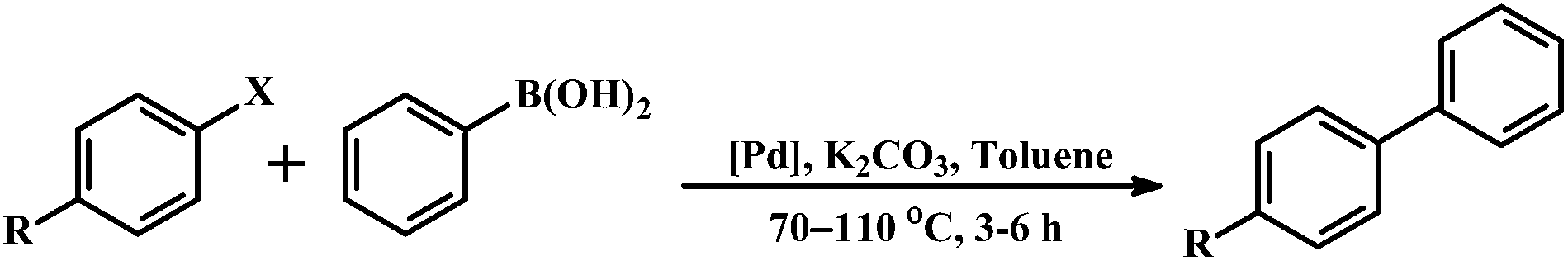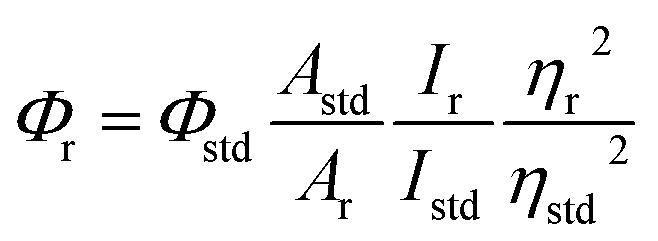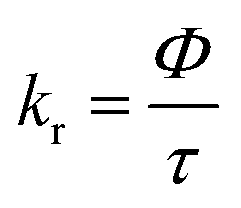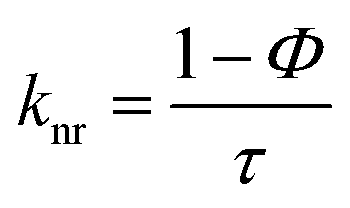DOI:
10.1039/C4RA16584E
(Paper)
RSC Adv., 2015,
5, 22544-22559
Insight into luminescent bisazoaromatic CNN pincer palladacycle: synthesis, structure, electrochemistry and some catalytic applications in C–C coupling†
Received
17th December 2014
, Accepted 16th February 2015
First published on 17th February 2015
Abstract
Chelating behavior of a potential flexidentate bisazoaromatic molecule 2-(phenylazo)azobenzene (PAAB) toward palladium have been examined. Activation of one instead of the two C(Ph)–H bonds of PAAB (HL) in the course of the metallation furnishes an exclusive unsymmetrical mode of ligation. Consequently, another member of the important class of CNN pincer palladacycles is achieved in almost quantitative yields. Crystallographic analysis authenticates the monoanionic terdentate ligation of the bisazoaromatic molecule and neutral [PdII(L)X] (X = acetate and halides) complexes with planar palladacycle and out-of-plane pendant phenyl ring. Unlike the N-donor pincers incorporating imine moieties, the introduction of two azo chromophores not only makes the PAAB ligand strongly π-acidic but also significantly modulates the FMOs in the palladacycles. An appreciable lowering of ligand-centered (LC) π* orbitals is apparent upon complexation which is in accordance with the electrochemical responses where large anodic shifts of LC LUMO and LUMO + 1 by 0.8–0.9 V have been noted. The significant modification of FMOs leads to interesting optoelectronic properties in the palladacycles. The cyclopalladated compounds are luminescent in solution at room temperature. This property is plausibly a result of the LC emissive states (azo π*). The optoelectronic features are interpreted by the Time-Dependant (TD) density functional theory (DFT) and natural transition orbital (NTO) analyses. In addition, scrutiny of Suzuki–Miyaura and Heck-type reactions indicates that the pincer palladacycles may be employed in the development of useful catalysts for C–C coupling reactions.
Introduction
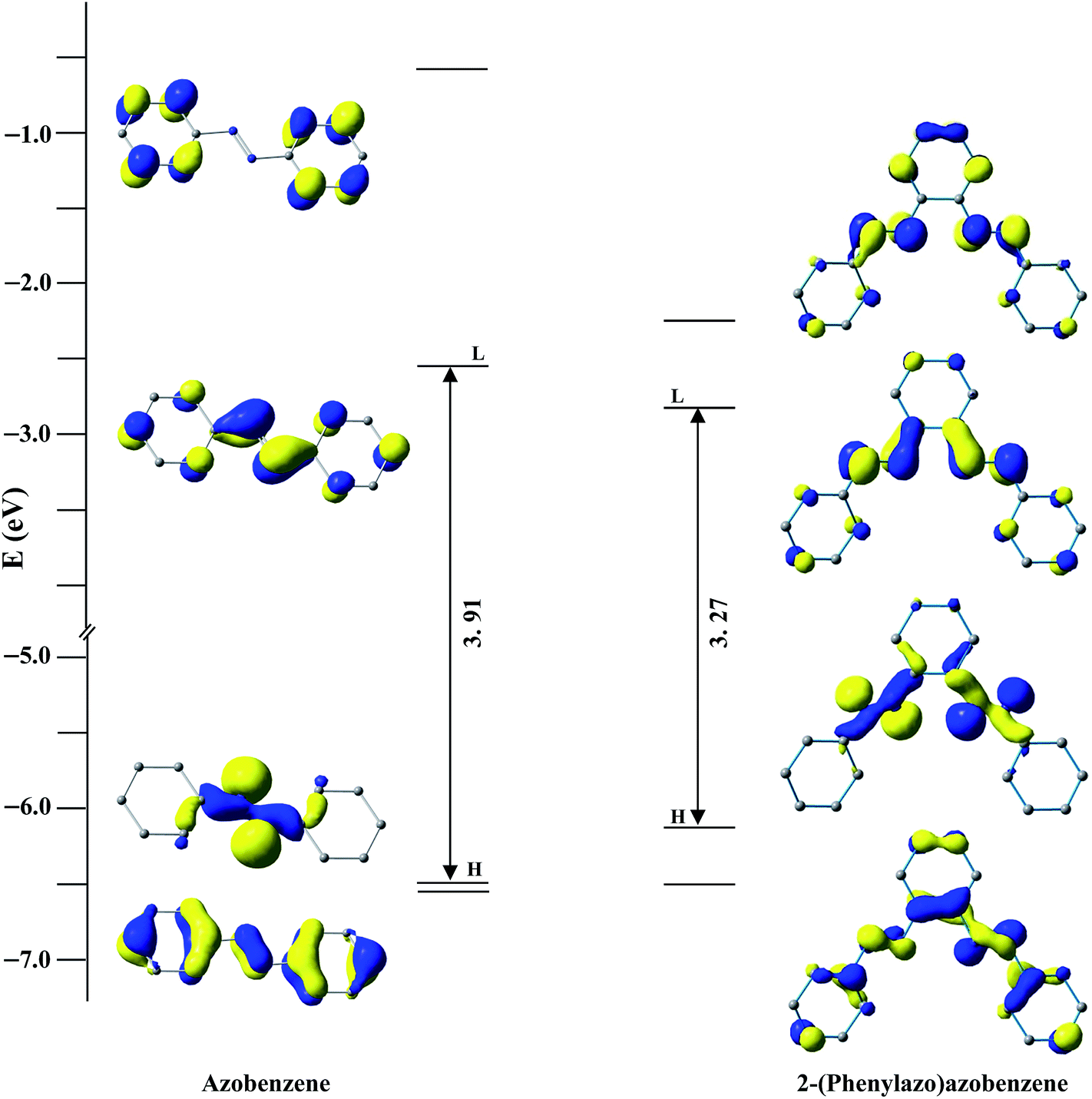
After the discovery of a [C,N] palladacycle via activation of the ortho C–H bond of azobenzene by Cope and coworkers1 in 1965, orthometallated palladium compounds have received increasing attention with azo, imine and other functionalities over the decades in coordination and organometallic chemistry.2 Among azobenzene complexes, two types of coordination modes namely monocyclopalladated3 and doubly cyclopalladated4 species have been documented (Scheme 1). Recently, another form of double cyclopalladation has been reported with 4-(phenylazo)azobenzene (Scheme 1).5 Transition metal complexes with the azo chromophore have been found to display interesting physicochemical properties such as photoluminescence (PL), photoconductivity, photoisomerization in addition to electrochemical and catalytic activities.6 Notably, the [C,N] palladacycles reported so far contain a single azo chromophore. Introduction of a higher homologue of azobenzene with multiple azo functionalities may appear as an attractive route to synthesize interesting azo compounds. We have employed a ligand incorporating two azo chromophores in conjugation with phenyl rings, 2-(phenylazo)azobenzene (PAAB) (Scheme 2). A comparison of frontier molecular orbitals (FMOs) of PAAB with that of azobenzene signifies superior σ-donor as well as π-acceptor behavior of the former (Scheme 2). The reason behind the choice of such multidentate azoaromatic compound lies in its orthometallating nature and azo-based enhanced π-acceptor ability, which causes strong d-orbital splitting and leads to significant stabilization of lower valence states of the metal. Substantial lowering of ligand-centered (LC) virtual FMOs in the bisazoaromatic system relative to that of metal-centered (MC) level may offer an easy way to modify the optoelectronic properties of the palladacycles. The complexes display luminescence in fluid solution at room temperature. The reports on luminescent mononuclear nonporphyrin palladacycles at room temperature are sparse and they usually display luminescence only in a rigid matrix at 77 K.7,8 Recently, Che and coworkers reported the most luminescent Pd(II) complexes till date in fluid solution using tetradentate ONCN ligand.9 The lack of room temperature luminescence for cyclopalladated compounds in solution is likely due to population of thermally accessible low-lying MC state (4dx2−y2) which leads to effective non-radiative decay through severe excited-state structural distortion.10
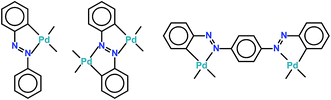 |
| | Scheme 1 Mono- and doubly cyclopalladation of azobenzene and 4-(phenylazo)azobenzene. | |
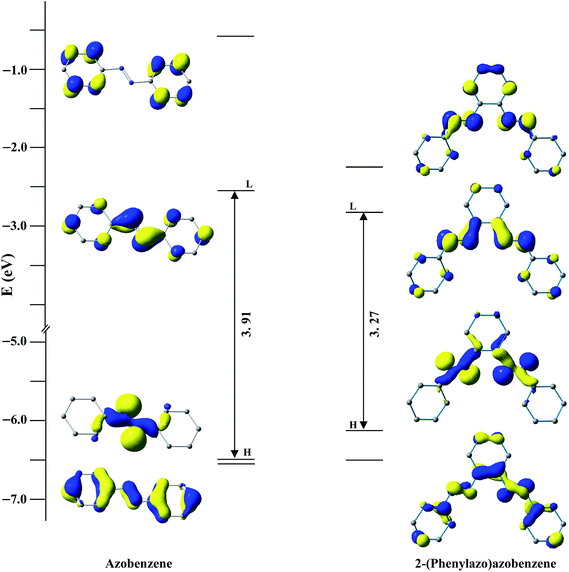 |
| | Scheme 2 Comparison of FMOs between azobenzene and 2-(phenylazo)azobenzene at B3LYP/(6-311+G(d,p) level. | |
Though a report of isolation and in part characterization of the ligand has been previously documented,11 we present a modified synthetic procedure with comprehensive characterization including crystal structure determination. We observe an exclusive pincer CPhNazoNazo type ligation of PAAB toward Pd(II) (coordination mode A), nonetheless the free ligand possesses four potential donors following the activation of two C–H bonds (Scheme 3). Accordingly, the ligand provides another member of the important class of unsymmetrical CNN pincer. The CNN pincer complexes incorporating Pd(II) are less common compared to that of Pt(II).7a,8e,12 Notably, pincer type palladacycles are one of the most well-known and scrutinized classes of orthometallated compounds.13 These attractive classes of non-phosphine palladacycles disclose a challenge to chemists not only in terms of their synthesis but also in terms of their structure, design, and the mode of coordination of the ligand to the metal. Significantly, unlike conjugated aromatic polyimine molecules, the polyazo ligands are sparse in the pincer chemistry and there are only two recent reports14 of bisazoaromatic ligands viz. 3-(phenylazo)azobenzene and 2,6-bis(phenylazo)pyridine with Rh(III) and Fe(II) respectively. It is worth mentioning that the variation of coordinating atoms in the anionic six-electron donor ligand may alter the nature of the metal center radically and make the palladacycles useful for much wider application in the organic synthesis and catalysis.15 In this context, deployment of superior π-accepting pincers are most enviable since the majority of catalytic cycles of Pd-catalyzed reactions are generally based on PdII/Pd0 shuttles.16 In this work, we report the high-yield synthesis and characterization of a new set of unsymmetrical CNN pincer palladacycles with 2-(phenylazo)azobenzene, as well as the discussion and rationalization of their structural and optoelectronic properties. Presence of low-lying LC virtual orbitals is appraised by means of electrochemical study (cyclic voltammetry). The experimental results are complemented by theoretical calculations in order to explain the nature of the electronic transitions in the palladacycles. Preliminary catalytic activity of the non-phosphine Pd(II) complexes has been explored toward C–C coupling reaction.
