
A tetrasulfate-resorcin[6]arene cavitand as the host for organic ammonium guests†
Carmine
Gaeta
*a,
Paolo
Della Sala
a,
Carmen
Talotta
*a,
Margherita
De Rosa
 a,
Annunziata
Soriente
a,
Giovanna
Brancatelli
b,
Silvano
Geremia
b and
Placido
Neri
a
a,
Annunziata
Soriente
a,
Giovanna
Brancatelli
b,
Silvano
Geremia
b and
Placido
Neri
a
aDipartimento di Chimica e Biologia “A. Zambelli”, Università degli Studi di Salerno, Via Giovanni Paolo II 132, I-84084 Fisciano, Salerno, Italy. E-mail: cgaeta@unisa.it; ctalotta@unisa.it
bCentro di Eccellenza in Biocristallografia, Dipartimento di Scienze Chimiche e Farmaceutiche, Università di Trieste, via L. Giorgieri 1, 34127 Trieste, Italy
First published on 8th August 2016
Abstract
The first example of a resorcin[6]arene cavitand in which sulfate bridges are introduced between the OH groups is here reported. In addition to their structural role, sulfate bridges behave as functional supramolecular interacting groups thus favoring the complexation of suitable organic ammonium guests.
In the last two decades bowl-shaped macrocycles1 have stimulated the imagination of chemists devoted to molecular recognition. In fact, thanks to the presence of a cavity, they are potentially able to accommodate appropriate guests. Among the bowl-shaped macrocycles, resorcin[4]arene-based cavitands2 are a family of hosts in which bridging elements are introduced between adjacent phenolic OH groups.
In the last decade, the search for supramolecular applications of cavitand hosts has produced interesting examples of molecular recognition,3 sensing,4 and catalysis.5 Among them, peculiar recognition abilities have been shown by tetraphosphonate resorcin[4]arene cavitands6 in which phosphorus bridges were introduced at the upper rim of the macrocycle. The molecular recognition properties of tetraphosphonate cavitands have been exploited in gas sensing7 and cantilever-based chemical sensors have been obtained.8 In these cases, a particular affinity was evidenced by tetraphosphonate hosts toward N-methylated ammonium guests, thanks to H-bonding and dipole–dipole interactions between P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bridging groups of the host and the ammonium moiety of the guest.8
O bridging groups of the host and the ammonium moiety of the guest.8
Very recently, we have reported9 an improved procedure for the synthesis of the large resorcin[6]arene macrocycle 1 and we have shown that in the solid state it adopts a pinched cone conformation very similar to that reported for p-tert-butylcalix[6]arene.10 The interesting recognition abilities showed by tetraphosphonate resorcin[4]arene hosts, prompted us to investigate the synthesis of a large resorcin[6]arene cavitand11 in which new interacting groups, such as sulfate, were used as bridging elements.
Our idea was inspired by a recent study by Shi12 and coworkers in which it is shown that the treatment of phenol derivatives with N,N′-sulfuryldiimidazole, in the presence of Cs2CO3 as the base, afforded diaryl sulfates in good yields.
Prompted by these results, we attempted the formation of sulfate-bridges between the OH groups of 1 under conditions modified with respect to those of Shi and coworkers.12,13 Thus, the treatment of 1 with 10 equiv. of N,N′-sulfuryldiimidazole [(Im)2SO2] (Scheme 1) and K2CO3 in dry DMA at 75 °C for 60 h, afforded derivative 2 in 30% yield, after column chromatography on silica gel.13
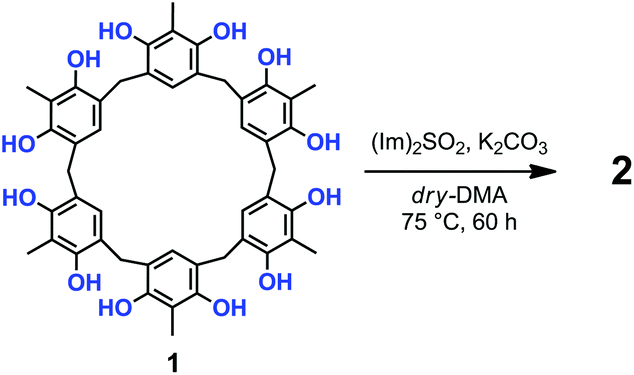 | ||
| Scheme 1 | ||
High-resolution MALDI Fourier-Transform ion cyclotron resonance (MALDI-FT-ICR) mass spectrometry showed a molecular ion peak at 1087.0895 m/z for 2 in accordance, with the molecular formula [M + Na]+ of a tetrasulfate-bridged derivative (calcd 1087.0888).13 The structure assignment of (SO4)4-bridged 2 was based on the spectral 1H and 13C NMR data. In fact, three possible (SO4)4-bridged derivatives are possible (Fig. 1), namely, 1,2,4,5-tetra-bridged, 1,2,3,4-tetra-bridged, and 1,2,3,5-tetra-bridged.
