
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of oxindoles via reductive CO2 fixation†‡
Toru
Amaya
*a,
Izumi
Kurata
a and
Toshikazu
Hirao
*ab
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: amaya@chem.eng.osaka-u.ac.jp
bThe Institute of Scientific and Industrial Research, Osaka University, Mihoga-oka, Ibaraki, Osaka 567-0047, Japan. E-mail: hirao@chem.eng.osaka-u.ac.jp
First published on 29th June 2016
Abstract
The synthesis of 3-aryl-3-hydroxy-2-oxindoles, which are a structural motif found in various natural products and pharmaceutically active compounds, was conducted via reductive coupling of (2-aminophenyl)(aryl)methanone derivatives and CO2 as a key step. The conditions employing Mg with chlorotrimethylsilane in DMA are the best for the reductive coupling, where the aryl halide moiety is intact. This reaction proceeds well without the protection of the amino group. The reductive coupling and acid-catalyzed lactam formation can be performed in a one-pot reaction to give the oxindoles.
Introduction
Carbon dioxide (CO2) is considered to be one of the causative agents for global warming. Therefore, the effective utilization of CO2 collected from plant emissions is important in view of sustainable chemistry. Utilization of CO2 as a C1 synthon for organic synthesis has been developed in this context.1Oxindoles are a structural motif found in various natural products and medicinally relevant molecules.2 In this study, the synthesis of 3-aryl-3-hydroxy-2-oxindoles 1 is focused on, as the skeleton is included in several pharmaceutical compounds such as SM-130686![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 and ECi8.4Scheme 1a shows the representative examples of the previously reported retrosynthesis for 1. Most commonly, oxindoles 1 are synthesized via 1,2-addition of an aryl anion equivalent to an isatin derivative.5 Intramolecular arylation to a ketone is also reported.6 Another approach involves aromatic nucleophilic substitution with an enolate of mandelic acid derivatives as a key step.7 In this context, our retrosynthesis was designed based on reductive CO2 fixation with (2-aminophenyl)(aryl)methanone derivatives 2, followed by lactam formation (Scheme 1b). So far, an enormous number of synthetic reports for 1 have been published (Scheme 1b). However, to the best of our knowledge, such an approach via CO2 fixation has not been reported.
3 and ECi8.4Scheme 1a shows the representative examples of the previously reported retrosynthesis for 1. Most commonly, oxindoles 1 are synthesized via 1,2-addition of an aryl anion equivalent to an isatin derivative.5 Intramolecular arylation to a ketone is also reported.6 Another approach involves aromatic nucleophilic substitution with an enolate of mandelic acid derivatives as a key step.7 In this context, our retrosynthesis was designed based on reductive CO2 fixation with (2-aminophenyl)(aryl)methanone derivatives 2, followed by lactam formation (Scheme 1b). So far, an enormous number of synthetic reports for 1 have been published (Scheme 1b). However, to the best of our knowledge, such an approach via CO2 fixation has not been reported.
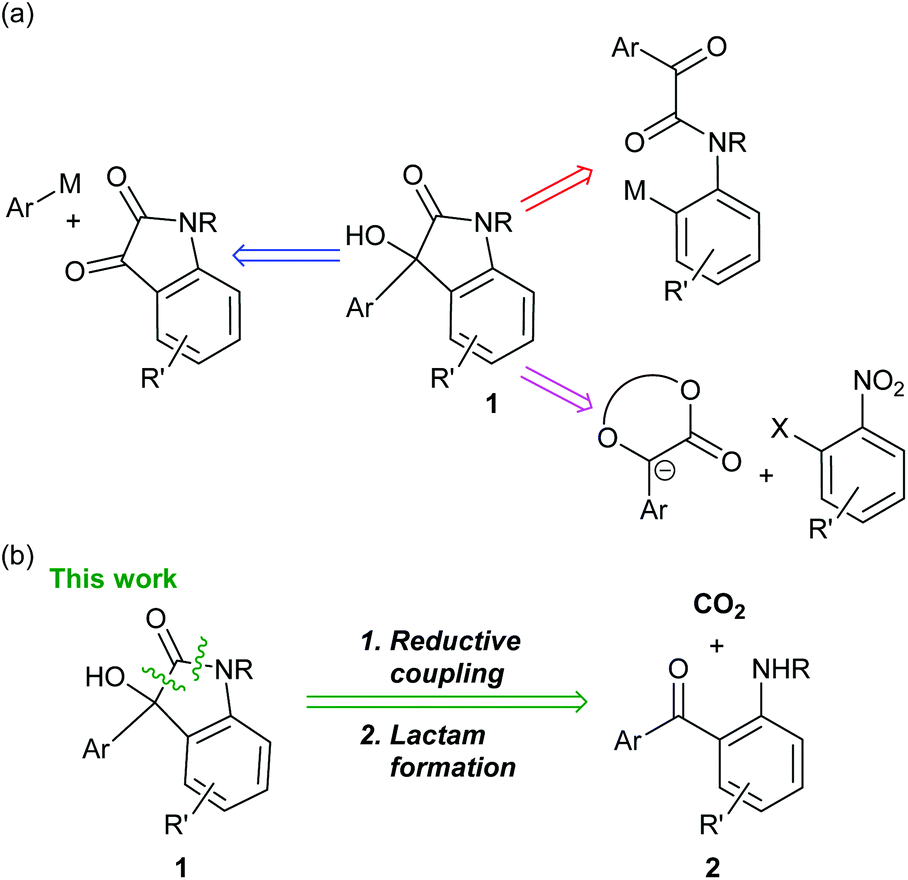 | ||
| Scheme 1 (a) Representative examples of the previously reported retrosynthesis for 3-aryl-3-hydroxy-2-oxindoles 1. (b) Our retrosynthesis for 1. | ||
Reductive CO2 fixation of diaryl ketones has been studied.8–11 Such a reaction can be roughly classified into four reduction patterns, reduction by (1) electrodes,8 (2) alkali metals,9 (3) low-valent transition or rare-earth metals10 and (4) light.11 Our group also has developed the reductive coupling of aldehydes, especially utilizing early-transition metals such as vanadium and titanium compounds as a catalyst in the presence of chlorotrimethylsilane.12 Here, we report the synthesis of 3-aryl-3-hydroxy-2-oxindoles 1via reductive coupling of CO2 and ketoamines 2 as a key step.
