DOI:
10.1039/C5PY00874C
(Paper)
Polym. Chem., 2015,
6, 6193-6201
Organocatalysis by hydrogen-bonding: a new approach to controlled/living polymerization of α-amino acid N-carboxyanhydrides†
Received
8th June 2015
, Accepted 13th July 2015
First published on 14th July 2015
Abstract
A new method, based on hydrogen-bonding organocatalysis, was developed to achieve living ring-opening polymerization of N-carboxyanhydride of α-amino acids using aminoalcohols as initiators in the presence of N,N′-bis[3,5-bis(trifluoromethyl)phenyl]thiourea (TU-S). The thiourea provides, through hydrogen bonding, simultaneous activation of NCA monomers/reversible deactivation of polymer chain-ends/silencing of the tertiary amine and thus allows the polymerization to proceed in a highly controllable mode. For example, by using N,N-dimethyl ethanolamine (DMEA), as an initiator in the presence of TU-S, a series of well-defined linear polypeptides with differently designed Mns (3.01 × 104–18.10 × 104) and low PDI values (1.02–1.05) were successfully synthesized. This general strategy was also extended to the synthesis of well-defined di- and multi-armed polypeptides by using di-, tri-, or tetra-aminoalcohol initiators (methyldiethanolamine (MDEA), triethanolamine (TEA) or N,N,N′,N′-tetrakis(2-hydroxyethyl)ethylenediamine (THEED)) in the presence of TU-S.
Introduction
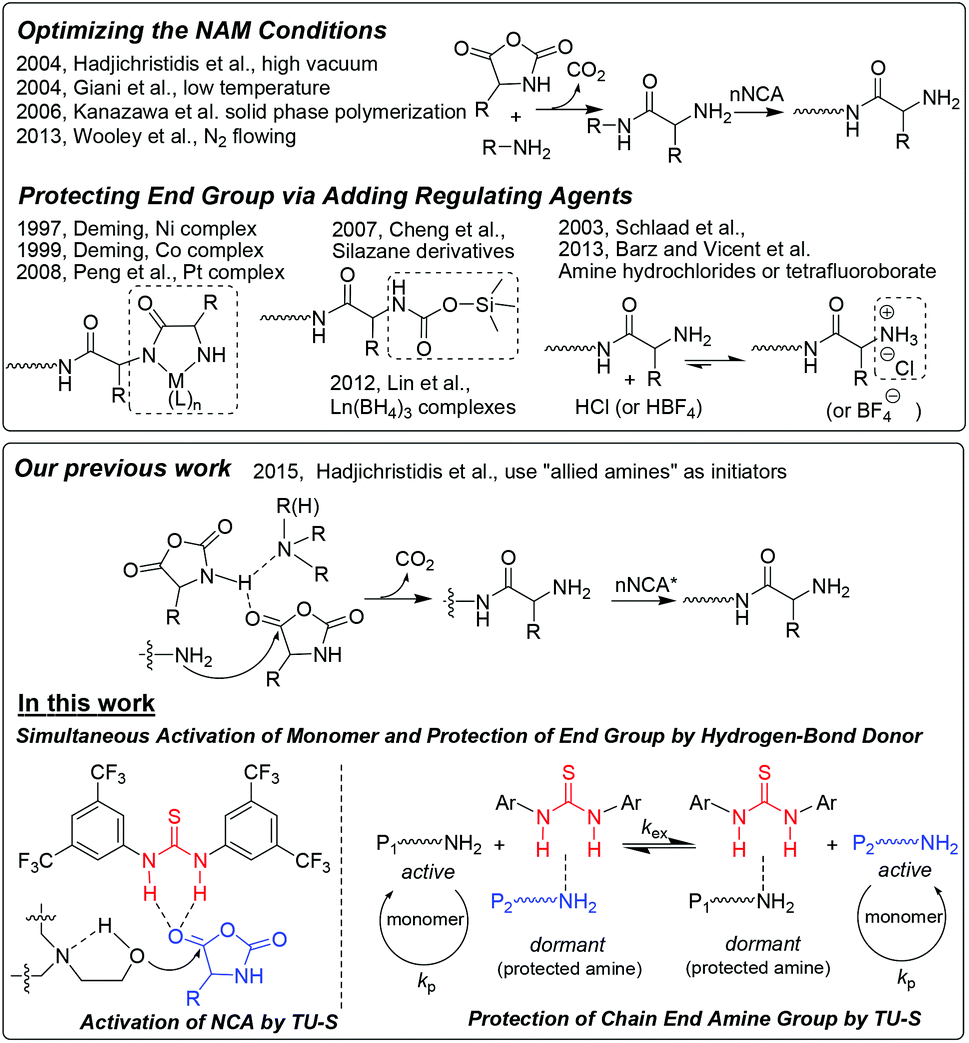
Well-defined synthetic polypeptides, as mimics of natural analogues, have found extensive applications in areas such as drug delivery, tissue engineering, sensing and catalysis.1 Among different methods leading to polypeptides, the ring-opening polymerization (ROP) of α-amino acid N-carboxyanhydrides (NCA) mediated by amines is the most efficient one. Although high molecular weight polypeptides can be readily synthesized using this method, typically ill-defined structures (uncontrolled molecular weight and broad molecular weight distribution) are obtained. Therefore, it is necessary and crucial to develop living ROP of NCAs to obtain well-defined polypeptides. In 1997, Deming reported the first example of living ROP of NCAs by using transition metal complexes as initiators.2 After that, a few controlled NCA polymerizations were reported either by employing new initiators (primary amine hydrochlorides or trifluoroboranes,3 silazane derivatives,4 Pt based5a and rare earth metal complexes5b,c) or by optimizing the primary amine initiated polymerizations (high vacuum, low temperature, nitrogen flowing and polymerization in solid phase).6 Very recently, we developed a new strategy for fast and living polymerization of NCAs at room temperature by employing a series of initiators including both primary and secondary or tertiary amines. In such polymerizations, the secondary or tertiary amines were found to activate the monomer, which resulted in a faster rate of polymerization (Scheme 1).7
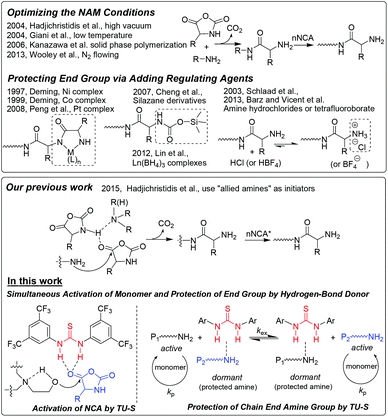 |
| | Scheme 1 Reported strategies for the living polymerization of α-amino acid NCAs and the new approach proposed in this work. | |
Herein, we investigate and report another new approach that also relies on monomer activation but unlike our previous work the catalyst is N,N′-bis[3,5-bis(trifluoromethyl)phenyl]thiourea (TU-S) and the initiation of the NCA polymerization is triggered by aminoalcohols. The TU-S simultaneously activates the NCA monomer, reversibly neutralizes the growing –NH2 chain ends and silences the tertiary amine which promotes the undesirable activated monomer mechanism, and thus affords a controlled polymerization as well as the synthesis of well-defined structures. This new strategy not only demonstrates the relevance of TU-S in NCA polymerization but also provides a novel approach towards the synthesis of well-defined polypeptide-based architectures.
Experimental section
General methods
All reactions were carried out under a dry and oxygen-free argon atmosphere by using Schlenk techniques or under an argon atmosphere in an MBraun glovebox. Solvents were purified using the MBraun SPS system. Anhydrous dimethylformamide (DMF) and dichloromethane (DCM) were further dried by passing through an activated alumina column. THF was dried vigorously with sodium–potassium alloy until a characteristic blue color was evident in the solvent. Anhydrous DCM-d2 was dried over P2O5 at room temperature under Ar overnight followed by distillation under reduced pressure. All other liquids were dried over activated 4 Å molecular sieves for a week and distilled before use, and solid materials were used as received. All purified reagents were stored over 4 Å molecular sieves in a glove box. H–Glu(OBn)–OH was purchased from Sigma-Aldrich and used as received. Glu-NCA was prepared and recrystallized four times according to published procedures.4N,N′-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea (abbreviated as TU-S) was synthesized and purified following the literature with slight modifications.8
Instruments and measurements
1H and 13C NMR spectra were recorded on a Bruker AV600 (FT, 500 MHz for 1H; 125 MHz for 13C) spectrometer. NMR assignments were confirmed by 1H–1H (COSY), 1H–13C (HMQC), and 13C NMR (DEPT) experiments when necessary. In situ IR study of NCA polymerization was carried out by using ReactIR 45m with an MCT detector from METTLER TOLEDO. AutoChem. DiComp probe (Diamond) was connected via AgX 9.5 mm × 1.5 m Fiber (Silver Halide). Spectra were taken from 2800 cm−1 to 650 cm−1 at 8 wavenumber resolution and the automatic sampling interval was 10 seconds. The real-time concentration of NCA was quantified by measuring the intensity of NCA's anhydride peak at 1792 cm−1 by FT-IR. The conversion of NCA was determined by comparing the NCA concentration during polymerization with the NCA concentration at t = 0. Polymer characterization was carried out by using the Agilent 1260 infinity SEC instrument equipped with a 1200 HPLC pump, an Optilab T-rEX RI detector, a ViscoStar-II viscometer and a DAWN HELEOS-II multiangle laser-light scattering (MALLS) detector at a wavelength of 690 nm (from Wyatt Technology). One guard column and three 7.8 × 300 mm columns (Styragel® HT 2 DMF, Styragel® HT 3 DMF and Styragel® HT 4 DMF) were used. HPLC-grade DMF (containing 0.1 M LiBr) was used as the mobile phase at a flow rate of 1.0 mL min−1. The whole system, including columns and detectors, was maintained at 60 °C. Polymer solutions with concentrations between 5.0 and 10.0 mg mL−1 were injected at an injection volume of 200 μL. ASTRA software from Wyatt Technology was used to collect and analyze the data from the detectors.