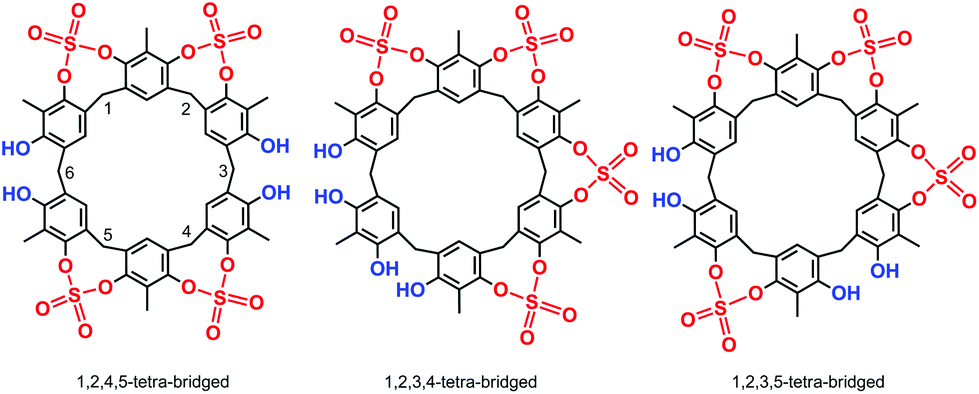 | ||
| Fig. 1 The three possible regioisomers of a tetrasulfate-bridged resorcin[6]arene. | ||
The 1H NMR spectrum of 2 at 403 K (Fig. 2, 400 MHz, TCDE) reveals the presence of two broad ArCH2Ar singlets at 4.05 and 3.94 ppm in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio,13 two ArCH3 singlets at 2.29 and 2.42 ppm (2:1 ratio),13 and finally two ArH signals in a 2:1 ratio at 7.30 and 7.33 ppm, which allowed the confidential assignment of the 1,2,4,5-bridging pattern (Fig. 1). In accordance, the 13C NMR spectrum13 of 2 in TCDE (75 MHz, 403 K) showed two ArCH3 signals at 9.3 and 10.3 ppm, two ArCH2Ar signals at 33.3 and 29.3 ppm, and 10 signals in the aromatic region.
:1 ratio,13 two ArCH3 singlets at 2.29 and 2.42 ppm (2:1 ratio),13 and finally two ArH signals in a 2:1 ratio at 7.30 and 7.33 ppm, which allowed the confidential assignment of the 1,2,4,5-bridging pattern (Fig. 1). In accordance, the 13C NMR spectrum13 of 2 in TCDE (75 MHz, 403 K) showed two ArCH3 signals at 9.3 and 10.3 ppm, two ArCH2Ar signals at 33.3 and 29.3 ppm, and 10 signals in the aromatic region.
 | ||
| Fig. 2 Methylene region of the 1H NMR spectrum (400 MHz, TCDE) of 2 at different temperatures. | ||
Surprisingly, cooling at 298 K caused dramatic changes in the 1H NMR spectrum of 2 (Fig. 2, TCDE, 400 MHz). In fact, two distinct sets of ArCH2Ar AX systems were identified by a 2D COSY spectrum (Fig. S4†) which are corresponding to two conformational isomers 2a and 2b (Fig. 3 and 4). The first set (red in Fig. 2), composed of three AX systems in a 1:1:1 ratio, is attributable to conformer 2a (Fig. 3 and 4d–f), while the second one (two AX systems in a 2:1 ratio, blue in Fig. 2) is related to conformer 2b (Fig. 3, 4a and b). Integration of the corresponding 1H NMR signals evidenced a 50/50 ratio for the two conformers 2a and 2b.
