
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold-catalyzed cycloisomerization of trifluoromethylated allenols: sustainability and mechanistic studies†‡
Linda
Lempke
,
Hülya
Sak
,
Michael
Kubicki
and
Norbert
Krause
*
Dortmund University of Technology, Organic Chemistry, Otto-Hahn-Strasse 6, D-44227 Dortmund, Germany. E-mail: norbert.krause@tu-dortmund.de
First published on 10th September 2016
Abstract
Trifluoromethyl-substituted α-allenols are cyclized to the corresponding 2,5-dihydrofurans in the presence of neutral or cationic gold catalysts. The catalyst can be recycled if ionic liquids (in particular [BMIM][PF6]) are used as reaction medium. The allene forms droplets in the ionic liquid generating a heterogeneous two-phasic system. Kinetic studies in organic solvents using electronically different gold catalysts allowed to identify the protodeauration of a σ-gold-species as rate-determining step of typical allene cyclizations. Only if a CF3 group is present at C-5, π-complex formation is rate-limiting.
Introduction
The importance of fluorinated substances is reflected in a wide range of applications in academia and industry.1 Numerous reports concentrate on selective fluorinations and the introduction of fluorinated substituents, emphasizing the rising interest in fluoroorganic chemistry.2 The dramatic influence of fluorine or fluorinated substituents on molecular properties renders them indispensable for the modulation of acidity, metabolic stability, lipophilicity, solubility, bioavailability, and binding affinity to target proteins.3In homogeneous gold catalysis, various methods are available for the formation of carbon–fluorine bonds.4 Frequently, these involve the use of electrophilic fluorinating agents like Selectfluor®.5 There is also a limited number of studies on gold-catalyzed transformations of fluorinated substrates which afford fluorine-tagged heterocycles.5a,6 Based on our experience in the gold-catalyzed activation of unsaturated molecules (mostly allenes) for regio- and stereoselective cyclization reactions,7 we now present the first results of a study on the effect of trifluoromethyl substituents on the cyclization of α-hydroxyallenes to 2,5-dihydrofurans. Main objectives of these investigations are the synthesis of fluorinated heterocycles under sustainable reaction conditions employing recyclable gold catalysts7d,8,9 in ionic liquids (ILs)9 as reaction medium, as well as, an improved mechanistic understanding of these transformations with the aid of kinetic studies.
Results
Synthetic procedures
For a systematic study of the influence of trifluoromethyl substituents on the reactivity in the gold-catalyzed cycloisomerization, three regioisomeric α-hydroxyallenes 1a–c were synthesized (see ESI‡ for details). For the first part of these investigations, the imidazolium-based ILs and gold catalysts shown in Scheme 1 were used.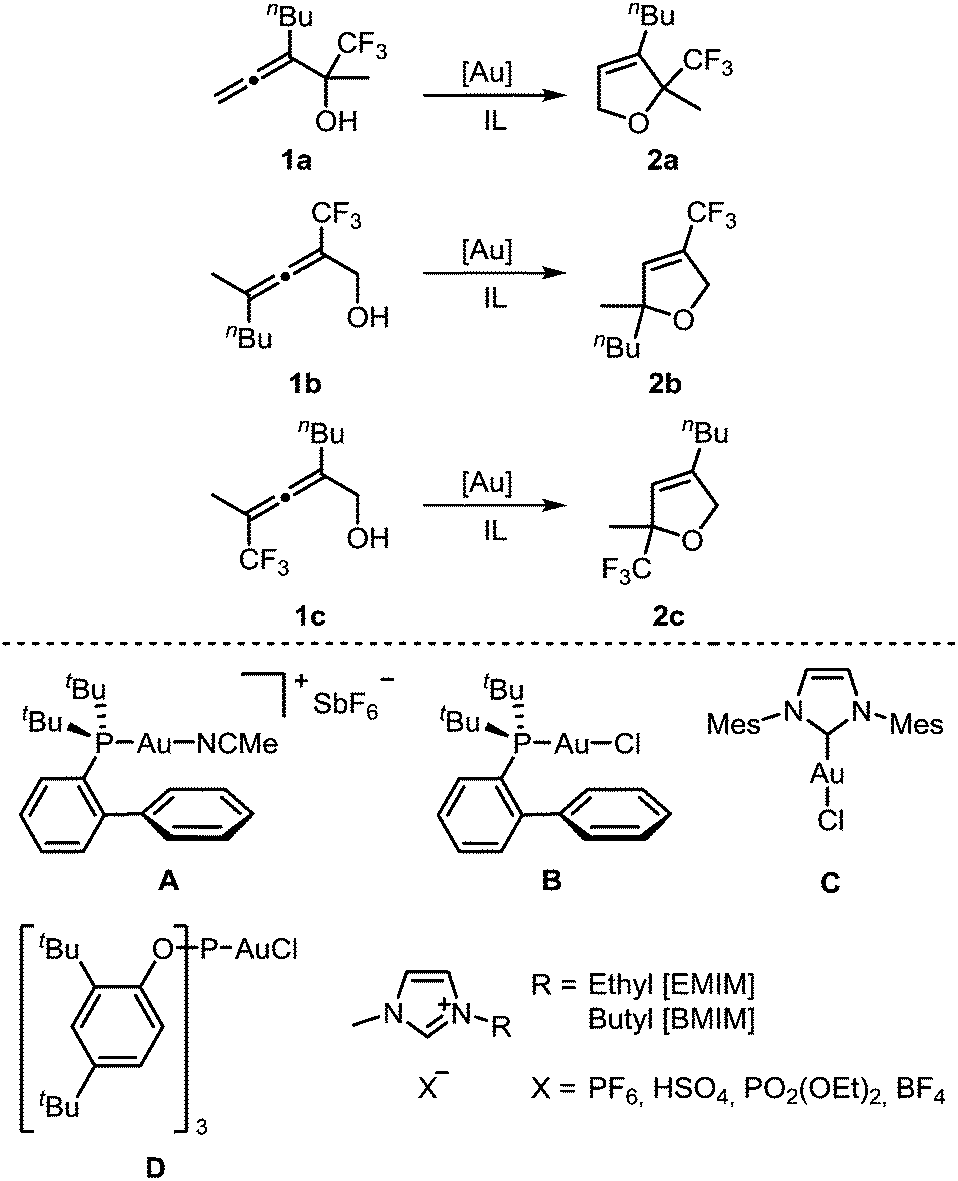 | ||
| Scheme 1 Gold-catalyzed cycloisomerization of trifluoromethylated allenes 1a–c using gold catalysts A–D and imidazolium-based ionic liquids as solvent. | ||
The optimization of the reaction conditions was carried out with allene 1a. Different ILs were tested in the presence of 5 mol% AuCl3 as catalyst (Table 1). Similar to earlier studies,9 [BMIM][PF6] gave the shortest reaction time of 4 h (entry 1), while the cyclization in [EMIM][PF6] is much slower (entry 2). The ionic liquids [EMIM][HSO4] and [BMIM][HSO4] were used at 60 °C to decrease their viscosity. In this manner, complete conversions were observed after 1–1.5 h (entries 4 & 5). When [BMIM][PF6] was used at the same temperature, complete consumption of the starting material was detected already after 15 min (entry 3). To rule out the possibility of an acid-catalyzed cyclization10 in the presence of [EMIM][HSO4] and [BMIM][HSO4], the reactions were repeated in the absence of a gold catalyst. No conversion was observed after 24 h (entries 6 & 7). With [EMIM][(PO2)OEt2], [BMIM][BF4], or [EMIM][BF4], unsatisfactory results were obtained (entries 8–10). The high reactivity observed in [BMIM][PF6] may be due to formation of L–Au–OPOF2 species by fluoride abstraction or hydrolysis of PF6− by adventitious water.11