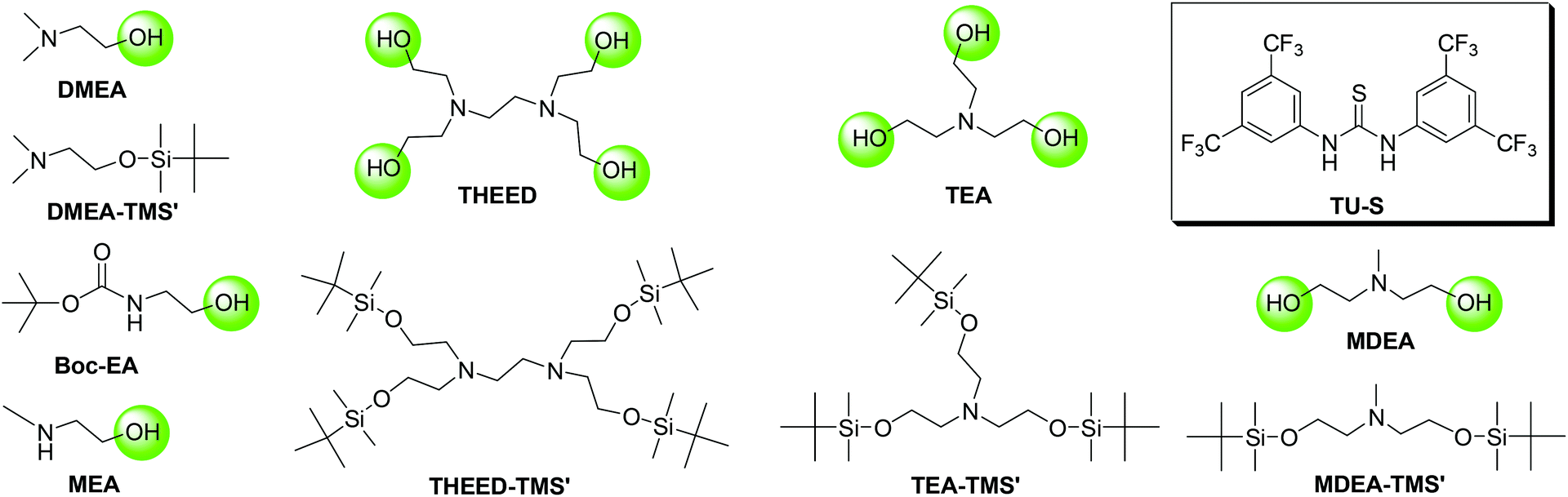
Synthesis of DMEA-TMS′
A solution of tert-butyldimethylchlorosilane (7.61 g, 0.05 mol) in N,N-dimethylformamide (DMF; 20 mL) was added dropwise to a mixture of N,N-dimethyl ethanolamine DMEA (5 g, 0.06 mol), imidazole (7.64 g, 0.11 mol), and DMF (20 mL) at 0 °C under nitrogen. The mixture was stirred for 6 h at room temperature, then washed with water, extracted with DCM and dried over anhydrous MgSO4. The solvents were removed in a vacuum to afford DMEA-TMS′. The crude product was purified by a silica gel column using hexane–dichloromethane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) as an eluent. Isolated yield: 90%. Purity: 99% (by gas chromatography). 1H NMR (CD2Cl2, 500 M, 25 °C): δH = 0.05 (s, 6H, –Si(CH3)2C(CH3)3), 0.88 (s, 9H, –Si(CH3)2C(CH3)3), 2.20 (s, 6H, –N(CH3)2), 2.38 (t, 2H, –NCH2CH2O–), 3.68 (t, 2H, –NCH2CH2O–). 13C NMR (CD2Cl2, 125 M, 25 °C): δC = −5.28 (2C, –Si(CH3)2C(CH3)3), 18.55 (1C, –Si(CH3)2C(CH3)3), 26.05 (3C, –Si(CH3)2C(CH3)3), 46.25 (2C, –N(CH3)2), 62.03 (1C, –NCH2CH2O–), 62.11 (4C, –NCH2CH2O–). 1H NMR and 13C NMR see the ESI.†
:1, v/v) as an eluent. Isolated yield: 90%. Purity: 99% (by gas chromatography). 1H NMR (CD2Cl2, 500 M, 25 °C): δH = 0.05 (s, 6H, –Si(CH3)2C(CH3)3), 0.88 (s, 9H, –Si(CH3)2C(CH3)3), 2.20 (s, 6H, –N(CH3)2), 2.38 (t, 2H, –NCH2CH2O–), 3.68 (t, 2H, –NCH2CH2O–). 13C NMR (CD2Cl2, 125 M, 25 °C): δC = −5.28 (2C, –Si(CH3)2C(CH3)3), 18.55 (1C, –Si(CH3)2C(CH3)3), 26.05 (3C, –Si(CH3)2C(CH3)3), 46.25 (2C, –N(CH3)2), 62.03 (1C, –NCH2CH2O–), 62.11 (4C, –NCH2CH2O–). 1H NMR and 13C NMR see the ESI.†
Synthesis of MDEA-TMS′
This compound was synthesized using the same procedure as for DMEA-TMS′ from tert-butyldimethylchlorosilane (18.97 g, 0.13 mol), imidazole (11.43 g, 0.17 mol), and methyldiethanolamine MDEA (5 g, 0.04 mol). Isolated yield: 90%. Purity: 99% (by gas chromatography). 1H NMR (CD2Cl2, 500 M, 25 °C): δH = 0.04 (s, 12H, –Si(CH3)2C(CH3)3), 0.88 (s, 18H, –Si(CH3)2C(CH3)3), 2.28 (s, 3H, –N(CH3)–), 2.52 (t, 4H, –NCH2CH2O–), 3.66 (t, 4H, –NCH2CH2O–). 13C NMR (CD2Cl2, 150 M, 25 °C): δC = −5.25 (4C, –Si(CH3)2C(CH3)3), 18.54 (2C, –Si(CH3)2C(CH3)3), 26.06 (6C, –Si(CH3)2C(CH3)3), 43.85 (1C, –N(CH3)–), 60.64(2C, –NCH2CH2O–), 62.10 (4C, –NCH2CH2O–). 1H NMR and 13C NMR see the ESI.†
Synthesis of TEA-TMS′
This compound was synthesized using the same procedure as for DMEA-TMS′ from tert-butyldimethylchlorosilane (20.20 g, 0.13 mol), imidazole (13.69 g, 0.20 mol), and triethanolamine TEA (5 g, 0.03 mol). Isolated yield: 92%. Purity: 99% (by gas chromatography). 1H NMR (CD2Cl2, 500 M, 25 °C): δH = 0.04 (s, 18H, –Si(CH3)2C(CH3)3), 0.88 (s, 27H, –Si(CH3)2C(CH3)3), 2.67 (t, 6H, –NCH2CH2O–), 3.62 (t, 6H, –NCH2CH2O–). 13C NMR (CD2Cl2, 150 M, 25 °C): δC = −5.24 (6C, –Si(CH3)2C(CH3)3), 18.54 (3C, –Si(CH3)2C(CH3)3), 26.08 (9C, –Si(CH3)2C(CH3)3), 58.32 (3C, –NCH2CH2O–), 62.58 (3C, –NCH2CH2O–). 1H NMR and 13C NMR see the ESI.†
Synthesis of THEED-TMS′
This compound was synthesized using the same procedure as for DMEA-TMS′ from tert-butyldimethylchlorosilane (15.94 g, 0.10 mol), imidazole (11.52 g, 0.17 mol), and N,N,N′,N′-tetrakis(2-hydroxyethyl)ethylenediamine THEED (5 g, 0.02 mol). Isolated yield: 95%. Purity: 99% (by gas chromatography). 1H NMR (CD2Cl2, 500 M, 25 °C): δH = 0.05 (s, 24H, –Si(CH3)2C(CH3)3), 0.88 (s, 36H, –Si(CH3)2C(CH3)3), 2.59 (s, 4H, –NCH2CH2N–), 2.62 (t, 8H, –NCH2CH2O–), 3.62 (t, 8H, –NCH2CH2O–). 13C NMR (CD2Cl2, 150 M, 25 °C): δC = −5.09 (8C, –Si(CH3)2C(CH3)3), 18.65 (4C, –Si(CH3)2C(CH3)3), 26.21 (12C, –Si(CH3)2C(CH3)3), 54.98 (2C, –NCH2CH2N–), 58.18 (4C, –NCH2CH2O–), 62.64 (4C, –NCH2CH2O–). 1H NMR and 13C NMR see the ESI.†
Polymerization procedure
A typical procedure for polymerization of NCA was performed in a 25 mL flask in a Braun Labmaster glovebox. To a vigorously stirred solution of DMEA and TU-S in 4 mL of solvent (DMF, THF or DCM) was added 0.4 g NCA monomer in 4 mL of solvent (DMF, THF or DCM). AutoChem. DiComp probe (Diamond) of ReactIR 45m (METTLER TOLEDO) was connected to the reaction flask via AgX 9.5 mm × 1.5 m Fiber (Silver Halide) to monitor the conversion of monomer. After a specific time, 0.2 ml of the reaction mixture was taken out from the system and diluted to 10 mg (PBLG or PZLL) mL−1 using DMF (containing 0.1 M LiBr). The solution was then analyzed by SEC to determine the molecular weight and PDI of the obtained polypeptides. The remaining final reaction mixture was precipitated with methanol, sonicated and centrifuged to remove the solvent. The obtained polymer was collected and dried under vacuum overnight after two repetitions of the sonication–centrifugation procedure.
