
C–H insertions in oxidative gold catalysis: synthesis of polycyclic 2H-pyran-3(6H)-ones via a relay strategy†
Zhitong
Zheng
and
Liming
Zhang
*
Department of Chemistry & Biochemistry, University of California, Santa Barbara, CA 93106, USA. E-mail: zhang@chem.ucsb.edu
First published on 14th October 2015
Abstract
An expedient synthesis of bicyclic/polycyclic 2H-pyran-3(6H)-ones is realized in a cascade cyclization triggered by oxidative gold catalysis and terminated by streamlining C–H insertions. In this reaction, an α-oxo gold carbene intermediate, initially formed upon gold-catalyzed oxidation of alkynes, could be trapped by a tethered C–C triple bond, thereby resulting in the formation of a putative vinyl cation intermediate. This intermediate of high electrophilicity is proposed to be responsible for intramolecular C–H insertions, the mostly concerted nature of which is established by the reactions of chiral substrates. The reaction provides a rapid approach to the construction of functionalized polycyclic systems from easily accessible bispropargyl ethers and with minimal structural prefunctionalization.
Functionalization of unactivated C(sp3)–H bonds is a topic of immense contemporary interest and of exceptional value in organic synthesis.1 A time-honored strategy is C–H insertions by in situ-generated, highly reactive intermediates, among which are carbenes/carbenoids,1c,d vinylidenes2 and their metal counterparts,3 and vinyl cations.4 In 2010, we5 reported a general strategy for generating highly electrophilic α-oxo gold carbenes via gold-catalyzed intermolecular oxidation of alkynes (Scheme 1A). Despite the development of various versatile transformations based on these in situ-generated intermediates,3d,6,7 their highly reactive nature has sparsely lent themselves to the implementation of new C–H insertion strategies.7d Some time ago we initiated two parallel programs aiming to explore the utilities of these oxidatively generated gold carbenes in intramolecular C–H insertions, namely, direct C–H insertions (e.g., Scheme 1B), and transformations into other reactive species capable of C–H insertions (the relay approach). For the direct approach, we have very recently realized such insertions into unactivated C(sp3)–H bonds by in situ-generated acceptor-/acceptor-substituted gold carbenes.6g For the relay approach,8 as shown in Scheme 1C, our design anticipated that with a diyne substrate, a terminal α-oxo gold carbene could be selectively generated upon oxidation of the terminal C–C triple bond. Subsequently, the gold carbene would be attacked by an appropriately tethered internal alkyne to afford a reactive vinyl cation intermediate, i.e., A. A would either undergo the reported C–H insertions4 to afford the cyclic product C, or