Results and discussion
Our investigation commences with the screening of the combination of an early-transition metal catalyst and Zn or Mg as a terminal reductant. First, Zn was used as a terminal reductant (Table 1). Ketoamine 2a reacted with CO2 (balloon) in the presence of TiCl4 (20 mol%), Zn (5 equivalents) and chlorotrimethylsilane (2 equivalents) in N,N-dimethylformamide (DMF) at room temperature, followed by treatment with an aqueous 3 M HCl solution. But, the desired oxindole 1a was not obtained, instead, indole 4 was formed (Table 1, entry 1). Indole 4 is considered to be formed via intramolecular McMurry coupling after formylation of the amine with DMF. The use of THF or DME gave the complex mixtures (Table 1, entries 2 and 3). When DMA was used as a solvent, the reactions with Cp2TiCl2, VCl3, VBr3 or Cp2VCl2 as a catalyst afforded the desired oxindole 1a in low yields (2–9%) with the alcohol 3 as a main product (Table 1, entries 4–7). The reaction without a catalyst also gave 1a in a low yield (Table 1, entry 8).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Yielda/% | ||
| Recovered 2ab | Oxindole 1a | Alcohol 3b | |||
a Yield was calculated by using the integral ratio of the peaks for each compound and the internal standard (1,3,5-trimethoxybenzene) in the 1H NMR spectrum of the crude mixture.
b Starting substrate 2a and the byproducts 3 and 5 were present as a HCl salt in the aqueous layer with a HCl solution after the treatment with an aqueous 3 M HCl solution. To extract them from the aqueous layer, a saturated aqueous NaHCO3 solution was added, and they were extracted with ethyl acetate.
c Reaction time was 20 h.
d The formation of indole 4 and deoxygenated compound 5 was observed as by-products in this entry.
e Not determined.
|
|||||
| 1c,d | TiCl4 | DMF | 65 | 0 | n.d.e |
| 2c | TiCl4 | THF | Complex mixture | ||
| 3c | TiCl4 | DME | Complex mixture | ||
| 4 | Cp2TiCl2 | DMA | 20 | 3 | 13 |
| 5 | VCl3 | DMA | 23 | 9 | 19 |
| 6 | VBr3 | DMA | 22 | 4 | 44 |
| 7 | Cp2VCl2 | DMA | 7 | 2 | 27 |
| 8 | — | DMA | 8 | 2 | 43 |
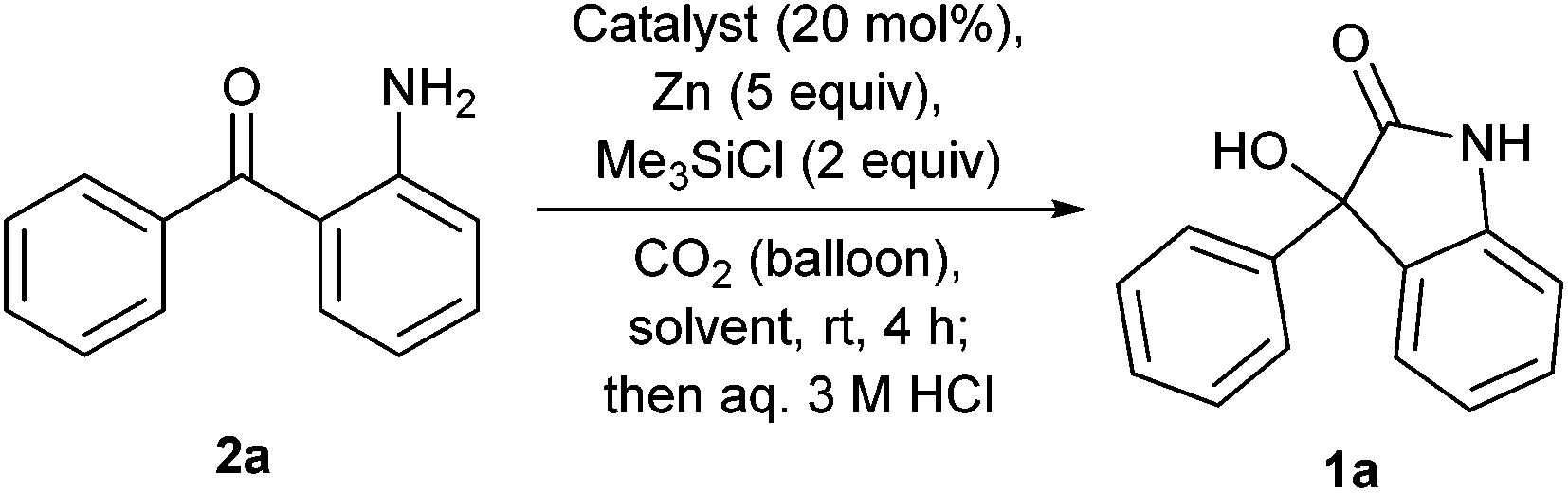
The screening was continued using Mg as a terminal reductant in DMA (Table 2). In this case, the desired oxindole 1a was obtained under the conditions with a catalyst shown in entries 1–6 in Table 2. But, the formation of alcohol 3 was observed in the presence of titanium or vanadium catalysts. Finally, the desired oxindole 1a was obtained in the absence of a catalyst in 95% yield (Table 2, entry 7).13 Decreasing the amount of chlorotrimethylsilane to 1 equivalent resulted in lowering of the yield