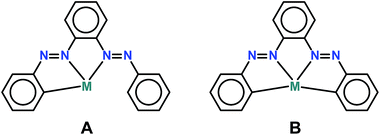 |
| | Scheme 3 Probable coordination modes of PAAB. | |
Results and discussion
Synthesis, characterization and electrochemical study
The PAAB ligand was synthesized by Baeyer–Mills coupling reaction17 of 2-(amino)azobenzene with nitrosobenzene in glacial acetic acid at elevated temperature followed by the extraction with n-hexane. The reaction of Pd(OAc)2 and PAAB in glacial acetic acid afforded [Pd(L)OAc] 1 in almost quantitative yield. The chloro derivative [Pd(L)Cl] 2 was obtained from K2[PdCl4] under similar reaction conditions in near stoichiometric yield. The ligand furnishes unsymmetrical terdentate CNN mode of coordination in the palladacycles following the activation of one C(Ph)–H bond despite of the presence of two such C–H bonds in the free ligand. Coordination mode A is favored over coordination mode B plausibly due to get rid of the structural strain within the rigid PAAB skeleton. The anionic six-electron donor PAAB furnishes two contiguous five-membered chelates and provides robust metal–organic conjugates, which limits their dissociation from the metal center. The other halo derivatives [Pd(L)Br] 3 and [Pd(L)I] 4 are obtained by substituting the OAc− of 1 with the respective excess of X− (X = Br, I) in acetone. All the complexes are soluble both in polar and nonpolar solvents such as toluene, dichloromethane, acetonitrile, ethanol, ether and furnishing a red solution. The acetato complex was obtained as H-bonded dimer, where the free O atoms of acetato groups are linked with one water molecule via H bonding. The presence of water of hydration was corroborated with thermogravimetric analysis (TGA). Thermogram (ESI, Fig. S1†) shows two decomposition steps within the temperature range 40–300 °C. The first step of decomposition, within the temperature range 40–150 °C, corresponds to the loss of water molecule of hydration with a mass loss of 2.2% (calcd 2.0%). The second step (150–300 °C) corresponds to the removal of the acetate group with a mass loss of 12.7% (calcd 13.1%). Sharp vibrations around 1486–1439 cm−1 in the infrared spectra of the free ligand were assigned to the νN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (ESI, Fig. S2†). The observed lowering of νNN values in complexes (1445–1430 cm−1) as compared to that of free ligand is consistent with the Pd(II) → π*(azo) back-bonding. An electrospray ionization (ESI) mass spectrum (MS) of the ligand in acetonitrile displays a peak at m/z 287 amu for [L + H]+ (ESI, Fig. S3†). The analogous spectra of complexes show the peaks for [M − X]+ and [2M − X]+ with the expected isotopic distribution pattern (ESI, Fig. S4†), indicating their substantial stability in solution. The NMR spectra of ligand and complexes are presented in ESI (Fig. S5 and S6†).
N (ESI, Fig. S2†). The observed lowering of νNN values in complexes (1445–1430 cm−1) as compared to that of free ligand is consistent with the Pd(II) → π*(azo) back-bonding. An electrospray ionization (ESI) mass spectrum (MS) of the ligand in acetonitrile displays a peak at m/z 287 amu for [L + H]+ (ESI, Fig. S3†). The analogous spectra of complexes show the peaks for [M − X]+ and [2M − X]+ with the expected isotopic distribution pattern (ESI, Fig. S4†), indicating their substantial stability in solution. The NMR spectra of ligand and complexes are presented in ESI (Fig. S5 and S6†).
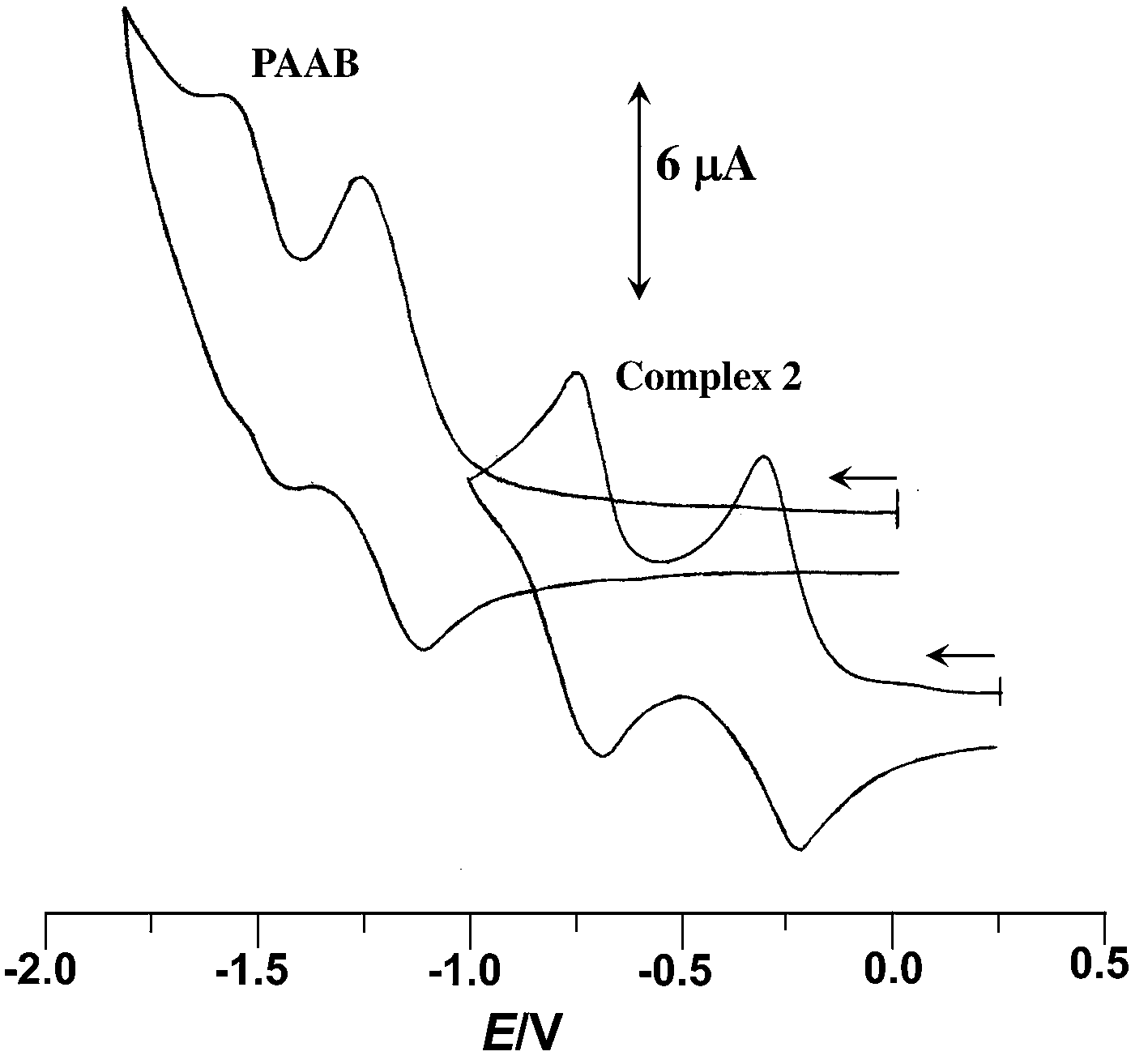
The redox behavior of the uncoordinated ligand and the cyclopalladated compounds has been scrutinized by cyclic voltammetry; the data collected in Table 1. The free ligand exhibits two successive one-electron quasi-reversible reductive responses, at −1.21 and −1.51 V (vs. Ag/AgCl) (Fig. 1), indicating the chronological filling of π* orbitals. The facile reductive nature of PAAB is consistent with its superior π-acceptor ability. A significant anodic shift of these redox couples, by ∼0.9 and ∼0.8 V respectively, was observed upon palladation (Fig. 1). The reductive responses near −0.25 and −0.75 V vs. Ag/AgCl may be attributed to the azo reductions in the complexes and this trend can be ascribed to remarkable stabilization of ligand-centered LUMO and LUMO + 1 in presence of electro-positive metal center.
Table 1 Electrochemical Dataa
| Compound |
E1/2/V (ΔEp/mV) |
| Solute concentration ≈10−3 mol dm−3 scan rate, 50 mV s−1; E1/2 = 0.5(Epa − Epc), where Epa and Epc are the anodic and cathodic peak potentials, respectively; ΔEp = Epa − Epc. |
| PAAB |
−1.21(160), −1.51(160) |
| [Pd(L)(OAc)] 1 |
−0.30(81), −0.76(76) |
| [Pd(L)Cl] 2 |
−0.28(80), −0.73(76) |
| [Pd(L)Br] 3 |
−0.26(82), −0.74(78) |
| [Pd(L)I] 4 |
−0.26(82), −0.73(77) |
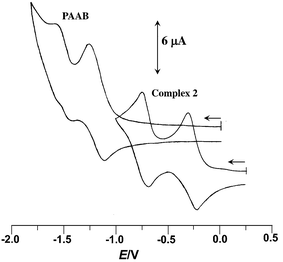 |
| | Fig. 1 Voltammogram of PAAB and 2 in acetonitrile at 50 mV s−1. | |
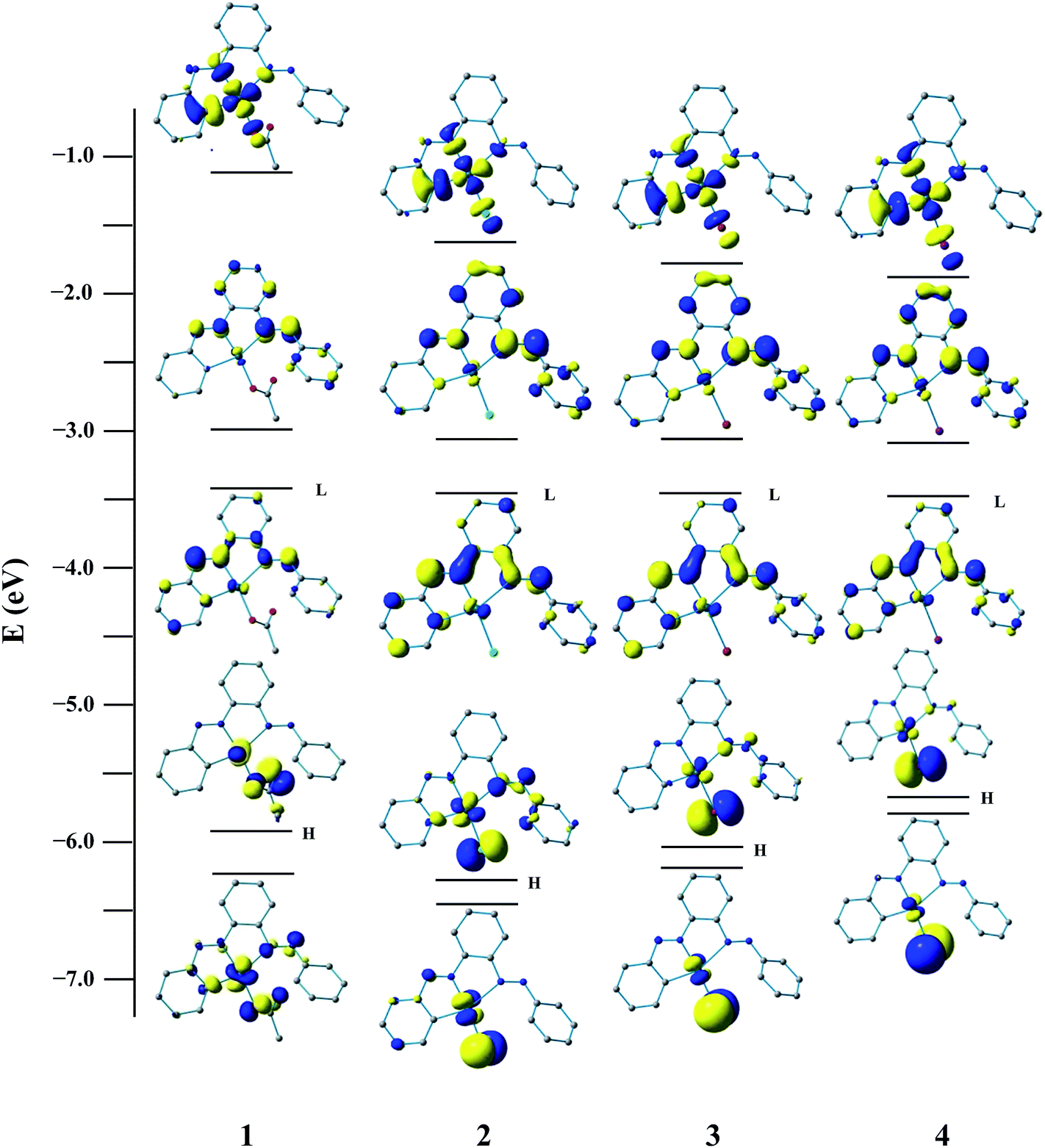
To gain insight into the nature of the redox orbitals, we have performed DFT calculation of the free ligand and complexes. The observed redox responses nicely substantiated with the theoretical study. Illustrations of the selected FMOs of the complexes are presented in Fig. 2. Calculation indicates that LUMO and LUMO + 1 of PAAB encompass exclusively the azo as well as phenyl moieties and remain symmetric along the C2 axis. A sizeable stabilization of both LUMO and LUMO +1 by ∼0.60 and ∼0.75 eV respectively upon complexation has been computed (ESI, Tables S1–S5†). In these complexes, a slight polarization between the coordinated and pendant phenyl rings has been observed for these virtual orbitals. The energies of these FMOs in the palladacycles remain almost insensitive with the nature of coligands, excluding their involvement in the redox processes. The components of LUMO and LUMO + 1 of all the complexes are essentially similar in nature and comprise exclusively azo-π* (50%) and phenyl-π* (50%) of coordinated bisazoaromatic ligand with practically no participation of metal orbitals (ESI, Tables S2–S5†). It is very likely that these electrochemical processes correspond to the reduction of the bisazoaromatic system, generating the metal-stabilized azo anion radicals (eqn (1) and (2)). Redox noninnocence behavior of the azo ligands is well documented in the literature.18
| | |
[PdII(L−I)X] + e− ↔ [PdII(L−II)X]−
| (1) |
| | |
[PdII(L−II)X]− + e− ↔ [PdII(L−III)X]2−
| (2) |
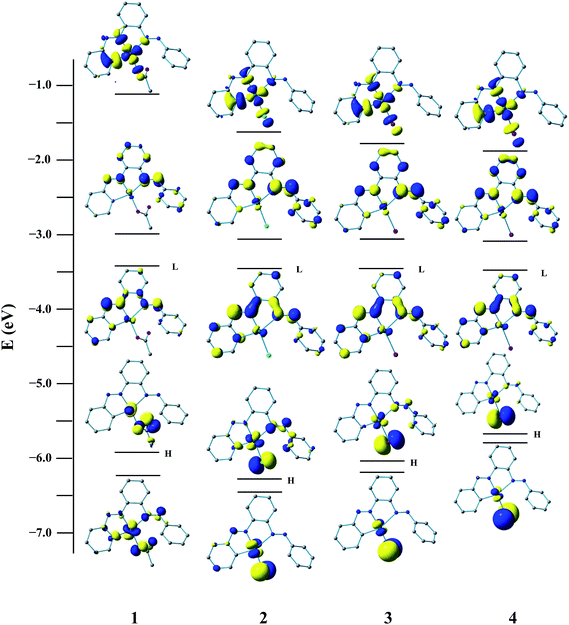 |
| | Fig. 2 Isodensity surface plots of some selected FMOs (HOMO − 1 to LUMO + 2) for the complexes 1, 2, 3 and 4 at their optimized S0 geometry in gas phase (isodensity value 0.05 e Bohr−3). | |
Crystal structures
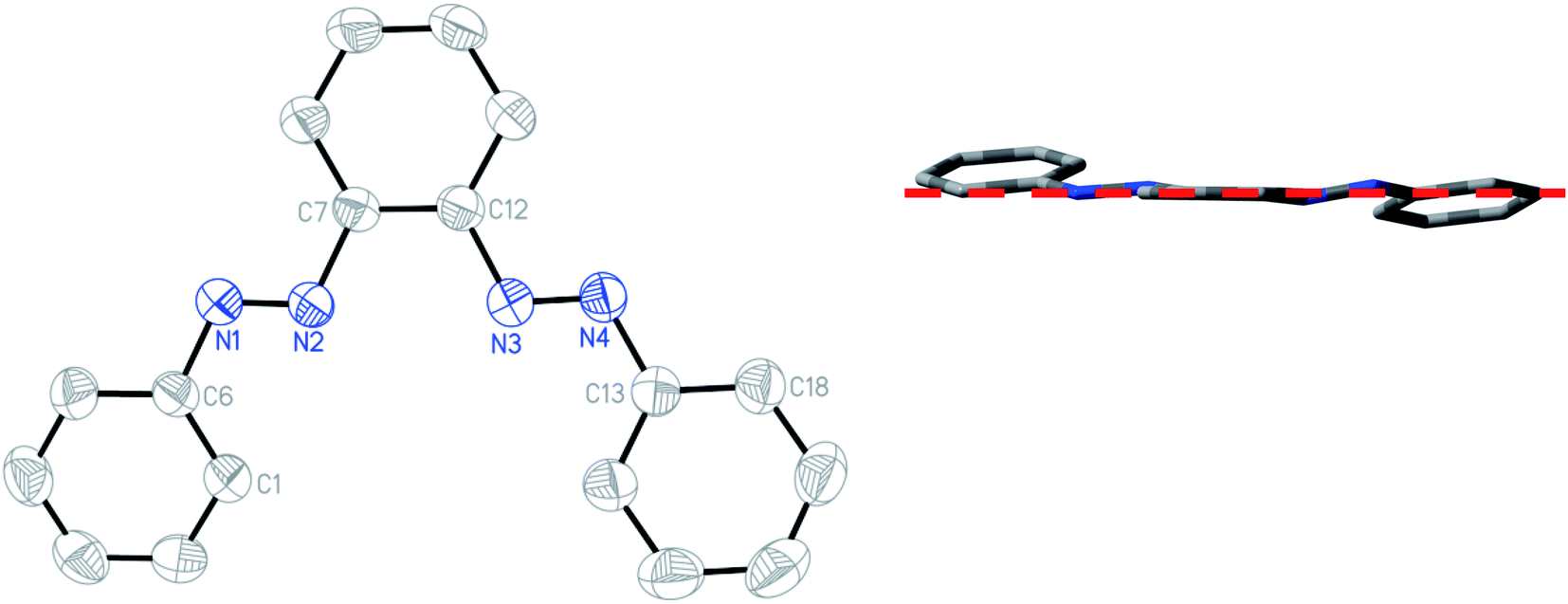
The structures of the free ligand and all four complexes have been authenticated by single-crystal X-ray diffraction. Crystallographic details are given in ESI (Table S6†), significant metrical parameters in Table 2. Rod-shaped crystals suitable for X-ray crystallography of the free ligand have been grown by slow evaporation of hexane solution of the compound. Fig. 3 shows the ORTEP and atom numbering scheme of PAAB. The X-ray crystallographic analysis reveals a symmetrical (C2) and almost planar structure (md = 0.165 Å), with practically identical dN–N values of 1.244(1) and 1.243(1) Å for two azo groups, respectively. Two phenylazo groups are lying opposite to each other with respect to the plane passing through the central benzene ring (inset of Fig. 3).