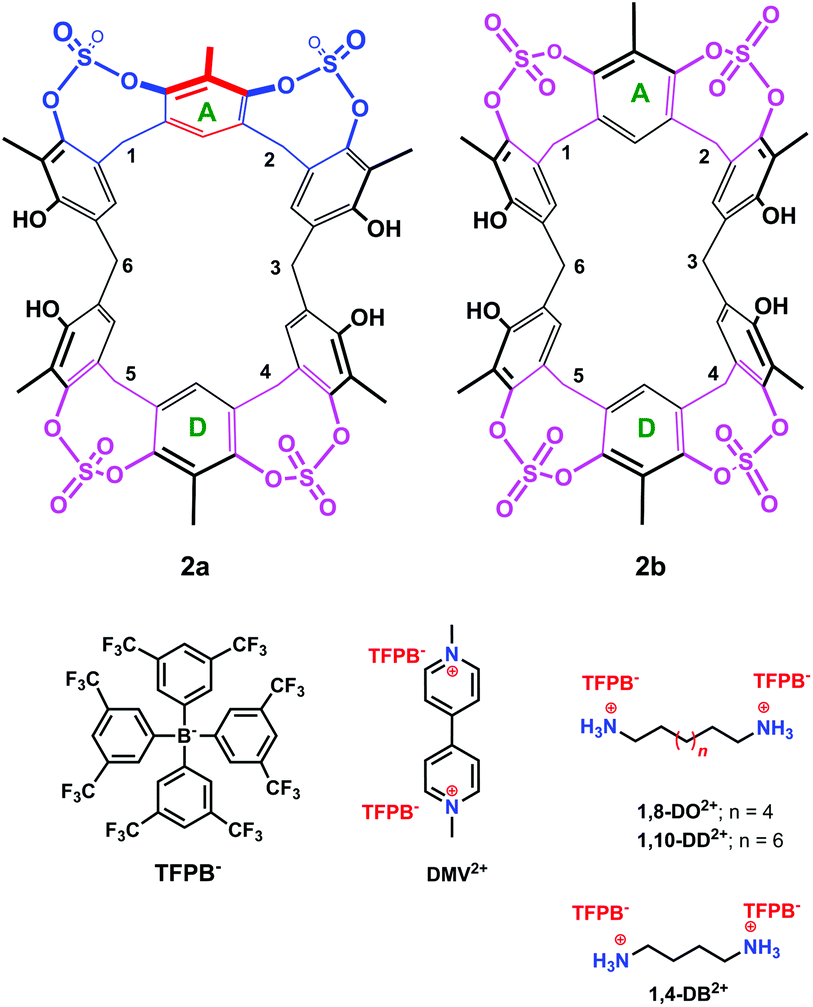 | ||
| Fig. 3 Chemical drawing of the two conformers 2a and 2b and of the guests studied in the present work. | ||
 | ||
| Fig. 4 Lowest energy conformations (Monte Carlo conformational search, MM3, CHCl3 as solvent) of: (a) top view of 2b; (b) side view of 2b; (c) particularly of the boat-chair conformation of the 8-membered ring bearing the sulfate bridge; (d) top view of 2a; (e) side view of 2a; (f) particularly of the distorted boat conformation of the 8-membered rings bearing sulfate bridge in 2a. | ||
Regarding conformer 2b, the presence of two ArCH2Ar AX systems at 4.51/3.70 (8H) and 3.90/3.14 (4H) ppm in a 2:1 ratio (COSY spectrum,13Fig. 2 at 298 K, signals in blue), two ArH singlets in a 2:1 ratio, and two ArCH3 singlets in a 2:1 ratio in its 1H NMR spectrum (TCDE, 298 K) pointed to a structure characterized by two orthogonal symmetry planes bisecting the aromatic rings A and D (Fig. 3) and the ArCH2Ar carbons 3 and 6. An insight into the conformation 2b was obtained by a Monte Carlo conformational search (MM3 force field, CHCl3 as solvent).