lead to the same product through the rearranged gold carbene intermediate B.9 Hashmi and co-workers have reported a single example of this type of C–H insertion using an O-phenylenedialkyne substrate,9 and another single case was recently reported.10 We surmised that this little explored relay strategy, if realized with more flexible and readily accessible systems, would permit rapid access to cyclic products with no/little prior functionalization.
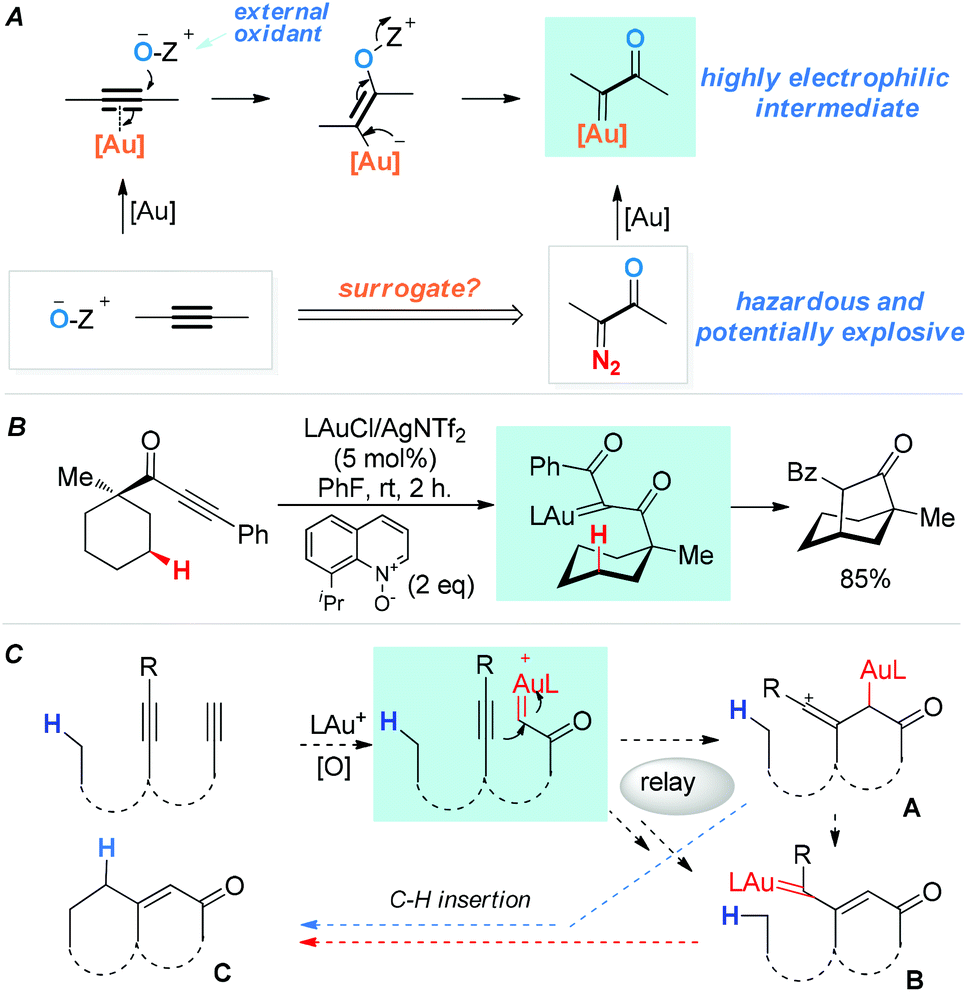 | ||
| Scheme 1 α-Oxo gold carbenes: (A) catalytic generation via oxidation of alkynes. (B) An example of direct insertion into unactivated C–H bonds. (C) A designed relay approach to C–H insertions. | ||
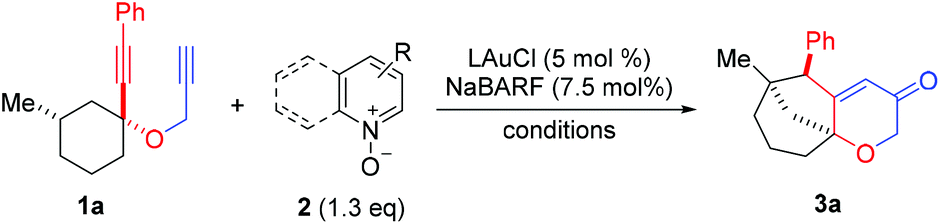
We set out to explore the relay approach by using the bispropargyl ether 1a as the substrate, which was readily prepared from 3-methylcyclohexanone in two steps and 40% yield. Our unpublished results of another oxidative catalysis suggest that this type of ethereal diyne has a tendency to undergo oxidative cyclization.10 While initial attempts with typical gold pre-catalysts including Ph3PAuCl, IPrAuCl and JohnPhosAuCl led to complex mixtures (Table 1, entries 1–3), a combination of BrettPhosAuCl,11 NaBARF and 2,6-dichloropyridine N-oxide (2a) yielded an encouraging 24% yield of the anticipated tricyclic 2H-pyran-3(6H)-one 3a (entry 3). An improved yield (40%) was achieved with the sterically highly demanding Me4tBuXPhos (entry 5) or the P,N-bidentate ligand Mor-DalPhos12 (entry 6) as the metal ligand. Due to the faster reaction with Me4tBuXPhosAuCl, it was chosen for further condition optimization. A brief screening of solvents (entries 5 and 7–8) revealed that fluorobenzene was the solvent of choice, and 3a was formed in 50% yield (entry 5). A series of N-oxides with different steric and electronic properties were also examined (Table 1, entries 9–12). 8-Isopropylquinoline N-oxide (2e) turned out to be the most effective, and 3a was formed in 70% yield (entry 12). To our surprise, the reaction efficiency was somehow independent of the reaction temperature, despite the large span of reaction times (entries 13–15). It is noteworthy that 3a was formed regioselectively, indicating significant preference for the methine C–H bond over the methylene counterpart in the C–H insertion step; moreover, the reaction was highly diastereoselective, and 2D NMR studies revealed that the shown endo-Ph isomer was formed with >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio over the unidentified exo isomer.
:1 ratio over the unidentified exo isomer.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | N-Oxide | Conditions | Yield |
a Reaction is run in a vial.
b A complicated mixture formed, and no desired product detected.