Table 2 Selected Metrical Parameters for PAAB, 1, 2, 3 and 4
| Bond parameter |
PAAB |
1 |
2 |
3 |
4 |
| Pd1–C1 |
|
1.974(2) |
1.971(3) |
1.978(3) |
1.984(5) |
| Pd1–N2 |
|
1.945(2) |
1.961(2) |
1.971(2) |
1.973(4) |
| Pd1–N3 |
|
2.182(2) |
2.229(2) |
2.227(2) |
2.207(4) |
| Pd1–O1 |
|
2.053(2) |
|
|
|
| Pd1–Cl1 |
|
|
2.289(1) |
|
|
| Pd1–Br1 |
|
|
|
2.412(1) |
|
| Pd1–I1 |
|
|
|
|
2.590(1) |
| N1–N2 |
1.244(1) |
1.274(2) |
1.282(3) |
1.280(3) |
1.272(5) |
| N3–N4 |
1.243(1) |
1.258(3) |
1.242(3) |
1.252(3) |
1.258(5) |
| C1–Pd1–N2 |
|
79.27(9) |
79.49(10) |
79.70(10) |
79.39(17) |
| N2–Pd1–N3 |
|
80.69(7) |
78.90(9) |
78.70(9) |
78.75(14) |
| O1S⋯O2A (H1SB⋯O2A) |
|
2.868 (2.036) |
|
|
|
| O1S⋯O4A (H1SB⋯O4A) |
|
2.869 (2.086) |
|
|
|
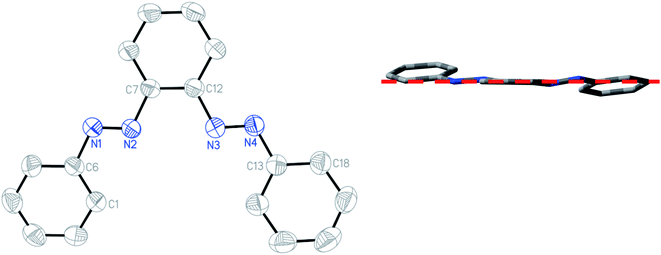 |
| | Fig. 3 ORTEP of PAAB with ellipsoids at the 50% probability level. Hydrogen atoms are omitted for clarity. Inset: Molecular view along the plane passing through the central benzene ring. | |
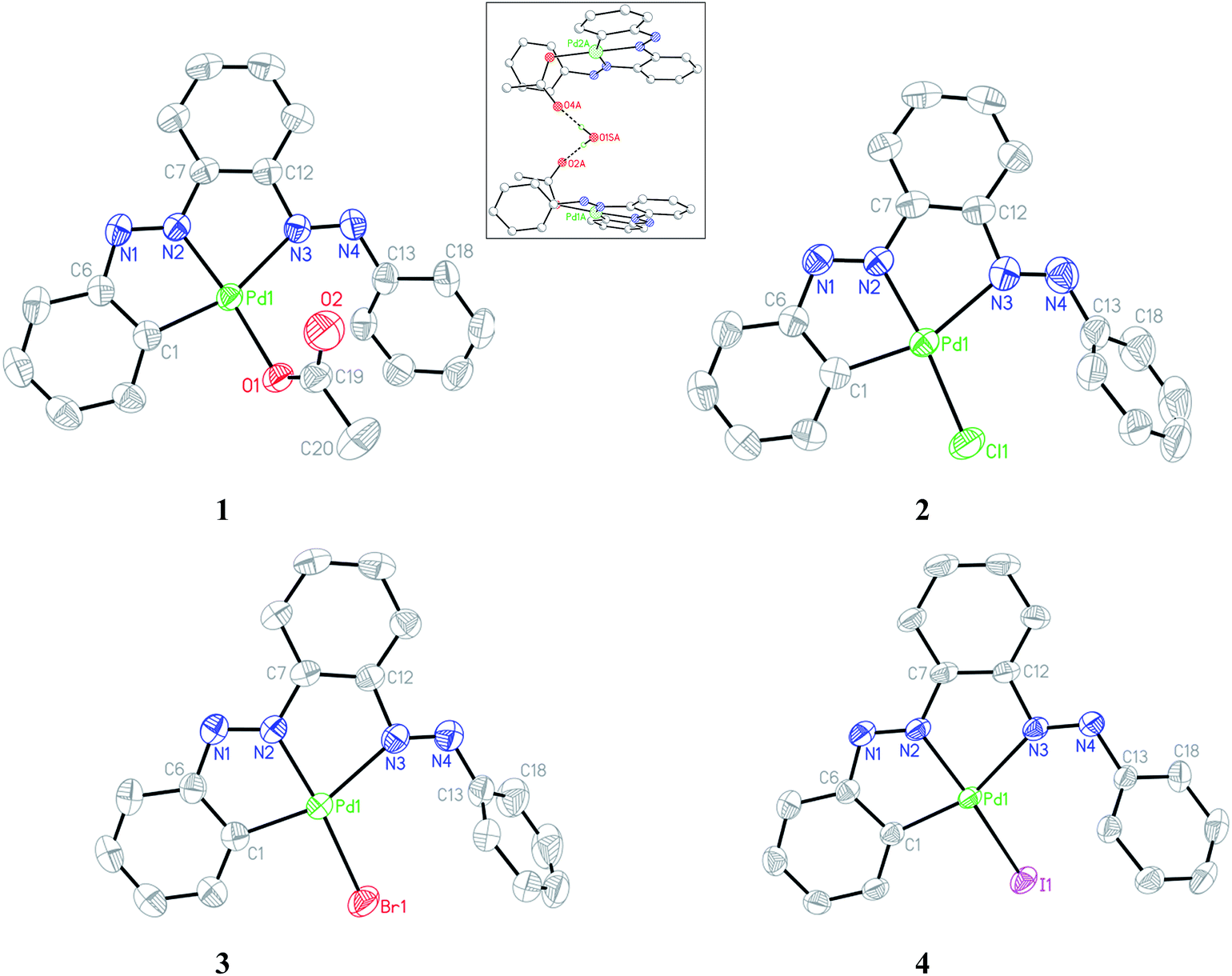
Single crystals of complexes 1–4 suitable for X-ray diffraction analysis were obtained by slow diffusion of their respective toluene solution into hexane solvent. The ORTEP and atom-numbering scheme are shown in Fig. 4. The bis azo ligand acts as monoanionic tridentate donor toward Pd(II) using one CPh and two Nazo atoms with the formation of two contiguous five-membered chelates, albeit it possesses four potential donor atoms; furnishing an unsymmetrical coordination mode. The coordination geometry of all the complexes is described as a square planar with two dissimilar Pd–N(azo) bond lengths differ by ∼0.25 Å. The Nazo atom closer to orthometallated CPh forms a stronger bond with the metal relative to the other Nazo atom. The appreciable difference in Pd–Nazo bond lengths is reflected in the corresponding dN–N distances. The lengthening of N1–N2 as compared to N3–N4 bonds by ∼0.03 Å is in accordance with the greater amount of Pd(d) → azo(π*) back donation in the former azo linkage. A systematic variation in the Pd–N2 bond was observed in the four complexes with the shortest length in 1 and the longest in 4. This observation is in accordance with the trans effect order: I− > Br− > Cl− > OAc−.
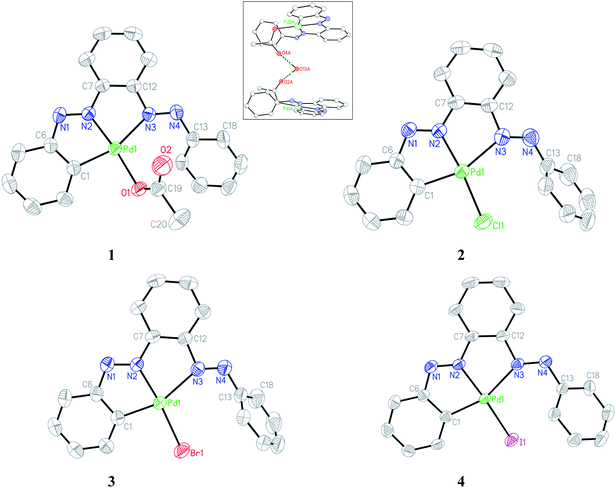 |
| | Fig. 4 ORTEP of 1–4 with ellipsoids at the 50% probability level. Hydrogen atoms are omitted for clarity. Inset: H-bonding interaction in acetato complex. | |
The Pd1–N3 bond length remains almost unaltered in the halo compounds while a slight decrease of ∼0.04 Å in 1 occurs possibly due to less steric crowding. The pendant phenyl rings (C13–C18) do not lie on the molecular plane in the complexes and makes an angle varying in the range 41–68° with the rest. Crystallographic analysis reveals that the two acetato complexes are joined via intervening water molecule with strong H-bonding (Oacetato⋯Owater 2.87(1) Å) as shown in the inset of Fig. 4. The chelate bite angle C1–Pd1–N2 is marginally larger than that of N2–Pd1–N3 by ∼1°.
Ground state geometries and frontier molecular orbital compositions
All the complexes are diamagnetic at room temperature indicating their singlet ground state. The geometry optimizations of the complexes were performed using their crystallographic coordinates at (R)B3LYP levels in gas phase in their singlet spin state without any ligand simplification. The optimized geometries of the synthesized complexes and the significant metrical parameters are given in ESI (Fig. S7 and S8 and Table S7† respectively). The optimized structural parameters of 1, 2, 3 and 4 are in general agreement with the experimental values and the slight discrepancy arises due to the crystal lattice distortion existing in real molecules. The calculated N–N(azo) distance of the free ligand (dN–N 1.253 Å) is found to be shorter, as compared to their coordinated forms (average dN–N 1.276 and 1.263 Å, respectively), indicating reasonable population of ligand π* in complexes. Isodensity plots of some selected FMOs of the Pd(II) complexes and energy levels (H − 1 to L + 2) are depicted in Fig. 2. The partial frontier molecular orbital compositions of the PAAB ligand and the Pd(II) complexes along with the HOMO–LUMO energy gap, are listed in ESI (Tables S1–S5†).