| Entry | IL | T [°C] | Time | Conv.b [%] |
|---|---|---|---|---|
| a Catalyst: AuCl3 (5 mol%). b Determined by GC-FID. c Reaction carried out without gold catalyst. d Allene 1a was reisolated. | ||||
| 1 | [BMIM][PF6] | 25 | 4 h | 100 |
| 2 | [EMIM][PF6] | 25 | 24 h | 14d |
| 3 | [BMIM][PF6] | 60 | 15 min | 100 |
| 4 | [BMIM][HSO4] | 60 | 1 h | 100 |
| 5 | [EMIM][HSO4] | 60 | 1.5 h | 100 |
| 6 | [BMIM][HSO4]c | 60 | 24 h | 0d |
| 7 | [EMIM][HSO4]c | 60 | 24 h | 0d |
| 8 | [EMIM][(PO2)OEt2] | 25 | 5 d | 9d |
| 9 | [BMIM][BF4] | 25 | 24 h | 30d |
| 10 | [EMIM][BF4] | 25 | 24 h | 0d |
Next, we investigated the reactivity of allenol 1a in [BMIM][PF6] in the presence of different gold catalysts at room temperature (Table 2). Compared to AuCl3, the activity of AuBr3 is much lower (entry 2 vs. 1). In contrast to this, cationic gold(I) complexes afforded full conversion of 1a with only 2 mol% catalyst loading (entries 3–8). The highest activity was found with catalyst A which required only 15 min for complete conversion (entry 4). In the case of neutral chlorogold(I) complexes B, C, D, and Ph3PAuCl, addition of AgSbF6 is mandatory for formation of the active cationic catalyst which gave reaction times of 1–6 h (entries 5–8). Without the silver additive no reaction takes place. AgSbF6 itself is a less reactive catalyst, leading to 25% conversion after 3 d (entry 9).
After optimization of the reaction conditions, we determined reactivity profiles of the regioisomeric trifluoromethyl-substituted α-hydroxyallenes 1a–c in [BMIM][PF6] at room temperature (Table 3). Here, catalysts of different reactivity and chemical nature were selected: AuCl3, Ph3PAuNTf2, A, and D/AgSbF6. Irrespective of the catalyst, the same reactivity order for the three allenols was found: 1a is the most reactive one, followed by 1c, whereas 1b displays the lowest reactivity. In the latter case, AuCl3 and D/AgSbF6 afforded incomplete conversion after 24 h at room temperature. Thus, compared to simple non-fluorinated allenes, introduction of a trifluoromethyl substituent causes a strong reactivity decrease.12 A consistent reactivity profile for the different gold catalysts was also observed. Catalyst A is the most active one, followed by Ph3PAuNTf2 (entries 2 & 3), whereas AuCl3 (entry 1) and D/AgSbF6 (entry 4) are less reactive.
| Allene: | 1a | 1b | 1c | |
|---|---|---|---|---|
| Entry | [Au] | 1a [Time h−1] | 1b [Time h−1] | 1c [Time h−1] |
| a Time required for full conversion (or conversion after 24 h) at room temperature with 2 mol% catalyst, determined by GC-FID. b 5 mol% catalyst loading. c 45% conversion. d 95% conversion. e 83% conversion. | ||||
| 1 | AuCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
4 | 24c | 24d |
| 2 | A | 0.25 | 4 | 1 |
| 3 | Ph3PAuNTf2 | 1.5 | 6 | 2.5 |
| 4 | D/AgSbF6 | 6 | 24e | 24 |
Previously, it was demonstrated that gold catalysts in ionic liquids are stable to water, air and organic solvents and thus can be recycled after extraction of the product.9 This technique can also be applied to trifluoromethylated allenols, as shown exemplarily for substrate 1a (Table 4). The dihydrofuran 2a was isolated by extraction with pentane in good yields13 over five runs, and no loss of the catalytic activity was observed.
