particles (180 nm) were prepared following the procedure used by van Blaaderen et al.55 In brief, 1.3 L of the resulting ethanolic dispersion with a silica content of 2.5 wt% was treated with 1.6 mL 3-methacryloxypropyltrimethoxysilane (MEMO). The mixture was heated to 60 °C and stirred for 1 h, subsequently ethanol and ammonia were removed under reduced pressure and replaced with a solution of 100 mg sodium dodecyl sulfate (SDS) in 400 mL water. The volume of the dispersion was then reduced to 300 mL. The obtained MEMO-functionalized silica dispersion revealed a solid content of 8.5 wt%.Synthesis of silica@PSAN core–shell particles
Under argon, a 250 mL flask equipped with a stirrer and a reflux condenser was filled with 170 mL of the MEMO-functionalized silica dispersion and heated to 75 °C. Then a solution of 30 mg SDS in 5 mL water was added. The polymerization was initiated by adding a solution of 150 mg of sodium persulfate in 5 mL of water. After 10 min a monomer emulsion consisting of 68 mg of SDS, 130 mg of KOH, 113 mg of Triton X-405 (solution 70%), 34 g of water, 15.47 g of acrylonitrile (AN), 6.63 g of styrene (St) and 2.21 g of 1-dodecanethiol was added continuously over a period of 4 h. After addition, the reaction was stirred for another 60 min.Silica@PSAN hybrid film preparation
For the preparation of hybrid colloidal crystal films, the latex was dried at 40 °C. The powder was then extruded at 180 °C using a lab microextruder (micro1, DSM Research). In order to produce thin films, the resulting polymer material was covered with PET foil (Mylar A 75, DuPont) and heated to 180 °C within the press plates of a laboratory press (300E, Dr Collin). Then, by applying a pressure of 250 bar for 3 min, melt flow was induced and a hybrid colloidal crystal film was received.3D-ordered carbon film preparation
For the preparation of 3D carbon materials, the samples were calcined at 240 °C to induce a stabilization of polyacrylonitrile. After stabilization of the matrix material, the silica cores were removed by etching with hydrofluoric acid (10% in water). After etching, the samples were washed with water several times and then dried. Samples of these pre-stabilized materials were further thermally treated under Ar at 400 °C and 1500 °C.4. Conclusion
In conclusion, we have fabricated porous carbonaceous films by using the melt-shear organization technique for novel tailor-made core–shell particles. As a precursor material, poly(styrene-co-acrylonitrile) (PSAN) shell silica core particles were prepared using Stöber protocols and seeded emulsion polymerization. Transduction of inorganic particles into an emulsion polymerization succeeded by adding an appropriate amount of the bifunctional reagent, i.e. methacryloxypropyl-trimethoxysilane (MEMO), which enabled further reaction of styrene and acrylonitrile monomers. The prepared hybrid core–shell particles were capable of forming highly ordered colloidal crystal films by applying melt-shear organization. No additional infiltration steps were necessary as the core–shell particles were used as single-source precursors. Thermal treatment protocols for efficient crosslinking and film stabilization at moderate temperatures due to the intramolecular ring formation of the nitrile moieties was carried out, followed by etching of the core particles. The evolution of the pore shrinkage was shown without the loss of the pore order after subsequent thermal treatment as evidenced through microscopy (SEM, TEM) studies. XPS measurements clearly revealed the successive conversion of PSAN into carbonaceous materials. The final material was proven to be conductive by using conductive atomic force microscopy (C-AFM) whereas from results obtained by Raman spectroscopy it could be concluded that a significant content of the graphitic material was obtained. With this convenient particle synthesis and processing of easy-scalable films, these porous carbonaceous materials are well-suited for the application in fields of batteries and sensing. Moreover, it is expected that this feasible approach can be used as an excellent platform for doping of such carbonaceous materials (during porous precursor preparation or for the final carbonaceous material) in order to fine-tune electronical, chemical, and mechanical properties of the carbon material.Acknowledgements
M. G. and S. V. thank the Evangelisches Studienwerk Villigst and Max-Buchner-Foundation for financial support of this work.Notes and references
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- G. M. Whitesides, Small, 2005, 1, 172–179 CrossRef CAS PubMed.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS PubMed.