In the ground state, the HOMO of 1 mainly consists of 2p(OAc) with little 4dz2(Pd) character, while that of 2 is constituted largely of 3p(Cl) with minor participation of π(Ph) and π(azo). The more stabilized HOMO – 1 in both the complexes are constituted of comparable metal participation (∼25%), whereas the involvement of the respective coligand OAc/Cl differs appreciably. The HOMO and HOMO − 1 are mostly 4p(Br)/5p(I) coligand character for 3 and 4. The LUMO and LUMO + 1 are essentially similar in all four complexes and composed of ∼50% π*(azo) and ∼50% π*(Ph) of the PAAB ligand. These two FMOs are substantially stabilized upon complexation. It is worth noting that the LUMO + 2 in these complexes are basically analogous in nature and alike to 4dx2−y2 of Pd (∼45%) with substantial contribution from π*(Ph) (∼35%). Computation, therefore, reveals that LUMO and LUMO + 1 (LC states) are appreciably stabilized relative to the LUMO + 2 (MC level) by ∼1.85 and ∼1.45 eV, respectively.
Photophysical studies
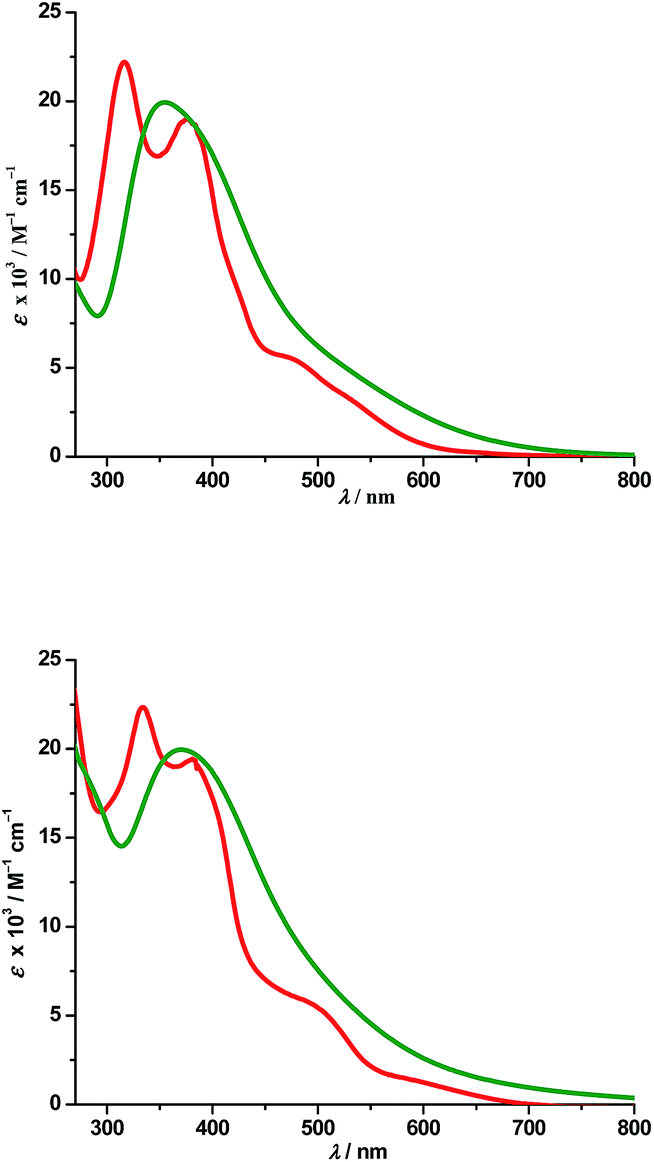
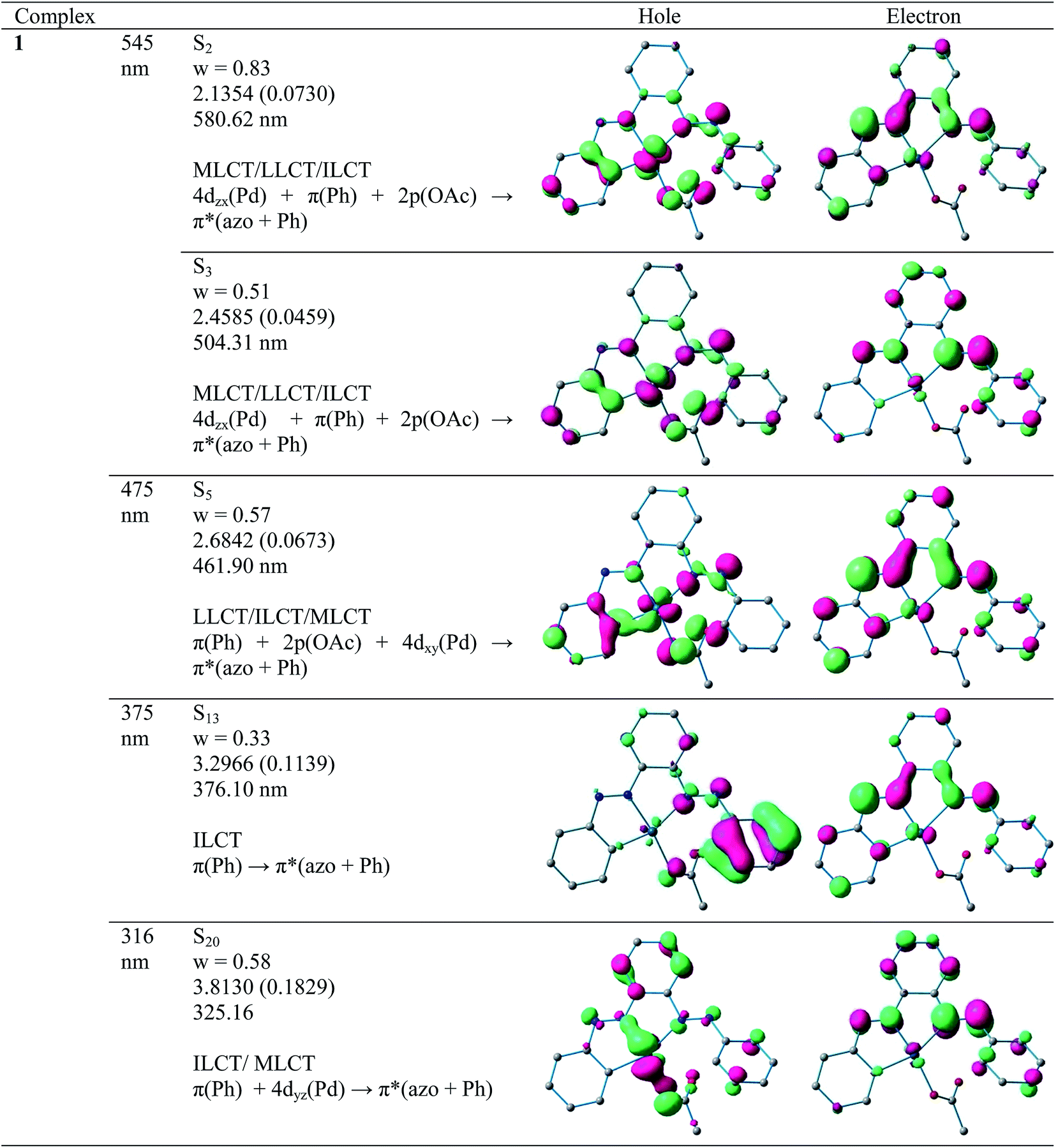
a. UV-Vis absorption spectra. Electronic spectra of compounds 2 and 4 were recorded in dichloromethane at room temperature and are depicted in Fig. 5 along with their respective theoretical spectra and that of compounds 1, 3 and PAAB are shown in ESI (Fig. S9†). More structured profile observed for palladacycles may result from the increased electronic asymmetry caused by the presence of the electron-accepting PAAB ligand. Multiple transitions are the characteristics of the spectra for these complexes (Table 3) and the excitations are attributed primarily to the charge-transfer transitions within the tri-coordinated bisazoaromatic ligand with varying amounts of anionic coligand-p orbital and metal-d orbital participation. To gain deeper insight into the electronic properties of the transitions involved in the optical absorption processes, we investigated all the synthesized complexes by means of time-dependent density functional theory (TD-DFT) in CH2Cl2 solvent using CPCM model. The most relevant transitions involved, along with their energy, character, oscillator strengths of all the compounds, are listed in ESI (Tables S8–S11†). In order to analyze the nature of absorption, we performed an NTO analysis based on the calculated transition density matrices.19 This method offers the most compact representation of the transition density between the ground and excited states in terms of an expansion into single-particle transitions (hole and electron states for each given excitation). Here we refer to the unoccupied and occupied NTOs as “electron” and “hole” transition orbitals. The NTOs for 1 are illustrated in Fig. 6 and that of 2, 3 and 4 are given in ESI (Fig. S10–S12†). Based on TD-DFT NTO analysis for 1, lower energy transition around 545 nm is computed at 580 nm (2.1354 eV, f = 0.0730) and 504 nm (2.4585 eV, f = 0.0459) as the 4dzx(Pd) + 2p(OAc) + π(Ph) → π*(azo + Ph) 1MLCT/1LLCT/1ILCT transition. The transition near 475 nm (2.6842 eV, f = 0.0673, λtheo = 462 nm) is computed as the 2p(OAc) + π(Ph) + 4dxy(Pd) → π*(azo + Ph) 1LLCT/1ILCT/1MLCT transitions. For 2, lower energy transitions around 530 nm and 480 nm are computed at 523 nm (2.3803 eV, f = 0.0700) and 488 nm (2.5421 eV, f = 0.0669) and both are attributed to the 4dzx(Pd) + 3p(Cl) → π*(azo + Ph) 1MLCT/1LLCT transition. For 3, the lower energy transition around 560 nm is computed at 590 nm (2.1002 eV, f = 0.0380) as 4p(Br) + 4dxy(Pd) + π(azo) → π*(azo + Ph) 1LLCT admixed with some 1MLCT/1ILCT character and at 524 nm (2.3660 eV, f = 0.0353) as 4dz2(Pd) → π*(azo + Ph) 1MLCT transition. The shoulder near 490 nm is calculated at 503 nm (2.4642 eV, f = 0.0685) as 4p(Br) + 4dz2(Pd) + π(Ph) → π*(azo + Ph) 1LLCT/1MLCT/1ILCT transition. For 4, lowest energy transition around 600 nm is computed at 653 nm (1.8970 eV, f = 0.0257) as predominantly 5p(I) → π*(azo + Ph) 1LLCT transition. The next lower energy transition near 500 nm is computed at 507 nm (2.4414 eV, f = 0.0347) and 503 nm (2.4631 eV, f = 0.0921). The first one can be exclusively ascribed to 4dz2(Pd) → π*(azo + Ph) 1MLCT transition and the latter one is assigned to π(Ph) + 4dzx(Pd) → π*(azo + Ph) 1ILCT/1MLCT transition. In the UV region two distinct excitations near 380 and 325 nm are observed. The former excitation can be assigned exclusively as π(Ph) → π*(azo + Ph) 1ILCT transition for 1 while incorporation of some 4dz2(Pd) → π*(azo + Ph) 1MLCT character is found in the halo analogues 2–4. In contrast, higher energy UV absorption at 316 nm of 1 is described mostly as π(Ph) → π*(azo) 1ILCT transition mixed with small 1MLCT nature. For 2, the corresponding peak at 323 nm is assigned as π(azo + Ph) + 4dz2(Pd) → π*(azo + Ph) 1ILCT transition admixed with small 1MLCT character. The analogous transition at 318 nm (3.7788 eV, f = 0.0296, λtheo = 312 nm) in 3 is computed as π(Ph + azo) + 4p(Br) → π*(azo + Ph) and can be attributed to 1ILCT/1LLCT. In 4, the related band (λexp = 334 nm, λtheo = 348 nm) can be ascribed largely as π(Ph) → π*(azo + Ph) 1ILCT transition admixed with some 1LLCT character originated from 5p(I).
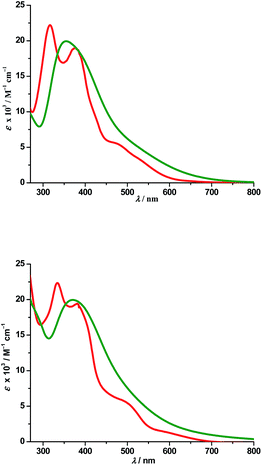 |
| | Fig. 5 Experimental (red) and theoretical (green) absorption spectra of 2 (top) and 4 (bottom). | |
Table 3 UV-vis and luminescence spectral dataa for 1, 2, 3 and 4 in dichloromethane at room temperature
| Compound |
λmax [nm] (ε [M−1 cm−1]) |
λem (nm) |
τ (ns) |
Φ (×10−3) |
kr, s−1 (×105) |
knr, s−1 (×108) |
| The compounds PAAB, 1, 2, 3 and 4 are excited at 324 nm. Shoulder. |
| PAAB |
306 (26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500), 343b (19900), 446b (1000) 500), 343b (19900), 446b (1000) |
384 |
7.06 |
4.507 |
6.384 |
1.410 |
| 1 |
316 (23700), 375 (20000), 475b (5700), 545b (2900) |
369 |
9.52 |
4.531 |
4.759 |
1.046 |
| 2 |
323 (22300), 381 (19000), 480b (5600), 530b (3200) |
352 |
9.13 |
3.018 |
3.306 |
1.092 |
| 3 |
318 (25500), 377 (21000), 490b (6000), 560b (2500) |
352 |
8.71 |
7.546 |
8.664 |
1.139 |
| 4 |
334 (22400), 382 (19400), 500b (5500), 600b (1300) |
352 |
9.23 |
9.358 |
10.139 |
1.073 |
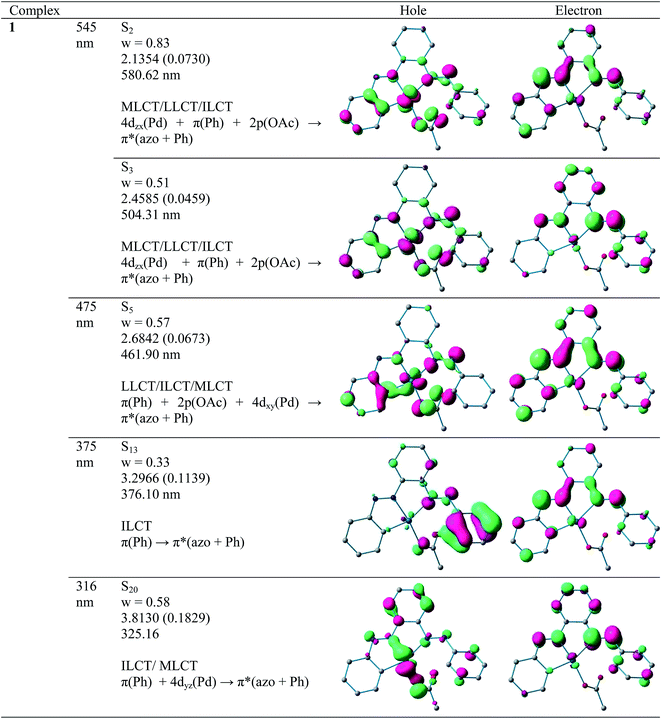 |
| | Fig. 6 Natural transition orbitals (NTOs) for complex 1 illustrating the nature of singlet excited states in the absorption bands in the range 250–700 nm. For each state, the respective number of the state, transition energy (eV), and the oscillator strength (in parentheses) are listed. Shown are only occupied (holes) and unoccupied (electrons) NTO pairs that contribute more than 25% to each excited state. | |
The lowest energy transitions in the complexes are affected largely by the nature of anionic coligand. Each of these excitations originates from energetically dissimilar occupied orbitals i.e., exclusive 5p orbital of iodine in 4, while the involvement of metal t2 becomes the dominating feature on going from 3 to 1. In contrast, almost similar unoccupied orbitals are found in all complexes and composed of mainly azo-π* admixed with little phenyl-π*. The bathochromic shift of longest wavelength transition from 1 to 4 is associated with the increasing order of polarizability of the anionic coligand. The major features in the 300–400 nm region in all the complexes can be attributed largely due to π–π* within the terdentate ligand.
