 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular assemblies of semiconductor quantum dots and a bis(bipyridinium) derivative: luminescence quenching and aggregation phenomena†
Marek
Oszajca
ab,
Christophe
Lincheneau
a,
Matteo
Amelia
a,
Massimo
Baroncini
a,
Serena
Silvi
a,
Konrad
Szaciłowski
bc and
Alberto
Credi
*a
aPhotochemical Nanosciences Laboratory, Dipartimento di Chimica “G. Ciamician”, Università di Bologna, Via Selmi 2, 40126 Bologna, Italy. E-mail: alberto.credi@unibo.it; Fax: +39 051 2099456; Tel: +39 051 2099540
bFaculty of Chemistry, Jagiellonian University, Ingardena 3, 30-060 Krakow, Poland
cFaculty of Non-Ferrous Metals, AGH University of Science and Technology, al. Mickiewicza 30, 30-060 Kraków, Poland
First published on 23rd June 2014
Abstract
We have synthesized CdSe and CdSe–ZnS core–shell luminescent nanocrystal quantum dots and studied their interaction with a ditopic bis(bipyridinium) compound in solution. The latter strongly quenches the luminescence of the quantum dots by a static mechanism, indicating that the nanocrystal and molecular components undergo association in the ground state. Photoexcitation of these inorganic–organic hybrids causes an electron-transfer process from the conduction band of the nanocrystal to the LUMO of the molecule. The ability of the bipyridinium-type species to trigger association of the quantum dots is evidenced by spectrofluorimetric titrations and DLS measurements in solution, and confirmed by TEM experiments on surfaces. The quantum dot–molecule complexes can be disassembled in solution by addition of a calixarene host capable of encapsulating the bipyridinium units of the molecular connector. Our results demonstrate that supramolecular chemistry offers convenient ways to control the aggregation of semiconductor nanocrystals, a crucial task for the generation of nanostructured arrays with well defined properties.
Introduction
Quantum dots (QDs) are semiconductor nanocrystals with diameters ranging between 1 and 15 nm and are characterized by the quantum confinement of the charge carriers.1 Quantum confinement confers peculiar optoelectronic properties2 to QDs – in particular, a large molar absorption coefficient in the UV-visible and an intense size-tunable luminescence band with a narrow spectral profile – that render them valid substitutes for molecular luminophores3 for a variety of technological applications.4 Moreover such properties, together with a high thermal and photochemical stability, and versatile surface functionalization, make QDs attractive components for the construction of photoactive nanocrystal-molecule assemblies.5,6The vast majority of QD-molecule nanohybrids reported to date are based on the covalent attachment of the molecular units to the nanocrystal surface.6 Such a strategy requires the synthesis of derivatives in which the molecule of interest is connected to a ligand moiety for the QD surface, and the optimization of exchange procedures in order to replace the native ligands of the nanocrystal with the functional ones. Several recent reports, however, have shown7,8 that bare semiconductor nanocrystals and functional molecular components can be self-assembled in an efficient manner taking advantage of supramolecular interactions in solution. This approach is advantageous because (i) no exchange of the capping ligands of the quantum dots is required, (ii) the molecular components do not need to have a surface docking unit, and (iii) the association process, relying on non-covalent interactions, is reversible under appropriate conditions.
In a previous investigation9 we showed that CdSe–ZnS core–shell QDs form remarkably stable supramolecular complexes with 1,1′-dialkyl-4,4′-bipyridinium salts in nonpolar solvents, most likely owing to a combination of van der Waals forces between the guest and the surface-bound molecular monolayer, and solvophobic effects experienced by the positively charged guest. These complexes may also be stabilized by electrostatic interactions between the bipyridinium units and the negatively charged ZnS surface.10 The formation of these complexes is signalled by the decrease of the luminescence intensity of the nanocrystals, which is quenched because of a photoinduced electron-transfer process from the QD conduction band to the LUMO of the nearby bipyridinium unit.9,11,12 The original luminescence intensity of the nanoparticles could be quantitatively restored upon addition of a calix[6]arene host able to compete with the QDs for the bipyridinium quenchers.9
On the basis of these results we designed and synthesized a new bis(bipyridinium) derivative 14+ (Chart 1), composed of two 4,4′-bipyridinium units linked together by means of a p-xylylene spacer and bearing decyl side chains. We then investigated the association of 14+ with either CdSe or CdSe–ZnS core–shell quantum dots in organic solution, using absorption and luminescence spectroscopy, dynamic light scattering, and transmission electron microscopy. The complexation of the bipyridinium units of 14+ by the calix[6]arene host 2 was also investigated and successively exploited to cause the detachment of 14+ from the nanocrystals by competitive binding.