- Y. Piao, A. Burns, J. Kim, U. Wiesner and T. Hyeon, Adv. Funct. Mater., 2008, 18, 3745–3758 CrossRef CAS.
- Y. Zhao and L. Jiang, Adv. Mater., 2009, 21, 3621–3638 CrossRef CAS.
- D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- K. R. Phillips, G. T. England, S. Sunny, E. Shirman, T. Shirman, N. Vogel and J. Aizenberg, Chem. Soc. Rev., 2015 10.1039/c5cs00533g.
- N. Fechler and M. Antonietti, Nano Today, 2015 DOI:10.1016/j.nantod.2015.07.003.
- F. Schüth and W. Schmidt, Adv. Mater., 2002, 14, 629–638 CrossRef.
- A. Stein, Adv. Mater., 2003, 15, 763–775 CrossRef CAS.
- T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401–1443 CrossRef CAS PubMed.
- A. Thomas, F. Goettmann and M. Antonietti, Chem. Mater., 2008, 20, 738–755 CrossRef CAS.
- M. Llusar and C. Sanchez, Chem. Mater., 2008, 20, 782–820 CrossRef CAS.
- J. K. Sahoo, M. N. Tahir, A. Yella, T. D. Schladt, S. Pfeiffer, B. Nakhjavan, E. Mugnaioli, U. Kolb and W. Tremel, Chem. Mater., 2011, 23, 3534–3539 CrossRef CAS.
- R. K. Joshi and J. J. Schneider, Chem. Soc. Rev., 2012, 41, 5285–5312 RSC.
- A. Stein, B. E. Wilson and S. G. Rudisill, Chem. Soc. Rev., 2013, 42, 2763–2803 RSC.
- L. Yang, Z. Wang, Y. Ji, J. Wang and G. Xue, Macromolecules, 2014, 47, 1749–1756 CrossRef CAS.
- D. Scheid, G. Cherkashinin, E. Ionescu and M. Gallei, Langmuir, 2014, 30, 1204–1209 CrossRef CAS PubMed.
- C. G. Schäfer, S. Vowinkel, G. P. Hellmann, T. Herdt, C. Contiu, J. J. Schneider and M. Gallei, J. Mater. Chem. C, 2014, 2, 7960 RSC.
- Z. Li and M. Jaroniec, J. Am. Chem. Soc., 2001, 123, 9208–9209 CrossRef CAS PubMed.
- J.-S. Yu, S. Kang, S. B. Yoon and G. Chai, J. Am. Chem. Soc., 2002, 124, 9382–9383 CrossRef CAS PubMed.
- S. Kang, J.-S. Yu, M. Kruk and M. Jaroniec, Chem. Commun., 2002, 1670–1671 RSC.
- S. B. Yoon, G. S. Chai, S. K. Kang, J.-S. Yu, K. P. Gierszal and M. Jaroniec, J. Am. Chem. Soc., 2005, 127, 4188–4189 CrossRef CAS PubMed.
- S. Zhang, H.-M. Kwon, Z. Li, A. Ikoma, K. Dokko and M. Watanabe, ChemElectroChem, 2015, 2, 1080–1085 CrossRef CAS.
- S. Wong, V. Kitaev and G. A. Ozin, J. Am. Chem. Soc., 2003, 125, 15589–15598 CrossRef CAS PubMed.
- A. Esmanski and G. A. Ozin, Adv. Funct. Mater., 2009, 19, 1999–2010 CrossRef CAS.
- P. Akcora, S. K. Kumar, V. García Sakai, Y. Li, B. C. Benicewicz and L. S. Schadler, Macromolecules, 2010, 43, 8275–8281 CrossRef CAS.