c Reaction was less clean. 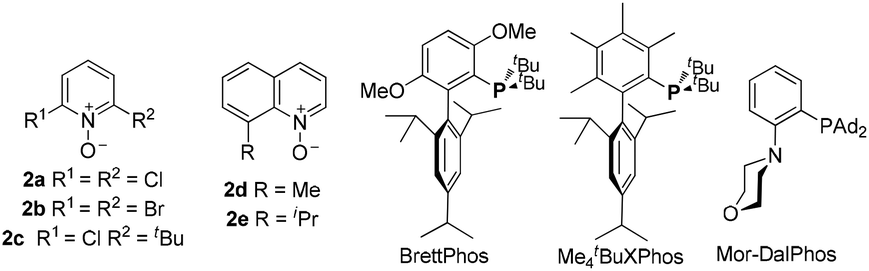
|
||||
| 1 | Ph3PAuCl | 2a | DCE, rt, 18 h | Traceb |
| 2 | IPrAuCl | 2a | DCE, rt, 18 h | Traceb |
| 3 | JohnPhosAuCl | 2a | DCE, rt, 18 h | Traceb |
| 4 | BrettPhosAuCl | 2a | DCE, rt, 3 h | 24% |
| 5 | Me4tBuXPhosAuCl | 2a | DCE, rt, 2 h | 40% |
| 6 | Mor-DalPhosAuCl | 2a | DCE, rt, 7 h | 41% |
| 7 | Me4tBuXPhosAuCl | 2a | PhMe, rt, 1 h | 46% |
| 8 | Me4tBuXPhosAuCl | 2a | PhF, rt, 1 h | 50% |
| 9 | Me4tBuXPhosAuCl | 2b | PhF, rt, 1 h | 49% |
| 10 | Me4tBuXPhosAuCl | 2c | PhF, rt, 1.5 h | 60% |
| 11 | Me4tBuXPhosAuCl | 2d | PhF, rt, 1 h | 58% |
| 12 | Me 4 t BuXPhosAuCl | 2e | PhF, rt, 1 h | 70% |
| 13 | Me4tBuXPhosAuCl | 2e | PhF, 50 °C, 5 min | 70%c |
| 14 | Me4tBuXPhosAuCl | 2e | PhF, 0 °C, 7 h | 66% |
| 15 | Me4tBuXPhosAuCl | 2e | PhF, −20 °C, 36 h | 65% |
With the optimal reaction conditions (cf. Table 1, entry 12) in hand, the scope of this relay chemistry was explored with substrates derived from substituted cyclohexanones, and the results are shown in Table 2. First, the phenyl group of 1a was varied by substitution of either an electron-donating 4-MeO (entry 1) or a slightly electron-withdrawing 4-Br (entry 2). In either case, the desired product was formed in a decent yield. The Me group on the cyclohexane ring of 1a was then modified. With an isopropyl (entry 3) or a phenyl group (entry 4) instead, the oxidative relay gold catalysis worked smoothly, affording the tricyclic ketone 3d or 3e in 64% or 65% yield, respectively. When the Me group of 1a was removed, the reaction still afforded 3f in a serviceable 50% yield (entry 5). This lower efficiency, as compared to that of 3a, is in accordance with the preference for tertiary C–H bonds over the secondary ones in the insertion step (vide supra). With the substrate prepared from 2-methylcyclohexanone, the isolated product 3g, albeit in a moderate 