
Ruthenium-catalysed hydrosilylation of carbon–carbon multiple bonds†
M.
Zaranek
 a,
B.
Marciniec
b and
P.
Pawluć
*a
a,
B.
Marciniec
b and
P.
Pawluć
*a
aFaculty of Chemistry, Adam Mickiewicz University in Poznań, Umultowska 89 B, 61-614 Poznań, Poland. E-mail: piotr.pawluc@amu.edu.pl
bCentre for Advanced Technologies, Adam Mickiewicz University in Poznań, Umultowska 81 C, 61-614 Poznań, Poland
First published on 9th August 2016
Abstract
Ruthenium-based complexes are generally considered to be efficient catalysts due to their high activity and electron transfer features. Although there are a limited number of highly selective olefin hydrosilylation protocols with ruthenium catalysts, recently a wide variety of the ruthenium complexes has been reported as unique catalyst precursors for the regiocontrolled hydrosilylation of alkynes. The ruthenium-catalysed hydrosilylation of alkynes represents one of the most efficient and straightforward methods for the synthesis of stereodefined vinylsilanes, which are particularly attractive scaffolds for further transformations including palladium-catalysed cross-coupling with organic halides or desilylative oxidation. The article highlights recent developments and covers the literature from the last two decades with respect to the ruthenium-catalysed hydrosilylation of alkenes and alkynes with particular emphasis on its application in organic synthesis.
 M. Zaranek | Maciej Zaranek is currently a Ph.D. student at the Faculty of Chemistry, Adam Mickiewicz University in Poznań, Poland. His research interests focus mostly on mechanisms in organometallic catalysis and optimisation of non-standard catalytic systems. |
 B. Marciniec | Bogdan Marciniec is a professor at the Faculty of Chemistry, Adam Mickiewicz University in Poznań (AMU), Poland, and the Director of Center for Advanced Technologies AMU, a member of the Polish Academy of Sciences (1994) and European Academy of Arts, Sciences and Humanities (2010). His research is focused on the synthesis of organosilicon (also boron and germanium) compounds on the basis of new reactions and new catalysts of known reactions. He has published over 400 scientific papers, including 24 book chapters, editor and co-author of 15 books, inter alia “Comprehensive Handbook on Hydrosilylation” (Pergamon Press, 1993) and “Hydrosilylation – A Comprehensive Review on Recent Advances” (Springer, 2009), 200 patents and over 17 technologies implemented in industry. |
 P. Pawluć | Piotr Pawluć received a Ph.D. (2004) and habilitation (2012) in chemistry from Adam Mickiewicz University in Poznań (Poland). He was a postdoctoral fellow under Professor Andre Mortreux at the Lille University (France). His research interests involve developing catalytic methodologies for the preparation of organosilicon building blocks and their application to organic synthesis. He is a co-author of the monograph: “Hydrosilylation – A Comprehensive Review on Recent Advances” (Springer 2009). More than 50 research publications, patents, and book chapters document his activity in the fields of organometallic chemistry, homogeneous catalysis and organic synthesis. |
Introduction
Hydrosilylation of alkenes and alkynes is the most fundamental and elegant method for laboratory and industrial synthesis of organosilicon compounds, which can be applied in organic synthesis, polymer chemistry and materials science. General reviews or books published in the last decade1–4 have provided general information on carbon–carbon multiple bonds hydrosilylation but the literature does not offer a more detailed review of the recent achievements in the field of ruthenium-catalysed reactions. The lack of a pertinent overview in this field, along with the recent reviews on catalysis by other metals,5–8 has prompted us to present the advances of ruthenium complex catalysed hydrosilylation reactions. The paper focuses on the most attractive results of the ruthenium-catalysed hydrosilylation of alkenes and alkynes published mostly in the last two decades with particular emphasis on its application in organic synthesis.Ruthenium catalysis in hydrosilylation of alkenes
Apart from silylene complexes, all other “classic” ruthenium complexes, especially the carbonyl ones, are well-known for their lack of selectivity, yielding vinylsilanes as by-products of competitive dehydrogenative silylation which in extreme situations can outnumber the product of desired hydrosilylation.9,10 However, the simplest [Ru3(CO)12] proved to be effective in hydrosilylation of 1-octene11 with triethoxysilane and, what is practically far more significant, allyl chloride with trimethoxysilane (Scheme 1).12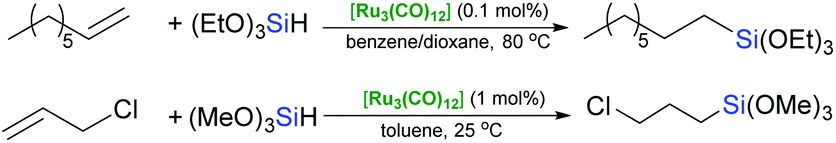 | ||
| Scheme 1 Hydrosilylation of olefins catalysed by Ru3(CO)12. | ||
So far, the two examples mentioned above have been the only ones of selective hydrosilylation in the presence of [Ru3(CO)12] which is an active catalyst for dehydrogenative silylation whenever a vinyl group involved is of an electron-deficient character. This was the case of styrene and its p-substituted derivatives (Scheme 2),13,14 pentafluorostyrene, and 3,3,3-trifluoropropene,15 undergoing reactions with trialkyl-, dialkylphenyl-, as well as triethoxysilane. A more complex compound, [Ru(CO)3(PPh3)2], was found to be effective in direct hydrosilylation of allylamine, giving (3-aminopropyl)triethoxysilane – one of the most widely used silane coupling agent.16
 | ||
| Scheme 2 Exemplary dehydrogenative silylation with triethylsilane. | ||
If an olefin has a hydrogen atom in the allylic position, dehydrogenative silylation is often accompanied by isomerisation giving a mixture of vinyl- and allylsilanes.9,14 The source of this behaviour can be traced back to the mechanism of alkene hydrosilylation by “classic” ruthenium complexes. It has been proposed that ruthenium catalysts act according to a novel mechanism involving σ-bond metathesis (SBM; Scheme 3).17,18
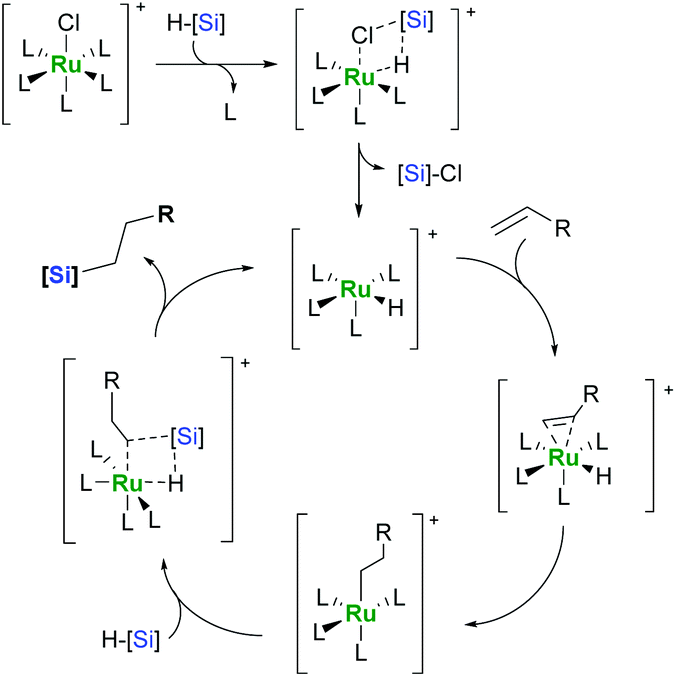 | ||
| Scheme 3 σ-Bond metathesis mechanism. | ||
This theory has been thoroughly investigated using the DFT method, however, simple carbonyl and phosphine complexes seem unable to undergo the proposed activation pathway due to the lack of ligands possible to eliminate along with a silyl group, which suggests rather a (modified) Chalk–Harrod mechanism proven true for the electron-rich complexes of platinum group metals (Scheme 4).19,20 It explains the occurrence of both dehydrogenative silylation and isomerisation, as in the latter case, the olefin can form a π-allyl complex.