A symmetrical lowest energy structure was found in which the resorcin[6]arene skeleton adopts a pinched cone conformation10 (Fig. 4a–c). Interestingly, in this structure the 8-membered rings (in magenta in Fig. 3) bearing the sulfate-bridge, adopt a boat-chair conformation (Fig. 4c), in analogy to the known 6H,12H-dibenzo[d,g][1,3]dioxocine ring.14
Regarding conformer 2a, the 1H NMR spectrum (TCDE, 298 K) evidenced the presence of three ArCH2Ar AX systems at 4.55/3.75, 3.62/4.22, and 4.14/3.28 ppm in a 1:1:1 ratio (COSY spectrum,13Fig. 2, 298 K, signals in red), three ArH singlets in a 1:1:1 ratio, and three 1:1:1 ArCH3 singlets, corresponding to a less symmetrical structure with respect to 2b. A Monte Carlo conformational search (MM3 force field, CHCl3 as solvent) gave the conformation 2a in Fig. 4d as the lowest energy structure compatible with these 1H NMR signal patterns. In particular, the resorcin[6]arene skeleton adopts again a pinched cone conformation but, differently from 2b, two adjacent 8-membered rings (i.e.: 1 and 2 in blue in Fig. 3), adopt a distorted-boat conformation (Fig. 4f). Such distorted-boat conformation was already observed in macrocyclic compounds containing 6H,12H-dibenzo[d,g][1,3]dioxocine rings.14,15 The above variable temperature 1H NMR experiments (TCDE, 400 MHz) (Fig. 2) clearly indicated that, notwithstanding the presence of sulphate bridges, the resorcin[6]arene skeleton of 2 still shows a residual conformational mobility, likely due to the presence of the four free OH groups.
In fact, by increasing the temperature (Fig. 2), a coalescence for ArCH2Ar AX systems of 2a was observed at 323 K, which led to an energy barrier of 14.6 kcal mol−1.16 In analogy with the data previously reported for calixarenes bearing 1,3-dioxocine moieties,14,15 this energy barrier should be associated with an interconversion process between the distorted-boat and the boat-chair conformation of the 8-membered rings of 2a to give 2b. By increasing the temperature, a second coalescence was ascertained at 363 K (400 MHz), corresponding to an energy barrier of 16.5 kcal mol−1, to give an averaged symmetrical spectrum (Fig. 2). This conformational interconversion is related to the pinched cone inversion of 2b to its inverted pinched-cone structure.
The conformer 2a/2b ratio is strongly influenced by the nature of the solvent. In fact, in CD3CN the symmetrical pinched conformation 2b is favoured (Fig. 5) with respect to 2a (at 298 K) with a 98/2 ratio (measured by 1H NMR signal integration). Probably, the regular pinched conformation 2b in solution is templated by acetonitrile solvent molecules, which, as shown by X-ray crystallography (see below, Fig. 6a and d), are able to occupy its two sub-cavities establishing C–H⋯π and C–H⋯OS interactions (Fig. 6a and d).