40% yield, exhibits an unexpected regiochemistry as the methylene C–H bond vicinal to the methyl group is selectively functionalized (entry 6). To our delight, the phenyl group at the alkyne terminus of 1a could be replaced by alkenyl groups such as 2-propenyl (entry 7) and cyclohexylvinyl (entry 8), and the tricyclic 2H-pyran-3(6H)-ones 3h and 3i were isolated in fair yields.
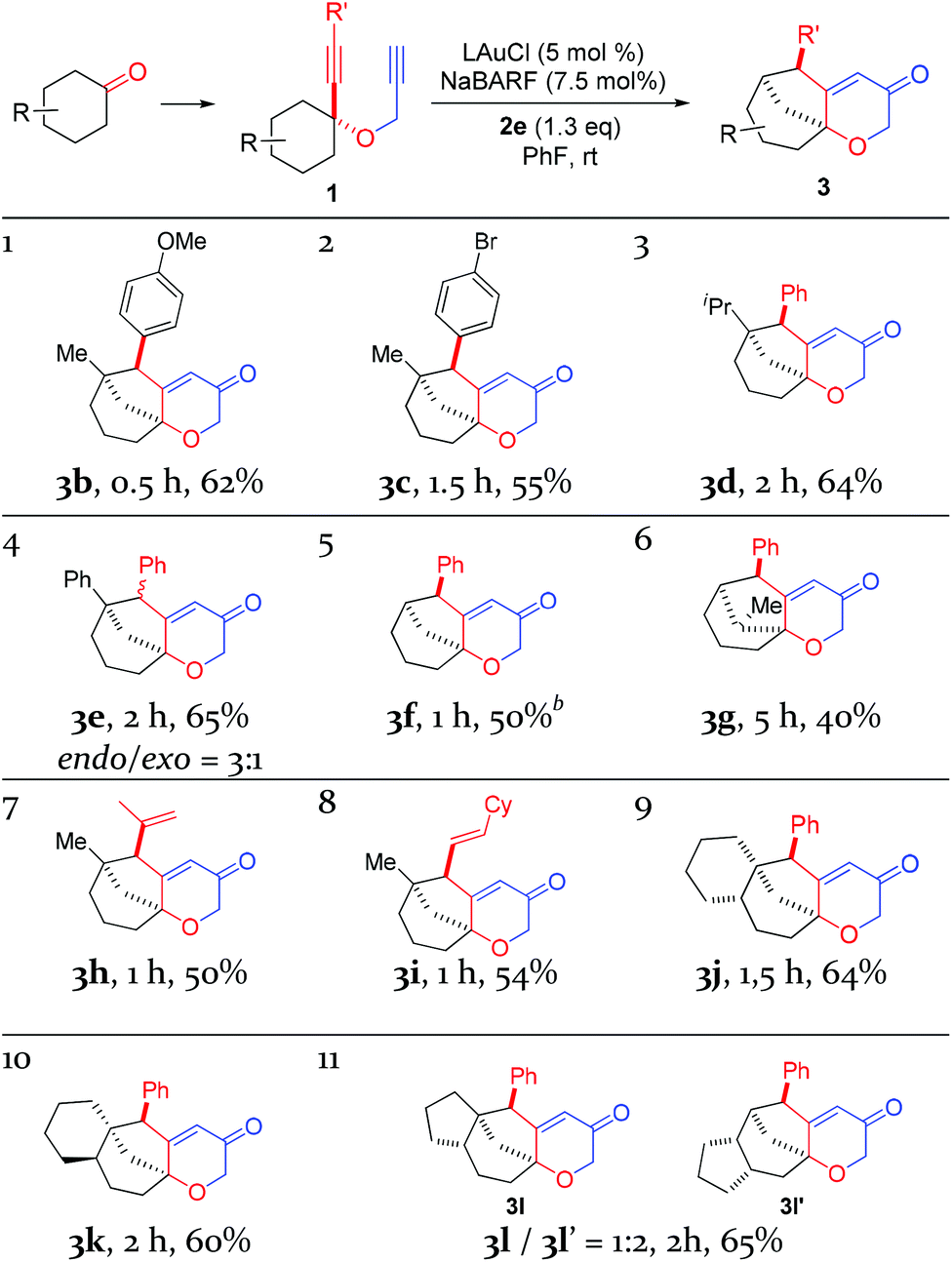
To probe the feasibility of this oxidative gold catalysis in rapid access to even more complex polycyclic structures, we prepared substrates from readily available cis- and trans-2-decalones. Both reacted smoothly to afford the tetracyclic ketones 3j and 3k in satisfactory yields, respectively (entries 9 and 10). On the other hand, the substrate derived from cis-hexahydro-1H-inden-5(6H)-one was found to form two regioisomeric products, i.e., 3l and 3l′, with a combined yield of 65% (entry 11). The major isomer 3l′ is the result of an insertion into the methylene C–H bond, while the minor isomer results from an insertion into the typically more preferred methine C–H bond. This seemingly inconsistent result is attributable to the likely impact of the cis-fused 5-membered ring on the requisite reaction conformation(s) of the cyclohexane ring. The importance of achieving appropriate reaction conformation(s) is even more apparent as the reaction of the diastereoisomer of 1a, where Me is cis to phenylethynyl, led to no desired products, despite full substrate consumption. In this case, the accessible insertion into the ring methylene C–H bond, as in the case of 3f, is hampered by the necessity of a minor reactive conformation with an axial methyl group and the steric hindrance it poses.
With the exception of 3e, all the other polycyclic pyranone products were formed as single diastereomers, in which the R′ group is positioned on the endo side of the bicyclo[3.2.1]octane skeleton.
