
Coordination and catalysis of Zn2+ in epoxy-based vitrimers†
A.
Demongeot
a,
S. J.
Mougnier
a,
S.
Okada
b,
C.
Soulié-Ziakovic
a and
F.
Tournilhac
*a
aLaboratoire Matière Molle et Chimie, UMR7167 CNRS-ESPCI Paris, PSL Research University, 10 rue Vauquelin, 75005 Paris, France. E-mail: francois.tournilhac@espci.fr
bDepartment of Chemistry, University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-0033, Japan
First published on 6th June 2016
Abstract
To elucidate the catalytic action of Zn(II) in the exchange reactions taking place in epoxy-acid vitrimers, extended X-ray absorption fine structure (EXAFS) spectroscopy has been used as well as infrared (IR) spectroscopy on vitrimers. The presence of Zn(II) in vitrimers is revealed by characteristic alterations in both the hydroxide groups and the ester carbonyl bond structures. An EXAFS analysis showed that the zinc cation is coordinated by four oxygen atoms, two of them being negatively charged, i.e. alkoxide groups to satisfy the electroneutrality, whereas IR analysis demonstrated the lengthening of the ester carbonyl bond. The catalytic effect of Zn2+ can be stated as follows: (1) shifting the alcohol/alkoxide equilibrium towards the more nucleophilic alkoxide form; (2) activating the carbonyl bonds by increasing the electrophilicity of the carbon atom; (3) bringing reacting centres close to each other. Zn2+ coordinated in the epoxy-based network therefore appears as a good option to catalyze exchange reactions by transesterification and afford vitrimer properties.
Introduction
Vitrimers1–15 are networks in which the topology can be changed under thermal activation while keeping the number of bonds and crosslinks constant. Consequently, even though they are crosslinked and insoluble, vitrimers are able to flow.1 Due to this combination of properties, vitrimers represent a third class of materials, aside from thermoplastics and thermosets. This unique feature lies in their chemical structure, characterized by the presence of exchangeable bonds, which make them also different from materials based on reversible bonds (either dynamic covalent or dynamic non-covalent systems). The concept is presented in Fig. 1. | ||
| Fig. 1 Basic view of an exchange reaction. | ||
Two partners, A and B, joined by an exchangeable bond do not thermally dissociate. However, if there is a third individual A′, chemically similar to A, there is a possibility that A′ takes the place of A, thereby changing the connection topology while keeping the number of A–B bonds constant. According to this picture, several types of chemical reactions may be recognized as exchange reactions,1–12,14–20 the thermodynamic feature that makes vitrimers different from systems with reversible covalent bonds21–29 is that temperature does not influence the number of open and closed links but only the rate of exchanges. Indeed in a system like that in Fig. 1, the initial state (before exchange) and the final state (after exchange) have the same energy. The difference between covalent adaptable networks, based on reversible bonds, and vitrimers, based on exchangeable bonds, has been well commented on.30 Existing chemical systems for vitrimers have also been the subject of a comprehensive review.31
Exchange reactions based on photoactivated radical processes were used by Bowman et al. to control stress relaxation in crosslinked materials.32 In vitrimer, the principle is to rely on thermoactivation and catalysis to control the rate of exchange while keeping the connectivity of the network. In first vitrimer prototypes1–3 the selected exchange reaction was transesterification, which follows an associative mechanism under basic conditions.33 Thus, as shown in Scheme 1, the nucleophilic attack of an alkoxide on the carbonyl of an ester leads to a tetrahedral intermediate that, in turn, re-dissociates into a new alkoxide and a new ester.
 | ||
| Scheme 1 The mechanism of a noncatalyzed transesterification reaction under basic conditions. | ||
In the studies published to date on transesterification based vitrimers, epoxy chemistry has been widely put to use owing to its technological relevance and its ability to easily produce β-hydroxy esters.1–8 Noticeably, it has been shown in these systems that the exchange kinetics can be controlled by organic2,5–7 or metal1–4,9 catalysis. More particularly zinc catalysis was selected for its superior thermal toughness in a more technologically oriented study on continuous fiber composites and commercial epoxy-based vitrimer formulations.5 For the further development of epoxy vitrimers, the rate of exchange has to be carefully controlled since, once properly catalyzed, vitrimer materials might be transformable in a tailor-made temperature range.
Even though metal catalysis fundamentally implies that the reactants get close together in an adequate orientation in the metal coordination shell, the mechanism of zinc-catalyzed transesterification in epoxy vitrimers is still to be determined.
In order to elucidate the action mode of metal catalysts in the transesterification reaction and, eventually, to correlate vitrimer performances with the nature of the catalytic system, we investigated vitrimers as well as model compounds. The curing of epoxy resins leads essentially to insoluble and amorphous systems. We selected spectroscopic methods adapted to insoluble materials. Infrared (IR) spectroscopy was used to control the achievement of curing reactions and to study the alteration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O stretching vibrations of ester groups upon complexation with Zn2+. Attenuated total reflection (ATR) was selected in order to produce quantitative measurements from thick elastomeric samples (typically 1 mm here). Extended X-ray absorption fine structure (EXAFS) spectroscopy was used to measure the coordination number and the Zn–O distances in the first coordination shell. Model zinc compounds, whose crystal structure is reported, were also recorded under the same conditions.
O and C–O stretching vibrations of ester groups upon complexation with Zn2+. Attenuated total reflection (ATR) was selected in order to produce quantitative measurements from thick elastomeric samples (typically 1 mm here). Extended X-ray absorption fine structure (EXAFS) spectroscopy was used to measure the coordination number and the Zn–O distances in the first coordination shell. Model zinc compounds, whose crystal structure is reported, were also recorded under the same conditions.