 | ||
| Scheme 4 Chalk–Harrod (CH) and modified Chalk–Harrod (mCH) mechanisms of olefin hydrosilylation along with dehydrogenative silylation (DS) and isomerisation pathways. | ||
In turn, the SBM mechanism appears applicable also to hydrosilylation of polyfluoroolefins with dimethoxy-methylsilane in the presence of [RuCl2{P(4-CF3C6H4)3}2] in supercritical carbon dioxide.21 On the other hand, [Ru(H)2(H2)2(PCy3)2] was found to be a good catalyst for dehydrogenative silylation of ethylene to vinylsilane.9
It is worth mentioning that a very simple system comprising ruthenium(III) chloride hydrate and iodine or copper(I) halide as a promoter is active in selective hydrosilylation of ethylene with triethoxysilane.22
Hydrosilylation catalysed by a pentamethylcyclopentadienyl phenylsilylene complex of ruthenium follows the mechanism proposed by Glaser and Tilley that is essentially unique for this system (Scheme 5).23,24
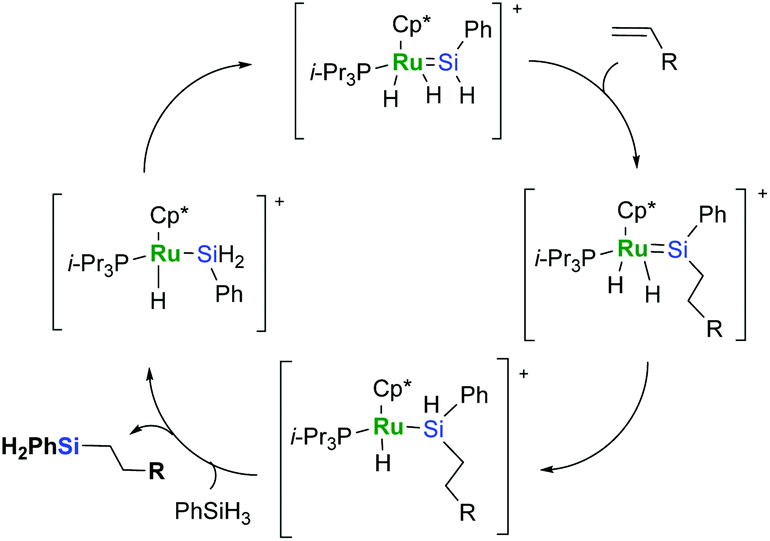 | ||
| Scheme 5 Glaser–Tilley mechanism of hydrosilylation with phenylsilane. | ||
It is very distinctive that in the Glaser–Tilley mechanism an insertion into the Si–H bond is strongly favoured over an insertion into Ru–H, and this is the source of the selectivity. Being apparently the most reliable and selective from among ruthenium-based catalytic systems, the use of this kind of catalyst is, however, considerably limited since it requires primary silanes to recover a silylene species and complete the catalytic cycle.
Known mostly as olefin metathesis catalysts, Grubbs complexes are active catalysts also for α-hydrosilylation of alkynes (vide infra). Nevertheless, it is not the limit of their capabilities in this field as they have been reported to catalyse ring-closing intramolecular α-hydrosilylation of alkenyl silyl ethers (Scheme 6).25 The 1st generation Hoveyda–Grubbs catalyst appeared to be the most active of the examined ones, apparently not leading to any product of metathesis. In-depth mechanistic experiments and calculations have been performed, allowing Jeon et al. to propose a comprehensive mechanism of this transformation in which a key step is the σ-bond metathesis leading to the formation of a silyl alkylidene complex.25
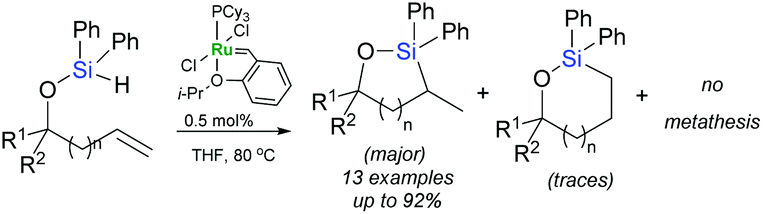 | ||
| Scheme 6 Intramolecular hydrosilylation using Hoveyda–Grubbs catalyst. | ||
Hydrosilylation of alkynes
Hydrosilylation of alkynes represents the most straightforward and atom economical approach toward vinylsilanes.26,27 The hydrosilylation of terminal and internal alkynes implies a synthetic challenge because of the potential product distribution. Nonselective hydrosilylation of terminal alkynes yields a mixture of three isomeric vinylsilanes: β-(E), β-(Z) and α (Scheme 7), whereas the hydrosilylation of internal alkynes would potentially give four isomeric products.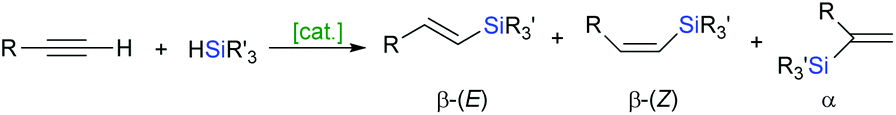 | ||
| Scheme 7 Hydrosilylation of terminal alkynes. | ||
As far as the terminal alkyne derivatives are concerned, both β-(E)- and β-(Z)-vinylsilanes are obtained via the addition of a hydrosilane across the carbon–carbon triple bond, where the silicon atom is attached to the terminal carbon. The addition of a hydrosilane to the same face of the triple bond (cis-addition) results in the formation of an (E)-product, while the trans-addition provides the respective (Z)-vinylsilane. The reversed addition of a hydrosilane to the terminal alkyne gives a geminal vinylsilane α as an internal adduct. The regio- and stereochemical outcome of the ruthenium-catalysed hydrosilylation depends on several factors including the coordination sphere of ruthenium (ligands), the substituents on both alkyne and hydrosilane used and the reaction parameters such as solvent, temperature or even on the order of addition of the reagents. The hydrosilylation of terminal alkynes catalysed by ruthenium complexes proceeds predominantly in a trans-fashion giving thermodynamically unfavourable (Z)-vinylsilanes as the major products.