Results and discussion
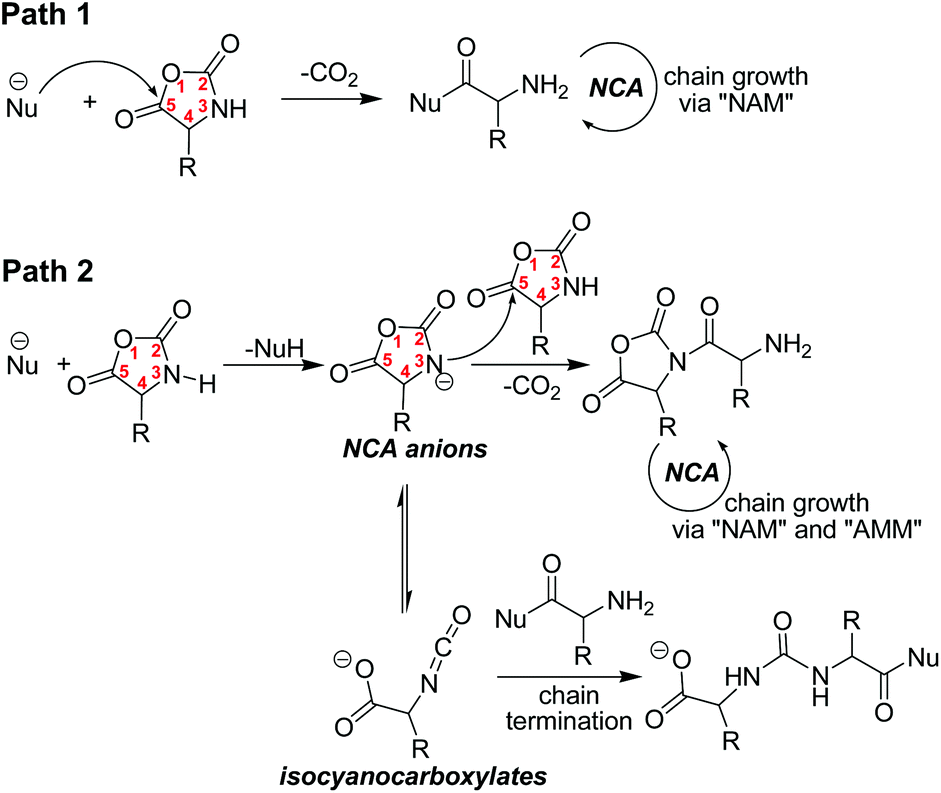
In α-amino acid NCAs, the carbonyl group (C5) is highly electrophilic and can easily be attacked by various nucleophiles, inducing ROP initiation (Scheme 2, path 1). In addition, the imine (N3) of NCA possesses a very active acidic N–H proton which can easily be abstracted under basic conditions. The resulting NCA anion can thus nucleophilically attack the carbonyl group (C5) of another NCA molecule and trigger the uncontrolled ROP of NCA in “activated monomer mechanism” (AMM), or can undergo rearrangement to isocyanocarboxylates and terminate the growing NH2-end active chains (Scheme 2, path 2). Highly nucleophilic initiators can thus induce the fast initiation that is required for living polymerization of NCA, but simultaneously abstract the N–H proton of NCA, resulting in the unwanted formation of NCA anions in the system. Consequently, it is challenging to find appropriate initiators for the living α-amino acid NCA polymerization.
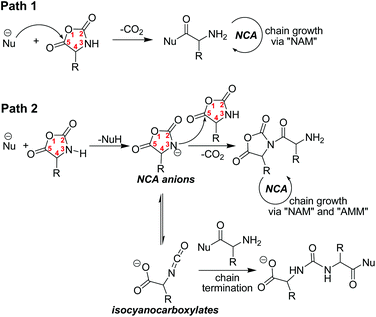 |
| | Scheme 2 Possible reactions in ROP of α-amino acid NCAs by nucleophilic initiators (“NAM” and “AMM” refer to “normal amine mechanism” and “activated monomer mechanism” respectively). | |
Inspired by the activation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in organic reactions by hydrogen bonding9 and the pioneering work of the Waymouth group in the ROP of cyclic esters,10 we selected a weak nucleophile, N,N-dimethyl ethanolamine (DMEA), as the initiator and a hydrogen-bonding donor compound, TU-S, as the activator of NCA monomers. Since DMEA is not a base, the possibility of abstracting the acidic N–H proton of NCA is very low and thus the formation of unwanted NCA anions can be excluded. On the other hand, TU-S is a widely used hydrogen-bonding donor that can dramatically increase the electrophilicity of carbon in carbonyls. Consequently, the TU-S/aminoalcohol combination should be an ideal and efficient catalytic system for NCA polymerization.
O bond in organic reactions by hydrogen bonding9 and the pioneering work of the Waymouth group in the ROP of cyclic esters,10 we selected a weak nucleophile, N,N-dimethyl ethanolamine (DMEA), as the initiator and a hydrogen-bonding donor compound, TU-S, as the activator of NCA monomers. Since DMEA is not a base, the possibility of abstracting the acidic N–H proton of NCA is very low and thus the formation of unwanted NCA anions can be excluded. On the other hand, TU-S is a widely used hydrogen-bonding donor that can dramatically increase the electrophilicity of carbon in carbonyls. Consequently, the TU-S/aminoalcohol combination should be an ideal and efficient catalytic system for NCA polymerization.
Polymerization of Glu-NCA by DMEA or TU-S
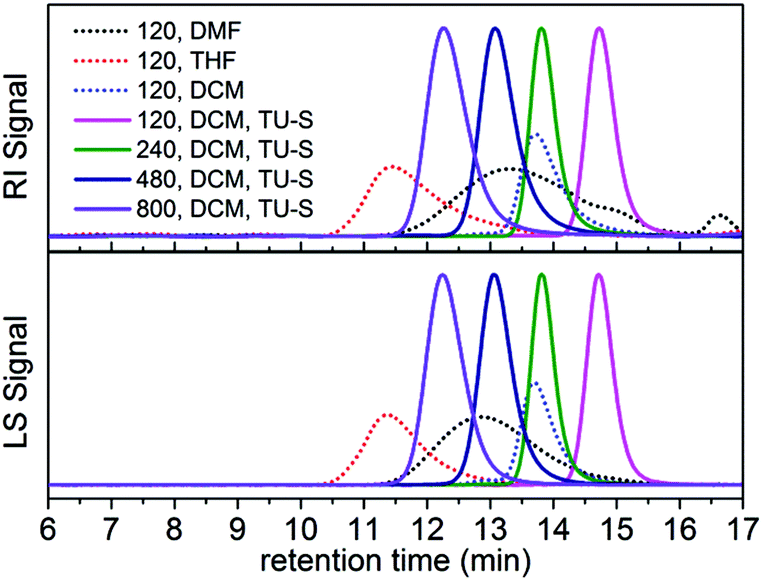
The polymerization of Glu-NCA initiated by DMEA was first evaluated in three different solvents N,N-dimethylformamide (DMF), tetrahydrofuran (THF) and dichloromethane (DCM) in the absence of TU-S (Table 1, runs 1–3). Under identical conditions (25 °C, [Glu-NCA] = 0.19 M and M/I = 120) the SEC trace of polypeptide obtained in DMF was bimodal (PDI = 1.58), indicating that the two mechanisms (NAM and AMM) occurred simultaneously during polymerization. When the polymerization was carried out in THF, a higher activity (100% monomer conversion in 3 h) compared to that in DMF was achieved but the molecular weight (Mn) (29.67 × 104) was much higher than the targeted one. The PDI of the obtained polypeptide was relatively high (1.28) and the SEC trace was asymmetric. When the solvent was changed to DCM, the polymerization was completed in 6 minutes and the Mn of the produced polypeptide (5.27 × 104) was two times that of the targeted Mn (2.63 × 104). The PDI of the final polymer was low (1.08), but the SEC chromatogram was still asymmetric (Fig. 1). These polymerization results indicate that, in a non-hydrogen bond forming solvent like DCM, DMEA promotes a faster ROP of Glu-NCA (vs. that in DMF or THF) and simultaneously exhibits a higher initiating efficiency (indicated by the value of Mn,calcd/Mn,SEC-LS). Therefore, most of the polymerizations of Glu-NCA initiated by DMEA were conducted in DCM.