of 1a to 70% (Table 2, entry 8). In the absence of both a catalyst and chlorotrimethylsilane, the product 1a was not detected, showing that chlorotrimethylsilane is essential (Table 2, entry 9). This is consistent with the previous results.12 Instead of chlorotrimethylsilane, the use of collidine HCl salt provided 1a in a low yield (Table 2, entry 10). Keeping the equivalents of Mg at three was not a problem (97%, Table 2, entry 11). But, the further decreasing to one equivalent gave rise to a decrease of the yield of 1a (74%, Table 2, entry 12).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Yielda/% | ||
| Recovered 2ab | Oxindole 1a | Alcohol 3b | ||
| a Yield was calculated by using the integral ratio of the peaks for each compound and internal standard (1,3,5-trimethoxybenzene) in the 1H NMR spectrum of the crude mixture. b Starting substrates 2a and 3 were present as a HCl salt in the aqueous layer with a HCl solution after the treatment with an aqueous 3 M HCl solution. To extract them from the aqueous layer, a saturated aqueous NaHCO3 solution was added, and they were extracted with ethyl acetate. c It was difficult to quantify. d Instead of chlorotrimethylsilane. e Isolated yield (containing a very small amount of ethyl acetate, see the ESI for the 1H NMR spectrum). | ||||
| 1 | TiCl4 | —c | 44 | 11 |
| 2 | VCl3 | 0 | 51 | 5 |
| 3 | Cp2TiCl2 | 0 | 68 | 2 |
| 4 | Cp2VCl2 | 0 | 80 | 16 |
| 5 | Yb(OTf)3 | 7 | 52 | 16 |
| 6 | VBr3 | 0 | 52 | 21 |
| 7 | — | 0 | 95 | 5 |
| 8 | — (1 equiv. of Me3SiCl) | 16 | 70 | 7 |
| 9 | — (Without Me3SiCl) | 27 | 0 | 48 |
| 10 | — (With collidine HCl salt)d | 51 | 11 | 11 |
| 11 | — (3 equiv. of Mg) | 0 | 97 (99)e | Trace |
| 12 | — (1 equiv. of Mg) | 19 | 74 | —c |
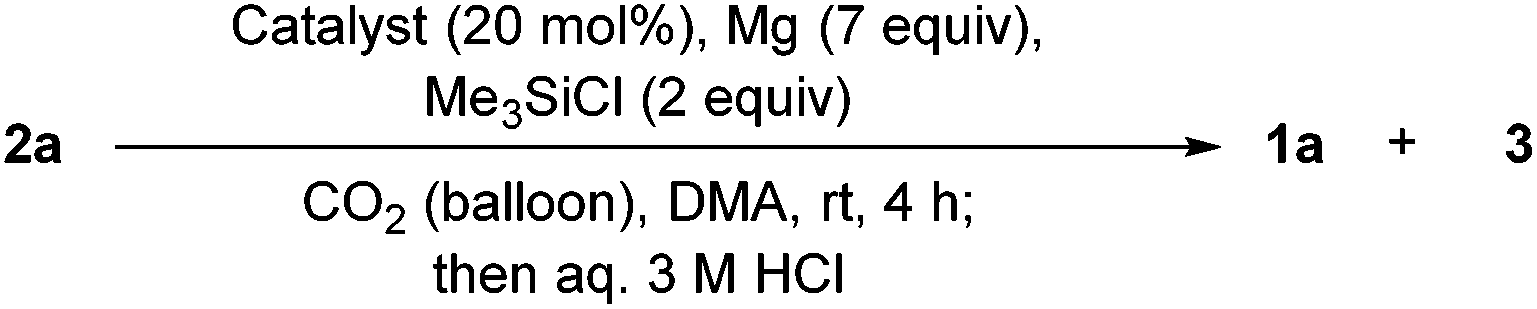
The scope and limitation of the substrates were investigated (Scheme 2). It is important to note that aryl halides such as fluoride, chloride and bromide can be used as a substrate to give oxindoles 1 in good yields in spite of the presence of Mg (96% for 1c, 93% for 1d, 88% for 1e, 76% for 1f and 96% for 1k). The presence of an electron donating group at the para position of the phenyl group was not a problem for this reaction (85% for 1g). The ester moiety was tolerated in this reaction to afford the corresponding product 1h in 83% yield. The product 1i with a furyl group was also synthesized in this reaction. N-Methylated substrates reacted well to give the corresponding products (76% for 1j and 96% for 1k). Instead of the aryl group, the methyl group at R3 did not provide a good result, and the yield of 1i was quite low (2%).