Experimental
Materials
3-Phenoxy-1,2-propanediol (95%), anhydrous pyridine (99%), acetyl chloride (99%), bisphenol A diglycidyl ether (DGEBA, DER 332), zinc acetate dihydrate (ACS Reagent, 98%) and chloroform (99%) were purchased from Sigma Aldrich and were used as received without further purification. Pripol® 1040 was kindly provided by Croda. It is a mixture of hydrogenated cycloaddition adducts of C18 fatty acid derivatives, containing about 23 wt% of C54 tricarboxylic acids and 77 wt% of C36 dicarboxylic acids (see the ESI†).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Typically, we used 15.7 g (46.9 mmol acid + 5.2 mmol Zn-carboxylate) of compound 1 and 9.06 g (52.1 mmol) of DGEBA. Initially biphasic, the mixture was heated to 130 °C until phase miscibility occurred, manually stirred, and then quickly poured into a 10 cm × 10 cm × 0.15 cm brass mold sandwiched between two anti-adhesive silicone papers. The mold was placed in a heating press and left for at least 6 h at 130 °C. Compound 2 bis was prepared similarly using an 1:1 acyl to epoxy ratio and a 5 mol% concentration of Zn2+.
:1. Typically, we used 15.7 g (46.9 mmol acid + 5.2 mmol Zn-carboxylate) of compound 1 and 9.06 g (52.1 mmol) of DGEBA. Initially biphasic, the mixture was heated to 130 °C until phase miscibility occurred, manually stirred, and then quickly poured into a 10 cm × 10 cm × 0.15 cm brass mold sandwiched between two anti-adhesive silicone papers. The mold was placed in a heating press and left for at least 6 h at 130 °C. Compound 2 bis was prepared similarly using an 1:1 acyl to epoxy ratio and a 5 mol% concentration of Zn2+.
3-Phenoxy-1,2-propanediol (compound 3 ) was purchased from Sigma Aldrich with a purity of 95%. This compound is a liquid with a boiling point of 315 °C. Our analysis revealed the presence of about 7 mol% (NMR) or 6 wt% (GC-MS) of dimerized ether derivatives (mixture of isomers) with a molecular weight of m/z = 318 g mol−1, together with the main product (racemic mixture) with m/z = 168 g mol−1. In GC-monitored kinetic experiments, these oligomers reproduce at longer retention times the characteristic reaction pattern of the monomer. The transesterification kinetic data were determined solely from the monomer signals.
Methods
GC-MS spectra were recorded using a Shimadzu GC-2010 gas chromatograph coupled to mass spectrometry (electron ionization at 70 eV). At any reaction time, the quantities of compound 3, compound 4 and their transesterification exchange products were identified by their characteristic MS molecular peak respectively at m/z = 168 g mol−1, m/z = 252 g mol−1 and m/z = 210 g mol−1. At higher retention times, the same scenario was observed for oligomers of compound 3 and compound 4 giving rise to characteristic m/z peaks at 318 g mol−1, 360 g mol−1 and 402 g mol−1.FT-IR spectra were recorded at 4 cm−1 resolution using a Bruker TENSOR 37 spectrometer fitted with a Specac Golden Gate ATR heating cell. Compound 2 samples were dried in a vacuum oven at 120 °C for 2 h before analysis. In ATR measurements the depth of penetration of the IR evanescent wave in a non-absorbing medium is a nearly linear function of the wavelength.34 On the OPUS software provided by Bruker, the transmitted signal, I/I0, is converted into ATR units via the following expression: ATR = −log(I/I0) × x/1000 where x is the wavenumber in cm−1. For any given band, the ATR signal thus calculated increases linearly with the amount of the corresponding vibrator.
1H NMR spectra were recorded from CDCl3 solutions using a Bruker Avance 400 spectrometer and referenced internally to tetramethylsilane as a standard.
EXAFS measurements were made at the Dutch-Belgian beam line (DUBBLE), at BM26A of the European Synchrotron Radiation Facility (ESRF), Grenoble, France. Zn-catalyzed epoxy-acid vitrimer plates with a thickness of 1.5 mm and reference powders, sandwiched between kapton tapes, were put into the X-ray beam. Spectra were acquired at room temperature in the transmission mode using a Si(111) double crystal monochromator above the Zn K-edge (9.659 keV). The EXAFS data analysis was processed using the IFEFFIT library of numerical XAS algorithms written in Perl programming language that utilizes the ab initio EXAFS code, FEFF 6.01.35 In the EXAFS fitting procedure, we used the parameters determined from the first shell analysis of anhydrous zinc acetate, keeping solely r and N as adjustable parameters. We used crystal structure data of anhydrous zinc acetate and zinc acetate dihydrate proposed respectively by Clegg et al.36 and Ishioka et al.37