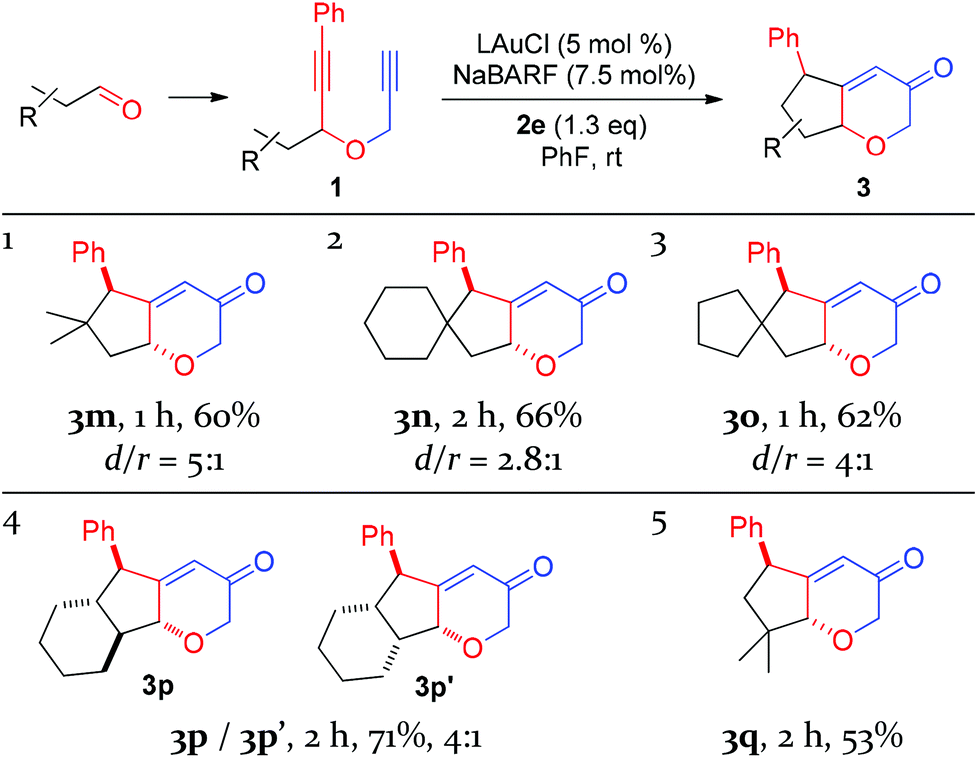
With the success of cyclohexanone-based substrates, we then investigated substrates prepared in one step from various aldehydes. The C–H insertion reaction worked well on a series of substrates bearing properly positioned methine C–H bonds, affording the desired products in fairly good yields and with modest diastereoselectivities (Table 3, entries 1–3). Secondary C–H bonds on a cyclohexane ring could also be inserted under the same conditions, and the linearly-fused tricyclic diastereomers 3p and 3p′ were formed in a good combined yield. Extensive 2D NMR spectroscopic studies revealed that their relative configurations differ at the ring fusion, where the trans-fused cyclohexane ring is moderately favored over the cis counterpart. Finally, the insertion into methyl C–H bonds could also be achieved as the substrate prepared from pivalaldehyde underwent the oxidative gold catalysis smoothly, affording 3q in a decent yield and with excellent diastereoselectivity. This last case highlights the high reactivity of the putative vinyl cation intermediate A or gold carbene B. In all these cases, the major diastereomers display trans relationships between the phenyl group and the ethereal oxygen.
| a Isolated yield reported. |
|---|
|
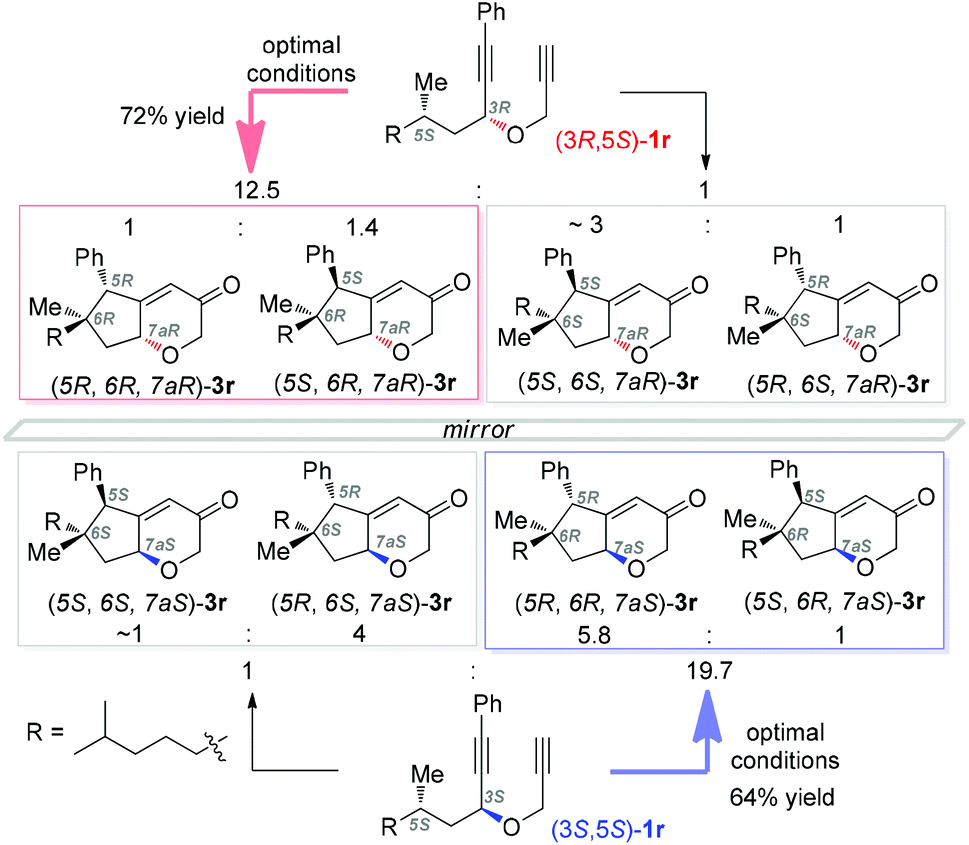
Supported by DFT calculations, Metzger4a in 2006 suggested that vinyl cations are inserted into C–H bonds in a concerted manner. Later, Gaunt4b in his Cu-catalyzed cascades to carbocycles largely confirmed the concerted nature by the conversion of a substrate of 99% ee into a product of 95% ee. To probe whether the C–H insertions in our system are concerted or not and thereby to shed light on the reaction mechanism, we prepared the separable diastereoisomers (3R,5S)-1r and (3S,5S)-1r from (S)-dihydrocitronellal (95% ee) and subjected each of them to the optimal conditions. As shown in Scheme 2, both reactions were efficient, and importantly in each case diastereomers with an unaltered configuration at the original dihydrocitronellal chiral center were formed predominantly. In the case of (3R,5S)-1r, the ratio of diastereomeric pairs of 12.5/1 