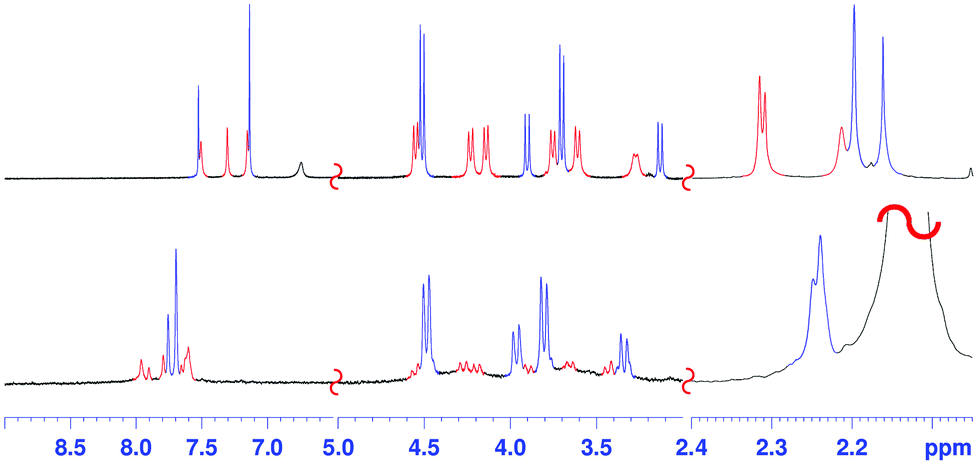 | ||
| Fig. 5 Significant portions of the 1H NMR spectra of: (top) 2 in TCDE (298 K, 600 MHz); (bottom) 2 in CD3CN (298 K, 600 MHz). Marked in blue and in red are the 1H NMR signals of the conformer 2b and 2a respectively. | ||
 | ||
| Fig. 6 X-ray structure of tetrasulfate-resorcin[6]arene cavitand 2. | ||
Crystals of tetrasulfate-cavitand 2 were obtained by slow evaporation of a CH3CN solution. The asymmetric unit of the monoclinic crystals of 2 consists of one resorcin[6]arene molecule and seven acetonitrile solvent molecules. In accordance with the results of MM3 calculations (structure 2b, Fig. 4a) in the solid state, 2 adopts a pinched cone conformation with a boat-chair conformation for all the sulfate 8-membered rings (Fig. 6). Interestingly, the free OH groups of 2, O18 and O19 (see ORTEP in the ESI†),13 acting as intramolecular H-bond acceptors from adjacent OH groups (Fig. 6b, O17 and O20 respectively, with an O⋯O distance of 2.7 Å), play the role of intermolecular H-bond donor groups toward acetonitrile solvent molecules (Fig. 6b, O⋯N distance of 2.7 Å). In the solid state the two three-membered sub-cavities of 2 are occupied by two acetonitrile molecules (Fig. 6a and d) in which the methyl groups are involved in C–H⋯π interactions with the aromatic rings of 2 (average distance C⋯πcentroid of 3.55 Å). In addition, weak C–H⋯OS hydrogen bonding17 interactions were detected between methyl groups of the acetonitrile guests inside the cavities and SO4 bridging groups of 2, with an average C–H⋯OS distance of 3.32 Å and an average C–H⋯OS angle of 147.5 Å.17
Surprisingly, the tetrasulfate-bridged 2 was the only isolated product even when 1 was reacted under more drastic conditions (20 equiv. of (Im)2SO2 for 120 h). In a similar way, a further treatment of 2 with (Im)2SO2 only gave an unreacted material. Probably, the introduction of further SO4-bridging elements between the free OH groups of 2 could be disfavoured by a consequent distortion of the pinched structure of the resorcin[6]arene macrocycle. In fact, a close inspection of the X-ray structure of 2‡ (Fig. 6) reveals a O⋯O distance between the free OH groups of 2.71 Å, significantly higher than that of SO4-bridged O-atoms (2.48 Å).