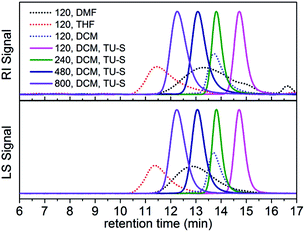 |
| | Fig. 1 SEC traces of polypeptides obtained in Table 1 (runs 1–3, and 7–10). | |
Table 1 Living polymerization of NCAs using alcohol/thiourea hydrogen-bonding organocatalytic system
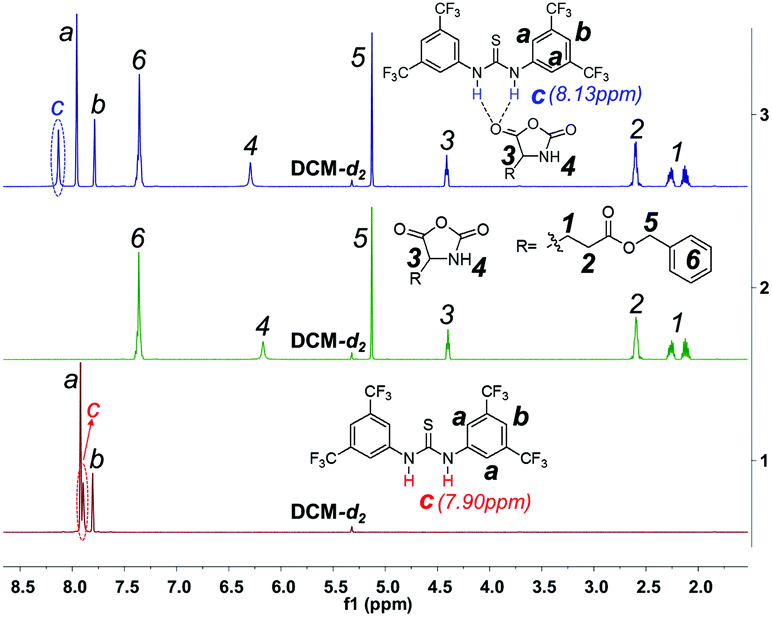
We also checked whether TU-S can itself initiate the polymerization of NCA and found that TU-S alone is unable to promote NCA polymerization. Under similar conditions to the previous case (25 °C, DCM, [Glu-NCA] = 0.19 M and M/I = 120), the monomer conversion was zero in 6 h (Table 1, run 4). The 1H NMR study of TU-S, Glu-NCA and TU-S/Glu-NCA mixture shows that TU-S cannot induce the ROP of NCA and consequently is only a hydrogen bonding donor to Glu-NCA. The signal of imide in TU-S is indeed deshielded from 7.90 to 8.13 ppm in the presence of Glu-NCA due to the hydrogen bonding between Glu-NCA and TU-S (Fig. 2 and S1 and S2†). According to the literature, hydrogen bonding between TU-S and the oxygen atom of carbonyl can dramatically enhance the activity of carbonyl.8
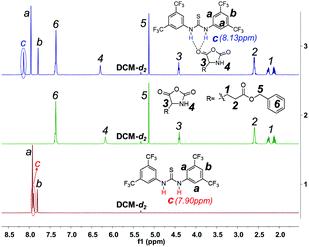 |
| | Fig. 2 Expanded 1H NMR spectra of TU-S, Glu-NCA and TU-S/Glu-NCA (1:1) mixture (500 M, 25 °C, CD2Cl2, for full spectra see Fig. S1 and S2†). | |
Polymerization of Glu-NCA by DMEA in the presence of TU-S
We then studied the polymerization promoted by DMEA in the presence of TU-S, expecting that the hydrogen-bonding donor TU-S can play a positive role in improving the performance of Glu-NCA polymerization. As expected, with a 0.8 mol% loading of TU-S relative to Glu-NCA (TU-S/DMEA = 1, M/I = 120), the polymerization proceeded very fast in DCM at room temperature and all the NCA monomer was converted into polypeptides in 12 min. Moreover, the obtained polymer exhibits a very narrow symmetric SEC trace (PDI = 1.05) and the Mn is close to the targeted one (Table 1, run 7). When the M/I ratio ([Glu-NCA]/[DMEA]) was gradually increased from 120 to 800 by decreasing the [DMEA], the Mn of the obtained polypeptide increased linearly as a function of M/I and the PDI: from 1.02 to 1.05, suggesting the absence of chain-breaking reactions during polymerization (Fig. S3†).11 The livingness of polymerization was further demonstrated by the linearity of Mnvs. conversion and the very low PDI values (Fig. S4†). Remarkably, when M/I ratio was increased to 800, 100% monomer conversion was obtained in 8 h, resulting in a polymer with the expected Mn and low PDI (18.10 × 104, 1.05). By varying the M/I ratios between 100 and 800, a series of polypeptides with different targeted Mns (from 3.01 × 104 to 18.10 × 104) and low PDI values (between 1.02 and 1.05) were successfully synthesized. Additionally, we compared the behavior of the TU-S/DMEA system in different polymerization solvents (DMF, THF and DCM). Well-defined polypeptides could only be obtained in the non-hydrogen bond forming solvent, DCM (Table 1, runs 5–7 and Fig. S5†). This indicates that hydrogen bonding is crucial for monomer activation and its subsequent polymerization, and that DMF and THF by their polar nature disrupt the interaction between the TU-S and the monomer.
Investigation of the initiation step
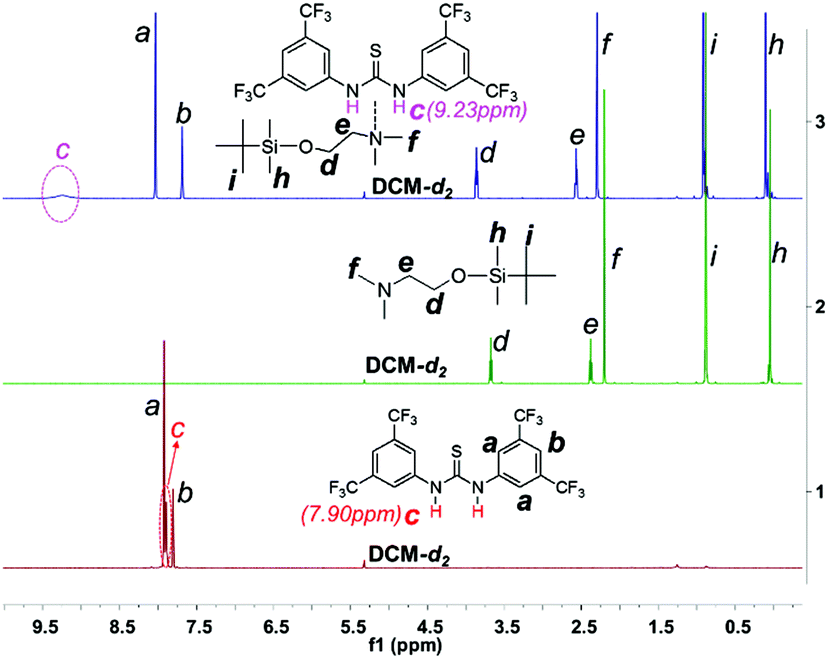
As DMEA contains both a primary alcohol and a tertiary amine, initiation may also occur through the latter group. In order to clarify this question, we protected the hydroxyl of DMEA by reacting with tert-butyldimethylsilane (resulting in DMEA-TMS′) and then used DMEA-TMS′ to initiate polymerization. We found that DMEA-TMS′ initiates the polymerization in the absence of TU-S but exhibits very low activity (24 h, 90%) and affords a polymer sample of very high Mn and PDI (Mn = 26.80 × 104, PDI = 1.61). In the presence of TU-S, DMEA-TMS′ showed no activity towards the ROP of Glu-NCA under the same conditions. 1H NMR indicates that TU-S is a hydrogen-bonding donor to the tertiary amine of DMEA-TMS′ and thus neutralizes the initiation capability of the tertiary amine. The signal of imide in TU-S shifts “downfield” from 7.90 to 9.23 ppm and the signals of DMEA-TMS′ (Hd, He, and Hf) also shift “downfield” (Fig. 3 and S6 and S7†). These results suggest that the tertiary amine of DMEA cannot trigger the polymerization of Glu-NCA in the presence of TU-S and the only initiating site is the hydroxyl group of DMEA. A further proof of the –OH initiating sites comes from the NMR spectra of low-Mn polypeptides where the initiator residuals can clearly be observed (see Fig. S8–S11†).