Results
Epoxy vitrimer synthesis
The method we used here is the same as the one previously reported.1 The starting point is a mixture of dicarboxylic and tricarboxylic fatty acids (Pripol® 1040), added in large excess to a given quantity of zinc acetate. A ligand exchange is performed as shown in Scheme 2A.1 Removing the acetic acid thereby formed shifts the equilibrium towards the formation of salts where Zn2+ is coordinated by two long chain carboxylate anions. Up to a concentration of 20 mol%, such salts are soluble in the fatty acid mixture. Thus the resulting material from this ligand exchange (compound 1) is a mixture of partially neutralized (typically 5–10 mol%) fatty polyacids wherein the counterion is zinc.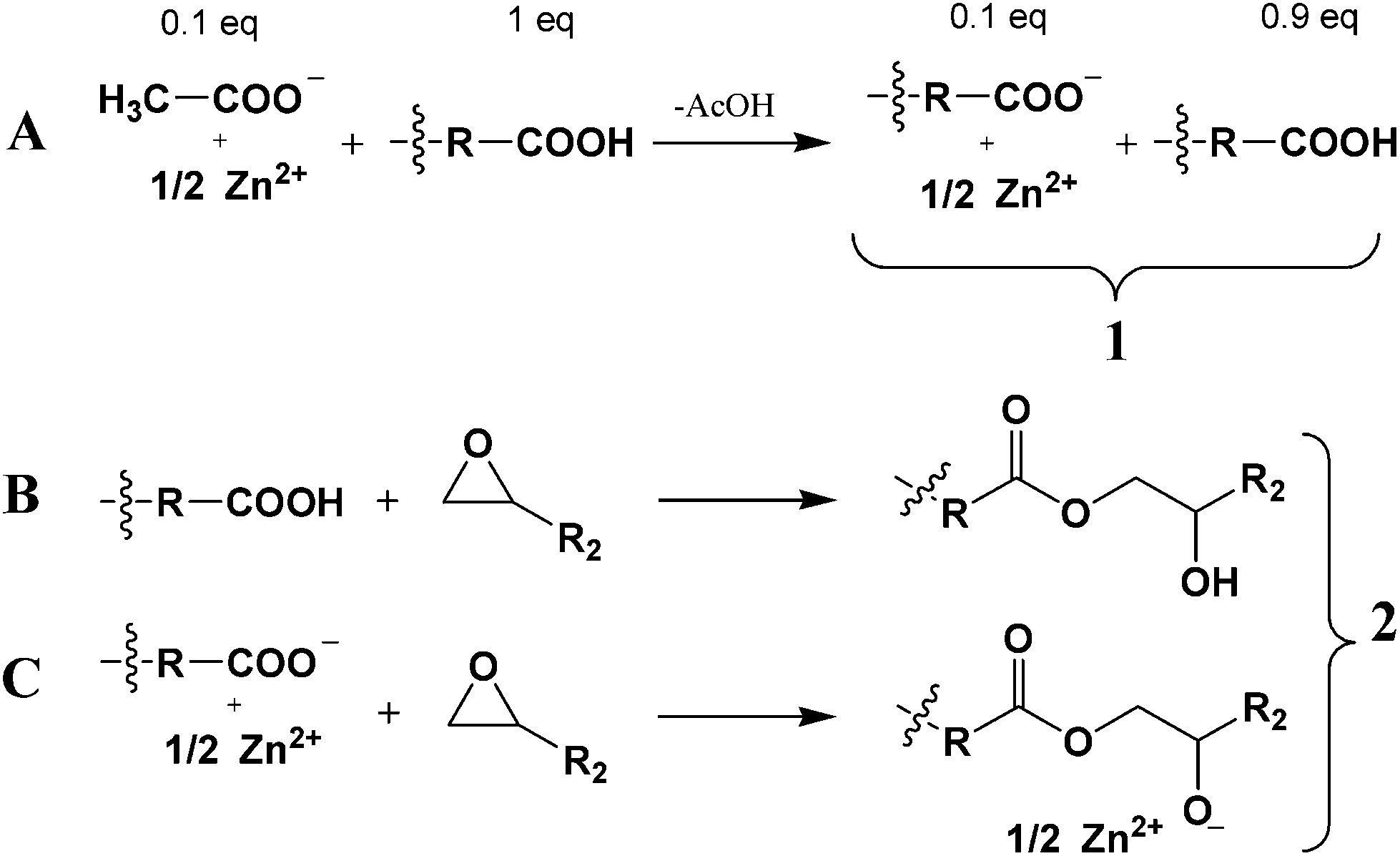 | ||
| Scheme 2 Preparation of Zn-catalyzed vitrimers. (A) First step: ligand exchange reaction between fatty polyacids and zinc diacetate; acetic acid is eliminated by vacuum stripping. (B) Second step, major reaction: generation of β-hydroxy-ester links by epoxy-acid curing. (C) Second step, minor reaction: generation of β-oxy-ester anions. (X1, X2) = (H, R2) or (R2, H). | ||
Compound 1 is then used to cure the epoxy resin that gives rise to the vitrimer network (compound 2). In this second step, the major reaction is the addition of a carboxylic acid to the epoxy ring to form the already mentioned β-hydroxy-ester link, constitutive of the epoxy vitrimer network (Scheme 2B). The number of epoxy functions is adjusted in order to consume all acylating species (carboxylic acid and carboxylate functions) and to leave, on average, one OH group per ester group formed. The time dependence of νCO stretching vibrations associated with carboxylic acid (1710 cm−1), carboxylate (1632 and 1550 cm−1) and ester (1737 cm−1) was followed by infrared spectroscopy (depicted in the ESI†) as well as the disappearance of δCOC of the epoxide (914 cm−1). It is evident from the spectra that after 60 min, the carboxylic acid, carboxylate and epoxide signals all disappear. Hence it logically follows that Zn2+ in epoxy vitrimers is no longer in the form of carboxylate at the end of the curing process. Consequently, besides the neutral β-hydroxy-ester groups, β-oxyester anions are necessary to ensure electroneutrality (Scheme 2C).
Catalysis
In previous studies,1,2 the catalytic action of zinc was evidenced by stress relaxation experiments of networks and GC titrations on model molecules. As a result of epoxy-acid chemistry to produce model molecules, 2-methylimidazole was often present in the systems where it could play the role of a co-catalyst. Here, we examine the efficiency of Zn2+ as a transesterification catalyst in model molecules not generated by epoxy-acid chemistry. The exchange reaction was studied by GC-MS. From 3-phenoxy-1,2-propanediol (compound 3), we synthesized 3-phenoxypropylene diacetate (compound 4). First a 1:1 mixture of compound 3 and compound 4 was prepared and the desired quantity of zinc acetate was added. Once heated to 130 °C, zinc salt rapidly dissolved and gaseous acetic acid evolution was observed. Then the mixture was continuously stirred and transesterification products appeared: two β-hydroxy-ester (isomers 5′ and 5′′) not distinguishable by GC. The total abundance of compound 5′ and compound 5′′, referred as compound 5, is plotted in Fig. 2 as a function of Zn2+ concentration. Without the addition of any metal catalyst, the equilibrium took around 75 h at 150 °C whereas the equilibrium is obtained in 1 h or 5 h when zinc acetate is added at a level of 5 mol% or 0.05 mol% respectively. Even a small amount of zinc catalyst accelerates the transesterification significantly. The rate of transesterification is a function of the metal concentration. In previous studies on epoxy vitrimers,1–3 Zn2+ was used at a molar concentration of 1–10%. Our data indicate that Zn2+ is efficient even at low concentration and in the absence of a co-catalyst. This result confirms the use of zinc as an excellent option to catalyze exchange reactions in epoxy vitrimers.