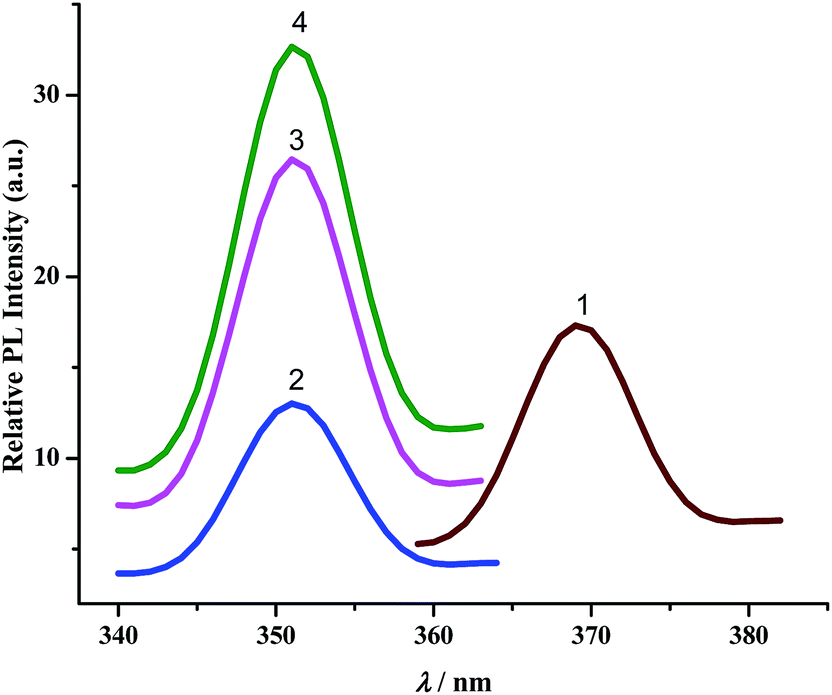
b. Luminescence spectra. The complexes exhibit broad luminescent maxima near 355 nm (Fig. 7) and these remain unaffected with the energy of excitation wavelengths. Time-resolved fluorescent spectra were recorded to understand the decay process and the emissive nature of the compounds (Fig. S13, ESI†). These are found to be moderate emitters (quantum yields (Φ) = 3.0–9.0 × 10−3). The luminescence spectrum and time-resolved fluorescence decay plot of PAAB are depicted in ESI (Fig. S14†). Table 3 summarizes the emission maxima (λem), quantum yield (Φ), lifetime (τ), radiative (kr) and nonradiative (knr) decay rate constants.
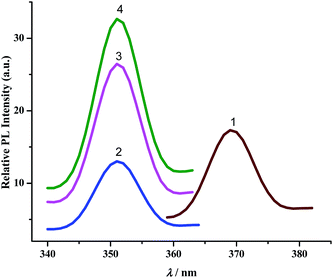 |
| | Fig. 7 Luminescence spectra of complexes. | |
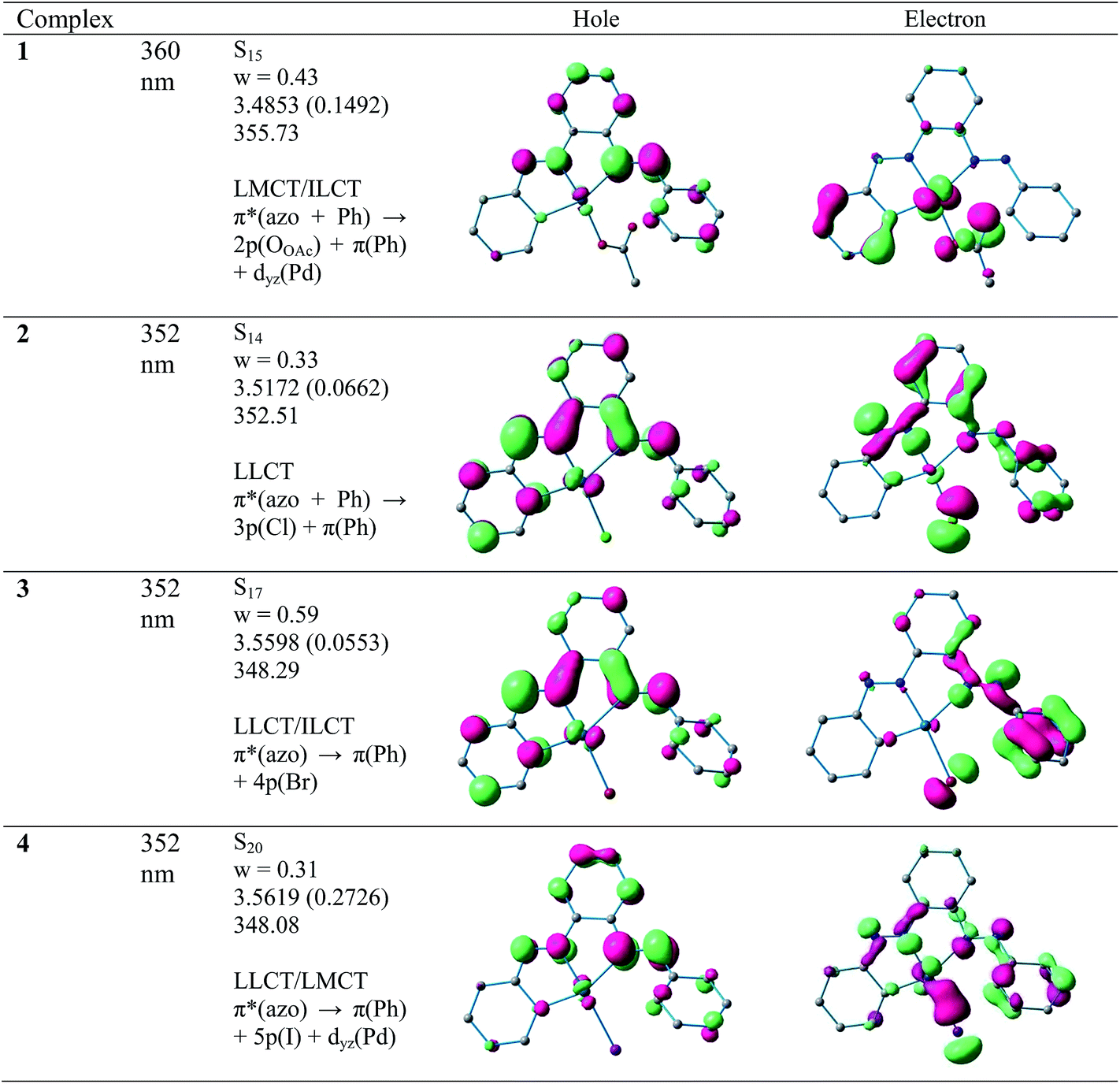
All CNN pincer complexes in this account are emissive in solution at room temperature which is an uncommon phenomenon for Pd(II) compounds.7 This is due to the presence of thermal accessible metal-centred (MC) excited states, the geometry of which is strongly distorted with respect to that of the ground state, leading to the rapid deactivation of the excited luminescent state.10 The emission spectra of 1–4 are very similar and comparable to the spectrum of free ligand. In order to analyze the nature of emission, NTO analysis was performed based on the calculated transition density matrices in their singlet states. The NTOs are illustrated in Fig. 8. The broad luminescent band of the 1 in the experimental spectrum at 360 nm is computed at 356 nm (3.4853 eV, f = 0.1492) with significant transition having π*(azo + Ph) → 2p(OAc)+ π(Ph) + dyz(Pd) and attributed to 1ILCT/1LLCT/1LMCT. The experimental emission spectrum (λem = 352 nm) of 2 is computed at 352 nm (3.5172 eV, f = 0.0662) and tentatively assigned to as π*(azo + Ph) → π(Ph) + 3p(Cl) 1ILCT/1LLCT transition. The emission spectrum (λem = 352 nm) of 3 is computed at 348 nm (3.5598 eV, f = 0.0553) and can be ascribed to as π*(azo) → π(Ph) + 4p(Br) 1ILCT/1ILCT transition. Similarly, the observed band at 352 nm for 4 is computed at 348 nm (3.5619 eV, f = 0.2726) and can be tentatively assigned to as mostly π*(azo) → π(Ph) + 5p(I) 1ILCT/1LLCT admixed with π*(azo) → dyz(Pd) 1LMCT transition. The photoluminescence property mainly originates from singlet state charge transfer transitions revealing the ligand-centered 1IL nature of the emissive state for all complexes. Our investigation has shown that the nature of the luminescent occupied (hole) transition orbitals of 1–4 and PAAB remains virtually similar and emerged as conjugated π* orbital encompassed over both the azo chromophores (Fig. 8). The luminescent unoccupied (electron) transition orbital appears exclusively as π(Ph) in the free ligand while that in the palladacycles is dependent upon the nature of anionic coligands. The corresponding orbital appears as the combination of π(Ph) with p(coligand) in chloro and bromo complexes whereas different amounts of metal-d participation is found in acetato and iodo analogues. The NTO analysis, therefore, suggest a ligand-dominated emissive states π*(azo + Ph) by virtue of their enhanced stability. The structurally similar luminescence spectra can be assigned as intraligand π–π* transitions within the planar bisazo chromophore perturbed by the anionic coligands and metal in varying extent. Notably, no metal contribution (dx2−y2) was found computationally in the emissive state of the present palladacycles. It should be noted that the reports of luminescent of azo complexes are less documented in the literarture.20
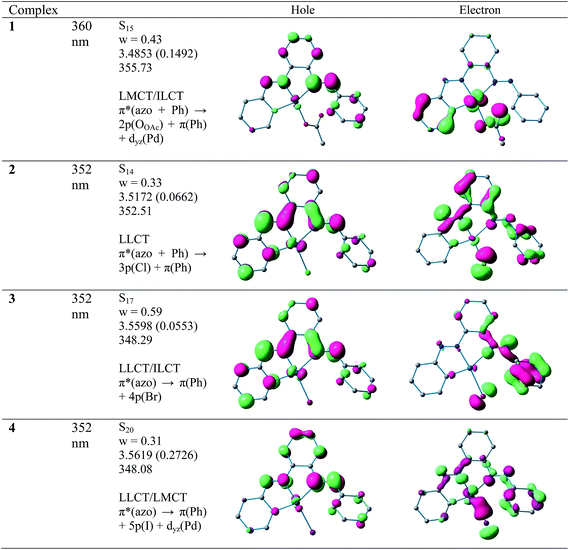 |
| | Fig. 8 Natural transition orbitals (NTOs) for the complexes 1, 2, 3 and 4 illustrating the nature of the singlet excited state in the emission band in the region 340–380 nm. For each state the transition energy (eV) and the oscillator strength (in parentheses) are listed. Shown are only occupied (holes) and unoccupied (electrons) NTO pairs that contribute more than 30% to each excited state. | |
Catalysis
Many of the N-donor ligands are inexpensive and stable in air unlike phosphine ligands. Facile synthesis, stability and easy handling of non-phosphine cyclometalated Pd complexes are accountable for their popularity and growing applications in catalysis. Since the C–C coupling reactions are of vital importance in organic synthesis and the palladacycles are a class of typical catalysts in these reactions,12a,15d,e,l the Suzuki–Miyaura cross-coupling reaction of aryl halides with phenylboronic acid was carried out to explore the catalytic activity of the non-phosphine pincers 1 and 4. Upon heating a mixture of arylbromide, phenylboronic acid and K2CO3 in toluene at 70 °C for 3 h in the presence of Pd complexes, the biaryls were isolated in excellent yields (Table 4). In general, the electron rich palladium center is beneficial toward Suzuki–Miyaura coupling reactions. The reported electron deficient palladacycles 1 and 4 show high turnover numbers with good yields for certain substituted bromobenzenes. As shown in Table 4, only the strong electron-withdrawing group on phenyl bromides can generate the corresponding products. Notably, we were unable to isolate any coupling product upon varying the substituents on phenyl bromides from strong electron-withdrawing to moderate electron-withdrawing groups e.g., carbaldehyde or electron-donating groups e.g., methoxy derivative under similar reaction conditions. To explore the versatility of the reported palladacycles, the coupling between aryl chlorides and phenyl boronic acid had also been performed. Aryl chlorides provide the corresponding biaryls in moderate to low yields (entries 7–9) with 0.01 mol % catalyst loading. Examples of Suzuki–Miyaura coupling with aryl chlorides are scarce with CN2 coordinated pincer palladacycles.15d
Table 4 Suzuki–Miyaura coupling reaction of 1 and 4 with aryl halides and phenylboronic acida
The report of the C–C cross coupling by Heck-type reactions catalyzed by the non-NHC CNN pincer Palladacycles are sparsely known.12e,g,h The high stability of the present pincer complexes may make themselves as promising candidates to achieve the high turnover numbers (TON's) under the harsh conditions of the Heck coupling reactions. In this context, investigation into the ability of easily obtainable complexes 1 and 4 to act as precatalysts has been performed for Heck-type coupling reactions. The results obtained are summarized in Table 5. Though catalysis was observed in both the cases, the acetato complex 1 shows somewhat better activity compared to that of the iodo analogue 4. All the reactions have been performed under argon with 0.01 mol% of catalysts in DMF at 135 °C with K2CO3 (1.8 equiv. of base). The use of 0.001 mol % catalyst loading merely affects the efficiency (entry 5). The complexes under investigation are thermally stable and inert toward air and moisture in the solid state. These properties allowed for catalytic experiments under aerobic conditions as well. The use of K2CO3 as base furnished better yield compared to that of NaOAc for the coupling reactions. Different para-functionalized aryl halides are used in the catalytic experiments. The palladacycles have shown good catalytic activity over 18 h at elevated temperature with activated substrates such as 4-bromobenzaldehyde, 4-bromonitrobenzene and 4-bromoacetophenone. A slightly lower activity was observed with the deactivated substrates like 4-bromoanisole. The coupling of bromobenzene with styrene proceeded as well but with low yield.