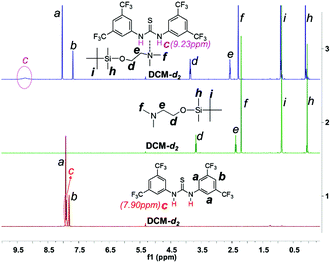 |
| | Fig. 3 Expanded 1H NMR spectra of TU-S, DMEA-TMS′ and TU-S/DMEA-TMS′ (1:1) mixture (500 M, 25 °C, CD2Cl2, for full spectra see Fig. S6 and S7†). | |
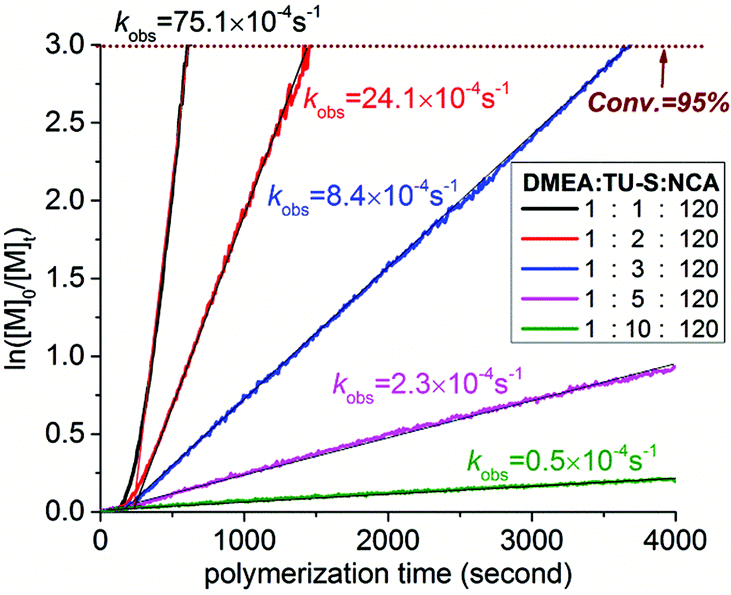
For the purpose of investigating the kinetics of polymerization, Glu-NCA in the presence of TU-S ([M]0 = 0.19 M, [M]0/[DMEA]0/[TU-S]0 = 120:1:1) was polymerized at 25 °C in DCM. The progress of the polymerization reaction was monitored in situ by IR at fixed time intervals (10 seconds) for a minimum of four half-live times. The polymerization exhibits a first-order dependence on the monomer concentration (Fig. 4, black line). The plot of ln([M]0/[M]t) vs. time gives two successive straight lines with different slopes. The difference in the slope is due to the secondary structure transition of PBLG from non-helices to α-helices (DP: 7–13). The linearity of the two lines suggests that the initiation is faster or comparable to chain propagation under the experimental conditions.12 After initiation, the active chain ends are primary amines and the whole polymerization exclusively follows the normal amine mechanism.
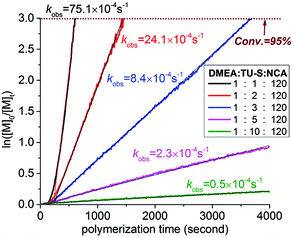 |
| | Fig. 4 Kinetics of the ROP of Glu-NCA promoted by DMEA/TU-S ([M] = 0.19 M, [Glu-NCA]/[DMEA] = 120, [TU-S]/[DMEA] = 1, 2, 3, 5, and 10, 25 °C, CH2Cl2, the automatic sampling interval of in situ IR is 10 seconds). | |
TU-S effect on the polymerization rate
We then increased the amount of TU-S in the reaction medium from 0.8 mol% (TU-S/DMEA = 1, M/I = 120, Table 1, run 7) to 1.6 mol% (TU-S/DMEA = 2, M/I = 120, Table 1, run 13) with a view to increasing the overall rate of polymerization. Instead of an increase, the rate of polymerization underwent a decrease and a longer time was required to complete polymerization (30 minutes). On increasing further the TU-S loading to 2.4 mol% (TU-S/DMEA = 3, M/I = 120, Table 1, run 14), the polymerization needed 90 minutes to achieve 100% conversion. A further increase to 8.0 mol% (TU-S/DMEA = 10, M/I = 120, Table 1, run 16) yielded only 57% monomer consumption after 4 h. The kinetic studies using the in situ IR showed that the observed polymerization rate kobs decreased from 75.1 × 10−4 to 0.5 × 10−4 s−1 upon increasing [TU-S] from 0.8 to 8.0 mol% in the system (Fig. 4). It is worth noticing that the incremental addition of TU-S into the medium did not affect the integrity of the samples. Good correlation between molecular weight values and initial [Glu-NCA]/[DMEA] value and narrow dispersity of polymers could still be obtained in all cases.
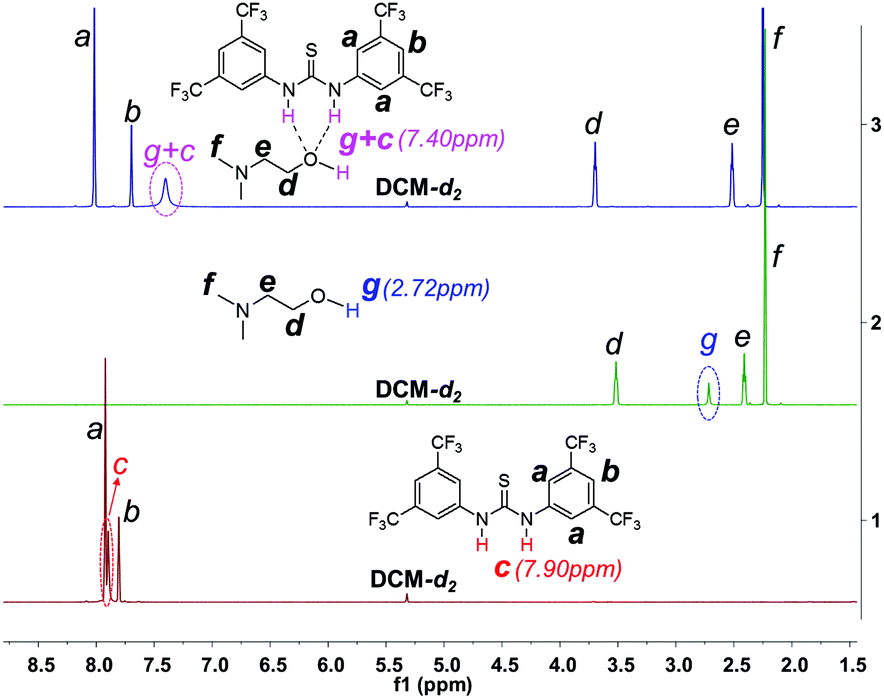
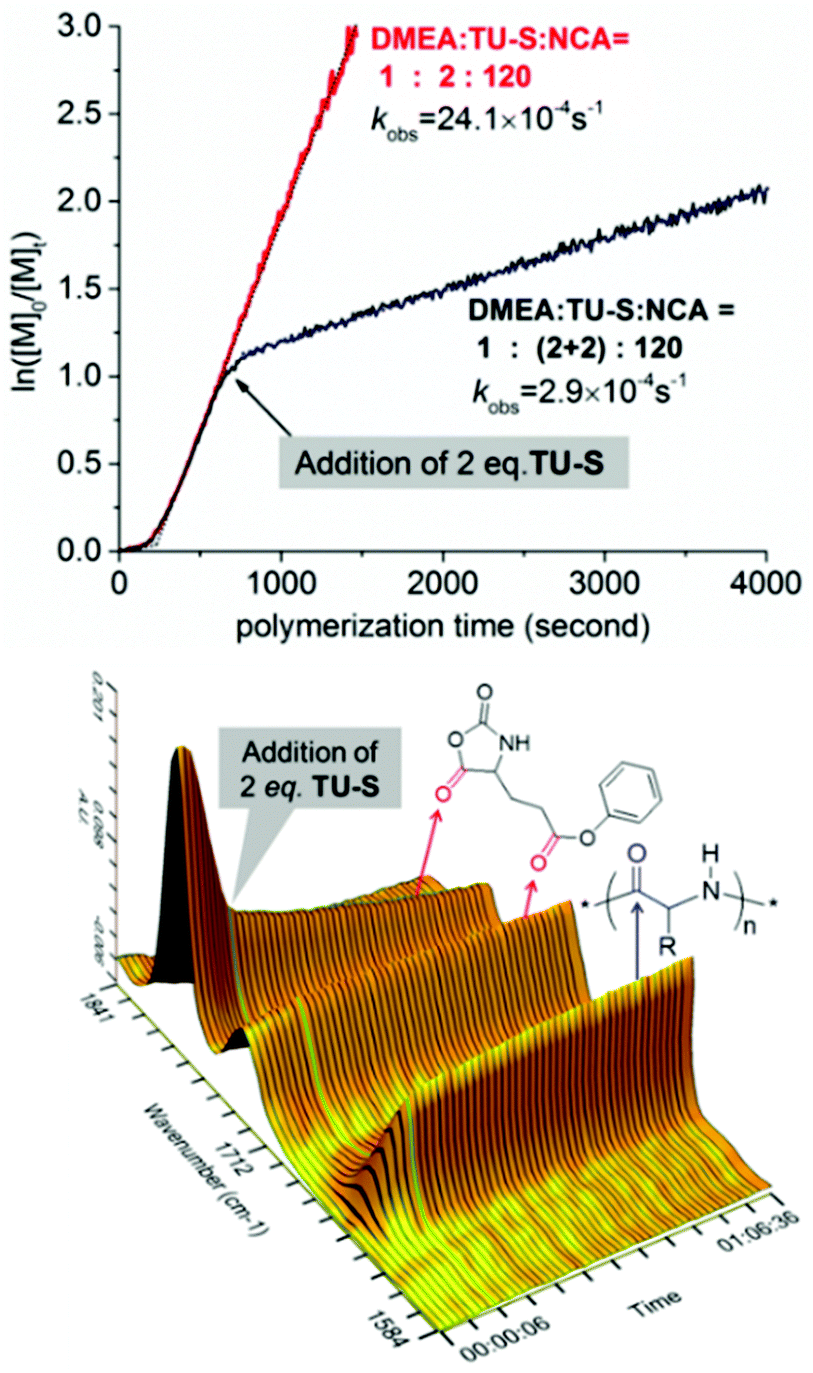
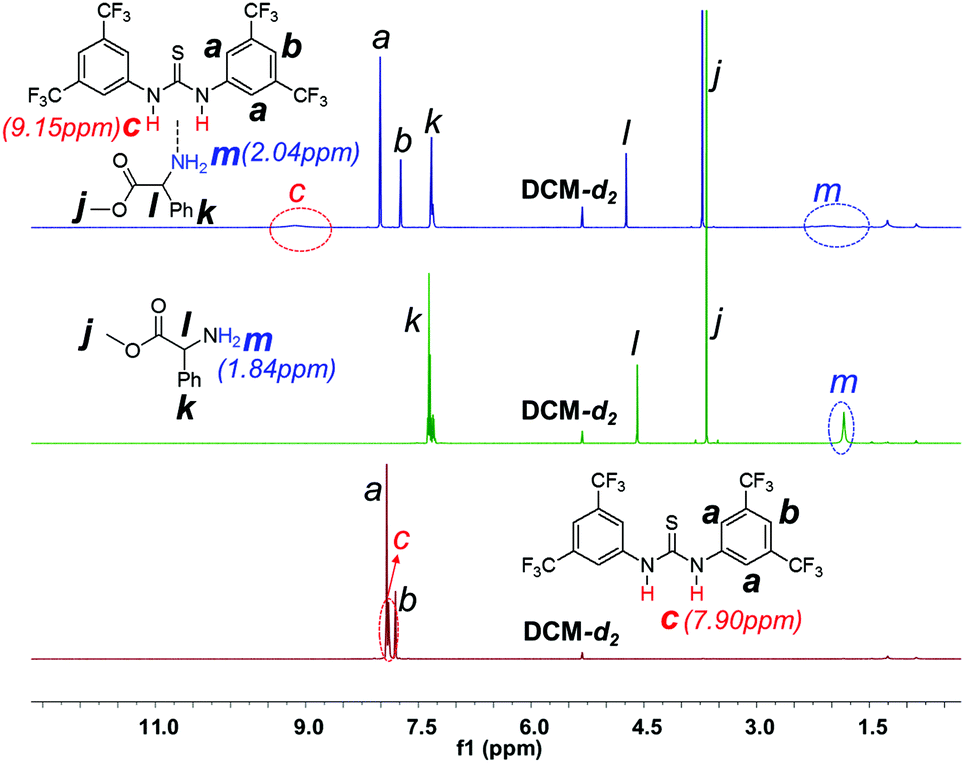