 | ||
| Fig. 2 Scheme of the transesterification of model molecules (top). Transesterification kinetics results (bottom) at 150 °C for the di-ester and di-hydroxy mix model molecules. Without metal catalyst added (triangle down, green), with added 0.05 mol% Zn(OAc)2 (triangle up, blue), 0.5 mol% Zn(OAc)2 (circle, red) and 5 mol% Zn(OAc)2 (square, black). | ||
EXAFS spectroscopy
We performed extended X-ray absorption fine structure spectroscopy (EXAFS) experiments at the Zn K-edge on vitrimer and model compounds. Special care was given to the choice of references whose atomic positions are known with accuracy. We analyzed anhydrous zinc diacetate (compound 6) and zinc diacetate dihydrate (compound 7), two well-known zinc salts whose crystal structures are available in the literature (Fig. 3). X-ray powder diffraction diagrams have been recorded to check the allomorphic purity of the samples (see the ESI†). In anhydrous zinc acetate, four oxygen atoms at an average Zn–O distance of 1.96 Å surround the zinc cation.36 The bidentate acetate groups in this compound are bridging ligands in a syn–anti arrangement (Fig. 3a). In contrast, in zinc acetate dihydrate, Zn2+ is hexacoordinated with an average Zn–O distance of 2.1 Å.37 In this case the bidentate acetate groups are chelating ligands as depicted in Fig. 3b. We took these compounds as references in the analysis of the EXAFS spectra obtained for our model vitrimer systems. The original X-ray absorption spectra, μ(E) plots, and the details of the analysis are reported in the ESI.†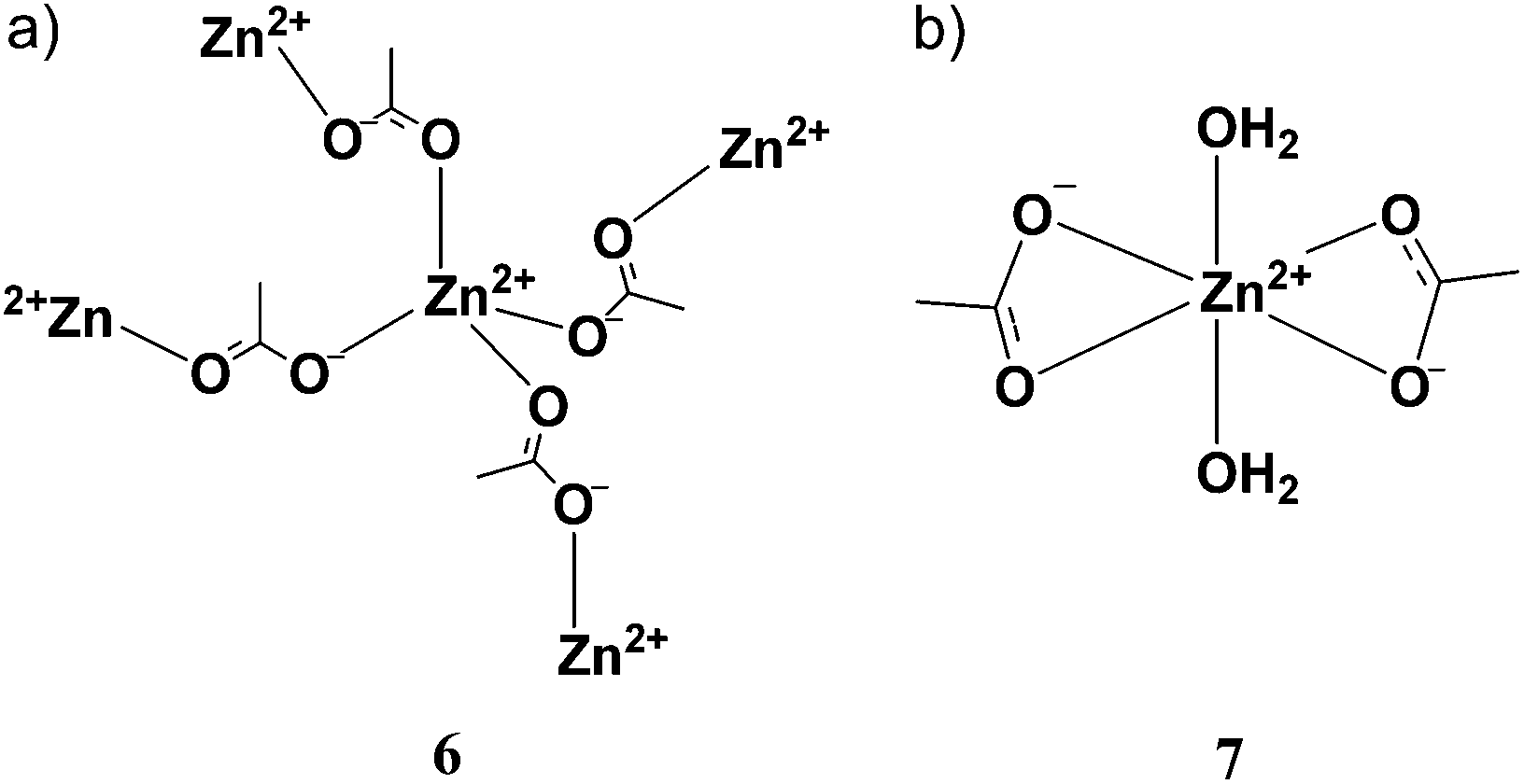 | ||
| Fig. 3 Reference compounds used in EXAFS studies. (a) In compound 6: (ZnAc2) Zn2+ is tetracoordinated with bridging acetate ligands. (b) In compound 7 (ZnAc2·2H2O) Zn2+ is hexacoordinated with bidentate acetate ligands and water. | ||
In Fig. 4a, we show the normalized X-ray absorption spectra χ(k) of zinc acetate dihydrate, anhydrous zinc acetate and the 10 mol% Zn-catalyzed epoxy-acid vitrimer system (compound 2) at room temperature. In this plot, it is already apparent that compound 2 and the anhydrous zinc salt have very similar χ(k) patterns whereas the spectrum of zinc acetate dihydrate shows quite different oscillations. In Fig. 4b, we present the results of EXAFS spectra analysis in the form of the radial structure function (A(R) plot) of the 10 mol% Zn-catalyzed epoxy-acid vitrimer system. In this representation, the maxima correspond to the presence of neighboring atoms at path length R. By fitting the whole χ(k) curve we determine the radial distance r and the coordination numbers N of each type of atom in the first shell. Note that the actual distance r is systematically larger than the position of the maximum in the A(R) plot due to the scattering phase shift. The general form of the 10 mol% Zn-catalyzed epoxy-acid vitrimer A(R) plot, and in particular the position and amplitude of the first shell maximum are similar to those of anhydrous zinc acetate whereas the whole A(R) plot is significantly different from that of zinc acetate dihydrate.