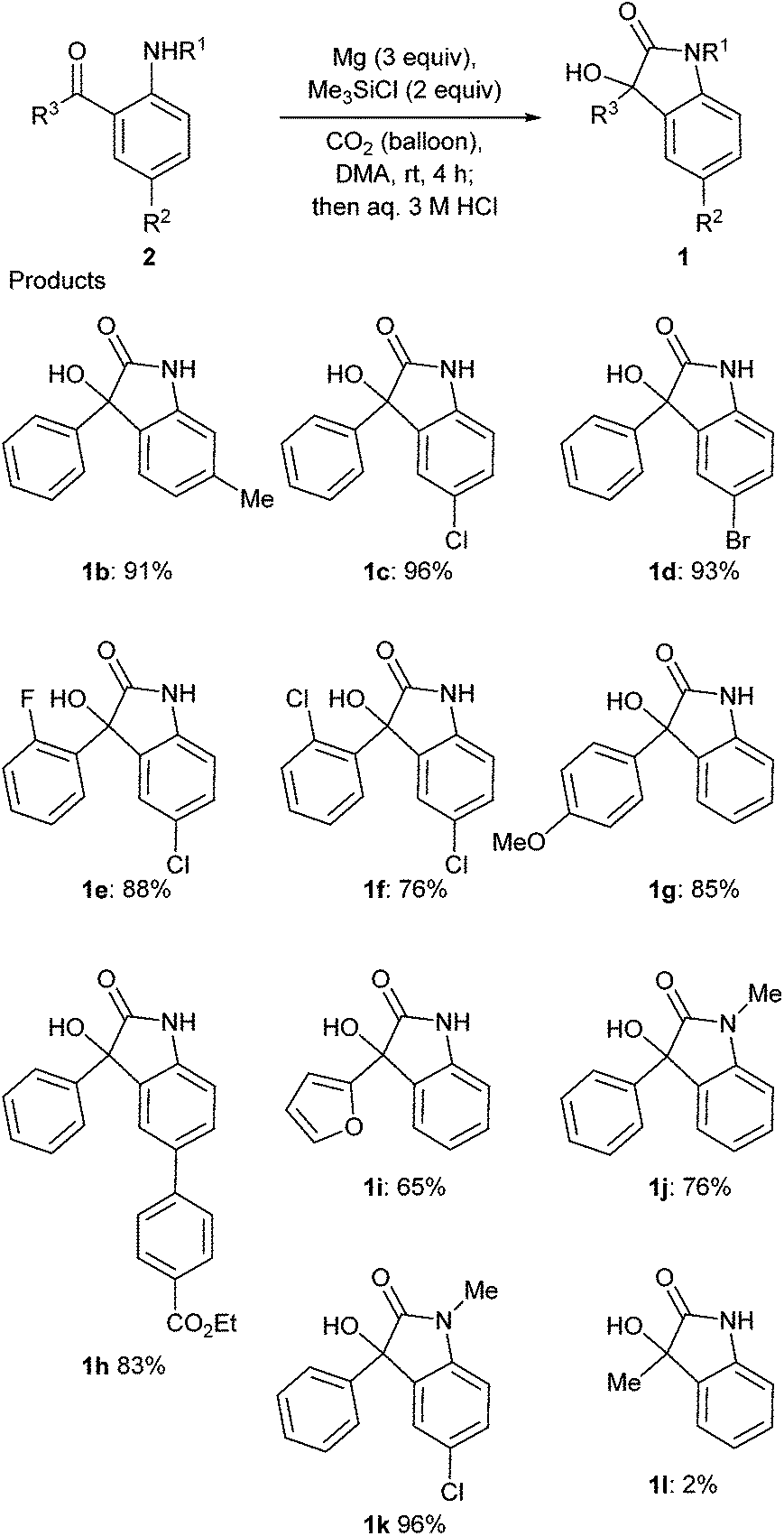 | ||
| Scheme 2 Scope and limitation of the synthesis of oxindoles 1via reductive coupling of 2 and CO2. | ||
To gain an insight into the reaction path, aqueous work-up with a basic solution (aqueous saturated NaHCO3 solution) instead of an aqueous 3 M HCl solution was carried out for the reaction of 2a. The aqueous solution was extracted with ethyl acetate. Both the organic and aqueous layers were separately treated with an aqueous 3 M HCl solution. From the aqueous layer (Scheme 3b), oxindole 1a was obtained in 64% yield, but 11% from the organic layer (Scheme 3a). These results indicate that the lactam formation in entry 7 in Table 2 and Scheme 2 mainly takes place in the process of the treatment with aqueous 3 M HCl solution.
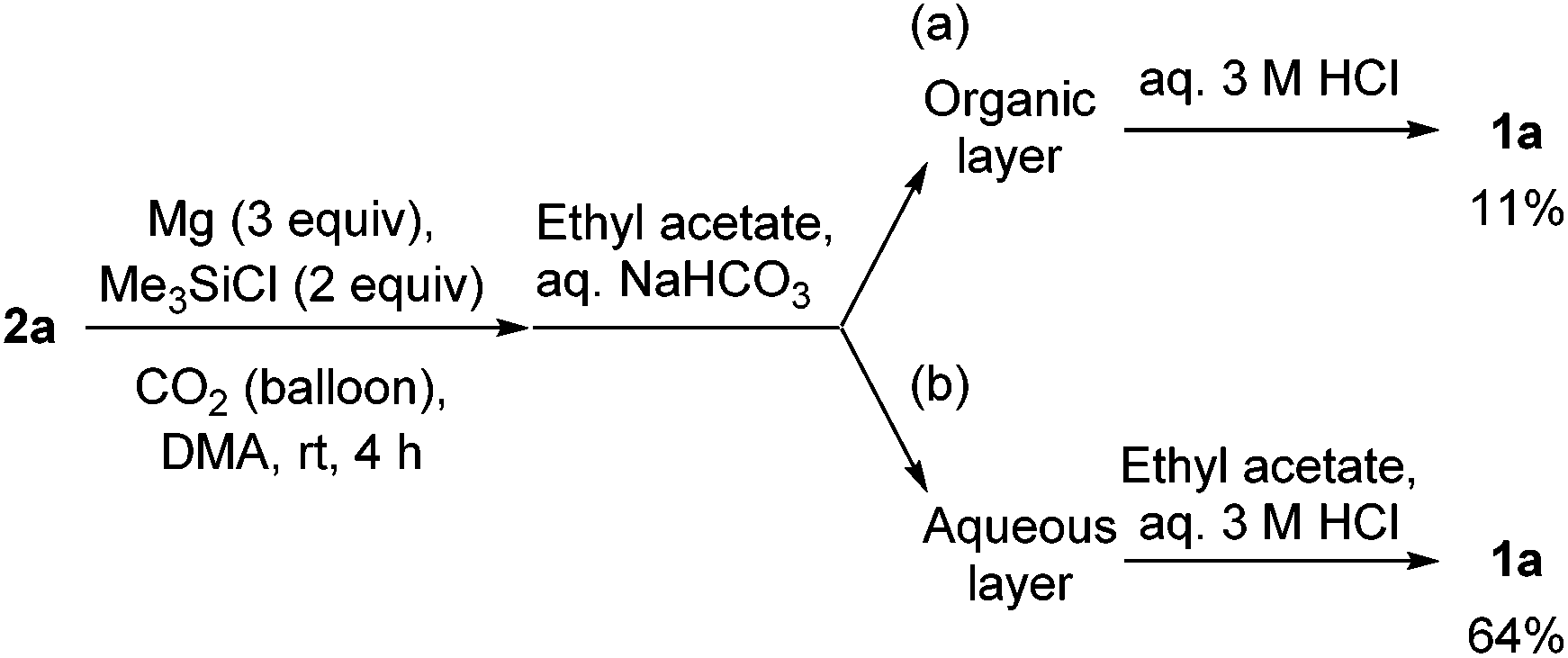 | ||
| Scheme 3 Aqueous work-up with a basic solution followed by acid treatment for the synthesis of oxindoles 1avia reductive coupling of 2a and CO2. | ||
Concerning the reductive coupling with CO2, there are two plausible paths (Scheme 4),13 (1) one-electron reduction of the ketone, addition of the resulting radical anion species to CO2, followed by one-electron reduction (path A) and (2) sequential two-electron reduction of the ketone and 1,2-addition of the carbanion (path B). The related papers presenting the mechanisms were also reported for both paths,9a,14,15 which cannot be excluded for this reaction at present.