Mechanistic studies
To improve the mechanistic understanding of gold-catalyzed transformations in ionic liquids and in organic solvents, several experiments were carried out. First, in order to gather information about the solubility of trifluoromethylated allenes in ionic liquids, we examined an ultrasonicated mixture of allenol 1a in [BMIM][PF6] under a light microscope. Droplets of the allene with diameters in the micrometer range were detected, which proves the presence of a heterogeneous two-phasic system where phase transfer processes may be important for the overall rate of the reactions (ESI, Fig. SI1‡). Accordingly, the gold-catalyzed cyclization should be faster if the droplets are smaller. Indeed, we found an accelerating effect when the reaction was performed under ultrasonic conditions (Table SI1‡).Another important question is the nature of the active gold catalyst species in an ionic liquid. Previous studies revealed a stabilizing effect of the IL9,14 and the formation of gold nanoparticles.14 In order to investigate whether the stabilization can be attributed to the formation of gold–NHC complexes, we used [BMMIM][PF6] which, compared to [BMIM][PF6], has an additional methyl substituent in 2-position and cannot form an N-heterocyclic carbene. Moreover, we applied the gold–NHC complexes E and F15 to the cyclization (Table 5).
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Au]a | IL | T [°C] | Time | Conv.b [%] |
| a Catalyst loading: 5 mol% (entries 1 & 2); 2 mol% (entries 3–9). b Determined by GC-FID. | |||||
| 1 | AuCl3 | [BMIM][PF6] | 60 | 15 min | 100 |
| 2 | AuCl3 | [BMMIM][PF6] | 60 | 20 min | 100 |
| 3 | A | [BMIM][PF6] | 60 | 5 min | 100 |
| 4 | A | [BMMIM][PF6] | 60 | 5 min | 100 |
| 5 | Ph3PAuNTf2 | [BMIM][PF6] | 60 | 15 min | 100 |
| 6 | Ph3PAuNTf2 | [BMMIM][PF6] | 60 | 20 min | 100 |
| 7 | E/AgSbF6 | [BMIM][PF6] | 25 | 4 h | 100 |
| 8 | E | [BMIM][PF6] | 25 | 24 h | 0 |
| 9 | F | [BMIM][PF6] | 25 | 24 h | 0 |
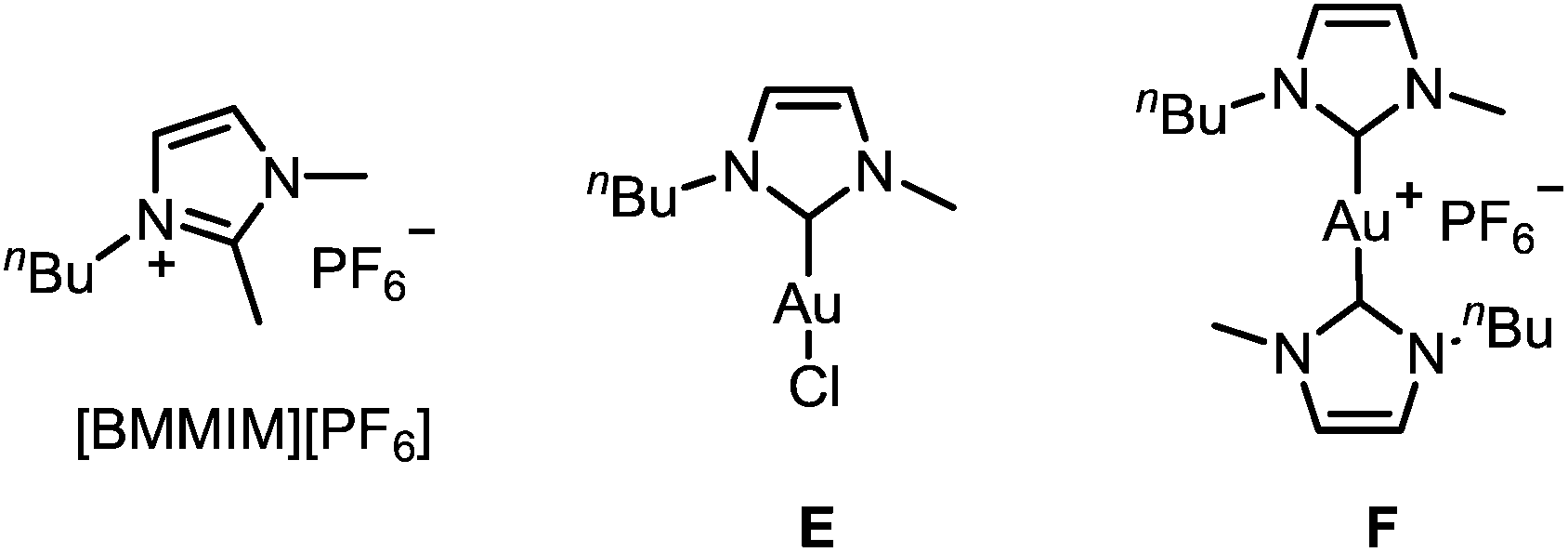
Both ILs were used at 60 °C to maintain a sufficiently low viscosity (Table 5). It turned out that there is virtually no reactivity difference between [BMIM][PF6] and [BMMIM][PF6] (entries 1 vs. 2; 3 vs. 4; 5 vs. 6). In all cases, a rapid cyclization took place within 5–20 min. Thus, the formation of gold–NHC complexes is not required for the catalytic activity, but it may contribute to the stability of the catalyst in ILs. Remarkably, complex E showed a good catalytic activity after activation with AgSbF6 (entries 7 and 8), while the ionic dimer F is completely unreactive (entry 9). Hence, formation of dimeric gold–NHC complexes of the type F may be a pathway leading to deactivation of the gold catalyst.
The mechanism of the gold-catalyzed endo-selective cycloisomerization of α-functionalized allenes has been studied extensively, leading to the catalytic cycle shown in Scheme 2.7,16 In the first step the π-complex II is generated by coordination of the catalytically active gold species to a C–C-double bond of allene I. The subsequent intramolecular attack of the nucleophile leads to the σ-gold species III. Finally, protodeauration affords the product IV and regenerates the gold catalyst. Due to the observation that the cycloisomerization of simple α-hydroxyallenes is accelerated in the presence of external proton donors, it is assumed that the protodeauration is the rate determining step of this reaction.