Table 5 Heck coupling of 1 and 4 with functionalized aryl halides and styrenea
|
| Entry |
Catalyst |
R |
X |
Amount of cat. (mol%) |
Product |
Yieldb (%) |
TONc |
| Reaction conditions: aryl halide (1.0 mmol), styrene (1.5 mmol), K2CO3 (1.8 mmol), Pd catalyst, dimethylformamide (7 mL), 135 °C, 18 h. Isolated yield. TON, turnover number {(mol of product)/(mol of catalyst)}. |
| 1 |
1 |
H |
I |
0.01 |
8 |
74 |
7400 |
| 2 |
4 |
H |
I |
0.01 |
8 |
70 |
7000 |
| 3 |
1 |
CHO |
Br |
0.01 |
9 |
84 |
8400 |
| 4 |
4 |
CHO |
Br |
0.01 |
9 |
86 |
8600 |
| 5 |
4 |
CHO |
Br |
0.001 |
9 |
83 |
83000 |
| 6 |
1 |
COCH3 |
Br |
0.01 |
10 |
72 |
7200 |
| 7 |
4 |
COCH3 |
Br |
0.01 |
10 |
68 |
6800 |
| 8 |
1 |
NO2 |
Br |
0.01 |
11 |
79 |
7900 |
| 9 |
4 |
NO2 |
Br |
0.01 |
11 |
76 |
7600 |
| 10 |
1 |
OCH3 |
Br |
0.01 |
12 |
63 |
6300 |
| 11 |
4 |
OCH3 |
Br |
0.01 |
12 |
58 |
5800 |
Conclusion
We have introduced a typical bisazobenzene, 2-(phenylazo)azobenzene, as the novel chelating ligand and have presented a high-yield preparation of a new class of unsymmetrical CPhNazoNazo pincer palladacycles with the symmetrical ligand. The reported palladacycles represent the first member of pincer compounds containing two Nazo atoms. A noteworthy feature of the ligand is the presence of two conjugated π-acidic azo chromophores, which makes it both a superior σ-donor as well as π-acceptor relative to azobenzene. Remarkable stabilization of azo-based virtual orbitals is also apparent upon palladation, where LC (LUMO and LUMO + 1) states are computed to be the lower energy orbitals relative to MC (LUMO + 2) level. Low-lying nature of LUMO and LUMO +1 in the free ligand as well as in all the palladacycles is nicely substantiated with the electrochemical study. In consequence, modification of FMOs leads to interesting optoelectronic property in the palladacycles. The cyclopalladated compounds exhibit luminescence in solution at room temperature. This property is plausibly as a result of the enhanced stability of LC emissive states (azo π*) which are hardly mixed by non-emissive MC state (dx2−y2). The similarity in luminescence spectra of the cyclometalated complexes suggest a ligand-dominated emissive state, which can be assigned essentially as intraligand π–π* transitions admixed with little coligands and/or metal orbitals. Among the palladacycles, the iodo complex exhibits the longest wave-length visible transition near 600 nm. The observed bathochromic shift of the lowest energy transition in chloro to iodo compounds is caused by the incorporation of π(halide) → π*(PAAB) LLCT character to a greater extent, consistent with the polarizability order of the halides. In contrast, analogous excitation in the chloro and acetato derivatives is dominated by 4dzx(Pd) → π*(PAAB) MLCT. The transitions in the UV region are almost entirely bisazoaromatic-based π–π* admixed with little participation of metal or coligand orbitals. In addition, the results of C–C cross-coupling reactions show that the CNN palladacycles may be employed in the advancement of effective catalysts for Suzuki–Miyaura and Heck-type reactions. Novel functionalized bisazoaromatic ligands and their cyclometalated complexes are currently under investigation.
Experimental section
General information
All manipulations were performed under an atmosphere of argon except where mentioned. Solvents were dried by standard methods and distilled under argon prior to use. Palladium chloride, palladium acetate and styrene were purchased from Aldrich Chemical Company. Other chemicals and solvents were purchased from Merck, India and dried before use according to standard methods.
Synthesis of ligand
2-(Phenylazo)azobenzene (PAAB). 2-Amino-azobenzene21 (5 g, 0.025 mol) was dissolved in 20 mL glacial acetic acid with heating. Solid nitrosobenzene (2.9 g, 0.027 mol) was added and the mixture was stirred for 24 h at 50 °C until the precipitation of a dark mass was obtained. The precipitate was dried in vacuo and subsequently dissolved in minimum volume of hot n-hexane. The deep red crystals were obtained from the hot hexane solution. An n-hexane extract was made with the filtrate, giving an additional crop of red crystals. Yield: 6.24 g, 86%. mp. 107–108 °C (lit. mp. 106−108 °C);11 FT-IR (cm−1): 1486–1439 (νNN); 1H NMR (300 MHz, CDCl3): δ 7.96–7.98 (m, 4H), 7.76 (q, 2H, J = 3.3 Hz), 7.51–7.58 (m, 8H). 13C NMR (300 MHz, CDCl3): δ 153.1, 148.2, 139.3, 130.9, 129.1, 123.3, 118.2. Elem. anal. calcd (%) for C18H14N4: C, 75.50; H, 4.93; N, 19.57. Found: C, 75.44; H, 4.87; N, 19.42. HRMS (ESI): m/z calcd for C18H15N4 287.1297; found 287.1293 (M + H)+.
Synthesis of complexes
[Pd(L)(OAc)] 1. To a solution of PAAB (317 mg, 1.1 mmol) in AcOH (25 mL) was added Pd(OAc)2 (226 mg, 1.0 mmol) under an argon atmosphere. The mixture was stirred at room temperature for 80 h. The solvent was removed under reduced pressure. Crystallization by slow diffusion of toluene solution of the complex into hexane afforded 1 as rod shaped crystals (445 mg, 98% yield). FT-IR (cm−1): 1446, 1430 (νNN), 3524 (νO–H). 1H NMR (300 MHz, CDCl3): δ 8.31 (d, 1H, J = 7.9 Hz), 8.10 (d, 1H, J = 6.0 Hz), 7.95 (d, 1H, J = 6.1 Hz), 7.82 (d, 1H, J = 7.0 Hz), 7.76–7.67 (m, 5H), 7.56 (d, 3H, J = 6.6 Hz), 7.28 (s, 1H), 1.72 (s, 3H). 13C NMR (300 MHz, CDCl3): δ 177.1, 165.6, 154.9, 152.3, 151.5, 134.3, 133.3, 132.1, 129.1, 128.7, 128.3, 126.9, 122.4, 117.9, 23.4. Elem. anal. calcd (%) for C20H16N4O2Pd: C, 53.29; H, 3.58; N, 12.43. Found: C, 53.21; H, 3.62; N, 12.36. HRMS (ESI): m/z calcd for C18H13N4Pd 391.0182; found 391.0180 (M − OAc)+.
[Pd(L)Cl] 2. To a solution of PAAB (193 mg, 0.674 mmol) in EtOH (25 mL) was added K2[PdCl4] (200 mg, 0.612 mmol) under an argon atmosphere. The mixture was stirred at room temperature for 80 h, until a dark red solution was formed. The solvent was removed under reduced pressure. Crystallization by slow diffusion of toluene solution of the complex into hexane afforded 2 as rod shaped crystals (251 mg, 96% yield). FT-IR (cm−1): 1446, 1430 (νNN). 1H NMR (300 MHz, CDCl3): δ 8.32 (d, 1H, J = 7.0 Hz), 8.11 (d, 1H, J = 6.9 Hz), 7.97 (d, 1H, J = 5.8 Hz), 7.83 (d, 1H, J = 7.0 Hz), 7.77–7.68 (m, 5H), 7.57 (d, 3H, J = 7.2 Hz), 7.29 (s, 1H). 13C NMR (300 MHz, CDCl3): δ 165.9, 152.7, 137.1, 133.7, 133.4, 132.6, 129.4, 128.7, 123.1, 118.4. Elem. anal. calcd (%) for C18H13N4ClPd: C, 50.61; H, 3.07; N, 13.12. Found: C, 50.53; H, 3.18; N, 12.97. HRMS (ESI): m/z for C18H13N4Pd 391.0182; found 391.0188 (M − Cl)+, calcd 819.0052; found 819.0059 (2M − Cl)+.
[Pd(L)Br] 3. To a solution of 1 (200 mg, 0.444 mmol) in acetone, KBr solution (10−2 molar solution in water) was added dropwise and the resulting solution was stirred for 1 h at room temperature. The dark residue was filtered and dried in vacuum (191 mg, 91% yield). FT-IR (cm−1): 1444, 1430 (νNN). 1H NMR (300 MHz, CDCl3): δ 8.31 (d, 1H, J = 6.9 Hz), 8.11 (d, 1H, J = 6.2 Hz), 7.95 (d, 1H, J = 5.6 Hz), 7.82 (d, 1H, J = 6.9 Hz), 7.76–7.68 (m, 5H), 7.56 (d, 3H, J = 7.5 Hz), 7.28 (s, 1H). 13C NMR (300 MHz, CDCl3): δ 162.3, 139.5, 133.7, 133.6, 133.4, 132.5, 129.5, 128.8, 126.8, 123.0, 118.5. Elem. anal. calcd (%) for C18H13N4BrPd: C, 45.84; H, 2.78; N, 11.88. Found: C, 45.71; H, 2.61; N, 11.75. HRMS (ESI): m/z for C18H13N4Pd 391.0182; found 391.0186 (M − Br)+, calcd 862.9543; found 862.9547 (2M − Br)+.
[Pd(L)I] 4. The complex 4 was prepared from 1 (200 mg, 0.444 mmol) with KI using similar procedure applied for the synthesis of complex 3 (212 mg, 92% yield). FT-IR (cm−1): 1435, 1383 (νNN). 1H NMR (300 MHz, CDCl3): 8.48 (d, 1H, J = 7.7 Hz), 8.36 (d, 1H, J = 7.3 Hz), 8.16 (d, 1H, J = 8.9 Hz), 8.01 (d, 1H, J = 7.4 Hz), 7.73–7.67 (m, 5H), 7.56 (d, 3H, J = 7.5 Hz), 7.22 (s, 1H). 13C NMR (300 MHz, CDCl3): δ 166.4, 144.0, 133.9, 133.8, 133.5, 132.6, 129.8, 129.0, 126.6, 123.2, 118.7. Elem. anal. calcd (%) for C18H13N4IPd: C, 41.68; H, 2.53; N, 10.80. Found: C, 41.49; H, 2.39; N, 10.65. HRMS (ESI): m/z for C18H13N4Pd 391.0182; found 391.0181 (M − I)+, calcd 910.9414; found 910.9412 (2M − I)+.
General procedure for Suzuki–Miyaura coupling reactions
A mixture of K2CO3 (2 mmol), aryl halide (1 mol) and phenyl boronic acid (1.3 mmol) were added in a flask and put under an atmosphere of argon followed by addition of 5 mL degassed toluene. After thermostating at an indicated temperature (typically at 70 °C) for 15 min, the catalyst was added against a positive stream of argon. The mixture was allowed to heat with stirring for the requisite time. After cooling to ambient temperature, the solvent was removed under vacuum and purified by column chromatography on silica gel, using 20:1 hexane/ethyl acetate as eluent. Products were identified by comparison of NMR data with those in the literature.
4-Acetylbiphenyl (5)22. 1H NMR: (300 MHz, CDCl3): δ 8.06 (d, J = 8.4 Hz, 2H), 7.71 (d, J = 8.7 Hz, 2H), 7.66 (d, J = 8.4 Hz, 2H), 7.53–7.42 (m, 3H), 2.66 (s, 3H). 13C NMR (300 MHz, CDCl3): δ 197.7, 145.8, 139.9, 135.9, 131.9, 129.9, 128.9, 128.2, 127.2, 26.6.
4-Nitro-biphenyl (6)22. 1H NMR (300 MHz, CDCl3): δ 8.23 (d, J = 8.7 Hz, 2H), 7.76 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 8.4 Hz, 2H), 7.47–7.55 (m, 3H). 13C NMR (300 MHz, CDCl3): δ 147.6, 147.1, 138.7, 129.2, 128.9, 127.8, 127.4, 124.1.
4-Phenylbenzonitrile (7)22. 1H NMR (300 MHz, CDCl3): δ 7.72 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 7.5 Hz, 2H), 7.59 (d, J = 8.1 Hz, 2H), 7.53–7.42 (m, 3H). 13C NMR (300 MHz, CDCl3): δ 145.6, 139.2, 132.6, 129.1, 128.7, 127.7, 127.2, 118.9, 110.9.
General procedure for Heck-coupling reactions
A mixture of K2CO3 (1.8 mmol), aryl halide (1 mmol) and styrene (1.5 mmol) were added in a flask and put under an atmosphere of argon followed by addition of 7 mL degassed DMF. After thermostating at 135 °C for 15 min the catalyst (0.01 mol%) was added against a positive stream of argon. The mixture was allowed to heat at 135 °C for 18 h. After cooling to ambient temperature, the solvent was removed under vacuum and purified by column chromatography on silica gel, using 20:1 hexane/ethyl acetate as eluent. Products were identified by comparison of NMR data with those in the literature.
(E)-1,2-Diphenylethene (8)22. 1H NMR (400 MHz, CDCl3): δ 7.51 (d, 4H, J = 7.6 Hz), 7.36 (t, 4H, J = 8.0 Hz), 7.26 (t, 2H, J = 7.2 Hz), 7.11 (s, 2H). 13C NMR (300 MHz, CDCl3): δ 137.4, 128.8, 127.9, 127.7, 126.6.