- V. Goel, J. Pietrasik, H. Dong, J. Sharma, K. Matyjaszewski and R. Krishnamoorti, Macromolecules, 2011, 44, 8129–8135 CrossRef CAS.
- R. Mangal, S. Srivastava and L. A. Archer, Nat. Commun., 2015, 6, 7198 CrossRef CAS PubMed.
- L. Cao and M. Kruk, Polymer, 2015, 72, 356–360 CrossRef CAS.
- S. Wenzel, T. Hara, J. Janek and P. Adelhelm, Energy Environ. Sci., 2011, 4, 3342 CAS.
- M. Oschatz, L. Borchardt, M. Thommes, K. A. Cychosz, I. Senkovska, N. Klein, R. Frind, M. Leistner, V. Presser, Y. Gogotsi and S. Kaskel, Angew. Chem., Int. Ed., 2012, 51, 7577–7580 CrossRef CAS PubMed.
- B. Fang, J. H. Kim, M.-S. Kim and J.-S. Yu, Acc. Chem. Res., 2013, 46, 1397–1406 CrossRef CAS PubMed.
- S. Porada, L. Borchardt, M. Oschatz, M. Bryjak, J. S. Atchison, K. J. Keesman, S. Kaskel, P. M. Biesheuvel and V. Presser, Energy Environ. Sci., 2013, 6, 3700 CAS.
- L. Ma, A. H. C. Hart, S. Ozden, R. Vajtai and P. M. Ajayan, Faraday Discuss., 2014, 173, 9–46 RSC.
- A. Morelos-Gómez, P. G. Mani-González, A. E. Aliev, E. Muñoz-Sandoval, A. Herrera-Gómez, A. A. Zakhidov, H. Terrones, M. Endo and M. Terrones, Adv. Funct. Mater., 2014, 24, 2612–2619 CrossRef.
- K. Sakaushi and M. Antonietti, Acc. Chem. Res., 2015, 48, 1591–1600 CrossRef CAS PubMed.
- R. Ciriminna, N. Zhang, M.-Q. Yang, F. Meneguzzo, Y.-J. Xu and M. Pagliaro, Chem. Commun., 2015, 51, 7090–7095 RSC.
- C. G. Schäfer, T. Winter, S. Heidt, C. Dietz, T. Ding, J. J. Baumberg and M. Gallei, J. Mater. Chem. C, 2015, 3, 2204–2214 RSC.
- D. Scheid, C. Lederle, S. Vowinkel, C. G. Schäfer, B. Stühn and M. Gallei, J. Mater. Chem. C, 2014, 2, 2583–2590 RSC.
- C. G. Schäfer, C. Lederle, K. Zentel, B. Stühn and M. Gallei, Macromol. Rapid Commun., 2014, 35, 1852–1860 Search PubMed.
- C. G. Schäfer, B. Viel, G. P. Hellmann, M. Rehahn and M. Gallei, ACS Appl. Mater. Interfaces, 2013, 5, 10623–10632 Search PubMed.
- C. G. Schäfer, M. Gallei, J. T. Zahn, J. Engelhardt, G. P. Hellmann and M. Rehahn, Chem. Mater., 2013, 25, 2309–2318 CrossRef.
- C. G. Schäfer, M. Biesalski, G. P. Hellmann, M. Rehahn and M. Gallei, J. Nanophotonics, 2013, 7, 070599 CrossRef.
- T. Ding, G. Cao, C. G. Schäfer, Q. Zhao, M. Gallei, S. K. Smoukov and J. J. Baumberg, ACS Appl. Mater. Interfaces, 2015, 7, 13497–13502 CAS.
- C. G. Schäfer, D. A. Smolin, G. P. Hellmann and M. Gallei, Langmuir, 2013, 29, 11275–11283 CrossRef PubMed.
- C. L. Renschler, A. P. Sylwester and L. V. Salgado, J. Mater. Res., 1989, 4, 452–457 CrossRef CAS.
- J. Pospisil, M. Samoc and J. Zieba, Eur. Polym. J., 1998, 34, 899–904 CrossRef CAS.