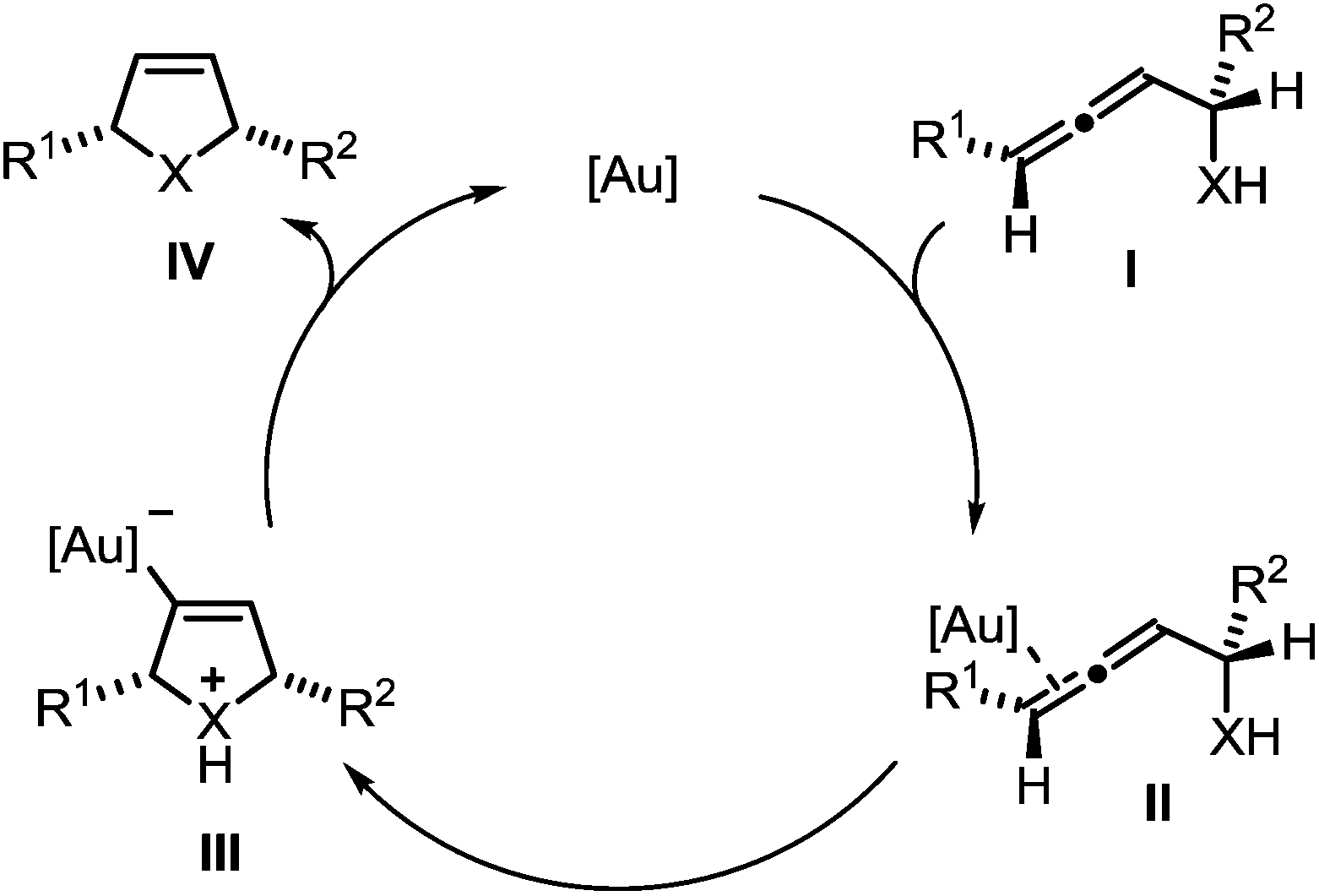 | ||
| Scheme 2 Proposed mechanism of the gold-catalyzed cycloisomerization of α-functionalized allenes (X = NR, O, S). | ||
In a recent seminal contribution, Wang, Hammond, and Xu17 have examined the influence of electronically different ligands on the rate of gold-catalyzed transformations. Whereas the activation of an unsaturated substrate (e.g., I → II) can be accelerated by electron-deficient ligands, the rate of the protodeauration of a σ-vinylgold species (e.g., III → IV) can be increased by electron-rich ligands. Thus, the opposing electron demand of the individual reactions can be used to determine the rate-determining step of the catalytic cycle. We first applied Hammond's method to the gold-catalyzed cycloisomerization of the exemplary non-fluorinated α-functionalized allenes 3a/b which afford the heterocycles 4a/b (Scheme 3).
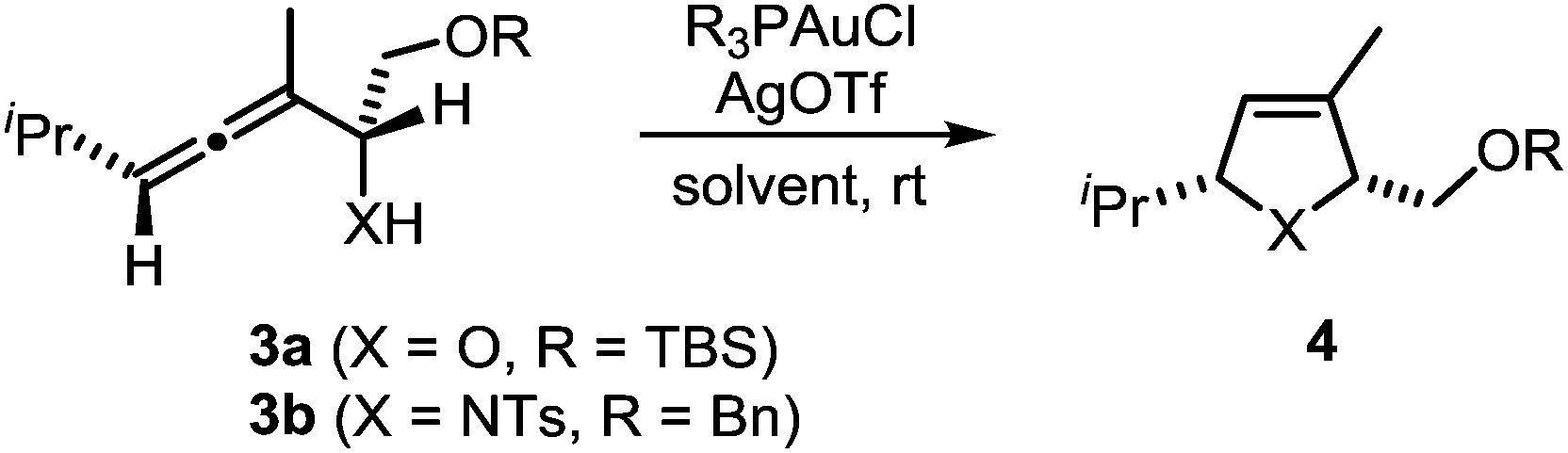 | ||
| Scheme 3 Gold-catalyzed cycloisomerization of allenes 3a/b. | ||
Six phosphine– or phosphite–gold complexes were used for this kinetic study; the ligands exhibit decreasing electron-donating ability in the following order: B > (4-MeOC6H4)3PAuCl > (4-MeC6H4)3PAuCl > Ph3PAuCl > (4-CF3C6H4)3PAuCl > D. The chlorocomplexes were activated with equimolar amounts of AgOTf. Different solvents and catalyst loadings were tested to find optimal reaction conditions. Reaction times between 1 and 3 h were required in order to gather a sufficient number of samples which were analyzed by gas chromatography. The results obtained for α-hydroxyallene 3a are shown in Fig. 1. They clearly show that the rate of the cyclization is rising with increasing electron-donating properties of the ligand. In the context of Hammond's results, this indicates that the protodeauration of the σ-vinylgold species III is the rate-determining step of the catalytic cycle.