(E)-1-Formyl-4-styrylbenzene (9)22. 1H NMR (400 MHz, CDCl3): δ 9.99 (s, 1H), 7.87 (d, 2H, J = 8.4 Hz), 7.66 (d, 2H, J = 8.4 Hz), 7.55 (d, 2H, J = 7.6 Hz), 7.39 (t, 2H, J = 8.0 Hz), 7.32 (t, 1H, J = 7.2 Hz), 7.27 (d, 1H, J = 16.4 Hz), 7.15 (d, 1H, J = 16.4 Hz). 13C NMR (300 MHz, CDCl3): δ 191.6, 143.4, 136.6, 135.4, 132.2, 130.2, 128.8, 128.5, 127.3, 126.9.
(E)-1-Acetyl-4-styrylbenzene (10)22. 1H NMR (400 MHz, CDCl3): δ 7.96 (d, 2H, J = 8.4 Hz), 7.59 (d, 2H, J = 8.4 Hz), 7.55 (d, 2H, J = 7.2 Hz), 7.39 (t, 2H, J = 8.0 Hz), 7.30 (t, 1H, J = 7.2 Hz), 7.24 (d, 1H, J = 17.2 Hz), 7.14 (d, 1H, J = 16.4 Hz), 2.62 (s, 3H). 13C NMR (300 MHz, CDCl3): δ 197.5, 142.0, 136.7, 136.0, 131.5, 128.9, 128.8, 128.3, 127.5, 126.8, 126.5, 26.6.
(E)-1-Nitro-4-styrylbenzene (11)22. 1H NMR (300 MHz, CDCl3): δ 8.22 (d, 2H, J = 8.7 Hz), 7.64 (d, 2H, J = 8.7 Hz), 7.55 (d, 2H, J = 7.1 Hz), 7.42–7.33 (m, 3H), 7.28 (d, 1H, J = 16.3 Hz), 7.14 (d, 1H, J = 16.5 Hz). 13C NMR (300 MHz, CDCl3): δ 146.8, 143.9, 136.2, 133.3, 128.9, 128.8, 127.0, 126.9, 126.3, 124.1.
(E)-1-Methoxy-4-styrylbenzene (12)22. 1H NMR (400 MHz, CDCl3): δ 7.49 (d, 2H, J = 7.6 Hz), 7.46 (d, 2H, J = 8.4 Hz), 7.35 (t, 2H, J = 7.6 Hz), 7.23 (t, 1H, J = 7.2 Hz), 7.07 (d, 1H, J = 16 Hz), 6.97 (d, 1H, J = 16.4 Hz), 6.90 (d, 2H, J = 8.8 Hz), 3.83 (s, 3H). 13C NMR (300 MHz, CDCl3): δ 159.4, 137.7, 130.2, 128.7, 128.3, 127.7, 127.2, 126.7, 126.3, 114.2, 55.3.
Physical measurements
The elemental analyses (C, H, N) were performed with a Perkin-Elmer model 2400 series II elemental analyzer. FT-IR spectra were recorded on Perkin-Elmer L1600300 spectrometer. The attenuated total reflectance method was used. NMR spectral measurement was carried out on Bruker FT 300 MHz and Bruker DPX-400 MHz spectrometers with TMS as an internal reference. The electrospray ionization mass spectra (ESI-MS positive) were measured in acetonitrile on Waters HRMS spectrometers (Model: QTOF Micro YA263). Thermogravimetric analysis (TGA) was carried out using a SDT Q600 V8.2 Built 100 thermal analyzer. The electronic spectra in dichloromethane solution were obtained using a Perkin-Elmer LAMDA 25 spectrophotometer with a solute concentration of about 10−4 M. Emission spectra were recorded on PTI QM40 spectrometer in deaerated dichloromethane solutions at room temperature. Emission quantum yields of the complexes were determined in deaerated solutions of the complexes by a relative method using anthracene in ethanol as the standard.23 The emission quantum yield (Φr) and radiative (kr) and nonradiative (knr) decay rate constants for complexes was calculated by the equations24 given below:| |
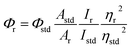 | (3) |
| |
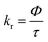 | (4) |
| |
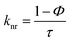 | (5) |
where Φr and Φstd are the quantum yields of unknown and standard samples (Φstd = 0.27 (at 298 K) in ethanol at λex = 341 nm),25 Ar and Astd (<0.1) are the solution absorbances at the excitation wavelength (λex), Ir and Istd are the integrated emission intensities, and ηr and ηstd are the refractive indices of the solvents.26 For all measurements, the same slit widths for excitation and emission were maintained. Time-correlated single photon counting (TCSPC) measurements were carried out for recording of the luminescence decay profile for the free ligand and Pd(II) complexes in dichloromethane solution using a picosecond diode laser (IBH Nanoled-07) in an IBH Fluorocube apparatus. Air-equilibrated solutions were used for recording of the luminescence lifetime. The fluorescence decays were collected on a Hamamatsu MCP photomultiplier (R3809). The fluorescence decays were analyzed using IBH DAS6 software. Electrochemical measurements were carried out at 27 °C with VersaStat II Princeton Applied Research potentiostat/galvanostat under argon atmosphere. The cell contained a Pt working electrode and a Pt wire auxiliary electrode. Tetraethylammonium perchlorate (NEt4ClO4) was used as a supporting electrolyte and the potentials are referenced to the Ag/AgCl electrode without junction correction.
Crystallographic studies
X-ray intensity data for all the compounds were measured at 296(2) K on Bruker AXS SMART APEX II equipped with a CCD diffractometer using graphite-monochromator (Mo Kα, λ = 0.71073 Å). Metal atoms were located by direct methods, and the rest of the non-hydrogen atoms emerged from successive Fourier synthesis. The structures were refined by full-matrix least squares procedures on F2. All the H atoms were included in calculated positions and treated as riding atoms using SHELXL default parameters (except those of water molecule in 1 which were found and isotopically refined). Calculations were performed using the SHELXTL v 6.14 program package.27 Thermal ellipsoids are drawn at the 30% probability level. Molecular structure plots were drawn using the Oak Ridge thermal ellipsoid plot ORTEP.28
Computational study
The molecular geometry of the singlet ground state (S0) of PAAB, 1, 2, 3 and 4 have been calculated by DFT method using the (R)B3LYP29 hybrid functional approach incorporated in GAUSSIAN 09 program package.30 The geometries of the complexes were fully optimized in gas phase without imposing any symmetry constraints. The nature of all the stationary points was checked by computing vibration frequencies, and all the species were found to be true potential energy minima, as no imaginary frequency were obtained (NImag = 0). The single crystal X-ray coordinates have been used as the initial input in all calculations. On the basis of the optimized geometries, the absorption and emission spectra properties in dichloromethane (CH2Cl2) media were calculated by the time-dependent density functional theory (TD-DFT)31 approach associated with the conductor-like polarizable continuum model (CPCM).32 We computed the lowest 100 singlet–singlet transitions in absorption and emission processes respectively. The electronic structure contributions to the differences in observed optoelectronic properties are compared by natural transition orbital (NTO) analysis. The computed vertical transitions were calculated at the equilibrium geometry of the S0 state and described in terms of one-electron excitations of molecular orbitals of the corresponding S0 geometry. The calculated transitions with moderate intensities (f ≥ 0.02) can be envisaged going from the lower to the higher energy region of the spectrum. The palladium atom and iodine were described by a double-ζ basis set with the effective core potential of Hay and Wadt (LANL2DZ)33 and the 6-311+G(d,p) basis set34 was used for the other elements present in the complexes to optimize the ground state geometries. The calculated electronic density plots for frontier molecular orbitals were prepared by using the GaussView 5.0 software. GaussSum program, version 2.2 (ref. 35) was used to calculate the molecular orbital contributions from groups or atoms.
Acknowledgements
We thank the Department of Science and Technology, New Delhi, for financial assistance (grant SR/S1/IC-75/2010). We are also thankful to the Department of Science and Technology, New Delhi, India for the data collection on the CCD facility setup (Jadavpur University). We also acknowledge CAS, Department of Chemistry, Jadavpur University and the DST-PURSE program for other facilities. Thanks to Dr S. Chakrabarti of University of Calcutta, Kolkata for computational assistance. Ms S. Roy thanks DST and Mr S. Pramanik and Mr T. Ghorui thank UGC, New Delhi for the research fellowship.
References
- A. C. Cope and R. W. Siekman, J. Am. Chem. Soc., 1965, 87, 3272–3273 CrossRef CAS
 .
. -
(a) A. Fernández, M. López-Torres, J. J. Fernández, D. Vázquez-García and J. M. Vila, J. Chem. Educ., 2012, 89, 156–158 CrossRef ;
(b) J.-P. Djukic, A. Hijazi, H. D. Flack and G. Bernardinelli, Chem. Soc. Rev., 2008, 37, 406–425 RSC ;
(c) J. Dupont and M. Pfeffer, Palladacycles: Synthesis, Characterization and Applications, Wiley-VCH Verlag GmbH & Co, 2008 Search PubMed ;
(d) M. Ghedini, I. Aiello, A. Crispini, A. Golemme, M. L. Deda and D. Pucci, Coord. Chem. Rev., 2006, 250, 1373–1390 CrossRef CAS PubMed ;
(e) M. Albrecht, Chem. Rev., 2010, 110, 576–623 CrossRef CAS PubMed ;
(f) V. V. Dunina, O. A. Zalevskaya and V. M. Potapov, Russ. Chem. Rev., 1988, 57, 250–269 CrossRef PubMed .
-
(a) N. Deibel, M. G. Sommer, S. Hohloch, J. Schwann, D. Schweinfurth, F. Ehret and B. Sarkar, Organometallics, 2014, 33, 4756–4765 CrossRef CAS ;
(b) O. A. Blackburn and B. J. Coe, Organometallics, 2011, 30, 2212–2222 CrossRef CAS ;
(c) A. R. Dick, M. S. Remy, J. W. Kampf and M. S. Sanford, Organometallics, 2007, 26, 1365–1370 CrossRef CAS ;
(d) I. Omae, Coord. Chem. Rev., 2004, 248, 995–1023 CrossRef CAS PubMed ;
(e) M. Ghedini, D. Pucci, A. Crispini, G. Barberio, I. Aiello, F. Barigelletti, A. Gessi and O. Francescangeli, Appl. Organomet. Chem., 1999, 13, 565–581 CrossRef CAS ;
(f) C. Sinha, D. Bandyopadhyay and A. Chakravorty, Inorg. Chem., 1988, 27, 1173–1178 CrossRef CAS ;
(g) A. K. Mahapatra, D. Bandyopadhyay, P. Bandyopadhyay and A. Chakravorty, Inorg. Chem., 1986, 25, 2214–2221 CrossRef CAS ;
(h) A. D. Ryabov, Synthesis, 1985, 233–252 CrossRef CAS ;
(i) I. Omae, Chem. Rev., 1979, 79, 287–321 CrossRef CAS ;
(j) M. I. Bruce, B. Goodall and G. A. Stone, J. Chem. Soc., Dalton Trans., 1978, 687–694 RSC ;
(k) M. I. Bruce, Angew. Chem., Int. Ed., 1977, 16, 73–86 CrossRef ;
(l) J. Dehand and M. Pfeffer, Coord. Chem. Rev., 1976, 18, 327–352 CrossRef CAS ;
(m) G. W. Parshall, Acc. Chem. Res., 1970, 3, 139–144 CrossRef CAS ;
(n) H. Takahashi and J. Tsuji, J. Organomet. Chem., 1967, 10, 511–517 CrossRef CAS .
-
(a) M. Juribašić, K. Užarević, D. Gracin and M. ćurić, Chem. Commun., 2014, 50, 10287–10290 RSC ;
(b) M. Juribašić, I. Halasz, D. Babić, D. Cinčić, J. Plavec and M. ćurić, Organometallics, 2014, 33, 1227–1234 CrossRef ;
(c) M. Juribašić, A. Budimir, S. Kazazić and M. CĆurić, Inorg. Chem., 2013, 52, 12749–12757 CrossRef PubMed ;
(d) N. Deibel, S. Hohloch, M. G. Sommer, D. Schweinfurth, F. Ehret, P. Braunstein and B. Sarkar, Organometallics, 2013, 32, 7366–7375 CrossRef CAS and references therein;
(e) D. Cinčić, M. Juribašić, D. Babić, K. Molčanov, P. Šket, J. Plavec and M. ćurić, Chem. Commun., 2011, 47, 11543–11545 RSC ;
(f) D. Babić, M. Ćurić, K. Molčanov, G. Ilc and J. Plavec, Inorg. Chem., 2008, 47, 10446–10454 CrossRef PubMed ;
(g) M. Ćurić, D. Babić, A. Višnjevac and K. Molčanov, Inorg. Chem., 2005, 44, 5975–5977 CrossRef PubMed .
- O. A. Blackburn, B. J. Coe and M. Helliwell, Organometallics, 2011, 30, 4910–4923 CrossRef CAS .
-
(a) I. Chakraborty, S. J. Carrington and P. K. Mascharak, Acc. Chem. Res., 2014, 47, 2603–2611 CrossRef CAS PubMed ;
(b) M. Han, T. Hiradeb and Y. Okuib, Dalton Trans., 2014, 43, 5929–5934 RSC ;
(c) T. Hirade, Y. Okui and M. Han, J. Mater. Chem. C, 2013, 1, 2672–2679 RSC ;
(d) M. E. Moustafa, M. S. McCready and R. J. Puddephatt, Organometallics, 2012, 31, 6262–6269 CrossRef CAS ;
(e) O. A. Blackburn, B. J. Coe, J. Fielden, M. Helliwell, J. J. W. McDouall and M. G. Hutchings, Inorg. Chem., 2010, 49, 9136–9150 CrossRef CAS PubMed ;
(f) M. J. Baena, P. Espinet, C. L. Folcia, J. Ortega and J. Etxebarrí, Inorg. Chem., 2010, 49, 8904–8913 CrossRef CAS PubMed ;
(g) M. Juribašić, M. Ćurić, K. Molčanov, D. Matković-Čalogović and D. Babić, Dalton Trans., 2010, 39, 8769–8778 RSC ;
(h) M. Han, T. Hirade and M. Hara, New J. Chem., 2010, 34, 2887–2891 RSC ;
(i) P. Pratihar, T. K. Mondal, A. K. Patra and C. Sinha, Inorg. Chem., 2009, 48, 2760–2769 CrossRef CAS PubMed ;
(j) M. Ghedini, I. Aiello, A. Crispini, A. Golemme, M. La Deda and D. Pucci, Coord. Chem. Rev., 2006, 250, 1373–1390 CrossRef CAS PubMed .