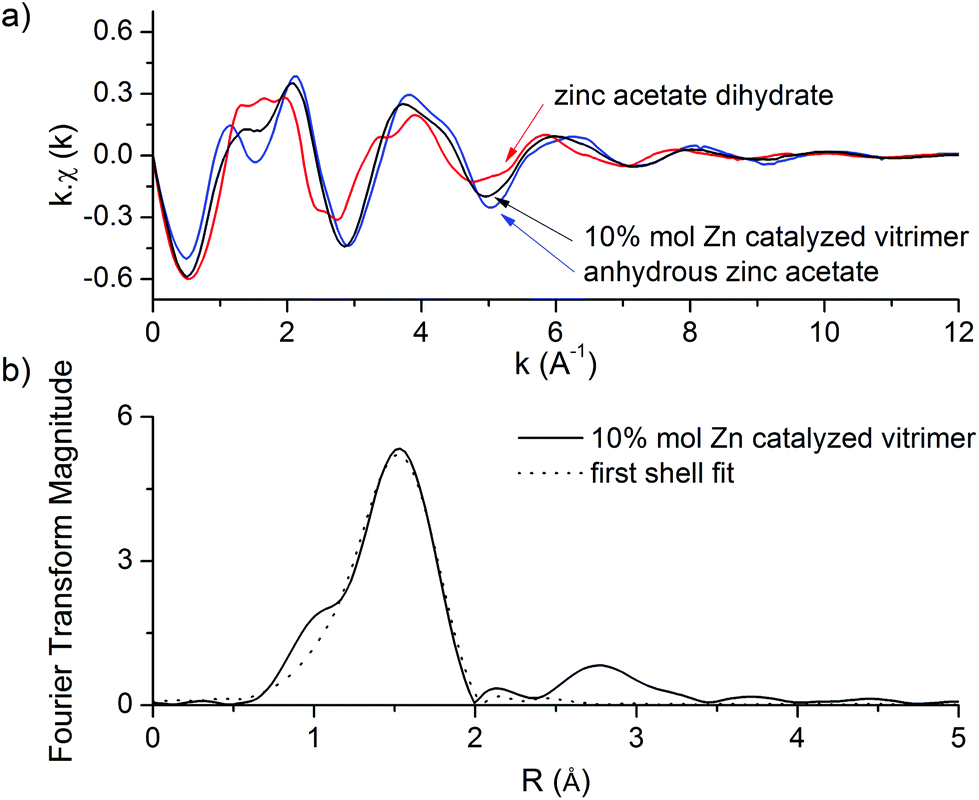 | ||
| Fig. 4 (a) EXAFS spectra of zinc acetate dihydrate (red), anhydrous zinc acetate (blue) and the 10 mol% Zn-catalyzed epoxy-acid vitrimer system (black). (b) Radial structure functions of the 10 mol% Zn-catalyzed epoxy-acid vitrimer system (solid, black) and the first shell fit associated (dot, black). | ||
The result of the first shell simulation is plotted as a dotted line in Fig. 4b. We determined by fitting a first shell coordination of N = 4.3 (±0.6) oxygen atoms and an average Zn–O distance of r = 1.98 (±0.01) Å at room temperature. At higher temperatures, i.e. 225 °C, this distance slightly decreases to a value of r = 1.95 (±0.01) Å while the coordination number also slightly decreases to N = 3.8 (±0.5). These distances and coordination numbers are close to those measured in anhydrous zinc acetate confirming that Zn2+ in vitrimers has a similar first shell environment i.e. the same coordination number and the same charges. In particular, it seems clear that the zinc cation in vitrimers is tetracoordinated with two neutral and two negatively charged oxygen atoms.
Concerning the more distant environment of the metallic cation, it is interesting to note that in bridged complexes such as compound 6, the Zn–Zn distance is about 4–5 Å. The existence of such bridges in vitrimers is also a question to consider. Unfortunately in all our data that were essentially collected at room temperature or above, we found it impossible to confirm that small peaks arising in this range, even in model compounds, actually correspond to a signal and not to noise.