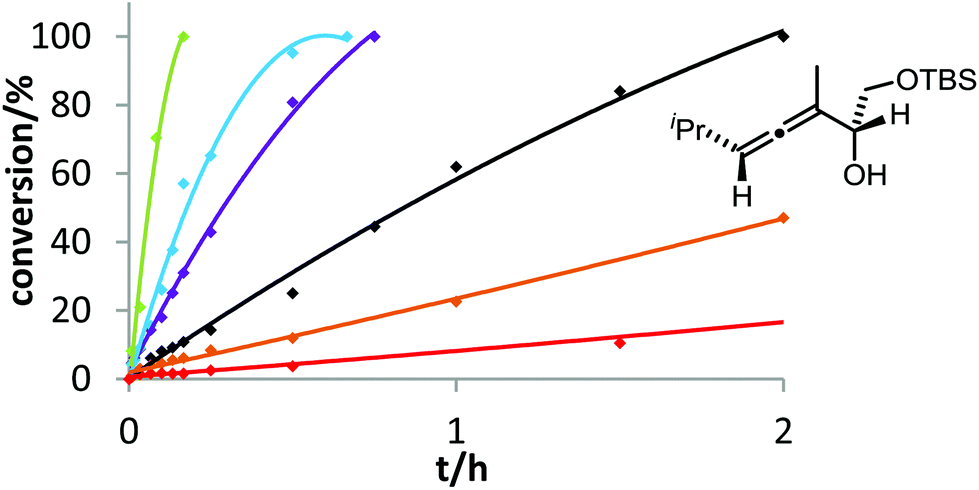 | ||
| Fig. 1 Ligand effect on the gold-catalyzed cycloisomerization of 3a. 4 mol% [Au] and 4 mol% AgOTf were used in DMF at room temperature. Decreasing reactivity was observed in the following order: B (green) > (4-MeOC6H4)3PAuCl (blue) > (4-MeC6H4)3PAuCl (violet) > Ph3PAuCl (black) > (4-CF3C6H4)3PAuCl (orange) > D (red). | ||
The corresponding kinetic study with α-aminoallene 3b was carried out with four phosphinegold complexes (Fig. 2). Again, electron-rich ligands lead to rapid cyclizations. Thus, also for aminoallene 3b, the protodeauration is the rate-determining step.
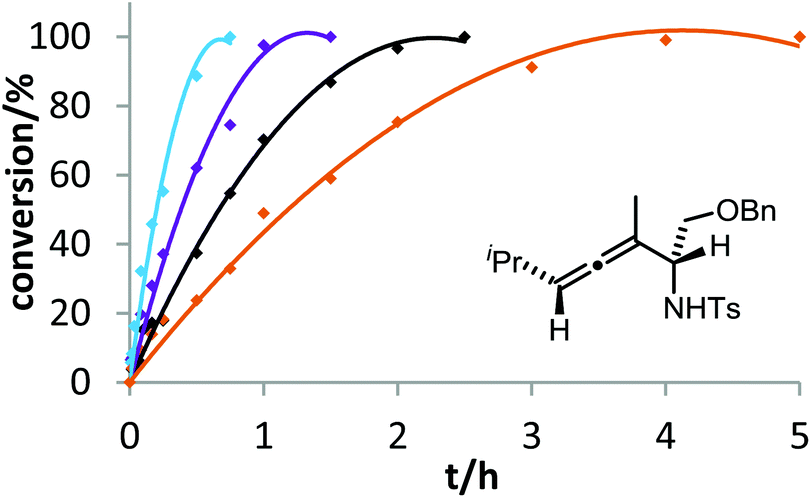 | ||
| Fig. 2 Ligand effect on the gold-catalyzed cycloisomerization of 3b. 2 mol% [Au] and 2 mol% AgOTf were used in THF at room temperature. Decreasing reactivity was observed in the following order: (4-MeOC6H4)3PAuCl (blue) > (4-MeC6H4)3PAuCl (violet) > Ph3PAuCl (black) > (4-CF3C6H4)3PAuCl (orange). | ||
Pushing the limits of the accelerating effect of electron-rich phosphine ligands is possible by changing the number and position of electron-releasing groups. This could be demonstrated for the cycloisomerization of allenol 3a with methoxy-substituted phosphinegold complexes (Fig. 3). It turned out that the accelerating effect of the methoxy group in 2-position is stronger than in 4-position. Accordingly, the catalyst formed from [2,4,6-(MeO)3C6H2]3PAuCl and AgOTf gave by far the fastest cyclization.