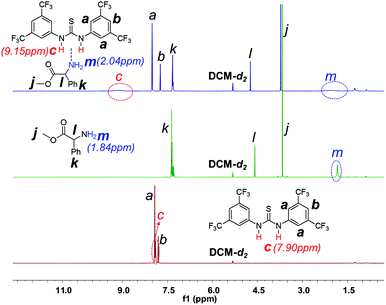
1H NMR investigation of TU-S, DMEA and TU-S/DMEA mixture in DCM-d2 revealed that hydrogen bonding was formed between TU-S and DMEA. In the presence of TU-S, the signal of the hydroxyl group in DMEA shifts “downfield” from 2.72 to 7.40 ppm and the signal of the imide protons in TU-S shifts “upfield” from 7.90 to 7.40 ppm (Fig. 5 and S12†), meaning that the nucleophilic character of the hydroxyl group in DMEA decreases upon formation of hydrogen-bonding with TU-S. Therefore, the presence of TU-S in the system resulted in a relative deactivation of the hydroxyl group. With the increase of TU-S, the concentration of TU-S-deactivated –OH increased, decreasing the rate of polymerization. However, all polymers from polymerization experiments with different amounts of TU-S possess the same targeted Mn and very low PDI, suggesting that all hydroxyl groups of DMEA (TU-S deactivated –OH and free –OH) trigger the polymerization of Glu-NCA through rapid exchange between TU-S deactivated –OH and free –OH. Since the rate of exchange is faster than that of the propagation, all hydroxyl groups have an equal chance to trigger the polymerization. After initiation, this exchange between dormant and active species still occurs between the amine end groups, which was confirmed by the results from in situ IR monitoring showing that the rate of polymerization slowed down after adding excess TU-S into the system during the polymerization (Fig. 6). This dormant/active exchange was further supported by the 1H NMR investigation of TU-S/2-amino-2-phenylacetate (MAP), a small model molecule with similar structure to that of the polymer chain end (Fig. 7 and S13†).
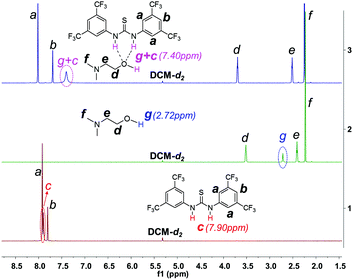 |
| | Fig. 5
1H NMR spectra of TU-S, DMEA and TU-S/DMEA (1:1) mixture (500 M, 25 °C, CD2Cl2, for full spectra see Fig. S12†). | |
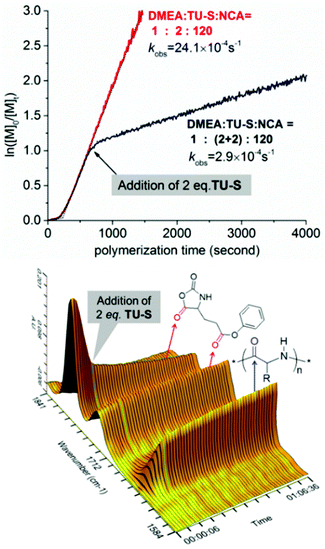 |
| | Fig. 6 Ln([NCA]0/([NCA]t) vs. time for the ROP of Glu-NCA initiated by DMEA/TU-S ([M] = 0.19 M, [Glu-NCA]/[TU-S]/[DMEA] = 120/(2 + 2)/1, 25 °C, CH2Cl2) and corresponding 3D kinetic behavior profile from in situ IR (the sampling interval of in situ IR is 10 seconds). | |
 |
| | Fig. 7
1H NMR spectra of TU-S, methyl 2-amino-2-phenylacetate (MAP) and TU-S/MAP mixture (500 M, 25 °C, CD2Cl2, for full spectrum of the mixture see Fig. S13†). | |
TU-S thus has a triple effect through its ability to donate hydrogen-bonding: (1) activation of the monomer; (2) reversibly deactivating the growing amines, which results in a slower rate of polymerization; (3) total silencing the tertiary amine in DMEA thus preventing proton abstraction of the monomers and the occurrence of AMM.