- Room temperature luminescent palladacycles in solution:
(a) P.-K. Chow, W.-P. To, K.-H. Low and C.-M. Che, Chem.–Asian J., 2014, 9, 534–545 CrossRef CAS PubMed ;
(b) S. Lentijo, J. A. Miguel and P. Espinet, Organometallics, 2011, 30, 1059–1066 CrossRef CAS ;
(c) S. M. Borisov, G. Zenkl and I. Klimant, ACS Appl. Mater. Interfaces, 2010, 2, 366–374 CrossRef CAS PubMed ;
(d) C. Bronner, S. A. Baudron, M. W. Hosseini, C. A. Strassert, A. Guenet and L. De Cola, Dalton Trans., 2010, 39, 180–184 RSC ;
(e) M. La Deda, M. Ghedini, I. Aiello, T. Pugliese, F. Barigelletti and G. Accorsi, J. Organomet. Chem., 2005, 690, 857–861 CrossRef PubMed ;
(f) C. S. Consorti, G. Ebeling, F. Rodembusch, V. Stefani, P. R. Livotto, F. Rominger, F. H. Quina, C. Yihwa and J. Dupont, Inorg. Chem., 2004, 43, 530–536 CrossRef CAS PubMed ;
(g) M. Ghedini, I. Aiello, M. La Deda and A. Grisolia, Chem. Commun., 2003, 2198–2199 RSC ;
(h) Y. Wakatsuki, H. Yamazaki, P. A. Grutsch, M. Santhanam and C. Kutal, J. Am. Chem. Soc., 1985, 107, 8153–8159 CrossRef CAS .
- Room temperature luminescent palladacycles in solid:
(a) D. V. Aleksanyan, V. A. Kozlov, Y. V. Nelyubina, K. A. Lyssenko, L. N. Puntus, E. I. Gutsul, N. E. Shepel, A. A. Vasil’ev, P. V. Petrovskii and I. L. Odinets, Dalton Trans., 2011, 40, 1535–1546 RSC ;
(b) J. Kuwabara, Y. Ogawa, A. Taketoshi and T. Kanbara, J. Organomet. Chem., 2011, 696, 1289–1293 CrossRef CAS PubMed ;
(c) V. A. Kozlov, D. V. Aleksanyan, Y. V. Nelyubina, K. A. Lyssenko, E. I. Gutsul, L. N. Puntus, A. A. Vasil’ev, P. V. Petrovskii and I. L. Odinets, Organometallics, 2008, 27, 4062–4070 CrossRef CAS ;
(d) M. Akaiwa, T. Kanbara, H. Fukumoto and T. Yamamoto, J. Organomet. Chem., 2005, 690, 4192–4196 CrossRef CAS PubMed ;
(e) F. Neve, A. Crispini, C. Di Pietro and S. Campagna, Organometallics, 2002, 21, 3511–3518 CrossRef CAS ;
(f) B.-C. Tzeng, S.-C. Chan, M. C. W. Chan, C.-M. Che, K.-K. Cheung and S.-M. Peng, Inorg. Chem., 2001, 40, 6699–6704 CrossRef CAS PubMed ;
(g) F. Neve, A. Crispini and S. Campagna, Inorg. Chem., 1997, 36, 6150–6156 CrossRef CAS .
- P. K. Chow, C. Ma, W.-P. To, G. S. M. Tong, S.-L. Lai, S. C. F. Kui, W.-M. Kwok and C.-M. Che, Angew. Chem., Int. Ed., 2013, 52, 11775–11779 CrossRef CAS PubMed .
-
(a) M. Maestri, V. Balzani, C. Deuschel-Cornioley and A. Von Zelewsky, Advanced Photochemistry, ed. D. Volman, G. Hammond and D. Neckers, JohnWiley & Sons, New York, 1992, pp. 1–68 Search PubMed ;
(b) G. R. Crosby, Acc. Chem. Res., 1975, 8, 231–238 CrossRef CAS .
- P. Ruggli and J. Rohner, Helv. Chim. Acta, 1942, 25, 1533–1542 CrossRef CAS .
-
(a) T. Wang, X.-Q. Hao, J.-J. Huang, K. Wang, J.-F. Gong and M.-P. Song, Organometallics, 2014, 33, 194–205 CrossRef CAS ;
(b) K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927–8955 CrossRef CAS PubMed ;
(c) A. Abellán-López, M. T. Chicote, D. Bautista and J. Vicente, Organometallics, 2012, 31, 7434–7446 CrossRef ;
(d) D. Vázquez-García, A. Fernández, M. López-Torres, A. Rodríguez, A. Varela, M. T. Pereira, J. M. Vila and J. J. Fernández, Organometallics, 2011, 30, 396–404 CrossRef ;
(e) T. Wang, X.-Q. Hao, J.-J. Huang, K. Wang, J.-F. Gong and M.-P. Song, Dalton Trans., 2011, 40, 8964–8976 RSC ;
(f) M. Bröring, C. Kleeberg and S. Köhler, Inorg. Chem., 2008, 47, 6404–6412 CrossRef PubMed ;
(g) B. L. Dietrich, J. Egbert, A. M. Morris and M. Wicholas, Inorg. Chem., 2005, 44, 6476–6481 CrossRef CAS PubMed ;
(h) K.-M. Wu, C.-A. Huang, K.-F. Peng and C.-T. Chen, Tetrahedron, 2005, 61, 9679–9687 CrossRef CAS PubMed ;
(i) C.-T. Chen, Y.-S. Chan, Y.-R. Tzeng and M.-T. Chen, Dalton Trans., 2004, 2691–2696 RSC ;
(j) S.-W. Lai, T.-C. Cheung, M. C. W. Chan, K.-K. Cheung, S.-M. Peng and C.-M. Che, Inorg. Chem., 2000, 39, 255–262 CrossRef CAS .
- D. Morales-Morales and C. M. Jensen, The Chemistry of Pincer Compounds, Elsevier, Amsterdam, 2007 Search PubMed .
-
(a) P. Ghosh, S. Samanta, S. K. Roy, S. Demeshko, F. Meyer and S. Goswami, Inorg. Chem., 2014, 53, 4678–4686 CrossRef CAS PubMed ;
(b) P. Majumder, S. Baksi, S. Halder, H. Tadesse, A. J. Blake, M. G. B. Drew and S. Bhattacharya, Dalton Trans., 2011, 40, 5423–5425 RSC .
-
(a) X.-Q. Hao, Y.-W. Zhao, J.-J. Yang, J.-L. Niu, J.-F. Gong and M.-P. Song, Organometallics, 2014, 33, 1801–1811 CrossRef CAS ;
(b) G. R. Rosa and D. S. Rosa, RSC Adv., 2012, 2, 5080–5083 RSC ;
(c) X.-Q. Hao, Y.-X. Xu, M.-J. Yang, L. Wang, J.-L. Niu, J.-F. Gong and M.-P. Song, Organometallics, 2012, 31, 835–846 CrossRef CAS ;
(d) Q.-L. Luo, J.-P. Tan, Z.-F. Li, W.-H. Nan and D.-R. Xiao, J. Org. Chem., 2012, 77, 8332–8337 CrossRef CAS PubMed ;
(e) K. Srinivas, P. Srinivas, P. S. Prathima, K. Balaswamy, B. Sridhar and M. M. Rao, Catal. Sci. Technol., 2012, 2, 1180–2118 RSC ;
(f) N. Selander and K. J. Szabó, Chem. Rev., 2011, 111, 2048–2076 CrossRef CAS PubMed ;
(g) V. A. Kozlov, D. V. Aleksanyan, Y. V. Nelyubina, K. A. Lyssenko, P. V. Petrovskii, A. A. Vasil’ev and I. L. Odinets, Organometallics, 2011, 30, 2920–2932 CrossRef CAS ;
(h) J.-L. Niu, X.-Q. Hao, J.-F. Gong and M.-P. Song, Dalton Trans., 2011, 40, 5135–5150 RSC ;
(i) R. Mosteiro, A. Fernández, D. Vázquez-García, M. López-Torres, A. Rodríguez-Castro, N. Gómez-Blanco, J. M. Vila and J. J. Fernández, Eur. J. Inorg. Chem., 2011, 1824–1832 CrossRef CAS ;
(j) L. Ray, M. M. Shaikh and P. Ghosh, Organometallics, 2007, 26, 958–964 CrossRef CAS ;
(k) J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527–2572 CrossRef CAS PubMed ;
(l) I. P. Beletskaya and A. V. Cheprakov, J. Organomet. Chem., 2004, 689, 4055–4082 CrossRef CAS PubMed ;
(m) M. Ćurić, D. Babić, Ž. Marinić, L. Paša-Tolić, V. Butković, J. Plavec and L. Tušek-Božić, J. Organomet. Chem., 2003, 687, 85–99 CrossRef PubMed .
-
(a) A. Kumar, G. K. Rao, S. Kumar and A. K. Singh, Organometallics, 2014, 33, 2921–2943 CrossRef CAS ;
(b) L. M. Mirica and J. R. Khusnutdinova, Coord. Chem. Rev., 2013, 257, 299–314 CrossRef CAS PubMed ;
(c) A. de Meijere and F. Diederich, Metal-Catalyzed Cross-coupling Reactions, Wiley-VCH, New York, 2004 Search PubMed .
- C. Mills, J. Chem. Soc., 1895, 925–933 RSC ; A. Baeyer, Chem. Ber., 1874, 7, 1638–1640 CrossRef CAS .
-
(a) N. D. Paul, U. Rana, S. Goswami, T. K. Mondal and S. Goswami, J. Am. Chem. Soc., 2012, 134, 6520–6523 CrossRef CAS PubMed ;
(b) W. Kaim, Inorg. Chem., 2011, 50, 9752–9765 CrossRef CAS PubMed ;
(c) D. Das, T. K. Mondal, S. M. Mobin and G. K. Lahiri, Inorg. Chem., 2009, 48, 9800–9810 CrossRef CAS PubMed ;
(d) K. Pramanik, M. Shivakumar, P. Ghosh and A. Chakravorty, Inorg. Chem., 2000, 39, 195–199 CrossRef CAS .
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS PubMed .
-
(a) S. Pramanik, S. Roy, T. Ghorui, S. Ganguly and K. Pramanik, Dalton Trans., 2014, 43, 5317–5334 RSC ;
(b) P. Mondal, A. Hens, S. Basak and K. K. Rajak, Dalton Trans., 2013, 42, 1536–1549 RSC ;
(c) L. Szabó, K. Herman, N. E. Mircescu, A. Fǎlǎmaş, L. F. Leopold, N. Leopold, C. Buzumurgǎ and V. Chiş, Spectrochim. Acta, Part A, 2012, 93, 266–273 CrossRef PubMed ;
(d) K. Pramanik and B. Adhikari, Polyhedron, 2010, 29, 1015–1022 CrossRef CAS PubMed .
- N. Maiti, S. Pal and S. Chattopadhyay, Inorg. Chem., 2001, 40, 2204–2205 CrossRef CAS .
-
(a) G.-Q. Kong, S. Ou, C. Zou and C.-D. Wu, J. Am. Chem. Soc., 2012, 134, 19851–19857 CrossRef CAS PubMed ;
(b) G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Mülhaupt, J. Am. Chem. Soc., 2009, 131, 8262–8270 CrossRef CAS PubMed .
- B. P. Sullivan, D. J. Salmon, T. J. Meyer and J. Peedrin, Inorg. Chem., 1979, 18, 3369–3374 CrossRef CAS .
- J. V. Houten and R. J. Watts, J. Am. Chem. Soc., 1976, 98, 4853–4858 CrossRef .
-
(a) W. H. Melhuish, J. Phys. Chem., 1961, 65, 229–235 CrossRef CAS ;
(b) W. R. Dawson and M. W. Windsor, J. Phys. Chem., 1968, 72, 3251–3260 CrossRef CAS and references therein.
-
(a) F. I. El-Dossoki, J. Chin. Chem. Soc., 2007, 54, 1129–1137 CAS ;
(b) P. Pacak, Chem. Pap., 1991, 45, 227–232 CAS ;
(c) R. A. MacRae, E. T. Arakawa and M. W. Williams, J. Chem. Eng. Data, 1978, 23, 189–190 CrossRef CAS .
- G. M. Sheldrick, SHELXTL Version 6.14, Bruker AXS Inc., Madison, WI, 2003 Search PubMed .
- C. K. Johnson, Report ORNL-5138, Oak Ridge National Laboratory, Oak Ridge, TN, 1976 Search PubMed .
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed ;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS .
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed .
-
(a) J. Autschbach, T. Ziegler, S. J. A. Gisbergen and E. J. Baerends, J. Chem. Phys., 2002, 116, 6930–6940 CrossRef CAS PubMed ;
(b) K. L. Bak, P. Jørgensen, T. Helgaker, K. Rund and H. J. A. Jenson, J. Chem. Phys., 1993, 98, 8873–8887 CrossRef CAS PubMed ;
(c) T. Helgaker and P. Jørgensen, J. Chem. Phys., 1991, 95, 2595–2601 CrossRef CAS PubMed ;
(d) E. K. U. Gross and W. Kohn, Adv. Quantum Chem., 1990, 21, 255–291 CrossRef CAS .
-
(a) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed ;
(b) M. Cossi and V. Barone, J. Chem. Phys., 2001, 115, 4708–4717 CrossRef CAS PubMed ;
(c) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS .
-
(a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS PubMed ;
(b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS PubMed .
-
(a) M. S. Gordon, J. S. Binkley, J. A. Pople, W. J. Pietro and W. J. Hehre, J. Am. Chem. Soc., 1982, 104, 2797–2803 CrossRef CAS ;
(b) J. S. Binkley, J. A. Pople and W. J. Hehre, J. Am. Chem. Soc., 1980, 102, 939–947 CrossRef CAS .
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed .
Footnote |
| † Electronic supplementary information (ESI) available: NMR, absorption, emission spectra and photoluminescence decay of ligand, Tables and figures for TD-DFT of complexes, crystallographic data and structure refinement parameters, coordinates of optimized geometries of complexes. CCDC 1031475–1031479. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra16584e |
|
| This journal is © The Royal Society of Chemistry 2015 |