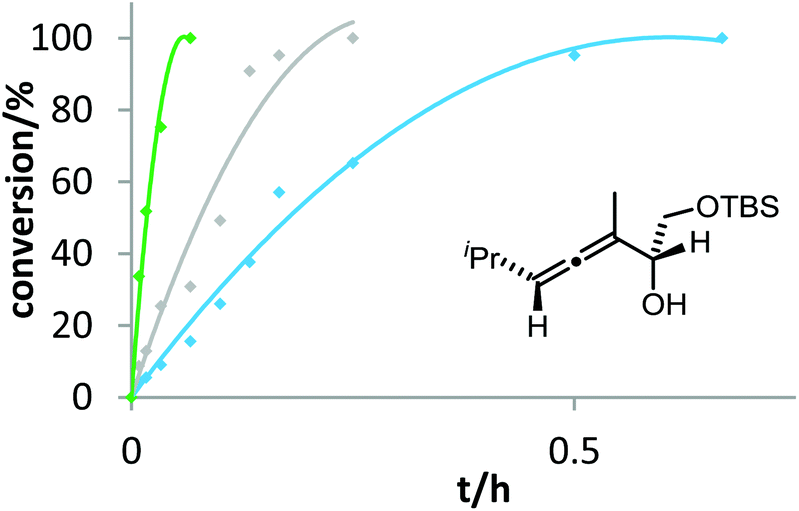 | ||
| Fig. 3 Effect of the number and position of electron-releasing substituents on the gold-catalyzed cycloisomerization of 3a. 4 mol% [Au] and 4 mol% AgOTf were used in DMF at room temperature. Decreasing reactivity was observed in the following order: [2,4,6-(MeO)3C6H2]3PAuCl (green) > (2-MeOC6H4)3PAuCl (grey) > (4-MeOC6H4)3PAuCl (blue). | ||
Recently, Hammond and co-workers demonstrated that additives with good hydrogen-bond acceptor ability can increase the rate of gold-catalyzed reactions if the protodeauration is the rate-limiting step.18 We examined this effect for the gold-catalyzed cycloisomerization of 3a with Ph3PAuCl/AgOTf in the presence of Bu4NOTf or pyridin-N-oxide (Fig. 4). As expected, both additives accelerate the conversion of allene 3a. This result confirms once more the protodeauration as rate-determining step.
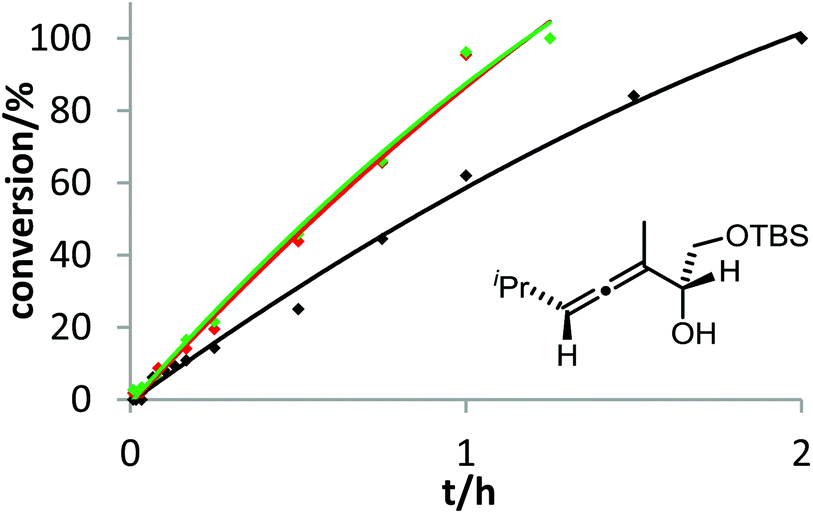 | ||
| Fig. 4 Effect of additives on the gold-catalyzed cycloisomerization of 3a. 4 mol% Ph3PAuCl, 4 mol% AgOTf, 5 mol% additive were used in DMF at room temperature. Decreasing reactivity was observed in the following order: Ph3PAuCl + pyridin-N-oxide (green) ≈ Ph3PAuCl + Bu4NOTf (red) > Ph3PAuCl (black). | ||
After demonstrating the viability of Hammond's method for the gold-catalyzed cyclization of allenes 3a/b, we extended our kinetic study to the trifluoromethylated counterparts 1a–c. As already mentioned above, introduction of the electron-withdrawing CF3 group causes a strong decrease of reactivity in the gold-catalyzed cyclization.12 Is this effect accompanied by a change of the rate-determining step? As can be seen in Fig. 5 and 6, this is not the case of allenols 1a and 1b. For both substrates, the electron-rich gold catalysts B and (4-MeOC6H4)3PAuCl gave faster reactions than the electron-poor phosphite–gold complex D. Thus, also for these allenes the protodeauration is rate-limiting.
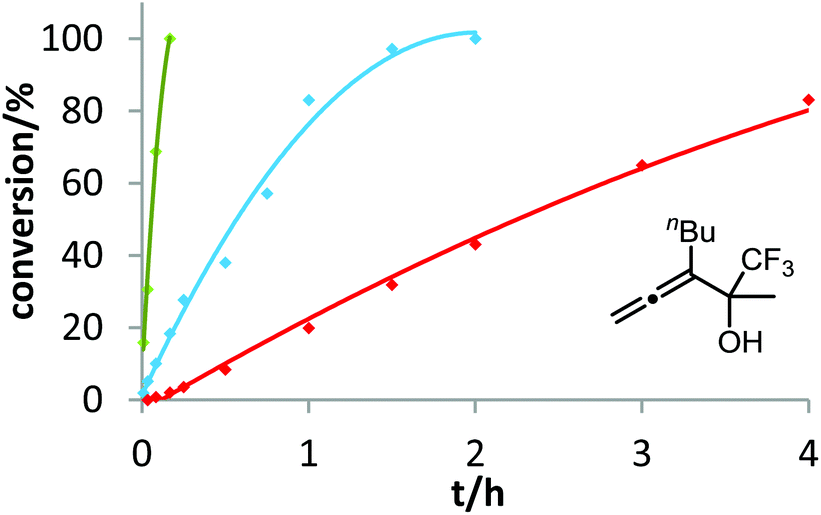 | ||
| Fig. 5 Ligand effect on the gold-catalyzed cycloisomerization of 1a. 5 mol% [Au] and 5 mol% AgOTf were used in THF at room temperature. Decreasing reactivity was observed in the following order: B (green) > (4-MeOC6H4)3PAuCl (blue) > D (red). | ||
 | ||
| Fig. 6 Ligand effect on the gold-catalyzed cycloisomerization of 1b. 2 mol% [Au] and 2 mol% AgOTf were used in DCM at room temperature. Decreasing reactivity was observed in the following order: B (green) > (4-MeOC6H4)3PAuCl (blue) > D (red). | ||
In contrast to the previous substrates, allenol 1c bearing a trifluoromethyl group at C-5 shows a different kinetic behavior (Fig. 7). Here, the electron-deficient phosphite–gold catalyst D induces the fastest reaction, the electron-rich phosphine–gold complex B the slowest. According to Hammond's investigations, this indicates that the activation of the allene with formation of π-complex II is the rate-determining step of the catalytic cycle.