- M. Peng, D. Li, L. Shen, Y. Chen, Q. Zheng and H. Wang, Langmuir, 2006, 22, 9368–9374 CrossRef CAS PubMed.
- M. Zhong, C. Tang, E. K. Kim, M. Kruk, E. B. Celer, M. Jaroniec, K. Matyjaszewski and T. Kowalewski, Mater. Horiz., 2014, 1, 121–124 RSC.
- M. Einert, C. Wessel, F. Badaczewski, T. Leichtweiß, C. Eufinger, J. Janek, J. Yuan, M. Antonietti and B. M. Smarsly, Macromol. Chem. Phys., 2015, 216, 1930–1944 CrossRef CAS.
- M. Kruk, B. Dufour, E. B. Celer, T. Kowalewski, M. Jaroniec and K. Matyjaszewski, J. Phys. Chem. B, 2005, 109 Search PubMed.
- A. Lu, A. Kiefer, W. Schmidt and F. Schüth, Chem. Mater., 2004, 16, 100–103 CrossRef CAS.
- T. Kowalewski, N. V. Tsarevsky and K. Matyjaszewski, J. Am. Chem. Soc., 2002, 124, 10632–10633 CrossRef CAS PubMed.
- C. Graf and A. van Blaaderen, Langmuir, 2002, 18, 524–534 CrossRef CAS.
- K. Landfester and M. Antonietti, Macromol. Rapid Commun., 2000, 21, 820–824 CrossRef CAS.
- S. Dalton, F. Heatley and P. M. Budd, Polymer, 1999, 40, 5531–5543 CrossRef CAS.
- M. Rahaman, A. F. Ismail and A. Mustafa, Polym. Degrad. Stab., 2007, 92, 1421–1432 CrossRef CAS.
- C. Weidenthaler, A.-H. Lu, W. Schmidt and F. Schüth, Microporous Mesoporous Mater., 2006, 88, 238–243 CrossRef CAS.
- J. R. Pels, F. Kaptejn, J. A. Moulijn, Q. Zhu and K. M. Thomas, Carbon, 1995, 33, 1641–1653 CrossRef CAS.
- H. Estrade-Szwarckopf, Carbon, 2004, 42, 1713–1721 CrossRef CAS.
- H. Darmstadt, C. Roy, S. Kaliaguine, S. J. Choi and R. Ryoo, Carbon, 2002, 40, 2673–2683 CrossRef CAS.
- A. Stein, Z. Wang and M. A. Fierke, Adv. Mater., 2009, 21, 265–293 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- J. Schwan, S. Ulrich, V. Batori, H. Ehrhardt and S. R. P. Silva, J. Appl. Phys., 1996, 80, 440–447 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Philos. Trans. R. Soc. London, Ser. A, 2004, 362, 2477–2512 CrossRef CAS PubMed.
- M. S. Dresselhaus, G. Dresselhaus, R. Saito and A. Jorio, Phys. Rep., 2005, 409, 47–99 CrossRef.
- M. S. Dresselhaus, G. Dresselhaus, A. Jorio, A. G. S. Filho and R. Saito, Carbon, 2002, 40, 2043–2061 CrossRef CAS.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51–88 CrossRef CAS.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- F. Tuinstra and J. L. Koening, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- A. C. Ferrari, Diamond Relat. Mater., 2002, 11, 1053–1061 CrossRef CAS.
- G. Irmer and A. Dorner-Reisel, Adv. Eng. Mater., 2005, 7, 694–705 CrossRef CAS.
- N. Larouche and B. L. Stansfield, Carbon, 2010, 48, 620–629 CrossRef CAS.
- A. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- G. A. Zickler, B. Smarsly, N. Gierlinger, H. Peterlik and O. Paris, Carbon, 2000, 44, 3239–3246 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: TGA, DLS measurements, XPS survey spectra, XPS photoelectron spectra, and AFM topography measurements. See DOI: 10.1039/c5tc03483c |
| This journal is © The Royal Society of Chemistry 2016 |