Proposed mechanism and extension of initiators to tri-, di- or mono-alcohols
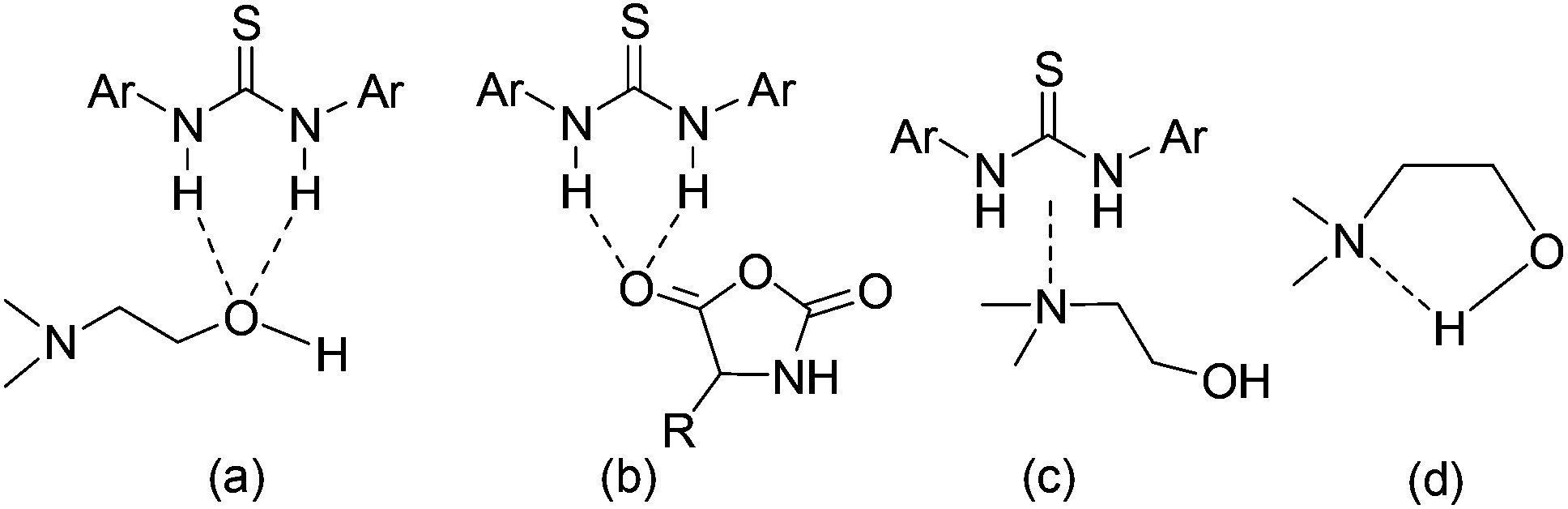
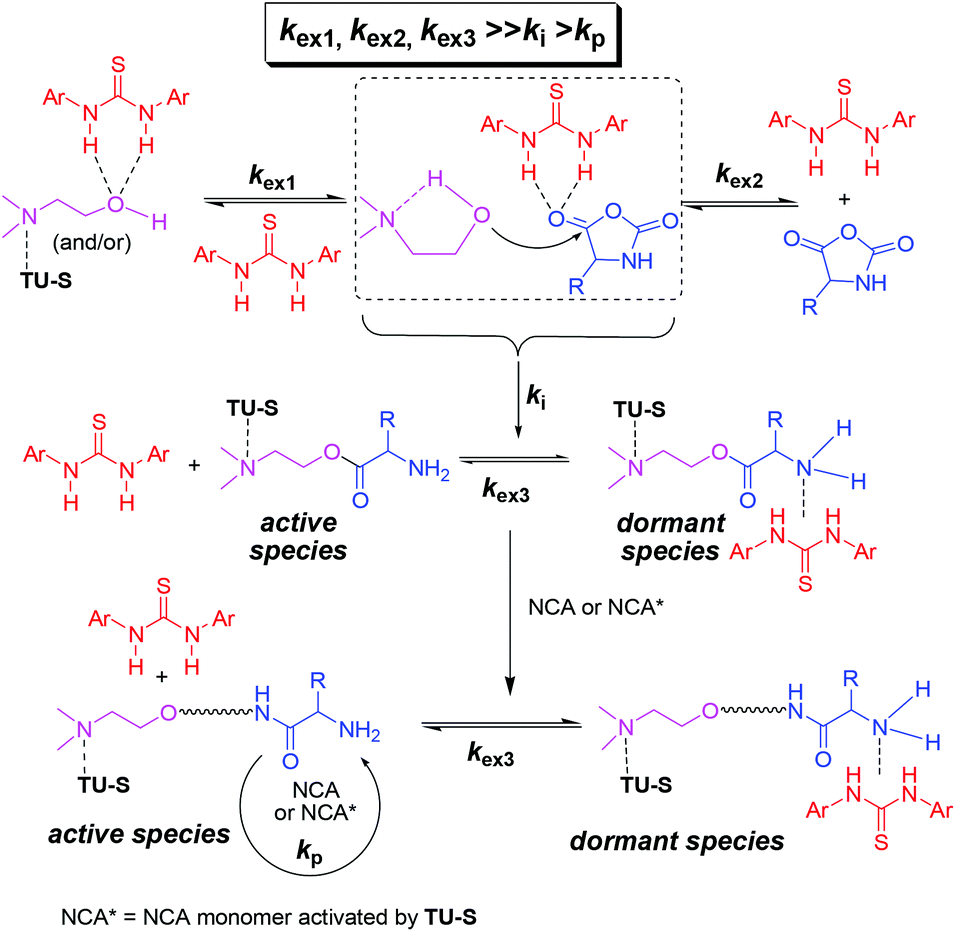
As mentioned before, the 1H NMR study indicates that TU-S not only interacts with the carbonyl of NCA monomer but also with the tertiary amine and the hydroxyl group of DMEA (Fig. 2, 3 and 5 and Scheme 3). To investigate the possible generality of the above observation, we used benzyl alcohol (BnOH) and tert-butyl 2-hydroxyethylcarbamate (Boc-EA) as an initiating system to promote the ROP of Glu-NCA in the presence of TU-S but failed to obtain the polymer. This indicates that the tertiary amine in DMEA plays an important role in the initiating step. The polymerizations respectively initiated by Boc-EA, MEA, or DMEA further supported this conclusion. In the case of MEA, the polymerization could be successfully triggered, but the polypeptide formed exhibits a high Mn and PDI due to the poor capability of the secondary amine to activate the hydroxyl group (vs. that of tertiary amine) (Table 1, run 43). So, a plausible polymerization pathway is shown in Scheme 4, in which multiple hydrogen-bonding equilibria involving the initiator (DMEA) and the catalyst (TU-S) exist in the ROP of the NCA monomer. By using tetra- tri-, or di- alcohols with tertiary amines (THEED, TEA, or MDEA), we successfully extended this strategy to multi-armed and well-defined PBLG with designed Mn and low PDI (Table 1, runs 23–26, 29–34 and 37–39). In addition, NMR and polymerization experiments like that for DMEA were carried out by using THEED as the initiator. Similar effects of TU-S on the polymerization rate and multiple hydrogen-bonding involving THEED, TU-S and NCA monomer were also observed (for the detailed results see Fig. S14–S21†). The lower viscosities in the Mark–Houwink–Sakurada plots (Fig. S22†) of PBLG prepared by THEED (four –OH initiator) rather than those prepared by DMEA (one –OH) and hexamethylene diamine HMDA (two –NH2) provide further proof that the –OH groups are the initiating species of the ROP of NCAs.
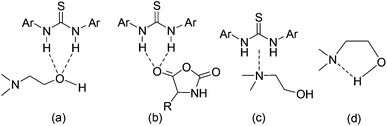 |
| | Scheme 3 Multiple hydrogen-bonding involving initiator (DMEA), catalyst (TU-S) and NCA monomer. | |
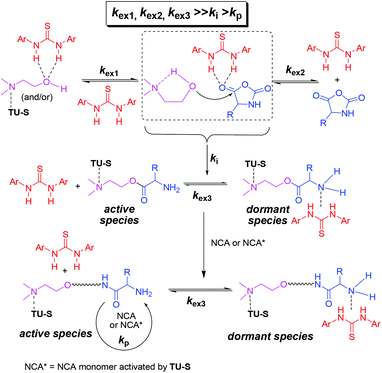 |
| | Scheme 4 Multiple hydrogen-bonding equilibria involving initiator (DMEA), catalyst (TU-S) and monomer during the ROP of NCA. | |
Polymerization of Lys-NCA and copolymerization of Glu- and Lys-NCA
Besides the homopolymerization of Glu-NCA, we also tried the homopolymerization of Lys-NCA and the copolymerization of Glu- and Lys-NCA. As shown in Table 2, all initiators (DMEA, MDEA, TEA, and THEED) could trigger controlled ROP of Lys-NCA in the presence of TU-S and give polypeptides with targeted Mn and very low PDI (1.02–1.05). In particular, by using THEED as the initiator, multi-armed PBLG-block-PZLL and PBLG-random-PZLL with targeted Mn and narrow PDI were easily prepared.