The presence of sulfate bridges should make the resorcin[6]arene cavitand 2 a host able to bind suitable guests by means of H-bonding and/or dipole–dipole interactions. Therefore, we undertook complexation studies by means of 1H NMR titrations18 with cationic guests (Fig. 3) such as 1,4-DiammoniumButane (1,4-DB2+), 1,8-DiammoniumOctane (1,8-DO2+), 1,10-DiammoniumDecane (1,10-DD2+), and DiMethylViologen (DMV2+). In order to minimize the ion-pairing effects, we decided to use the weakly-coordinating Tetrakis[3,5-bis(triFluoromethyl)Phenyl]Borate TFPB− (Fig. 3) as the counter-anion.19 In fact, in the last years, we have shown that the TFPB anion is able to induce the recognition of ammonium guests by scarcely preorganized calixarene hosts.20
1H NMR titrations were carried out in CD3CN, where the regular pinched 2b was the most abundant (98/2 ratio 2b/2a, see Fig. 5), by increasing the [guest]/[host] ratio while keeping constant the host concentration at about 0.001 mol L−1.18 In all cases, the host signals were observed as averaged single resonances because of the fast exchange on the NMR timescale between the free and bound states. The addition of TFPB salt of 1,8-diammoniumoctane 1,8-DO2+ to a solution of host 2 caused a downfield shift13 of the ArCH2Ar NMR signals of 2. The Job plot for 2 and 1,8-DO2+ showed a maximum at a mole fraction of 0.5, thus indicating a 1:1 binding stoichiometry. The titration data13 were analyzed by a nonlinear regression analysis18 and a value of 600 M−1 was calculated for the formation of a 1,8-DO2+ ⊂ 2 complex. Molecular mechanics calculations suggest that in the 1,8-DO2+ ⊂ 2 complex (Fig. 7, top) the bis-ammonium guests are linked to 2via a two-point double-hydrogen bonding between NH3+ and SO4 groups, while the host assumes a regular pinched cone conformation. When 1,10-DD2+·2TFPB− was added to a CD3CN solution of 2 no appreciable interaction has been detected by NMR spectroscopy, and analogous results were observed during the complexation experiments between 2 and 1,4-DB2+. Probably, in this case the Cn-chains (n = 10 and 4) are respectively too long and too short to allow the two-point double-hydrogen bonding between terminal ammonium groups and sulfate bridges as proposed for the 1,8-DO2+ ⊂ 2 complex (Fig. 7). 1H NMR spectroscopy and high-resolution MS evidenced13 that tetrasulfate cavitand 2 is able to interact with the DMV2+ guest (Fig. 8) with a 1:1 stoichiometry.13 A value of 1200 M−1 was found by 1H NMR titration for the association constant of the DMV2+ ⊂ 2 complex. A close inspection of the energy-minimized (OPLS force field) structure of the DMV2+ ⊂ 2 complex (Fig. 8, bottom) suggests that the positive N-atoms of the guest are in close contact with SO4 bridges of 2 (average distance N+⋯OS of 3.5 Å) to establish ion–dipole interactions. The formation of the DMV2+ ⊂ 2 complex was unequivocally confirmed by the base peak at 625.1068 m/z (calcd 625.1070) in the MALDI-FT-ICR mass spectrum (Fig. 8) in accordance with the molecular formula of the complex.
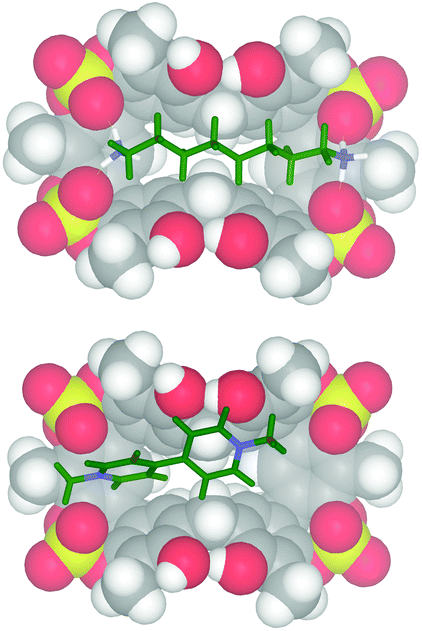 | ||
| Fig. 7 Minimized structures (molecular mechanics calculations, OPLS force field) of: (bottom) DMV2+ ⊂ 2; (top) 1,8-DO2+ ⊂ 2. | ||
 | ||
| Fig. 8 Significant portion of the HR MALDI FT-ICR mass spectrum of the DMV2+ ⊂ 2 complex (calcd 625.1070), showing the isotopic envelop with a Δ spacing of 0.5 m/z. | ||
In fact, the Δ spacing of 0.5 m/z between the peaks in the isotopic envelop (Fig. 8) solely accounts for a doubly charged species of 1,8-DO2+ ⊂ 2, excluding the supramolecular structure with higher charges.