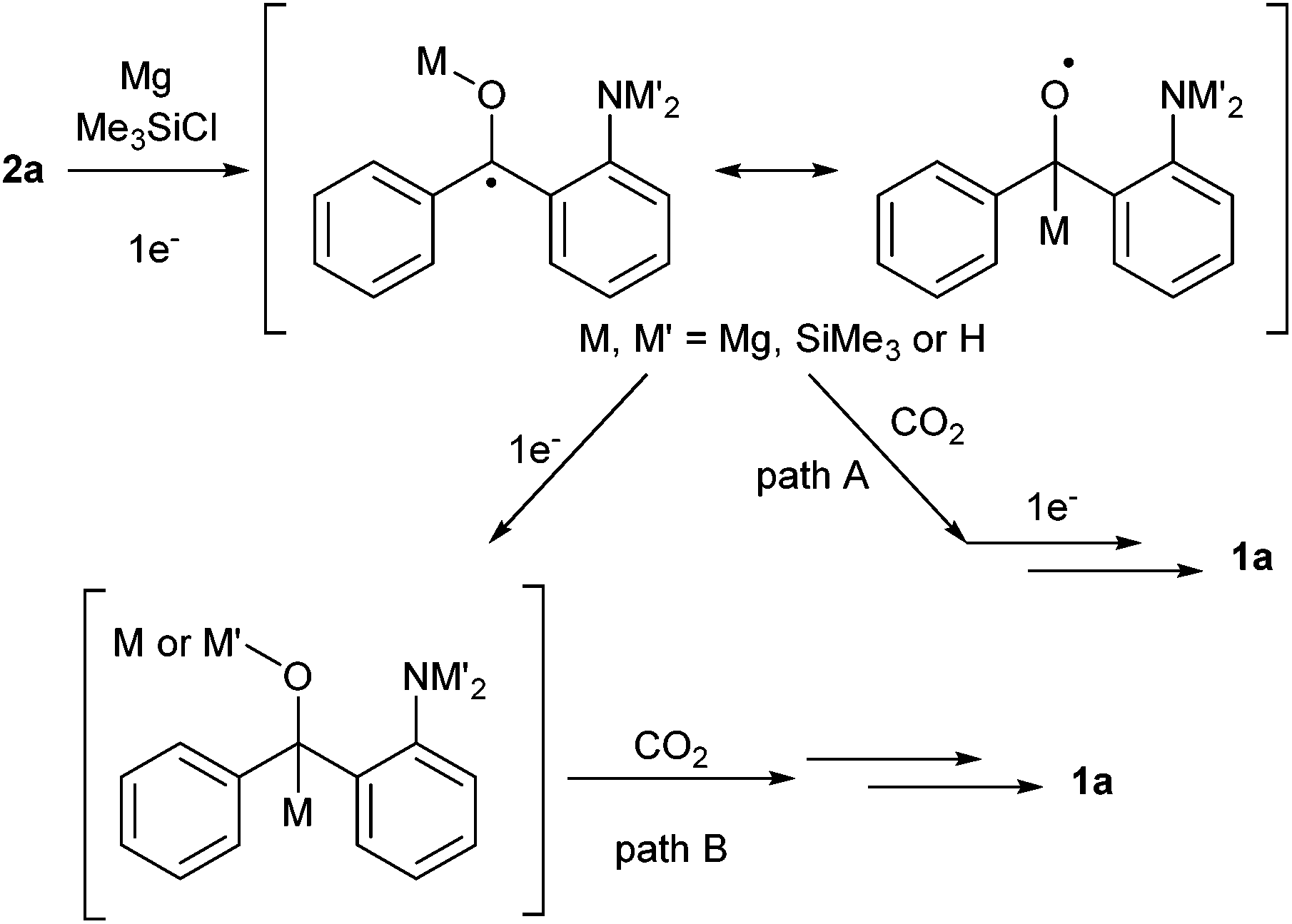 | ||
| Scheme 4 Plausible paths for the synthesis of oxindoles 1avia reductive coupling of 2a and CO2. | ||
Conclusions
We have demonstrated the synthesis of 3-aryl-3-hydroxy-2-oxindoles 1via reductive coupling of ketoamines 2 and CO2 as a key step. This synthetic approach is new although the synthesis of oxindoles 1 has been well studied. The conditions employing Mg with chlorotrimethylsilane in DMA are the best for the reductive coupling, where protection of the amino group is not required. In spite of using Mg, an aryl halide moiety such as aryl fluoride, chloride and bromide is intact in this reaction. Further studies including enantioselective synthesis and application to the synthesis of pharmaceutically active compounds are now underway.Experimental
General
NMR spectra were recorded on a JEOL JNM-ECS 400 spectrometer. Chemical shifts were reported in ppm on the δ scale relative to a residual solvent (DMSO-d6: δ = 2.50 for 1H NMR and 39.52 ppm for 13C NMR) or tetramethylsilane (δ = 0 ppm for 1H and 13C NMR) as an internal standard. Infrared spectra were recorded on a JASCO FT/IR-6200. Mass spectra were recorded on a JEOL JMS-700 spectrometer using the fast atom bombardment (FAB) or electron impact (EI) mode. Substrates 2a–f, k are available from commercial sources. Substrates 2g,162i17 and 2j18 were prepared according to the literature. Substrate 2h was prepared via the Suzuki–Miyaura coupling reaction of 2d with 1.2 equivalents of 4-(ethoxycarbonyl)phenylboronic acid in the presence of 2 mol% Pd(PPh3)4 and aqueous Na2CO3 in toluene and ethanol at 80 °C overnight. Recrystallized 2h (CH2Cl2–hexane) after silica-gel column chromatography was used for the reaction.General procedure
To a round-bottomed flask was added Mg (36.5 mg, 1.5 mmol). The atmosphere in the flask was exchanged with argon by repeating evacuation and purge. Then, DMA (4.1 mL, dried with MS4A before use) was added to the flask. The atmosphere in the flask was exchanged with CO2 using a balloon by repeating evacuation and purge. Chlorotrimethylsilane (127 μL, 1.0 mmol, distilled over CaH2 before use) and the 0.554 M DMA solution of 2 (0.9 mL, 0.5 mmol) were added to the mixture at room temperature. After the reaction mixture was stirred for 4 h, a 3 M aqueous HCl solution (5 mL) was added. The mixture was