To explain the predominant appearance of an unusual trans-addition product (Z-vinylsilane) which does not fit right in the Chalk–Harrod mechanism, Crabtree28 and Ojima29 proposed an alternative mechanism based on silylmetallation in the migratory insertion step followed by E/Z isomerisation (Scheme 8). The unique feature of this mechanism (originally proposed for Rh and Ir catalysis) is the exclusive insertion of an alkyne into the M–Si bond, forming a (Z)-silylvinylidene complex. Because of the steric repulsion between the silyl group and the substituted metal atom, the (Z)-silylvinylidene intermediate isomerises to a thermodynamically favourable (E)-silylvinylidene complex through either zwitterionic carbene species29 or a metallacyclopropene intermediate.28 Since the reductive elimination is the rate-determining step, (Z)-vinylsilanes are formed as the kinetic products.
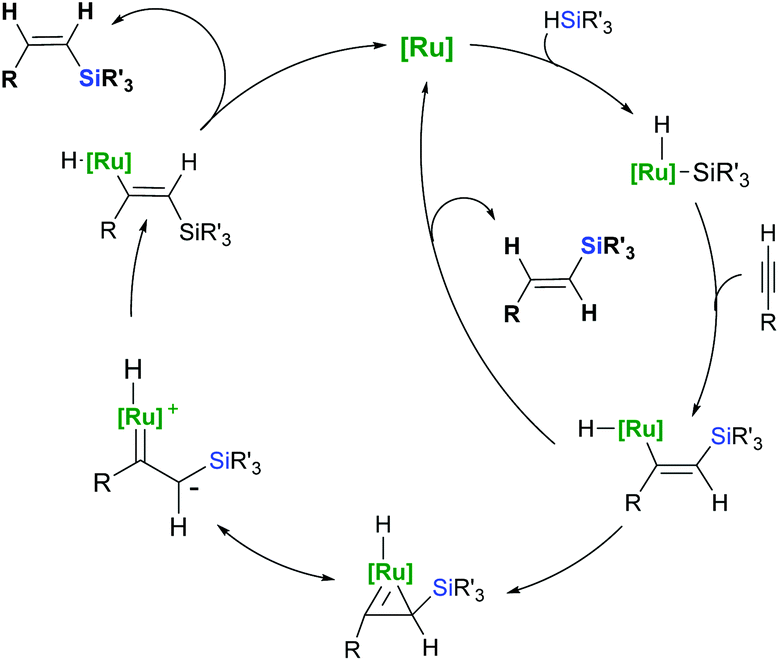 | ||
| Scheme 8 The Crabtree–Ojima mechanism for Z-selective alkyne hydrosilylation. | ||
Ozawa et al. have reported that hydrosilylation of alkynes with hydrosilanes containing aryl groups in the presence of the ruthenium-hydride catalyst [RuHCl(CO)(PPh3)3] led to the formation of the respective (E)-vinylsilane in over 99% selectivities. On the other hand, the same substrates can be effectively converted into isomeric (Z)-vinylsilanes with up to 99% selectivities using a ruthenium–silyl complex: [RuCl(SiMe2Ph)(CO)(Pi-Pr3)2].30,31
Extension of these protocols to 1,4-diethynylarenes led to stereoselectively isomeric 1,4-bis(β-silylethenyl)arenes – precursors for poly(arylenevinylene)s.32 The same dependency of the selectivity on the phosphine ligands used has been also demonstrated by Oro and co-workers in a set of their reports.33–36 Moreover, [RuHCl(CO)(Pi-Pr3)2] has been found to be a highly active catalyst for (Z)-selective hydrosilylation of arylalkynes,37 and [RuHCl(CO)(PCy3)2]-catalysed hydrosilylation of terminal alkynes led to formation of (Z)-vinylsilanes without yielding any significant amounts of trans- and gem-products when sterically undemanding alkynes are employed, in contrast to (E)-vinylsilanes that resulted from terminal alkynes containing bulky substituents (Scheme 9).38
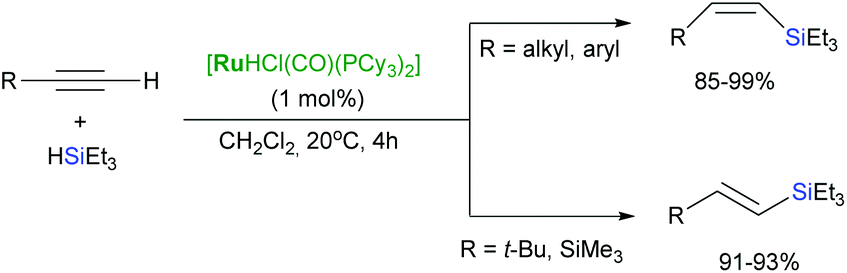 | ||
| Scheme 9 Hydrosilylation of terminal alkynes in the presence of [RuHCl(CO)(PCy3)2]. | ||
The [RuCl2(PPh)3]-catalysed hydrosilylation of p-substituted phenylacetylenes with HSiMeCl2 led to formation of the corresponding β-(Z)-silylstyrenes.39 [{Ru(p-cymene)Cl2}2] has been also demonstrated to be an active catalyst for the highly Z-selective hydrosilylation of a wide range of functionalised alkynes, except those having a hydroxyl group at the β-position to the triple bond (Scheme 10).40 In such a case, the regioisomeric α-vinylsilanes were generated with excellent selectivity.40
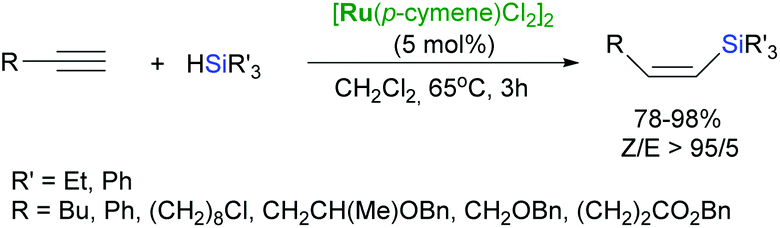 | ||
| Scheme 10 Z-Selective hydrosilylation of terminal alkynes. | ||
Grubbs’ 1st generation catalyst [Cl2(PCy3)2Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh] efficiently catalyses the hydrosilylation of terminal alkynes.41,42 The product selectivity depends on both alkyne and silane employed. The hydrosilylation of alkyl- and arylalkynes by trialkylsilanes led to the (Z)-stereoisomers, whereas the reaction of propargyl alcohols with alkyl- and alkoxy-substituted silanes predominantly gave the α-vinylsilanes.41,42 The conditions for α-selective hydrosilylation of alkynes without directing groups mediated by Grubbs’ 1st generation catalyst have also been established by Cossy and co-workers.43