Infrared spectroscopy
Epoxy-acid networks, cured in the presence of a catalyst or in the absence of any catalyst, have different chemical structures.38,39 Thus, to investigate vibrational changes induced by the presence of Zn2+, we took as the control a sample containing a lower concentration of Zn2+ (rather than a catalyst-free one) to ensure that the structure of the network is the same. Two epoxy vitrimer networks, with respectively 5 and 10 mol% of zinc, were analyzed by infrared (IR) spectroscopy in the attenuated total reflection (ATR) mode (Fig. 5a). In both the ATR-IR spectra of compound 2 (vitrimer with 10 mol% Zn catalyst) and compound 2 bis (vitrimer with 5 mol% Zn catalyst), one can recognize several characteristic bands: νCO of esters at 1737 cm−1, νC–C of aromatic fragments at 1604, 1579 and 1508 cm−1, δC–H of CH2 at 1458 cm−1, δC–H of C(CH3)2 at 1378 and 1363 cm−1, γ of CH2 at 1292 cm−1, several stretching vibrations of the C–O single bonds νC–O between 1276 and 1018 cm−1 and C–H deformation of aromatic fragments at 827 cm−1. To distinguish the characteristic vibrations associated with the presence of the metal from those of the matrix, we plotted in Fig. 5b the difference spectrum ATR(10%) − ATR(5%). In this plot, a positive (resp. negative) ΔATR signal for a given wavenumber indicates an increase (resp. a decrease) of the corresponding vibrator concentration. In the difference spectrum, basically five characteristic features appear: a strong negative band at 1737 cm−1, a large negative pattern between 1300 and 1100 cm−1 with a minimum at 1167 cm−1 and three positive signals centered at 1569, 1436 and 1026 cm−1. These observations show that the presence of the metal modifies the environment of C–O and CO vibrators. The negative bands correspond respectively to standard νCO and νC–O stretching vibrations of organic oxygenated compounds. In contrast, the two positive bands centered at 1569 and 1436 cm−1 are in the intermediate zone between νCO and νC–O stretching vibration wavenumbers and the positive band centered at 1026 cm−1 is at the edge of the classical νC–O stretching vibration region while all of them are clearly different from all the signals of starting materials (see the ESI†).
 | ||
| Fig. 5 (a) ATR-IR spectra of 5 mol% Zn-catalyzed epoxy vitrimer (red) and 10 mol% Zn-catalyzed vitrimer (black). (b) ATR-IR spectrum difference A(10%) − A(5%). | ||
Discussion
Zinc being a transition metal, it follows naturally that neutral and negatively charged oxygen atoms are present in its first coordination sphere, i.e. attached directly to the metal, as clearly observed in the EXAFS spectrum and the IR spectrum. The structures of oxygenated Zn(II) complexes are reported in the literature.36,37,40–44 Zinc is usually tetracoordinated, pentacoordinated or hexacoordinated, but tetrahedral geometry is the most frequently encountered. From EXAFS measurements, we established that the coordination number of Zn2+ in the epoxy-acid vitrimer system is very close to 4. The positive band centered at 1026 cm−1, observed in the IR difference spectrum, is characteristic of the vibration νC–O–M from a metallic alkoxide,44–46 represented as a type III complex in Fig. 6. This confirms the presence of Zn-bound alkoxide groups, already inferred from the reaction sequence exposed in Scheme 2C. The Zn–O mean distance in the epoxy-acid vitrimer system is the same as that in anhydrous zinc acetate. It suggests a similar environment of oxygen atoms, i.e. two neutral oxygen atoms and two negatively charged oxygen atoms. The characteristic C–O and CO vibrations of neutral oxygen atoms modified by the presence of zinc appear in the IR spectrum as shown in Fig. 5b.
 | ||
| Fig. 6 Possible complexation modes of a β-oxy ester group with a metal M. Experimental data suggest the presence of type I and type III complexation. | ||
New bands observed in the IR subtraction spectrum differ completely from the bands of the carboxylate observed in compound 4. The two negative bands correspond to the stretching frequencies of the ester group at 1737 cm−1 for CO and in the 1100–1300 cm−1 range for C–O. With the addition of zinc, these two bands diminish and two positive bands appear. The new stretching frequencies of the ester group in the presence of zinc fall outside the tabulated ranges of a free or H-bonded ester (1750–1735 cm−1 for CO, 1300–1000 cm−1 for C–O). The important shifts of these bands suggest that the ester group is involved in complexation. Metal complexes wherein the ligand is an ester have been widely reported in the literature.47–54 The two possible complexation modes of the ester group with a metal (type I and II) are presented in Fig. 6. The CO stretching frequency, as low as 1569 cm−1, reveals a decrease of the bond order, consistent with a partial electron transfer onto an oxygen–metal bond, the metal cation having a withdrawing effect thus strongly supports the complexation of type I. In the literature such metal complexation always induces a 50 to 150 cm−1 shift of the CO band towards lower frequencies. Oppositely, the withdrawing effect of the metal cation shortens the C–O bond whose stretching mode is shifted to higher frequencies. These IR-bands shifts, confirmed by computational simulations (see the ESI†), are in agreement with the presence of Zn2+ complexes of type I thus, interpretation as type I and type III complexes, supported by data seem the most appropriate. In Type II, the CO stretching vibration is shifted towards higher frequencies, it was clearly not observed but we cannot exclude that other categories of sp3 oxygen atoms (ether and hydroxyle) could be present in the first shell of Zn2+. Hydroxyles in particular are certainly present. Indeed β-diol structures (like compound 3) are produced by transesterification and the conjugated base, a β-hydroxy-alkoxide anion, is very likely to form a chelate with Zn2+ (see the ESI†).
Thus, the surroundings of Zn2+ in the epoxy-acid vitrimer system as revealed by the above analysis can be represented, as tentatively done in Fig. 7. Geometrically the best first shell approximation is the structure already shown in Fig. 3a for compound 6: two negatively charged oxygen atoms from alkoxides and two neutral oxygen atoms of sp2 or sp3 type are placed in the immediate neighborhood of the zinc atom. The sp2 oxygen atoms from carbonyl get activated by the electron withdrawing effect towards Zn2+. When trying to estimate the distances from the data of compound 6, we find at equilibrium an interatomic distance of about 3.5 Å between the carbonyl sp2 carbon and the alkoxide oxygen atoms. Thus the exchange reaction is very likely to occur between these two sites whenever Zn2+ complexation brings them close to one another. Therefore, the action of Zn2+, as a transesterification catalyst, can be summarized as follows: (1) by shifting the acid–base alcohol/alkoxide balance toward the more nucleophilic species alkoxide, (2) by activating the carbonyl via CO link polarization, increasing the partial charge of the sp3 carbon atom, and (3) by bringing the reacting centers close to each other.