stirred for at least 30 min. The mixture was transferred to a separatory funnel. The product 1 was extracted with ethyl acetate twice. The organic layer was washed with brine, dried over Na2SO4 and filtered through filter paper. The filtrate was evaporated to give the crude product. To calculate the yield, 1,3,5-trimethoxybenzene (25 mg, 0.15 mmol) was added as an internal standard. The 1H NMR spectra of the mixture were recorded. The yield was calculated by the integral ratio of the peaks for the product 1 and internal standard. Identification of the products 1 was conducted by comparison with the 1H NMR data reported previously: 1a,71b,5c1c,191g,201j6a and 1k.5c:2 for 1h and 1:3 for 1i).
Characterization data for the compounds whose 1H NMR data are not reported previously
1d: 1H NMR (400 MHz, DMSO-d6) δ = 10.58 (s, 1 H), 7.44 (dd, J = 8.2, 1.8 Hz, 1 H), 7.38–7.257 (m, 5 H), 7.21 (d, J = 1.8 Hz, 1 H), 6.88 (d, 8.2 Hz, 1 H), 6.79 (s, 1 H) ppm; 13C NMR (100 MHz, DMSO-d6) δ = 177.97, 141.24, 140.82, 136.13, 131.98, 128.28, 127.71, 127.40, 125.30, 113.65, 112.03, 77.33 ppm; IR(ATR) ν = 3369, 3199, 2358, 1704, 1473, 1182, 821, 737 cm−1; HRMS(FAB) m/z: [M]+ calcd for C14H10BrNO2: 302.9895; found: 302.9889.1e: 1H NMR (400 MHz, DMSO-d6) δ = 10.67 (s, 1 H), 7.91 (ddd, 7.9, 7.9, 1.8 Hz, 1 H), 7.42–7.25 (m, 3 H), 7.06 (ddd, J = 11.5, 8.1, 1.1 Hz, 1 H), 7.00 (s, 1 H), 6.90 (d, J = 9.9 Hz, 1 H), 6.91 (s, 1 H) ppm; 13C NMR (100 MHz, DMSO-d6) δ = 177.05, 158.39 (d, J = 245.4 Hz), 141.25, 134.29, 129.88 (d, J = 7.7 Hz), 129.27, 128.16 (d, J = 12.5 Hz), 128.01 (d, J = 3.8 Hz), 125.68, 124.30 (d, J = 2.9 Hz), 124.09, 115.16 (d, J = 21.1 Hz), 111.29, 74.25 ppm; IR(ATR) ν = 3410, 3237, 2367, 2328, 1713, 1485, 1049, 823, 755 cm−1; HRMS(FAB) m/z: [M]+ calcd for C14H9ClFNO2: 277.0306; found: 277.0308.
1f: 1H NMR (400 MHz, DMSO-d6) δ = 10.70 (s, 1 H), 8.06 (dd, J = 7.8, 1.8 Hz, 1 H), 7.49 (ddd, J = 7.8, 7.8, 1.8 Hz 1 H), 7.41–7.32 (m, 2 H), 7.29 (dd, J = 8.2, 2.3 Hz, 1 H), 7.03 (s, 1 H), 6.89 (d, J = 8.2 Hz, 1 H), 6.76 (d, J = 2.3 Hz, 1 H) ppm; 13C NMR (100 MHz, DMSO-d6) δ = 176.45, 142.21, 137.89, 133.54, 130.28, 129.72 × 2, 129.29, 129.02, 126.89, 125.51, 123.82, 111.19, 76.07 ppm; IR(ATR) ν = 3420, 3237, 2361, 2342, 1711, 1483, 1470, 1437, 1036, 826, 746, 732 cm−1; HRMS(FAB) m/z: [M]+ calcd for C14H9Cl2NO2: 293.0010; found: 293.0008.