CHPh] efficiently catalyses the hydrosilylation of terminal alkynes.41,42 The product selectivity depends on both alkyne and silane employed. The hydrosilylation of alkyl- and arylalkynes by trialkylsilanes led to the (Z)-stereoisomers, whereas the reaction of propargyl alcohols with alkyl- and alkoxy-substituted silanes predominantly gave the α-vinylsilanes.41,42 The conditions for α-selective hydrosilylation of alkynes without directing groups mediated by Grubbs’ 1st generation catalyst have also been established by Cossy and co-workers.43
Trost et al. have developed a mild and flexible protocol for hydrosilylation of terminal alkynes (trans-addition) in the presence of a cationic cyclopentadienyl ruthenium(II) complex: [Cp*Ru(MeCN)3]PF6 to give α-vinylsilanes in excellent yields (Scheme 11).44,45 The reaction is tolerant to a wide range of functional groups, including halogens, unprotected alcohols, esters, amines etc.
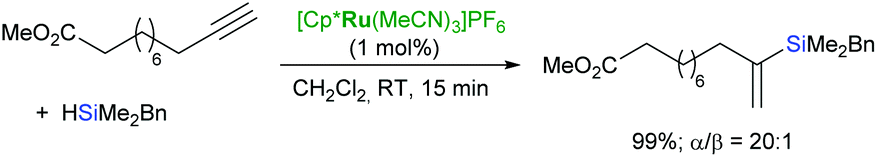 | ||
| Scheme 11 α-Selective hydrosilylation of terminal alkynes. | ||
Hydrosilylation of internal alkynes in the presence of [Cp*Ru(MeCN)3]PF6 allows the regioselective formation of trisubstituted (Z)-vinylsilanes.45–48 Hydrosilylation of 2-alkynes results in the formation of (Z)-alkenes with the silyl substituent occupying the less sterically-demanding position, whereas, for the other internal alkynes, the silyl group substitutes the more sterically-demanding position in the (Z)-alkene. During the addition of hydrosilanes to propargylic alcohols, alkynyl ketones and esters, the silyl group selectively occupies the distal position to the hydroxyl functionality of the (Z)-alkene, however, the hydrosilylation of unprotected propargylic alcohols by alkoxysilanes led to formation of cyclic alkenylsilanes.
Fürstner et al. have reported that the use of ruthenium-chloride precursors: [Cp*RuCl]4 or [Cp*RuCl(cod)] as catalysts leads to increased selectivity of the trans-hydrosilylation of unsymmetrical internal alkynes bearing protic functional groups e.g. alcohols and carboxylic acids.49 Ruthenium complexes [Cp*Ru(MeCN)3]PF6 and [CpRu(MeCN)3]PF6 have been also applied in the hydrosilylation of silylalkynes to form isomeric bis(silyl)alkenes with the opposite regioselectivity (Scheme 12).50
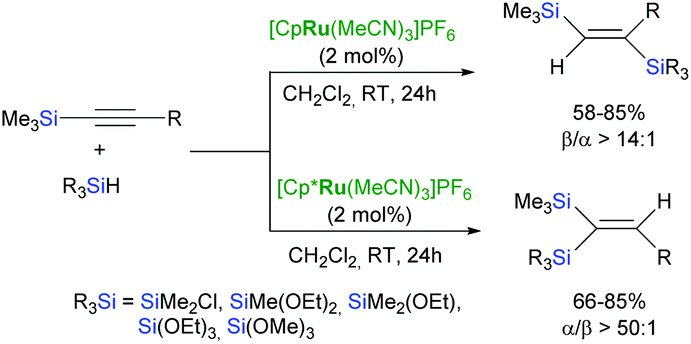 | ||
| Scheme 12 Regioselective hydrosilylation of silylalkynes. | ||
Hydrosilylation of thioalkynes with tris(trimethylsiloxy)-silane in the presence of the [Cp*Ru(MeCN)3]PF6 catalyst led to the formation of the corresponding α-addition products under mild conditions (Scheme 13).51
 | ||
| Scheme 13 α-Selective hydrosilylation of thioalkynes. | ||
The [Cp*Ru(MeCN)3]PF6 complex has been also successfully applied in the double trans-hydrosilylation of 1,4-diaryl-1,3-butadiynes by 9-silafluorene to afford 2,5-diarylsiloles.52 The rutheniumhydride complex [Cp*RuH3(PPh3)] has been shown to catalyse hydrosilylation of 1-alkynes by chlorosilanes to afford α-adducts in good yields.53
Intramolecular hydrosilylation of functionalised alkynes has gained increasing attention since it enables the synthesis of stereodefined silacyclic compounds, which can serve as platforms for subsequent oxidation or palladium-catalysed cross-coupling with organic halides.54–57 The challenge with intramolecular hydrosilylation lies in controlling both the selectivity of the reaction since it provides three different types of unsaturated silacyclic compounds (exo–cis, exo–trans and endo–trans) depending on the mode of cyclisation, and the addition of an Si–H bond. The course of intramolecular exo-hydrosilylation is dependent upon the catalyst used: platinum