reflects that ∼5% of the products experienced stereochemical inversion at the C–H insertion site, which is more significant than that reported by the Gaunt group (∼2%);4b however, with (3S,5S)-1r as the substrate, the ratio of 19.5/1 reflects a comparable 2.3% stereochemical erosion. These results, indicative of mostly concerted C–H insertions in our system, are consistent with the intermediacy of vinyl cations of type A. While at this point the involvement of gold carbene intermediates of type B could not be ruled out, previous studies of related donor-/acceptor-substituted gold carbenes reveal their relative stability and little tendency for C–H insertion.7a,b,q,13 Hence, it is more likely that the C–H insertions are achieved by the vinyl cation intermediates.
 | ||
| Scheme 2 Results supporting a mostly concerted C–H insertion mechanism. | ||
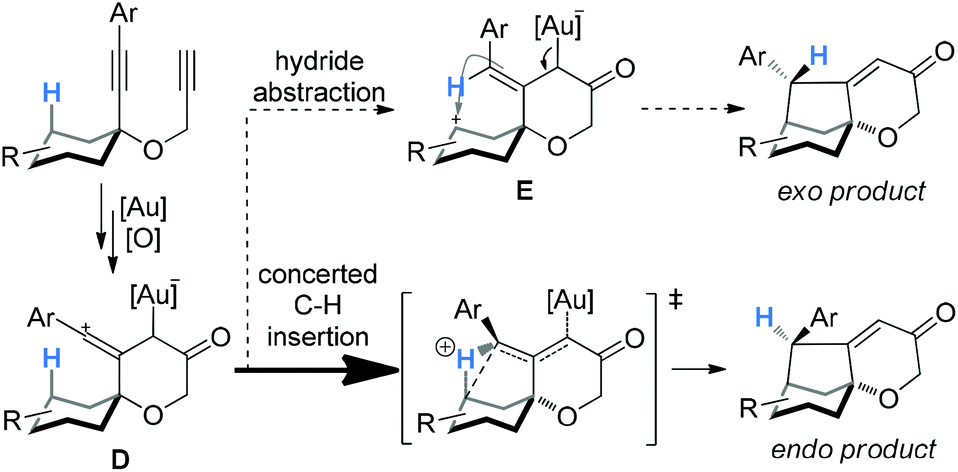
As mentioned previously, almost all the reactions of cyclohexanone-derived substrates exhibited excellent endo diastereoselectivities (>20:1), and with the exception of 3e, the thermodynamically more stable exo-products were not detectable. These stereoselectivities are consistent with the concerted C–H insertion mechanism by vinyl cation intermediates and can be readily rationalized. As shown in Scheme 3, the vinyl cation intermediate D formed upon the relay of the initially formed electrophilic gold carbene would have its linear vinyl cation moiety largely bisecting the cyclohexane ring. As such, a concerted C–H insertion would place the H atom away from the developing endo face of the bicyclo[3.2.1]octane system in the transition state,4a thereby affording the observed endo product. If hydride abstraction followed by cyclization were in operation, the carbenium intermediate E, with the expected double bond geometry, would lead to a significant/preferred formation of the unobserved exo product due to the approach of the carbenium moiety to the back side of the alkene moiety. This rationale can also readily account for the observation of the thermodynamic isomer in the case of 3e, where the benzylic hydrogen is much more prone to be abstracted as a hydride.