Conclusions
In conclusion, we have reported here the first example of a resorcin[6]arene cavitand in which sulfate bridges were introduced between the OH groups. Such bridging elements have the double role of structural scaffolds and of functional supramolecular interacting groups. Consequently, the obtained tetrasulfate cavitand is able to interact in solution with the 1,8-diammoniumoctane cation via a two-point double-hydrogen bonding between the ammonium groups and the sulfate bridges. The resorcin[6]arene cavitand is also able to complex the dimethylviologen cation by means of ion–dipole interactions. The resorcin[6]arene cavitand described here can be considered one the first examples of supramolecular application of larger resorcinarene macrocycles which could show further intriguing supramolecular properties.Notes and references
- K. Rissanen, Angew. Chem., Int. Ed., 2005, 44, 3652 CrossRef CAS PubMed.
- D. J. Cram, Science, 1983, 38, 456 Search PubMed; D. J. Cram and J. M. Cram, in Container Molecules and Their Guests, Monographs in Supramolecular Chemistry, ed. J. F. Stoddart, Royal Society of Chemistry, Cambridge, 1994, ch. 5 CrossRef CAS; L. M. Tunstad, J. A. Tucker, E. Dalcanale, J. Weiser, J. A. Bryant, J. C. Sherman, R. C. Helgeson, C. B. Knobler and D. J. Cram, J. Org. Chem., 1989, 54, 1305 CrossRef CAS.
- I. Pochorovski and F. Driedrich, Acc. Chem. Res., 2014, 47, 2096 CrossRef CAS PubMed; S. M. Biros and J. Rebek Jr., Chem. Soc. Rev., 2007, 36, 93 RSC; S. Mosca, D. Ajami and J. Rebek Jr., Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11181 CrossRef PubMed; N. K. Beyeh, M. Cetina and K. Rissanen, Chem. Commun., 2014, 50, 1959 RSC; R. Pinalli, G. Brancatelli, A. Pedrini, D. Menozzi, D. Hernandez, P. Ballester, S. Geremia and E. Dalcanale, J. Am. Chem. Soc., 2016, 138, 8569 CrossRef PubMed; N. K. Beyeh, M. Cetina and K. Rissanen, Chem. Commun., 2014, 50, 1959 RSC; E. Busseron, J. Lux, M. Degardin and J. Rebek Jr., Chem. Commun., 2013, 49, 4842–4844 RSC; O. Perraud, V. Robert, A. Martinez and J.-P. Dutasta, Chem. – Eur. J., 2011, 17, 13405 CrossRef PubMed.
- R. Pinalli and E. Dalcanale, Acc. Chem. Res., 2013, 46, 399 CrossRef CAS PubMed; M. R. Ams, D. Ajami, S. L. Craig, J.-S. Yang and J. Rebek Jr., J. Am. Chem. Soc., 2009, 131, 13190 CrossRef PubMed; I. Alessandri, E. Biavardi, A. Gianoncelli, P. Bergese and E. Dalcanale, ACS Appl. Mater. Interfaces, 2016, 22, 14944 Search PubMed; J. Ampurdanes, G. A. Crespo, A. Maroto, M. A. Sarmentero, P. Ballester and F. X. Rius, Biosens. Bioelectron., 2009, 25, 344 CrossRef PubMed; S. Neri, R. Pinalli, E. Dalcanale and L. J. Prins, ChemNanoMat, 2016, 2, 489 CrossRef; D. Masseroni, E. Biavardi, D. Genovese, E. Rampazzo, L. Prodi and E. Dalcanale, Chem. Commun., 2015, 51, 12799 RSC.