catalysts (e.g., H2PtCl6, (dvtms)Pt(t-Bu3P)) react with alkynes to give (E)-alkenylsilacycles through a cis-addition,58 whereas ruthenium-based catalysts usually promote trans-addition to afford isomeric (Z)-alkenyl silacyclic products.59 Intramolecular trans-hydrosilylation has been developed by Denmark et al. by the use of a ruthenium-arene complex: [{Ru(C6H6)Cl2}2] as a catalyst.59 First generation Grubbs’ catalyst has been also shown to act as an intramolecular trans-hydrosilylation catalyst since silylated homopropargylic alcohols are transformed into exo–cis products (Scheme 14).42
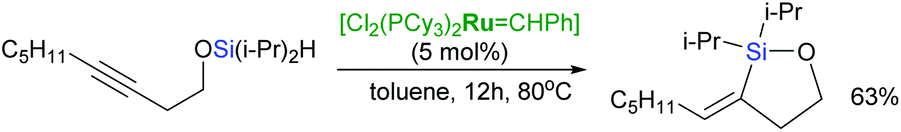 | ||
| Scheme 14 Intramolecular trans-hydrosilylation in the presence of Grubbs catalyst. | ||
Trost et al. have reported an unprecedented endo-dig trans-hydrosilylation in the presence of a ruthenium catalyst.60,61 The cationic ruthenium complex: [Cp*Ru(MeCN)3]PF6 induces intramolecular hydrosilylation of silylated homopropargylic and bishomopropargylic alcohols to yield oxasilacycles of unique stereochemistry under mild conditions (Scheme 15).61
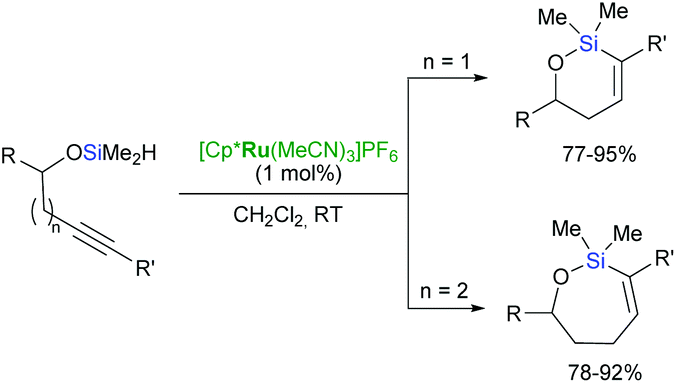 | ||
| Scheme 15 Intramolecular trans-hydrosilylation of homopropargylic alcohols. | ||
The intriguing stereoselectivity of such intramolecular endo-dig hydrosilylation have prompted a re-examination of the mechanistic pathways providing trans-hydrosilylation reactions, based on the concerted oxidative addition and alkyne insertion into a ruthenacyclopropene intermediate (rearrangement mechanism – Scheme 16).61
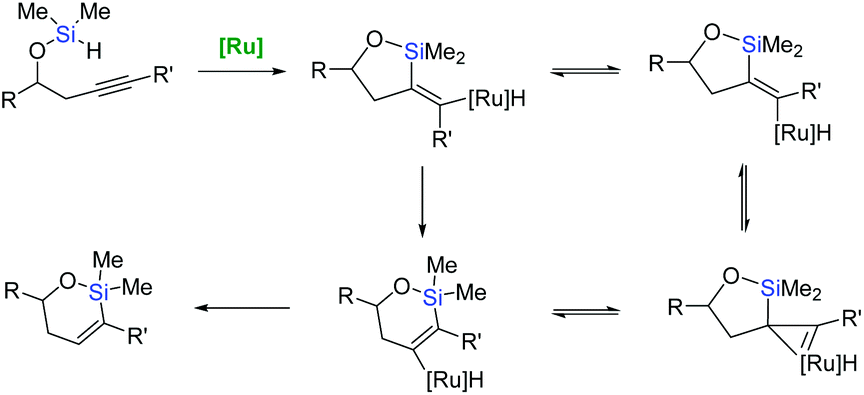 | ||
| Scheme 16 Intramolecular endo-dig hydrosilylation mechanism. | ||
Applications in organic synthesis
Growing interest in the development of sequential processes, including ruthenium-catalysed hydrosilylation as the initial step, stems from the ability to assemble complex molecules from simple starting materials through organosilicon intermediates in a convergent and atom-economical manner. The breakthrough in the application of ruthenium-catalysed hydrosilylation in organic synthesis was the discovery of the catalytic activity of a cationic ruthenium complex: [Cp*Ru(MeCN)3]PF6 by Trost and co-workers.44,45 Thanks to the compatibility of this reaction with the sequential desilylation reactions such as protodesilylation, desilylative oxidation or cross-coupling, this useful and universal process has been applied as a direct method for alkyne functionalisation.Sequential ruthenium catalysed hydrosilylation–oxidative desilylation of propargylic alcohols and their homologues has been widely used for the synthesis of hydroxy ketone derivatives (Scheme 17).45–47 The one-pot hydrosilylation/oxidation of propargyl alcohols gave the corresponding β-hydroxy ketones, and provided a regioselective introduction of a carbonyl group at the alkyne β-carbon to the alcohol.46,47 Followed by the treatment with TBAF and Tamao oxidation, diastereoselective epoxidation of (Z)-vinylsilanes provide additional complexity at the α-carbon resulting in production of the syn-diols with excellent selectivity.46,47
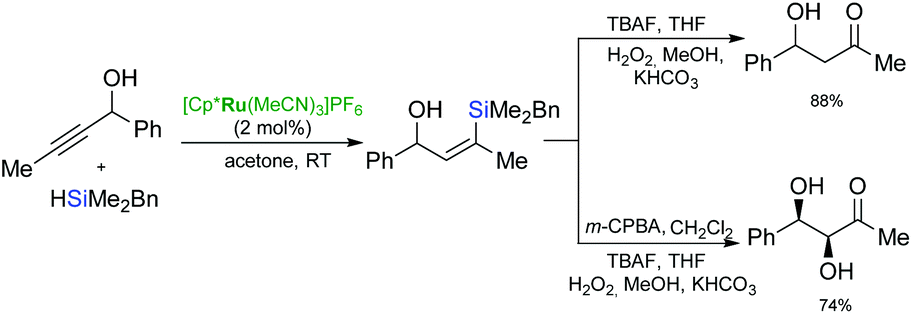 | ||
| Scheme 17 Synthesis of β-hydroxy ketones. | ||
endo-dig-Hydrosilylation of silylated homopropargyl alcohols catalysed by [Cp*Ru(MeCN)3]PF6 and the subsequent oxidation of the resulting silacycles, led to γ-hydroxy ketones (Scheme 18).37,61 The epoxidation/oxidation sequence of the endo-cyclic hydrosilylation intermediates can furnish the hydroxyl ketone functionality to give diastereomerically pure syn-α,γ-dihydroxy ketones.46 The intramolecular endo-dig hydrosilylation–epoxidation–oxidation pathway applied to (bis)homopropargyl alcohols afforded the α,δ-dihydroxy ketones.46