 | ||
| Scheme 3 Rationale for the observed diastereoselectivity. | ||
Conclusions
We have achieved an expedient synthesis of bicyclic/polycyclic 2H-pyran-3(6H)-ones from readily accessible bispropargyl ethers with minimal structural prefunctionalizations. This cascade cyclization is triggered by an oxidative gold catalysis and features streamlining insertions into unactivated C–H bonds. In the reaction, an α-oxo gold carbene intermediate is initially formed upon gold-catalyzed regioselective oxidation of the terminal C–C triple bond and is subsequently trapped by the tethered internal C–C triple bond, thereby affording a putative vinyl cation intermediate and realizing a relay of electrophilic sites. This intermediate of high electrophilicity is likely responsible for the intramolecular C–H insertions, the mostly concerted nature of which is established by the reactions of chiral substrates. This relay strategy opens a new avenue for the application of oxidative gold catalysis in the development of novel and synthetically streamlining C–H insertions.Acknowledgements
We acknowledge support from the NSF grant CHE-1301343 and the NIH shared instrument grant S10OD012077 for a 400 MHz NMR.Notes and references
- (a) Alkane C-H Activation by Single-Site Metal Catalysis, ed. P. J. Perez, [In: Catal. Met. Complexes, 2012; 38], Springer, 2012, 269pp Search PubMed; (b) C-H Activation, ed. J.-Q. Yu and Z. Shi, [In: Top. Curr. Chem., 2010; 292], Springer GmbH, 2010, 384pp Search PubMed; (c) M. P. Doyle, Y. Liu and M. Ratnikov, Org. React., 2013, 80, 1–131 CAS; (d) H. M. L. Davies and A. R. Dick, Top. Curr. Chem., 2010, 292, 303–345 CrossRef CAS.
- W. Kirmse, Angew. Chem., Int. Ed. Engl., 1997, 36, 1164–1170 CrossRef CAS PubMed.
- (a) L. Ye, Y. Wang, D. H. Aue and L. Zhang, J. Am. Chem. Soc., 2012, 134, 31–34 CrossRef CAS PubMed; (b) M. Wieteck, Y. Tokimizu, M. Rudolph, F. Rominger, H. Ohno, N. Fujii and A. S. K. Hashmi, Chem. – Eur. J., 2014, 20, 16331–16336 CrossRef CAS PubMed; For related studies, see: (c) A. S. K. Hashmi, I. Braun, P. Nösel, J. Schädlich, M. Wieteck, M. Rudolph and F. Rominger, Angew. Chem., Int. Ed., 2012, 51, 4456–4460 CrossRef CAS PubMed; (d) T. Wang, S. Shi, M. M. Hansmann, E. Rettenmeier, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2014, 53, 3715–3719 CrossRef CAS PubMed.
- (a) U. Biermann, R. Koch and J. O. Metzger, Angew. Chem., Int. Ed., 2006, 45, 3076–3079 CrossRef CAS PubMed; (b) F. Zhang, S. Das, A. J. Walkinshaw, A. Casitas, M. Taylor, M. G. Suero and M. J. Gaunt, J. Am. Chem. Soc., 2014, 136, 8851–8854 CrossRef CAS PubMed; (c) T. Jin, M. Himuro and Y. Yamamoto, J. Am. Chem. Soc., 2010, 132, 5590–5591 CrossRef CAS PubMed.
- (a) L. Ye, L. Cui, G. Zhang and L. Zhang, J. Am. Chem. Soc., 2010, 132, 3258–3259 CrossRef CAS PubMed; (b) L. Ye, W. He and L. Zhang, J. Am. Chem. Soc., 2010, 132, 8550–8551 CrossRef CAS PubMed; (c) B. Lu, C. Li and L. Zhang, J. Am. Chem. Soc., 2010, 132, 14070–14072 CrossRef CAS PubMed.
- (a) L. Zhang, Acc. Chem. Res., 2014, 47, 877–888 CrossRef CAS PubMed; (b) K. Ji, Y. Zhao and L. Zhang, Angew. Chem., Int. Ed., 2013, 52, 6508–6512 CrossRef CAS PubMed; (c) Y. Wang, K. Ji, S. Lan and L. Zhang, Angew. Chem., Int. Ed., 2012, 51, 1915–1918 CrossRef CAS PubMed; (d) Y. Luo, K. Ji, Y. Li and L. Zhang, J. Am. Chem. Soc., 2012, 134, 17412–17415 CrossRef CAS PubMed; (e) L. Ye, W. He and L. Zhang, Angew. Chem., Int. Ed., 2011, 50, 3236–3239 CrossRef CAS PubMed; (f) W. He, C. Li and L. Zhang, J. Am. Chem. Soc., 2011, 133, 8482–8485 CrossRef CAS PubMed; (g) Y. Wang, Z. Zheng and L. Zhang, J. Am. Chem. Soc., 2015, 137, 5316–5319 CrossRef CAS PubMed; (h) K. Ji, Z. Zheng, Z. Wang and L. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1245–1249 CrossRef CAS PubMed.