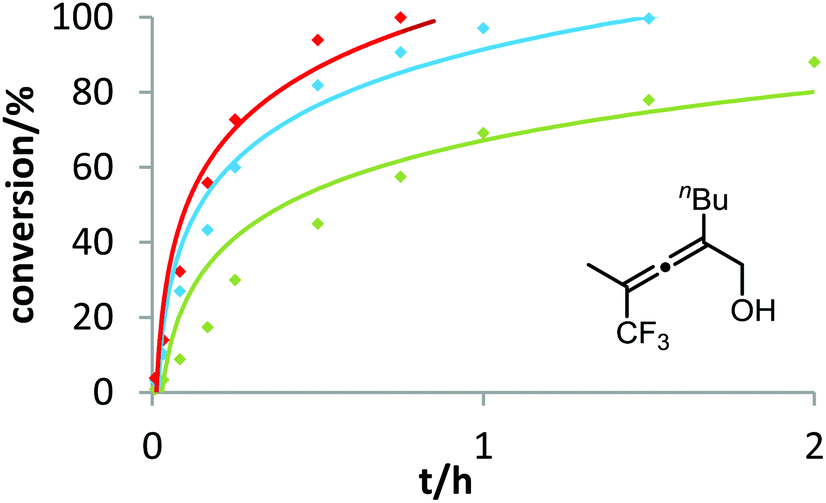 | ||
| Fig. 7 Ligand effect on the gold-catalyzed cycloisomerization of 1c. 3 mol% [Au] and 3 mol% AgOTf were used in toluene at room temperature. Decreasing reactivity was observed in the following order: D (red) > (4-MeOC6H4)3PAuCl (blue) > B (green). | ||
With our kinetic study, we could show for the first time that the rate-determining step of a gold-catalyzed transformation can be changed by variation of the electronic properties of the substrate. It appears that in the case of allenol 1c, the electron-withdrawing effect of the CF3 group at C-5 causes a strong decrease of electron density at the adjacent C–C double bond. As a consequence, formation of π-complex II is slowed down to such an extent that this step becomes rate-limiting. Unfortunately, due to the heterogeneous nature of the reaction mixture in ILs (see above), it is not possible to conduct these kinetic studies in ionic liquids. In the future, we will include steric effects,19 as well as the influence of the counterion of cationic gold catalysts,20 in these mechanistic studies.
Conclusions
In this study, we have demonstrated that trifluoromethyl-substituted α-allenols can be cyclized to the corresponding 2,5-dihydrofurans in the presence of neutral or cationic gold catalysts. When ionic liquids (in particular [BMIM][PF6]) are used as reaction medium, the sustainability of these transformations is improved since a recycling of the gold catalyst becomes possible. Distinct reactivity differences between the allenols 1a–c and the gold catalysts were noted. The allenes form droplets in ionic liquids generating a heterogeneous two-phasic system. As a result, diffusion and interface exchange processes may have a strong influence on the rate of the reaction. The formation of gold–NHC complexes is not required for the catalytic activity, but it may contribute to the stability of the catalyst in ILs. Extensive kinetic studies in organic solvents using phosphine- or phosphite–gold catalysts with different electronic properties allowed to identify the protodeauration of σ-gold-species III as rate-determining step of the catalytic cycle for fluorinated allenols 1a/b, as well as for the non-fluorinated substrates 3a/b. In contrast, activation of the allene with formation of π-complex II is the rate-limiting step for substrate 1c. This mechanistic change (which has been determined for the first time for a gold-catalyzed transformation) is probably caused by the electron-withdrawing effect of the CF3 group at C-5 which strongly decreases the electron density of the adjacent C–C double bond. Our study underlines the importance of sustainable reaction conditions, as well as, mechanistic investigations for the improvement of catalytic processes. Further work along these lines is in progress.Notes and references
- (a) W. R. Dolbier Jr., J. Fluorine Chem., 2005, 126, 157 CrossRef; (b) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, 2013 Search PubMed.
- (a) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (b) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (c) M. F. Kuehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 52, 3328 CrossRef PubMed; (d) P. R. Savoie and J. T. Welch, Chem. Rev., 2015, 115, 1130 CrossRef CAS PubMed.
- (a) H.-J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637 CrossRef PubMed; (b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (c) D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071 CrossRef; (d) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed.
- (a) A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657 CrossRef CAS PubMed; (b) G. Liu, Org. Biomol. Chem., 2012, 10, 6243 RSC.