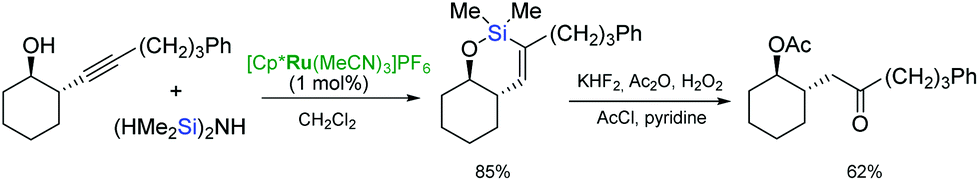 | ||
| Scheme 18 Synthesis of γ-hydroxy ketone derivatives. | ||
The regioselective [Cp*Ru(MeCN)3]PF6-catalysed trans-hydrosilylation of alkynyl-substituted ketones and esters followed by one-pot TBAF-promoted C–C bond formation and silane oxidation provides a strategy for the synthesis of β-carbonyl-substituted tertiary alcohols (Scheme 19).48
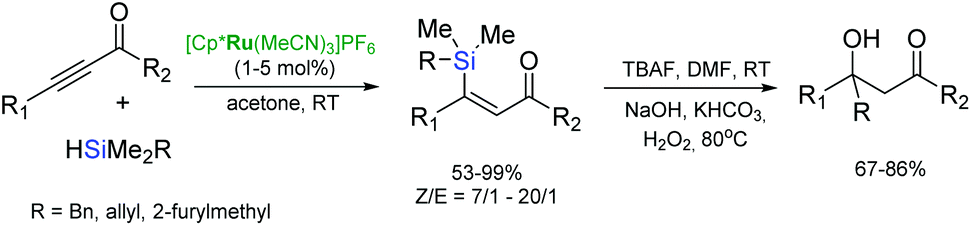 | ||
| Scheme 19 Synthesis of β-carbonyl-substituted tertiary alcohols. | ||
The ruthenium-catalysed trans-hydrosilylation/desilylative oxidation methodology exploiting the synthetic potential of stereodefined organosilicon intermediates has been applied as a key step in the total syntheses of natural products such as piperidine alkaloid – (+)-spectaline,46 mycobacterial lipids component – phthiocerol,62 or the C13–C29 fragment of marine macrocycle – amphidinolide N.63
The hydrosilylation/protodesilylation protocol is a useful method for stereoselective alkyne reduction to (E)-alkenes.64,65 Trost et al. have applied the one-pot combination of [Cp*Ru(MeCN)3]PF6-catalysed internal alkyne hydrosilylation and CuI-induced protodesilylation, which allows a net trans-alkyne reduction for a wide variety of functionalised alkynes including esters, ketones and propargyl alcohols.64 Fürstner et al. have reported a two-step protocol for the selective synthesis of macrocyclic (E)-cycloalkenes from cycloalkynes (obtained via ring-closing alkyne metathesis) using trans-selective hydrosilylation catalysed by [Cp*Ru(MeCN)3]PF6 and following AgF-mediated protodesilylation (Scheme 20).65,66
 | ||
| Scheme 20 Synthesis of macrocyclic (E)-cycloalkenes. | ||
The feasibility of this method was also demonstrated in the stereoselective synthesis of (E,E)-dienes.66,67 The hydrosilylation–protodesilylation methodology can also be successfully applied for stereoselective reduction of functionalised alkynyl ketones to give (E)-α,β-unsaturated carbonyl compounds.68
The Pd-catalysed cross-coupling of the α-vinylsilanes containing trialkoxy- or benzyldimethylsilyl groups with aryl or alkenyl iodides proceeds smoothly to afford geminal-substituted alkenes in a regioselective manner (Scheme 21).44,69
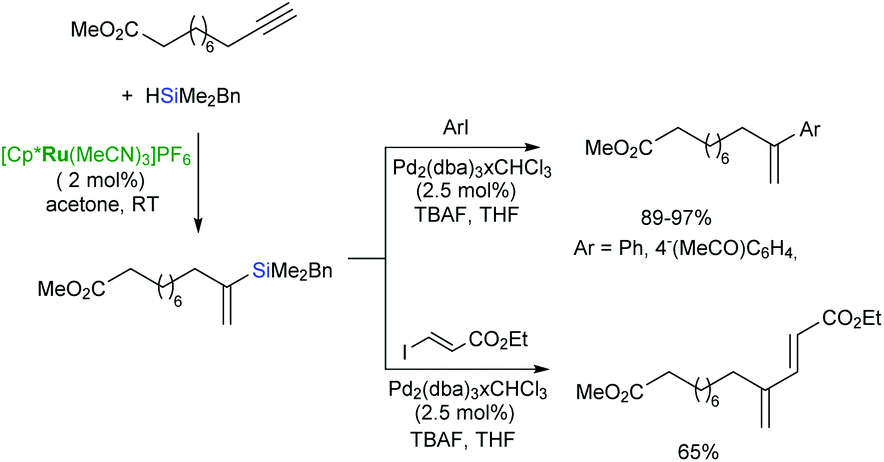 | ||
| Scheme 21 Sequential Ru-catalysed hydrosilylation/Pd-catalysed cross-coupling. | ||
The synthetic potential of the consecutive Ru-catalysed intramolecular hydrosilylation of (homo)propargyl alcohols and Pd-catalysed cross-coupling reactions has been proved in the stereoselective synthesis of a wide variety of (E)- and (Z)-homoallylic alcohols (Scheme 22).58,70
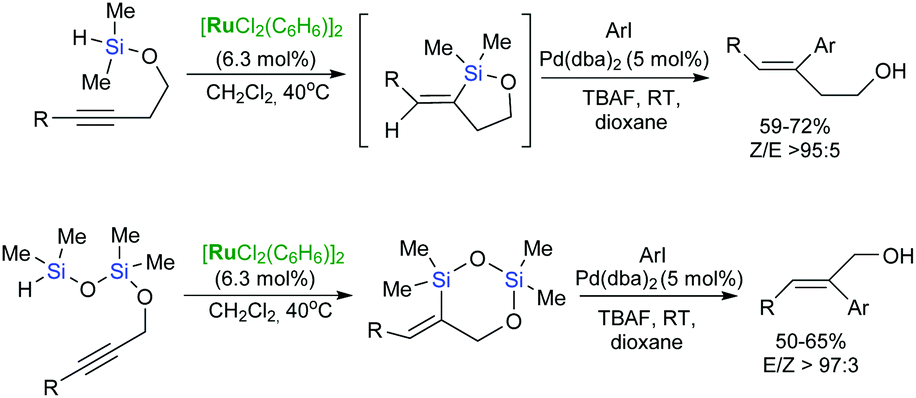 | ||
| Scheme 22 Synthesis of (E)- and (Z)-homoallylic alcohols. | ||
(Z)-Selective hydrosilylation of dialkynylarenes by HSiMe2Ph in the presence of a ruthenium complex bearing a diphosphinidenecyclobutene ligand, followed by NBS-mediated bromodesilylation of the hydrosilylation products afforded (Z,Z)-bis(2-silylethenyl)arenes (Scheme 23).71,72
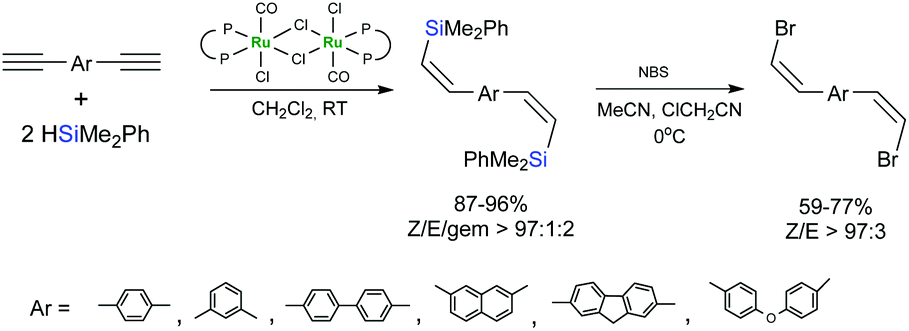 | ||
| Scheme 23 Hydrosilylation/bromodesilylation sequence. | ||
Conclusions