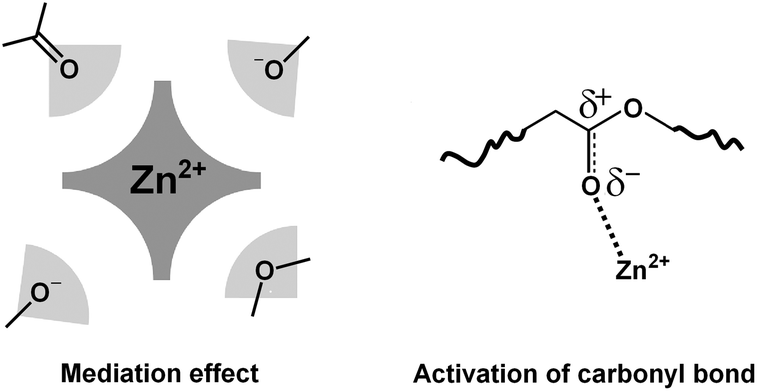 | ||
| Fig. 7 The two main actions of zinc as a catalyst in an epoxy-acid vitrimer network: mediation effect and ester activation designed from EXAFS and IR spectroscopy observations. | ||
Last, bringing reacting centers close to each other is the intrinsic role of transition metal catalysts ruled by their coordination number. Contrary to alkali or earth-alkaline metal catalysts, coordination bonds of transition metals are directional. Ligand positions in the coordination sphere are thus fixed and determine their proper orientation to react. The feature revealed here is that Zn2+ is chemically bonded to the vitrimer backbone, without intervention of another ligand. Thus, even though the coordination number does not exceed 4, there is a probability to find exchangeable functions, alkoxide and ester, in the immediate environment of the zinc cation.
In the literature, binuclear zinc complexes are described,51,55,56 and frequently observed in natural55,57,58 and synthetic catalytic processes.51,54,56,59 Coates’ group showed that not only bimetallic complexes are more stable but also monometallic complexes are unreactive during the copolymerization of epoxides and CO2.60 According to their observations, zinc centers do not act independently but rather in a cooperative bimetallic mode. Last, Hammes et al. observed that zinc catalyzed transesterification did not occur when the complex was mononuclear.51 The existence of such structures in vitrimers is an open question but the present data cannot resolve this point.
Conclusion
Metal-catalyzed transesterification is a key reaction in polymer chemistry. Extensively used to make polyesters or seen as an undesirable side reaction in ring-opening polymerizations,61 it was also recently recognized as an original route to obtain sequence-controlled copolymers.62 In this article, the catalytic role and the action modes of the zinc cation in epoxy-acid vitrimer networks have been investigated. Kinetic studies revealed that zinc catalyzed the transesterification reaction even at low concentration. EXAFS and IR studies revealed that zinc cations are tetracoordinated by oxygen atoms of the β-hydroxy-ester groups inherited from the epoxy-acid chemistry. Thus, Zn2+ is structurally bonded to the vitrimer network and this fact should be taken into account in the future for understanding the stress relaxation mechanism. Indeed, as the catalyst is not present everywhere, it has to diffuse to activate reactions throughout the sample. Chemical factors are analyzed here. The conclusions suggest that physical factors like the diffusivity of coordinated Zn2+, not yet investigated, could play a prominent role in the overall efficiency of the catalyst. As for chemical factors, Zn2+ acts as a catalyst by activating ester carbonyl, stabilizing alkoxide groups and moving them close to each other. It is known for organic catalysts that simultaneous activation of ester and alcohol strongly increases the rate of transesterification.63 So, apart from the fact zinc is an environmentally friendly metallic atom, also present in many natural processes, it appears as a very attractive multifunctional option to catalyze exchange reactions in epoxy vitrimers.Acknowledgements
We thank L. Leibler for inspiration of this work and much advice. We gratefully acknowledge helpful discussions with J. G. P. Goossens from Eindhoven University of Technology. We acknowledge ESRF (European Synchrotron Radiation Facility) for the provision of X-ray beam time and thank Dr D. Banerjee for his help with Dutch-Belgian Beamline (DUBBLE). We acknowledge D. Montarnal and R. Fournier for preliminary work. S.-J. Mougnier acknowledges the financial support from Arkema.References
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965 CrossRef CAS PubMed.
- M. Capelot, M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789 CrossRef CAS.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664 CrossRef CAS PubMed.
- E. Chabert, J. Vial, J. P. Cauchois, M. Mihaluta and F. Tournilhac, Soft Matter, 2016, 12, 4838 RSC.
- Z. Y. Pei, Y. Yang, Q. Chen, E. M. Terentjev, Y. Wei and Y. Ji, Nat. Mater., 2014, 13, 36 CrossRef CAS PubMed.
- Y. Yang, Z. Pei, X. Zhang, L. Tao, Y. Wei and Y. Ji, Chem. Sci., 2014, 5, 3486 RSC.
- Z. Pei, Y. Yang, Q. Chen, Y. Wei and Y. Ji, Adv. Mater., 2015, 28, 156 CrossRef PubMed.
- F. I. Altuna, V. Pettarin and R. J. Williams, Green Chem., 2013, 15, 3360 RSC.
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 607 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019 CrossRef CAS PubMed.
- Y. X. Lu, F. Tournilhac, L. Leibler and Z. J. Guan, Am. Chem. Soc., 2012, 134, 8424 CrossRef CAS PubMed.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451 CrossRef CAS.
- F. Romano and F. Sciortino, Phys. Rev. Lett., 2015, 114, 78104 CrossRef PubMed.
- F. Smallenburg, L. Leibler and F. Sciortino, Phys. Rev. Lett., 2013, 111, 188002 CrossRef PubMed.
- P. Zheng and T. J. McCarthy, J. Am. Chem. Soc., 2012, 134, 2024 CrossRef CAS PubMed.