- (a) M. Schuler, F. Silva, C. Bobbio, A. Tessier and V. Gouverneur, Angew. Chem., Int. Ed., 2008, 47, 7927 CrossRef CAS PubMed; (b) W. Wang, J. Jasinski, G. B. Hammond and B. Xu, Angew. Chem., Int. Ed., 2010, 49, 7247 CrossRef CAS PubMed; (c) J. Qian, Y. Liu, J. Zhu, B. Jiang and Z. Xu, Org. Lett., 2011, 13, 4220 CrossRef CAS PubMed; (d) Y. Liu, J. Zhu, J. Qian and Z. Xu, J. Org. Chem., 2012, 77, 5411 CrossRef CAS PubMed; (e) Z. Jin, R. S. Hidinger, B. Xu and G. B. Hammond, J. Org. Chem., 2012, 77, 7725 CrossRef CAS PubMed; (f) S. Li, Z. Li, Y. Yuan, Y. Li, L. Zhang and Y. Wu, Chem. – Eur. J., 2013, 19, 1496 CrossRef CAS PubMed; (g) A. Arcadi, E. Pietropaolo, A. Alvino and V. Michelet, Org. Lett., 2013, 15, 2766 CrossRef CAS PubMed; (h) A. Arcadi, E. Pietropaolo, A. Alvino and V. Michelet, Beilstein J. Org. Chem., 2014, 10, 449 CrossRef PubMed; (i) Y. Jeong, B.-I. Kim, J. K. Lee and J.-S. Ryu, J. Org. Chem., 2014, 79, 6444 CrossRef CAS PubMed.
- (a) R. Surmont, G. Verniest and N. De Kimpe, Org. Lett., 2009, 11, 2920 CrossRef CAS PubMed; (b) Y. Li, K. A. Wheeler and R. Dembinski, Adv. Synth. Catal., 2010, 352, 2761 CrossRef CAS; (c) Y. Li, K. A. Wheeler and R. Dembinski, Eur. J. Org. Chem., 2011, 2767 CrossRef CAS; (d) Y. Li, K. A. Wheeler and R. Dembinski, Org. Biomol. Chem., 2012, 10, 2395 RSC; (e) S. Li, Z. Li, Y. Yuan, D. Peng, Y. Li, L. Zhang and Y. Wu, Org. Lett., 2012, 14, 1130 CrossRef CAS PubMed; (f) S. Fustero, I. Ibáñez, P. Barrio, M. A. Maestro and S. Catalán, Org. Lett., 2013, 15, 832 CrossRef CAS PubMed.
- (a) N. Krause, V. Belting, C. Deutsch, J. Erdsack, H.-T. Fan, B. Gockel, A. Hoffmann-Röder, N. Morita and F. Volz, Pure Appl. Chem., 2008, 80, 1063 CrossRef CAS; (b) N. Krause, Ö. Aksin-Artok, V. Breker, C. Deutsch, B. Gockel, M. Poonoth, Y. Sawama, Y. Sawama, T. Sun and C. Winter, Pure Appl. Chem., 2010, 82, 1529 CrossRef CAS; (c) N. Krause, Ö. Aksin-Artok, M. Asikainen, V. Breker, C. Deutsch, J. Erdsack, H.-T. Fan, B. Gockel, S. Minkler, M. Poonoth, Y. Sawama, Y. Sawama, T. Sun, F. Volz and C. Winter, J. Organomet. Chem., 2012, 704, 1 CrossRef CAS; (d) B. Wagner, K. Belger, S. Minkler, V. Belting and N. Krause, Pure Appl. Chem., 2016, 88, 391 CrossRef CAS.
- (a) S. R. K. Minkler, B. H. Lipshutz and N. Krause, Angew. Chem., Int. Ed., 2011, 50, 7820 CrossRef CAS PubMed; (b) S. R. K. Minkler, N. A. Isley, D. J. Lippincott, N. Krause and B. H. Lipshutz, Org. Lett., 2014, 16, 724 CrossRef CAS PubMed; (c) S. Handa, D. J. Lippincott, D. H. Aue and B. H. Lipshutz, Angew. Chem., Int. Ed., 2014, 53, 10658 CrossRef CAS PubMed; (d) L. Lempke, A. Ernst, F. Kahl, R. Weberskirch and N. Krause, Adv. Synth. Catal., 2016, 358, 1491 CrossRef CAS; (e) K. Belger and N. Krause, Eur. J. Org. Chem., 2015, 220 CrossRef CAS; (f) K. Belger and N. Krause, Org. Biomol. Chem., 2015, 13, 8556 RSC.
- (a) Ö. Aksin and N. Krause, Adv. Synth. Catal., 2008, 350, 1106 CrossRef; (b) L. Lempke, T. Fischer, J. Bell, W. Kraus, K. Rurack and N. Krause, Org. Biomol. Chem., 2015, 13, 3787 RSC.
- (a) N. Krause, M. Laux and A. Hoffmann-Röder, Tetrahedron Lett., 2000, 41, 9613 CrossRef CAS; (b) A. Hoffmann-Röder and N. Krause, Org. Lett., 2001, 3, 2537 CrossRef.
- S. M. Kim, J. H. Park and Y. K. Chung, Chem. Commun., 2011, 47, 6719 RSC.
- Under the conditions of Table 3, the corresponding non-fluorinated allenols require only 5–10 min at room temperature for full conversion to the 2,5-dihydrofuran.
- Isolated yields of dihydrofurans 2b/c under the conditions of Table 4: 91% (2b), 90% (2c).
- A. Corma, I. Domínguez, T. Ródenas and M. J. Sabater, J. Catal., 2008, 259, 26 CrossRef CAS.
- L. Messori, L. Marchetti, L. Massai, F. Scaletti, A. Guerri, I. Landini, S. Nobili, G. Perrone, E. Mini, P. Leoni, M. Pasquali and C. Gabbiani, Inorg. Chem., 2014, 53, 2396 CrossRef CAS PubMed.
- N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS PubMed.
- W. Wang, G. B. Hammond and B. Xu, J. Am. Chem. Soc., 2012, 134, 5697 CrossRef CAS PubMed.
- W. Wang, M. Kumar, G. B. Hammond and B. Xu, Org. Lett., 2014, 16, 636 CrossRef CAS PubMed.
- R. E. Ebule, D. Malhotra, G. B. Hammond and B. Xu, Adv. Synth. Catal., 2016, 358, 1478 CrossRef CAS.
- M. Jia and M. Bandini, ACS Catal., 2015, 5, 1638 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Barry M. Trost on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental details and supporting characterization data. See DOI: 10.1039/c6qo00423g |
| This journal is © the Partner Organisations 2016 |