Ruthenium complexes represent probably the most diverse group from among all groups of catalysts for hydrosilylation of alkenes and, more importantly, alkynes. They act according to many mechanisms, which can cause problems with selectivity, however, the hydrosilylation of alkynes catalysed by ruthenium complexes proceeds predominantly in a trans-fashion. Thus, improvements in catalyst design have rendered the hydrosilylation of alkynes an efficient and mild method for the synthesis of unusual trans-addition products: (Z)-vinylsilanes and α-vinylsilanes. In particular, the [Cp*Ru(MeCN)3]PF6 complex has been shown to catalyse the hydrosilylation of a wide variety of alkynes to afford α-vinylsilane products with good regioselectivity. The reaction is tolerant to a wide range of functional groups, including halogens, ketones, alcohols, esters, amines etc. The relatively low and stable price of ruthenium is undeniably a great advantage over other noble metals and a factor determining continuous interest in the development of new catalytic systems. Ruthenium-based catalysts are now well-established in some sequential synthetic protocols, especially leading to unsaturated organic products, which was intended to be shown in this paper.Acknowledgements
Financial support from the National Science Centre (Poland); grant no. 2011/03/B/ST5/01034 is gratefully acknowledged.References
- Hydrosilylation: A Comprehensive Review on Recent Advances, ed. B. Marciniec, Springer, Berlin, 2009 Search PubMed.
- A. K. Roy, in Advances in Organometallic Chemistry, ed. R. West, A. F. Hill and M. J. Fink, Academic Press, 2007, vol. 55, pp. 1–59 Search PubMed.
- D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS.
- Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC.
- M. D. Greenhalgh, A. S. Jones and S. P. Thomas, ChemCatChem, 2015, 7, 190–222 CrossRef CAS.
- M. D. Greenhalgh, D. J. Frank and S. P. Thomas, Adv. Synth. Catal., 2014, 356, 584–590 CrossRef CAS.
- J. Sun and L. Deng, ACS Catal., 2016, 6, 290–300 CrossRef CAS.
- I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- B. Marciniec, Coord. Chem. Rev., 2005, 249, 2374–2390 CrossRef CAS.
- B. Marciniec and C. Pietraszuk, in Ruthenium Catalysts and Fine Chemistry, ed. C. Bruneau and P. H. Dixneuf, Springer, Berlin, Heidelberg, 2004, pp. 197–248 Search PubMed.
- H. S. Hilal, S. Khalaf and W. Jondi, J. Organomet. Chem., 1993, 452, 167–173 CrossRef CAS.
- M. Tanaka, T. Hayashi and Z.-Y. Mi, J. Mol. Catal., 1993, 81, 207–214 CrossRef CAS.
- A. N. Nesmeyanov, R. K. Freidlina, E. C. Chukovskaya, R. G. Petrova and A. B. Belyavsky, Tetrahedron, 1962, 17, 61–68 CrossRef CAS.
- Y. Seki, K. Takeshita, K. Kawamoto, S. Murai and N. Sonoda, J. Org. Chem., 1986, 51, 3890–3895 CrossRef CAS.
- I. Ojima, T. Fuchikami and M. Yatabe, J. Organomet. Chem., 1984, 260, 335–346 CrossRef CAS.
- K. Takatsuna, K. Shiozawa and Y. Okumura, US4927953(A), 1990 Search PubMed.
- T. Tuttle, D. Wang, W. Thiel, J. Köhler, M. Hofmann and J. Weis, Organometallics, 2006, 25, 4504–4513 CrossRef CAS.
- T. Tuttle, D. Wang, W. Thiel, J. Köhler, M. Hofmann and J. Weis, J. Organomet. Chem., 2007, 692, 2282–2290 CrossRef CAS.
- A. J. Chalk and J. F. Harrod, J. Am. Chem. Soc., 1965, 87, 16–21 CrossRef CAS.
- M. A. Schroeder and M. S. Wrighton, J. Organomet. Chem., 1977, 128, 345–358 CrossRef CAS.
- L.-N. He, J.-C. Choi and T. Sakakura, Tetrahedron Lett., 2001, 42, 2169–2171 CrossRef CAS.
- L. Liu, X. Li, H. Dong and C. Wu, J. Organomet. Chem., 2013, 745–746, 454–459 CrossRef CAS.
- P. B. Glaser and T. D. Tilley, J. Am. Chem. Soc., 2003, 125, 13640–13641 CrossRef CAS PubMed.
- M. A. Rankin, D. F. MacLean, G. Schatte, R. McDonald and M. Stradiotto, J. Am. Chem. Soc., 2007, 129, 15855–15864 CrossRef CAS PubMed.
- A. Bokka, Y. Hua, A. S. Berlin and J. Jeon, ACS Catal., 2015, 5, 3189–3195 CrossRef CAS.
- D. Lim and E. Anderson, Synthesis, 2012, 983–1010 CAS.
- B. M. Trost and Z. T. Ball, Synthesis, 2005, 853–887 CrossRef CAS.
- C.-H. Jun and R. H. Crabtree, J. Organomet. Chem., 1993, 447, 177–187 CrossRef CAS.