1h: 1H NMR (400 MHz, DMSO-d6) δ = 10.58 (s, 1 H), 7.97 (d, J = 8.7 Hz, 2 H), 7.72 (d, J = 8.7 Hz 2 H), 7.68 (dd, J = 8.2, 2.3 Hz, 1 H), 7.46 (d, J = 1.8 Hz, 1 H), 7.36–7.24 (m, 5 H), 7.04 (d, J = 8.2 Hz, 1 H), 6.75 (s, 1 H), 4.31 (q, J = 7.3 Hz, 2H), 1.32 (t, J = 7.3 Hz, 3H) ppm; 13C NMR (100 MHz, DMSO-d6) δ = 178.55, 165.63, 144.38, 142.47, 141.27, 134.76, 132.85, 129.87, 128.25, 128.13, 127.61, 126.25, 125.47, 123.18, 110.57, 77.41, 60.73, 14.23 ppm; IR(KBr) ν = 3308, 3061, 2980, 2933, 1728, 1713, 1606, 1484, 1278, 1175, 1103, 771 cm−1; HRMS(EI) m/z: [M]+ calcd for C23H19NO4: 373.1314; found: 373.1310.
1i: 1H NMR (400 MHz, DMSO-d6) δ = 10.44 (s, 1 H), 7.59 (brd, J = 1.8 Hz, 1 H), 7.30 (d, J = 7.3 Hz, 1 H), 7.24 (ddd, J = 7.8, 7.8, 0.9 Hz, 1 H), 6.98 (dd, J = 7.3, 7.3 Hz, 1 H), 6.85 (d, J = 7.3, 1 H), 6.73 (s, 1H), 6.40 (dd, J = 3.2, 1.8 Hz, 1 H), 6.30 (d, J = 3.2 Hz, 1 H) ppm; 13C NMR (100 MHz, DMSO-d6) δ = 176.18, 153.01, 143.19, 141.69, 130.78, 129.56, 124.99, 121.87, 110.27, 109.88, 107.65, 73.48 ppm; IR(KBr) ν = 3310, 2822, 2361, 1709, 1684, 1624, 1474, 1311, 1112, 763 cm−1; HRMS(EI) m/z: [M]+ calcd for C12H9NO3: 215.0582; found: 215.0583.
2h: 1H NMR (400 MHz, CDCl3) δ = 8.02 (d, J = 8.7 Hz, 2 H), 7.75 (d, J = 2.3 Hz 1 H), 7.72–7.68 (m, 2 H), 7.62 (dd, J = 8.7, 2.3 Hz, 1 H), 7.56 (tt, J = 7.3, 1.4 Hz, 1 H), 7.51–7.46 (m, 4 H), 6.85 (d, J = 8.7 Hz, 1 H), 6.20 (brs, 2 H) ppm; 13C NMR (100 MHz, CDCl3) δ = 198.89, 166.48, 150.78, 144.54, 139.77, 132.99, 132.88, 131.46, 130.13, 129.24, 128.42, 128.29, 127.25, 125.79, 118.34, 117.67, 60.88, 14.35 ppm; IR(KBr) ν = 3487, 3352, 3066, 2986, 2902, 1704, 1639, 1729, 1247, 1107, 775 cm−1; HRMS(EI) m/z: [M]+ calcd for C22H19NO3: 345.1365; found: 345.1360.
Acknowledgements
Financial support from JST (ACT-C) is acknowledged.Notes and references
- (a) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed; (b) Carbon Dioxide as Chemical Feedstock, ed. M. Aresta, Wiley-VCH, Weinheim, 2010 Search PubMed.
- Reviews: (a) B. M. Trost and M. K. Brennan, Synthesis, 2009, 3003 CrossRef CAS; (b) F. Zhou, Y.-L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381 CrossRef CAS; (c) R. Dalpozzo, G. Bartoli and G. Bencivenni, Chem. Soc. Rev., 2012, 41, 7247 RSC.
- (a) T. Tokunaga, W. E. Hume, T. Umezome, K. Okazaki, Y. Ueki, K. Kumagai, S. Hourai, J. Nagamine, H. Seki, M. Taiji, H. Noguchi and R. Nagata, J. Med. Chem., 2001, 44, 4641 CrossRef CAS PubMed; (b) T. Tokunaga, W. E. Hume, J. Nagamine, T. Kawamura, M. Taiji and R. Nagata, Bioorg. Med. Chem. Lett., 2005, 15, 1789 CrossRef CAS PubMed.
- Y. Shen, J. Liu, G. Estiu, B. Isin, Y.-Y. Ahn, D.-S. Lee, A.-L. Barabási, V. Kapatral, O. Wiest and Z. N. Oltvai, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1082 CrossRef CAS PubMed.
- Selected examples: (a) S. J. Angyal, E. Bullock, W. G. Hanger, W. C. Howell and A. W. Johnson, J. Chem. Soc., 1957, 1592 RSC; (b) Ref. 3a; ; (c) R. Shintani, M. Inoue and T. Hayashi, Angew. Chem., Int. Ed., 2006, 45, 3353 CrossRef CAS PubMed.
- Selected examples: (a) Y.-X. Jia, D. Katayev and E. P. Kündig, Chem. Commun., 2010, 46, 130 RSC; (b) L. Yin, M. Kanai and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 7620 CrossRef CAS PubMed.
- S. Barroso, G. Blay, L. Cardona, I. Fernández, B. García and J. R. Pedro, J. Org. Chem., 2004, 69, 6821 CrossRef CAS PubMed.