Table 2 Homo- and co-polymerization of Lys-NCA and Glu-NCA
| Runa |
Initiator |
[M]0/[I]0 |
Timeb (min) |
M
n,calcd × 10−4c |
M
n,exp × 10−4d |
PDId |
|
Polymerization was performed in DCM at 25 °C with [TU-S] = 7.6 mM and [NCA]0 = 0.19 M.
In situ IR was used to determine the time when >99% monomer conversion was achieved.
Calculated by [NCA]/[I] × (MNCA − 44) × X (X = Conv.).
Determined by SEC combined with MALS-VISC-DRI triple detection in 0.1 M LiBr in DMF at 60 °C. dn/dc(PBLG-co-PZLL) = 0.114 mL g−1, dn/dc(PBLG) = 0.104 mL g−1 and dn/dc(PZLL) = 0.123 mL g−1.
|
| 1 |
DMEA
|
120/1 |
53 |
3.15 |
2.90 |
1.02 |
| 2 |
DMEA
|
240/1 |
160 |
6.30 |
6.42 |
1.04 |
| 3 |
MDEA
|
120/1 |
45 |
3.15 |
2.64 |
1.04 |
| 4 |
MDEA
|
240/1 |
130 |
6.30 |
6.51 |
1.03 |
| 5 |
TEA
|
120/1 |
39 |
3.15 |
3.02 |
1.05 |
| 6 |
TEA
|
240/1 |
120 |
6.30 |
6.43 |
1.04 |
| 7 |
THEED
|
120/1 |
35 |
3.15 |
3.05 |
1.05 |
| 8 |
THEED
|
240/1 |
100 |
6.30 |
6.30 |
1.04 |
| 9 |
THEED
|
480/1 |
300 |
12.60 |
13.03 |
1.05 |
| 10 |
THEED (block) |
(120 + 120)/1 |
155 + 25 |
5.78 |
5.91 |
1.07 |
| 11 |
THEED (random) |
(120 + 120)/1 |
200 |
5.78 |
5.83 |
1.08 |
Conclusions
In summary, we have demonstrated, for the first time, that aminoalcohols in the presence of N,N′-bis[3,5-bis(trifluoromethyl)phenyl]thiourea (TU-S) catalyst promote the fast controlled/living ROP of Glu-NCA under mild conditions. It has been proven that TU-S has a triple effect through its ability to donate hydrogen-bonding: (1) activation of the monomer; (2) reversibly deactivating the growing amines, which results in a protection of active polymer chain ends; (3) totally silencing the tertiary amine in DMEA thus preventing proton abstraction of the monomers and the occurrence of AMM. This general strategy was successfully extended to the synthesis of well-defined di- and multi-armed polypeptides by using di-, tri-, or tetra-aminoalcohol initiators. Further study will be carried out to extend this strategy to other NCA monomers and to the synthesis of functionalized polypeptides.
Acknowledgements
Research reported in this publication was supported by the King Abdullah University of Science and Technology.
Notes and references
-
(a) T. J. Deming, Adv. Drug Delivery Rev., 2002, 54, 1145 CrossRef CAS;
(b) X. Y. Wang, H. J. Kim, C. Wong, C. Vepari, A. Matsumoto and D. L. Kaplan, Mater. Today, 2006, 9, 44 CrossRef CAS;
(c) S. Dos Santos, A. Chandravarkar, B. Mandal, R. Mimna, K. Murat, L. Saucede, P. Tella, G. Tuchscherer and M. Mutter, J. Am. Chem. Soc., 2005, 127, 11888 CrossRef CAS PubMed;
(d) R. J. Mart, R. D. Osborne, M. M. Stevens and R. V. Ulijn, Soft Matter, 2006, 2, 822 RSC;
(e) C. Deng, J. Wu, R. Cheng, F. Meng, H. Klok and Z. Zhong, Prog. Polym. Sci., 2014, 39, 330 CrossRef CAS PubMed;
(f) H. Lu, J. Wang, Z. Song, L. Yin, Y. Zhang, H. Tang, C. Tu, Y. Lin and J. Cheng, Chem. Commun., 2014, 50, 139 RSC.
-
(a) T. J. Deming, Nature, 1997, 390, 386 CrossRef CAS PubMed;
(b) T. J. Deming, J. Am. Chem. Soc., 1998, 120, 4240 CrossRef CAS.
-
(a) I. Dimitrov and H. Schlaad, Chem. Commun., 2003, 2944 RSC;
(b) I. Conejos-Sánchez, A. Duro-Castano, A. Birke, M. Barz and M. Vicent, Polym. Chem., 2013, 4, 3182 RSC;
(c) A. Birke, D. Huesmann, A. Kelsch, M. Weilbächer, J. Xie, M. Bros, T. Bopp, C. Becker, K. Landfester and M. Barz, Biomacromolecules, 2014, 15, 548 CrossRef CAS PubMed.
- H. Lu and J. Cheng, J. Am. Chem. Soc., 2007, 129, 14114 CrossRef CAS PubMed.
-
(a) Y. Peng, S. Lai and C. Lin, Macromolecules, 2008, 41, 3455 CrossRef CAS;
(b) H. Peng, J. Ling and Z. Shen, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1076 CrossRef CAS PubMed;
(c) H. Peng, J. Ling, Y. Zhu, L. You and Z. Shen, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3016 CrossRef CAS PubMed.
-
(a) T. Aliferis, H. Iatrou and N. Hadjichristidis, Biomacromolecules, 2004, 5, 1653 CrossRef CAS PubMed;
(b) W. Vayaboury, O. Giani, H. Cottet, A. Deratani and F. Schue, Macromol. Rapid Commun., 2004, 25, 1221 CrossRef CAS PubMed;
(c) W. Vayaboury, O. Giani, H. Cottet, S. Bonaric and F. Schue, Macromol. Chem. Phys., 2008, 209, 1628 CrossRef CAS PubMed;
(d) D. L. Pickel, N. Politakos, A. Avgeropoulos and J. M. Messman, Macromolecules, 2009, 42, 7781 CrossRef CAS;
(e) G. J. M. Habraken, K. H. R. M. Wilsens, C. E. Koning and A. Heise, Polym. Chem., 2011, 2, 1322 RSC;
(f) J. Zou, J. Fan, X. He, S. Zhang, H. Wang and K. L. Wooley, Macromolecules, 2013, 46, 4223 CrossRef CAS PubMed;
(g) H. Kanazawa, A. Inada and N. Kawana, Macromol. Symp., 2006, 242, 104 CrossRef CAS PubMed.
-
(a) W. Zhao, Y. Gnanou and N. Hadjichristidis, Chem. Commun., 2015, 51, 3663 RSC;
(b) W. Zhao, Y. Gnanou and N. Hadjichristidis, Biomacromolecules, 2015, 16, 1352 CrossRef CAS PubMed.
- C. B. Tripathi and S. Mukherjee, J. Org. Chem., 2012, 77, 1592 CrossRef CAS PubMed.
-
(a) A. Wittkopp and P. R. Schreiner, Chem. – Eur. J., 2003, 9, 407 CrossRef CAS PubMed;
(b) P. R. Schreiner, Chem. Soc. Rev., 2003, 32, 289 RSC;
(c) P. R. Schreiner and A. Wittkopp, Org. Lett., 2002, 4, 217 CrossRef CAS PubMed;
(d) Z. Zhang, Z. Bao and H. Xing, Org. Biomol. Chem., 2014, 12, 3151 RSC.
-
(a) A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 13798 CrossRef CAS PubMed;
(b) N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 7, 5813 CrossRef PubMed;
(c) D. Bourissou, S. Moebs-Sanchez and B. Martin-Vaca, C. R. Chim., 2007, 10, 775 CrossRef CAS PubMed;
(d) M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093 CrossRef CAS;
(e) A. P. Dove, ACS Macro Lett., 2012, 1, 1409 CrossRef CAS.
- O. Webster, Science, 1991, 251, 887 CrossRef CAS PubMed.
-
(a) R. D. Lundberg and P. Doty, J. Am. Chem. Soc., 1957, 79, 3961 CrossRef CAS;
(b) M. Idelson and E. R. Blout, J. Am. Chem. Soc., 1957, 79, 3948 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data, NMR spectra and SEC traces. See DOI: 10.1039/c5py00874c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.