- I. Ojima, N. Clos, R. J. Donovan and P. Ingallina, Organometallics, 1990, 9, 3127–3133 CrossRef CAS.
- H. Katayama, K. Taniguchi, M. Kobayashi, T. Sagawa, T. Minami and F. Ozawa, J. Organomet. Chem., 2002, 645, 192–200 CrossRef CAS.
- Y. Maruyama, K. Yamamura, I. Nakayama, K. Yoshiuchi and F. Ozawa, J. Am. Chem. Soc., 1998, 120, 1421–1429 CrossRef CAS.
- H. Katayama, M. Nagao, R. Moriguchi and F. Ozawa, J. Organomet. Chem., 2003, 676, 49–54 CrossRef CAS.
- M. Martín, E. Sola, F. J. Lahoz and L. A. Oro, Organometallics, 2002, 21, 4027–4029 CrossRef.
- M. A. Esteruelas, J. Herrero and L. A. Oro, Organometallics, 1993, 12, 2377–2379 CrossRef CAS.
- M. A. Esteruelas, A. M. López, L. A. Oro and J. Tolosa, J. Mol. Catal. A: Chem., 1995, 96, 21–23 CrossRef CAS.
- M. A. Esteruelas, L. A. Oro and C. Valero, Organometallics, 1991, 10, 462–466 CrossRef CAS.
- Y. Nakao, H. Imanaka, J. Chen, A. Yada and T. Hiyama, J. Organomet. Chem., 2007, 692, 585–603 CrossRef CAS.
- R. Gao, D. R. Pahls, T. R. Cundari and C. S. Yi, Organometallics, 2014, 33, 6937–6944 CrossRef CAS.
- S. I. M. Paris and F. R. Lemke, Inorg. Chem. Commun., 2005, 8, 425–428 CrossRef CAS.
- Y. Na and S. Chang, Org. Lett., 2000, 2, 1887–1889 CrossRef CAS PubMed.
- C. S. Aricó and L. R. Cox, Org. Biomol. Chem., 2004, 2, 2558–2562 Search PubMed.
- S. V. Maifeld, M. N. Tran and D. Lee, Tetrahedron Lett., 2005, 46, 105–108 CrossRef CAS.
- C. Menozzi, P. I. Dalko and J. Cossy, J. Org. Chem., 2005, 70, 10717–10719 CrossRef CAS PubMed.
- B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2001, 123, 12726–12727 CrossRef CAS PubMed.
- B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2005, 127, 17644–17655 CrossRef CAS PubMed.
- B. M. Trost, Z. T. Ball and K. M. Laemmerhold, J. Am. Chem. Soc., 2005, 127, 10028–10038 CrossRef CAS PubMed.
- B. M. Trost, Z. T. Ball and T. Jöge, Angew. Chem., Int. Ed., 2003, 42, 3415–3418 CrossRef CAS PubMed.
- B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2004, 126, 13942–13944 CrossRef CAS PubMed.
- S. M. Rummelt, K. Radkowski, D.-A. Roşca and A. Fürstner, J. Am. Chem. Soc., 2015, 137, 5506–5519 CrossRef CAS PubMed.
- S. Ding, L.-J. Song, L. W. Chung, X. Zhang, J. Sun and Y.-D. Wu, J. Am. Chem. Soc., 2013, 135, 13835–13842 CrossRef CAS PubMed.
- S. Ding, L.-J. Song, Y. Wang, X. Zhang, L. W. Chung, Y.-D. Wu and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 5632–5635 CrossRef CAS PubMed.
- T. Matsuda, S. Kadowaki and M. Murakami, Chem. Commun., 2007, 2627–2629 RSC.
- Y. Kawanami, Y. Sonoda, T. Mori and K. Yamamoto, Org. Lett., 2002, 4, 2825–2827 CrossRef CAS.
- G. Varchi and I. Ojima, Curr. Org. Chem., 2006, 10, 1341–1362 CrossRef CAS.
- S. Bracegirdle and E. A. Anderson, Chem. Soc. Rev., 2010, 39, 4114–4129 RSC.
- K. Tamao, Proc. Jpn. Acad., Ser. B, 2008, 84, 123–133 CrossRef CAS PubMed.
- H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845–1866 RSC.
- S. E. Denmark and W. Pan, Org. Lett., 2001, 3, 61–64 CrossRef CAS PubMed.
- S. E. Denmark and W. Pan, Org. Lett., 2002, 4, 4163–4166 CrossRef CAS PubMed.
- L. W. Chung, Y.-D. Wu, B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2003, 125, 11578–11582 CrossRef CAS PubMed.
- B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2003, 125, 30–31 CrossRef CAS PubMed.
- E. Casas-Arce, B. ter Horst, B. L. Feringa and A. J. Minnaard, Chem. – Eur. J., 2008, 14, 4157–4159 CrossRef CAS PubMed.
- B. M. Trost and J. Rey, Org. Lett., 2012, 14, 5632–5635 CrossRef CAS PubMed.
- B. M. Trost, Z. T. Ball and T. Jöge, J. Am. Chem. Soc., 2002, 124, 7922–7923 CrossRef CAS PubMed.
- A. Fürstner and K. Radkowski, Chem. Commun., 2002, 2182–2183 RSC.
- F. Lacombe, K. Radkowski, G. Seidel and A. Fürstner, Tetrahedron, 2004, 60, 7315–7324 CrossRef CAS.
- A. Fürstner, M. Bonnekessel, J. T. Blank, K. Radkowski, G. Seidel, F. Lacombe, B. Gabor and R. Mynott, Chem. – Eur. J., 2007, 13, 8762–8783 CrossRef PubMed.
- B. M. Trost and M. L. Crawley, Chem. – Eur. J., 2004, 10, 2237–2252 CrossRef CAS PubMed.
- B. M. Trost, M. R. Machacek and Z. T. Ball, Org. Lett., 2003, 5, 1895–1898 CrossRef CAS PubMed.
- S. E. Denmark and W. Pan, Org. Lett., 2003, 5, 1119–1122 CrossRef CAS PubMed.
- M. Nagao, K. Asano, K. Umeda, H. Katayama and F. Ozawa, J. Org. Chem., 2005, 70, 10511–10514 CrossRef CAS PubMed.
- H. Katayama, M. Nagao, T. Nishimura, Y. Matsui, K. Umeda, K. Akamatsu, T. Tsuruoka, H. Nawafune and F. Ozawa, J. Am. Chem. Soc., 2005, 127, 4350–4353 CrossRef CAS PubMed.
Footnote |
| † On the occasion of 75th birthday of Professor Barry M. Trost. |
| This journal is © the Partner Organisations 2016 |