- (a) C.-W. Li, G.-Y. Lin and R.-S. Liu, Chem. – Eur. J., 2010, 16, 5803–5811 CrossRef CAS PubMed; (b) R. B. Dateer, K. Pati and R.-S. Liu, Chem. Commun., 2012, 48, 7200–7202 RSC; (c) S. Ghorpade, M.-D. Su and R.-S. Liu, Angew. Chem., Int. Ed., 2013, 52, 4229–4234 CrossRef CAS PubMed; (d) S. Bhunia, S. Ghorpade, D. B. Huple and R.-S. Liu, Angew. Chem., Int. Ed., 2012, 51, 2939–2942 CrossRef CAS PubMed; (e) D. Qian and J. Zhang, Chem. Commun., 2012, 48, 7082–7084 RSC; (f) D. Qian and J. Zhang, Chem. Commun., 2011, 47, 11152–11154 RSC; (g) P. W. Davies, Pure Appl. Chem., 2010, 82, 1537–1544 CrossRef CAS; (h) P. W. Davies, A. Cremonesi and N. Martin, Chem. Commun., 2011, 47, 379–381 RSC; (i) G. Henrion, T. E. J. Chavas, X. Le Goff and F. Gagosz, Angew. Chem., Int. Ed., 2013, 52, 6277–6282 CrossRef CAS PubMed; (j) M. Xu, T.-T. Ren and C.-Y. Li, Org. Lett., 2012, 14, 4902–4905 CrossRef CAS PubMed; (k) S. Shi, T. Wang, W. Yang, M. Rudolph and A. S. K. Hashmi, Chem. – Eur. J., 2013, 19, 6576–6580 CrossRef CAS PubMed; (l) T. Wang, S. Shi, M. M. Hansmann, E. Rettenmeier, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2014, 53, 3715–3719 CrossRef CAS PubMed; (m) T. Wang, S. Shi, M. Rudolph and A. S. K. Hashmi, Adv. Synth. Catal., 2014, 356, 2337–2342 CrossRef CAS PubMed; (n) T. Wang, L. Huang, S. Shi, M. Rudolph and A. S. K. Hashmi, Chem. – Eur. J., 2014, 20, 14868–14871 CrossRef CAS PubMed; (o) F. Pan, S. Liu, C. Shu, R.-K. Lin, Y.-F. Yu, J.-M. Zhou and L.-W. Ye, Chem. Commun., 2014, 50, 10726–10729 RSC; (p) L. Wang, X. Xie and Y. H. Liu, Angew. Chem., Int. Ed., 2013, 52, 13302–13306 CrossRef CAS PubMed; (q) L. Li, C. Shu, B. Zhou, Y.-F. Yu, X.-Y. Xiao and L.-W. Ye, Chem. Sci., 2014, 5, 4057–4064 RSC; (r) M. Chen, Y. Chen, N. Sun, J. Zhao, Y. Liu and Y. Li, Angew. Chem., Int. Ed., 2015, 1200–1204 CrossRef CAS PubMed.
- For other diyne-relays in homogeneous gold catalysis, see: (a) T. Lauterbach, T. Higuchi, M. W. Hussong, M. Rudolph, F. Rominger, K. Mashima and A. S. K. Hashmi, Adv. Synth. Catal., 2015, 357, 775–781 CrossRef CAS PubMed; (b) T. Lauterbach, M. Ganschow, M. W. Hussong, M. Rudolph, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2014, 356, 680–686 CrossRef CAS PubMed; (c) T. Lauterbach, S. Gatzweiler, P. Nösel, M. Rudolph, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2013, 355, 2481–2487 CrossRef CAS PubMed; (d) T. Lauterbach, S. Arndt, M. Rudolph, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2013, 355, 1755–1761 CrossRef CAS PubMed.
- P. Nösel, L. N. dos Santos Comprido, T. Lauterbach, M. Rudolph, F. Rominger and A. S. K. Hashmi, J. Am. Chem. Soc., 2013, 135, 15662–15666 CrossRef PubMed.
- A related study using bispropargylic ethers was reported. For reference, see: K. Ji, X. Liu, B. Du, F. Yang and J. Gao, Chem. Commun., 2015, 51, 10318–10321 RSC.
- B. P. Fors, D. A. Watson, M. R. Biscoe and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 13552–13554 CrossRef CAS PubMed.
- R. J. Lundgren, B. D. Peters, P. G. Alsabeh and M. Stradiotto, Angew. Chem., Int. Ed., 2010, 49, 4071–4074 CrossRef CAS PubMed.
- (a) Y. Xi, Y. Su, Z. Yu, B. Dong, E. J. McClain, Y. Lan and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 9817–9821 CrossRef CAS PubMed; (b) Z. Yu, B. Ma, M. Chen, H.-H. Wu, L. Liu and J. Zhang, J. Am. Chem. Soc., 2014, 136, 6904–6907 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00308c |
| This journal is © the Partner Organisations 2015 |