- (a) S. Wawzonek and A. Gundersen, J. Electrochem. Soc., 1960, 107, 537 CrossRef CAS; (b) Y. Ikeda and E. Manda, Bull. Chem. Soc. Jpn., 1985, 58, 1723 CrossRef CAS; (c) S.-F. Zhao, H. Wang, Y.-C. Lan, X. Liu, J.-X. Lu and J. Zhang, J. Electroanal. Chem., 2012, 664, 105 CrossRef CAS PubMed; (d) R. Liu, G. Yuan, C. L. Joe, T. E. Lightburn, K. L. Tan and D. Wang, Angew. Chem., Int. Ed., 2012, 51, 6709 CrossRef CAS PubMed; (e) S.-F. Zhao, M. Horne, A. M. Bond and J. Zhang, Phys. Chem. Chem. Phys., 2015, 17, 19247 RSC.
- (a) Y. V. Kurbatov and A. S. Kurbatova, Russ. J. Org. Chem., 2001, 37, 1344 CrossRef CAS; (b) A. Häuβermann, F. Rominger and B. F. Straub, Chem. – Eur. J., 2012, 18, 14174 CrossRef PubMed.
- (a) Z. Hou, K. Takamine, O. Aoki, H. Shiraishi, Y. Fujiwara and H. Taniguchi, J. Chem. Soc., Chem. Commun., 1988, 668 RSC; (b) Z. Hou, K. Takamine, O. Aoki, H. Shiraishi, Y. Fujiwara and H. Taniguchi, J. Org. Chem., 1988, 53, 6077 CrossRef CAS; (c) J. J. Eisch, P. O. Fregene and J. N. Gitua, J. Organomet. Chem., 2007, 692, 4647 CrossRef CAS; (d) J. J. Eisch and P. O. Fregene, Eur. J. Org. Chem., 2008, 4482 CrossRef CAS.
- (a) M. Kanemoto, H. Ankyu, Y. Wada and S. Yanagida, Chem. Lett., 1992, 2113 CrossRef CAS; (b) T. Ogata, K. Hiranaga, S. Matsuoka, Y. Wada and S. Yanagida, Chem. Lett., 1993, 983 CrossRef CAS.
- (a) T. Hirao, T. Hasegawa, Y. Muguruma and I. Ikeda, J. Org. Chem., 1996, 61, 36 CrossRef; (b) For a review: T. Hirao, Chem. Rev., 1997, 97, 2707 CrossRef CAS PubMed; (c) T. Hirao, M. Asahara, Y. Muguruma and A. Ogawa, J. Org. Chem., 1998, 63, 2812 CrossRef CAS; (d) T. Hirao, B. Hatano, M. Asahara, Y. Muguruma and A. Ogawa, Tetrahedron Lett., 1998, 39, 5247 CrossRef CAS; (e) B. Hatano, A. Ogawa and T. Hirao, J. Org. Chem., 1998, 63, 9421 CrossRef CAS; (f) T. Hirao, B. Hatano, Y. Imamoto and A. Ogawa, J. Org. Chem., 1999, 64, 7665 CrossRef CAS; (g) T. Hirao, A. Ogawa, M. Asahara, Y. Muguruma and H. Sakurai, Org. Synth., 2005, 81, 26 CrossRef CAS; (h) X. Xu and T. Hirao, J. Org. Chem., 2005, 70, 8594 CrossRef CAS PubMed; (i) A. Miyasaka, T. Amaya and T. Hirao, Chem. – Eur. J., 2014, 20, 1615 CrossRef CAS PubMed.
- To check the possibility of intramolecular reductive coupling of trimethylsilyl arylcarbamate of 2a, an NMR experiment in DMSO-d6 was carried out in the absence of Mg. As a result, the formation of trimethylsilyl arylcarbamate was not observed. Therefore, this path is excluded here.
- Related conditions are reported for the reductive coupling of imines and CO2: A. A. Sathe, D. R. Hartline and A. T. Radosevich, Chem. Commun., 2013, 49, 5040 RSC.
- (a) I. Nishiguchi, M. Sakai, H. Maekawa, T. Ohno, Y. Yamamoto and Y. Ishino, Tetrahedron Lett., 2002, 43, 635 CrossRef CAS; (b) T. Uchida, Y. Kita, H. Maekawa and I. Nishiguchi, Tetrahedron, 2006, 62, 3103 CrossRef CAS.
- (a) Synthesis: S. A. Chandler, P. Hanson, A. B. Taylor, P. H. Walton and A. W. Timms, J. Chem. Soc., Perkin Trans. 2, 2001, 214 RSC; (b) Characterization: K. Kobayashi, S. Fujita, S. Fukamachi and H. Konishi, Synlett, 2009, 3378 CAS.
- V.-H. Nguyen, L. Vendier, M. Etienne, E. Despagnet-Ayoub, P.-A. R. Breuil, L. Magna, D. Proriol, H. Olivier-Bourbigou and C. Lorber, Eur. J. Inorg. Chem., 2012, 97 CrossRef CAS.
- (a) Synthesis: J. Huang, C. Wan, M.-F. Xu and Q. Zhu, Eur. J. Org. Chem., 2013, 1876 CrossRef CAS; (b) Characterization: D. Susanti, L. L. R. Ng and P. W. H. Chan, Adv. Synth. Catal., 2014, 356, 353 CrossRef CAS.
- J. Gui, G. Chen, P. Cao and J. Liao, Tetrahedron: Asymmetry, 2012, 23, 554 CrossRef CAS.
- Z.-K. Xiao, H.-Y. Yin and L.-X. Shao, Org. Lett., 2013, 15, 1254 CrossRef CAS PubMed.
Footnotes |
| † This paper is dedicated to Professor Barry M. Trost on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available: 1H NMR spectrum for 1a, and 1H and 13C NMR spectra for 1d, 1e, 1f, 1h, 1i and 2h. See DOI: 10.1039/c6qo00107f |
| This journal is © the Partner Organisations 2016 |