- A. Galan and P. Ballester, Chem. Soc. Rev., 2016, 45, 1720 RSC; L. Marchetti and M. Mevine, ACS Catal., 2011, 1, 362 Search PubMed; A. Mirabaud, J.-C. Mulatier, A. Martinez, J.-P. Dutasta and V. Dufaud, Catal. Today, 2016 DOI:10.1016/j.cattod.2016.03.050; R. J. Hooley, J. Richard and J. Rebek Jr., Chem. Biol., 2009, 16, 255 CrossRef CAS PubMed; D. Masseroni, S. Mosca, M. P. Mower, D. G. Blackmond and J. Rebek Jr., Angew. Chem., Int. Ed., 2016, 55, 8290 CrossRef PubMed; W. Nai-Wei and J. Rebek Jr., J. Am. Chem. Soc., 2016, 138, 7512 CrossRef PubMed; A. Mirabaud, J.-C. Mulatier, A. Martinez, J.-P. Dutasta and V. Dufaud, ACS Catal., 2015, 5, 6748 CrossRef; J. Hong, K. E. Djernes, I. Lee, R. J. Hooley and F. Zaera, ACS Catal., 2013, 3, 2154 CrossRef.
- P. Delangle and J.-P. Dutasta, Tetrahedron Lett., 1995, 36, 9325 CrossRef CAS.
- C. Tudisco, P. Betti, A. Motta, R. Pinalli, L. Bombaci, E. Dalcanale and G. G. Condorelli, Langumir, 2012, 28, 1782 CrossRef CAS PubMed.
- M. Dionisio, G. Oliviero, D. Menozzi, S. Federici, R. M. Yebeutchou, F. P. Schmidtchen, E. Dalcanale and P. Borgese, J. Am. Chem. Soc., 2012, 134, 2392 CrossRef CAS PubMed.
- P. Della Sala, C. Gaeta, W. Navarra, C. Talotta, M. De Rosa, G. Brancatelli, S. Geremia and P. Neri, J. Org. Chem., 2016, 81, 5726 CrossRef CAS PubMed.
- G. Brancatelli, S. Geremia, C. Gaeta, P. Della Sala, C. Talotta, M. De Rosa and P. Neri, CrystEngComm, 2016, 18, 5045 RSC.
- For an example of a resorcin[6]arene-based cavitand in which methylene bridges were introduced between the OH groups, see: C. Naumann, E. Román, C. Peinador, T. Ren, B. O. Patrick, A. E. Kaifer and J. C. Sherman, Chem. – Eur. J., 2001, 7, 1637 CrossRef CAS.
- B.-T. Guan, X.-Y. Lu, Y. Zheng, D.-G. Yu, T. Wu, K.-L. Li, B.-J. Li and Z.-J. Shi, Org. Lett., 2010, 12, 396 CrossRef CAS PubMed.
- See ESI† for further details.
- J. Wöhnert, J. Brenn, M. Stoldt, O. Aleksiuk, F. Grynszpan, I. Thondorf and S. E. Biali, J. Org. Chem., 1998, 63, 3866 CrossRef.
- F. Troisi, C. Gaeta and P. Neri, Tetrahedron Lett., 2005, 46, 8041 CrossRef CAS.
- R. J. Kurland, M. B. Rubin and M. B. Wise, J. Chem. Phys., 1964, 40, 2426 CrossRef CAS.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- K. Hirose, in Analytical Methods in Supramolecular Chemistry, ed. C. A. Schalley, Wiley-VCH, Weinheim, 2007, pp. 17–54 Search PubMed.
- S. H. Strauss, Chem. Rev., 1993, 93, 927 CrossRef CAS.
- C. Talotta, C. Gaeta and P. Neri, Org. Lett., 2012, 14, 3104 CrossRef CAS PubMed; C. Gaeta, C. Talotta, F. Farina, M. Camalli, G. Campi and P. Neri, Chem. – Eur. J., 2012, 18, 1219 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1485235. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00336b |
| ‡ Diffraction data were collected at the XRD1 beam-line of the Elettra synchrotron (Trieste, Italy). Crystallographic information. CCDC 1485235.13 Crystal data for 2: monoclinic, P21/n, Z = 4, a = 13.909(4) Å, b = 20.264(2) Å, c = 22.762(2) Å, α = 90, β = 91.41(3)°, V = 6414(2) Å3, T = 100 K, Dx = 1.173 g cm−3, μcalc = 0.217 mm−1. |
| This journal is © the Partner Organisations 2016 |