- A. Rekondo, R. Martin, A. Ruiz de Luzuriaga, G. Cabanero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237 RSC.
- A. R. de Luzuriaga, R. Martin, N. Markaide, A. Rekondo, G. Cabañero, J. Rodríguez and I. Odriozola, Mater. Horiz., 2016, 3, 241 RSC.
- M. Pepels, I. Filot, B. Klumperman and H. Goossens, Polym. Chem., 2013, 4, 4955 RSC.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536 CrossRef CAS.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, J. Am. Chem. Soc., 2015, 137, 6078 CrossRef CAS PubMed.
- W. G. Skene and J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8270 CrossRef CAS PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698 CrossRef CAS PubMed.
- R. Nicolaÿ, J. Kamada, A. Van Wassen and K. Matyjaszewski, Macromolecules, 2010, 43, 4355 CrossRef.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975 CrossRef CAS PubMed.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 Search PubMed.
- S. Telitel, Y. Amamoto, J. Poly, F. Morlet-Savary, O. Soppera, J. Lalevee and K. Matyjaszewski, Polym. Chem., 2014, 5, 921 RSC.
- C. R. Fenoli, J. W. Wydra and C. N. Bowman, Macromolecules, 2014, 47, 907 CrossRef CAS.
- C. Yuan, M. Q. Zhang and M. Z. Rong, J. Mater. Chem. A, 2014, 2, 6558 CAS.
- K. Imato, T. Ohishi, M. Nishihara, A. Takahara and H. Otsuka, J. Am. Chem. Soc., 2014, 136, 11839 CrossRef CAS PubMed.
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272 CrossRef CAS PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30 RSC.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615 CrossRef CAS PubMed.
- R. V. Kudryavtsev and D. N. Kursanov, Zh. Obshch. Khim., 1957, 27, 1686 CAS.
- N. J. Harrick, Phys. Rev. Lett., 1960, 4, 224 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS PubMed.
- W. Clegg, I. Little and B. P. Straughan, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 1701 CrossRef.
- T. Ishioka, Y. Shibata, M. Takahashi, I. Kanesaka, Y. Kitagawa and T. Nakamura, Spectrochim. Acta, Part A, 1998, 54, 1827 CrossRef.
- D. Montarnal, F. Tournilhac, M. Hidalgo and L. Leibler, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1133 CrossRef CAS.
- F. Sordo, S.-J. Mougnier, N. Loureiro, F. Tournilhac and V. Michaud, Macromolecules, 2015, 48, 4394 CrossRef CAS.
- J. N. van Niekerk, F. R. L. Schoening and J. H. Talbot, Acta Crystallogr., 1953, 6, 720 CrossRef CAS.
- T. Ishioka, Y. Shibata, M. Takahashi and I. Kanesaka, Spectrochim. Acta, Part A, 1998, 54, 1811 CrossRef.
- Y. Marcus, Chem. Rev., 1988, 88, 1475 CrossRef CAS.
- I. L. Alberts, K. Nadassy and S. J. Wodak, Protein Sci., 1998, 7, 1700 CrossRef CAS PubMed.
- H. D. Kaesz and F. G. A. Stone, Spectrochim. Acta, 1959, 15, 360 CrossRef.
- F. K. Butcher, W. Gerrard, E. F. Mooney, R. G. Rees and H. A. Willis, Spectrochim. Acta, 1964, 20, 51 CrossRef CAS.
- C. T. Lynch, K. S. Mazdiyasni, J. S. Smith and W. J. Crawford, Anal. Chem., 1964, 36, 2332 CrossRef CAS.
- D. S. Bytrov, J. Struct. Chem., 1964, 4, 638 CrossRef.
- M. F. Lappert, J. Chem. Soc., 1962, 542 RSC.
- D. S. Bystrov, J. Struct. Chem., 1963, 4, 501 CrossRef.
- M. F. Lappert, J. Chem. Soc., 1961, 817 RSC.
- B. S. Hammes, X. Luo, M. W. Carrano and C. Carrano, J. Inorg. Chim. Acta, 2002, 341, 33 CrossRef CAS.
- I. Sóvágó, C. Kállay and K. Várnagy, Coord. Chem. Rev., 2012, 256, 2225 CrossRef.
- B. Schreiner and W. Z. Beck, Anorg. Allg. Chem., 2010, 636, 499 CrossRef CAS.
- B. S. Hammes, X. Luo, B. S. Chohan, M. W. Carrano and C. J. Carrano, J. Chem. Soc., Dalton Trans., 2002, 17, 3374 RSC.
- D. E. Wilcox, Chem. Rev., 1996, 96, 2435 CrossRef CAS PubMed.
- A. M. Baruah, A. Karmakar and J. B. Baruah, Inorg. Chim. Acta, 2008, 361, 2777 CrossRef CAS.
- J. A. Cricco, E. G. Orellano, R. M. Rasia, E. A. Ceccarelli and A. J. Vila, Coord. Chem. Rev., 1999, 190, 519 CrossRef.
- N. Sträter, W. N. Lipscomb, T. Klabunde and B. Krebs, Angew. Chem., Int. Ed. Engl., 1996, 35, 2024 CrossRef.
- S. K. Papageorgiou, E. P. Kouvelos, E. P. Favvas, A. A. Sapalidis, G. E. Romanos and F. K. Katsaros, Carbohydr. Res., 2010, 345, 469 CrossRef CAS PubMed.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911 CrossRef CAS PubMed.
- M. Bouyahyi and R. Duchateau, Macromolecules, 2014, 47, 517 CrossRef CAS.
- K. Nakatani, Y. Koda, T. Terashima and M. Sawamoto, J. Am. Chem. Soc., 2012, 134, 4373 CrossRef CAS PubMed.
- J. Kadota, D. Pavlović, J.-P. Desvergne, B. Bibal, F. Peruch and A. Deffieux, Macromolecules, 2010, 43, 8874 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py00752j |
| This journal is © The Royal Society of Chemistry 2016 |