 | ||
| Chart 1 Structural formulas of the investigated molecular components. | ||
The general motivation of this work is to identify simple and efficient strategies for assembling quantum dot and molecular components, and to increase our knowledge of nanocrystal–molecule interactions. More specifically, the present study is aimed at understanding whether the ditopic structure of the 14+ guest can be exploited to induce the aggregation of the nanocrystals in solution, and if such a phenomenon is chemically reversible. Using supramolecular complexation to control aggregation of semiconductor nanocrystals13 is a promising route for the construction of colloidal superparticles14 and ordered two- and three-dimensional nanocrystal arrays (superlattices and artificial solids)15 that could exhibit useful chemical and optoelectronic properties.16
Experimental
Materials and methods
Cadmium oxide (CdO, 99.99%), 1-hexadecylamine (HDA, 98%), tri-n-octylphosphine oxide (TOPO, 99%), 1-n-octadecene (ODE, 90%), oleic acid (OA, 99%) and n-octadecylamine (ODA, 99%) were purchased from Aldrich; elemental selenium (99.999%, 200 mesh), and tri-n-butylphosphine (97%) were from Alfa Aesar. 1,4-Bis(1-decyl-4,4′-bipyridinium-1′-methyl)benzene tetrachloride (1·4Cl) was synthesized according to a previously published procedure.17 Commercially available compounds were reagent grade quality and were used without further purification. The tris(N-phenylureido)calix[6]arene 2 was a gift from Profs A. Arduini and A. Secchi, Università di Parma, and was available from previous investigations.9,181H And 13C NMR spectra were recorded at 295 K with a Varian Mercury 400 spectrometer with the deuterated solvent as the lock and the residual solvent as the internal standard. All chemical shifts are quoted using the δ scale, and all coupling constants (J) are expressed in Hertz (Hz). Melting points were determined using a Büchi 510 series apparatus and are uncorrected. Air equilibrated chloroform and acetonitrile (Merck Uvasol™) were used as the solvents for the spectroscopic measurements.Absorption and luminescence spectra were recorded in either air-equilibrated or deoxygenated (argon purged) solutions with a Perkin Elmer Lambda 45 spectrophotometer and a Perkin-Elmer LS50B spectrofluorimeter equipped with a Hamamatsu R928 phototube, respectively. Luminescence lifetimes were measured with an Edinburgh Instruments FLS920 time-correlated single-photon counting spectrofluorimeter, exciting the sample at 405 nm with a pulsed laser.
Titration experiments were carried out by adding small aliquots of a concentrated stock solution of 14+ to a known volume of a dilute nanocrystal solution. In all instances the absorbance at the excitation wavelength was small enough to avoid inner filter effects and take advantage of the linear relationship between absorbed and emitted light.19 The experimental errors are ±1 nm for the wavelength values, ±10% for the luminescence intensities and lifetimes, and ±20% for the stability constants.
The nanoparticle size distribution was determined by transmission electron microscopy (TEM) and dynamic light scattering (DLS) measurements. TEM experiments were carried out with a Philips CM 100 transmission electron microscope operating at 80 kV, while HRTEM measurements were carried out with a FEI Tecnai Osiris S/TEM system operating at 200 kV. A drop of the nanocrystal solution diluted with hexane was deposited on a 400 mesh copper coated with formvar support grid (TAAB Ltd.), which was then dried up under vacuum to remove any solvent trace. DLS experiments were performed with a Malvern Nano ZS instrument equipped with a 633 nm laser diode. The measurements were carried out at 20 °C in air-equilibrated solutions placed in 1 cm quartz cuvettes. The samples were filtered twice through 0.1 μm PTFE membrane filters prior to the measurements. The resulting size is the average of at least 20 independent experiments, and the standard deviation is taken as the experimental error.
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol–hexane mixture.
:1 methanol–hexane mixture.
Results and discussion
CdSe nanocrystals
The investigated CdSe core nanocrystals in CHCl3 exhibit a broad UV-visible absorption and the characteristic low energy exciton peak (λmax = 560 nm) arising from quantum confinement of the charge carriers.1–3 The diameter of the nanocrystals, determined from the position of their low-energy absorption peak according to a published procedure,23 resulted to be 3.3 nm, while the molar absorption coefficient of the exciton peak is εmax = 134000 M−1 cm−1. TEM Images revealed that the particles are spherical and well monodispersed, as expected from the applied synthetic methodology. The luminescence spectrum of the QDs in air equilibrated CHCl3 at room temperature shows the intense and narrow luminescence band (λmax = 578 nm, fwhm = 30 nm) typical of radiative exciton recombination. Such an emission exhibit a decay that could be satisfactorily fitted with a double exponential function, yielding an average lifetime of 33 ns.†
Because the bis(bipyridinium) compound 1·4PF6 is insoluble in CHCl3, its properties were examined in CHCl3/CH3CN 9:1 (v/v). The absorption spectrum of 14+ shows an intense band in the near UV (λmax = 255 nm, εmax = 51500 M−1 cm−1) and no luminescence, in line with the behaviour of 1,1′-dibenzyl-4,4′-bipyridinium.24
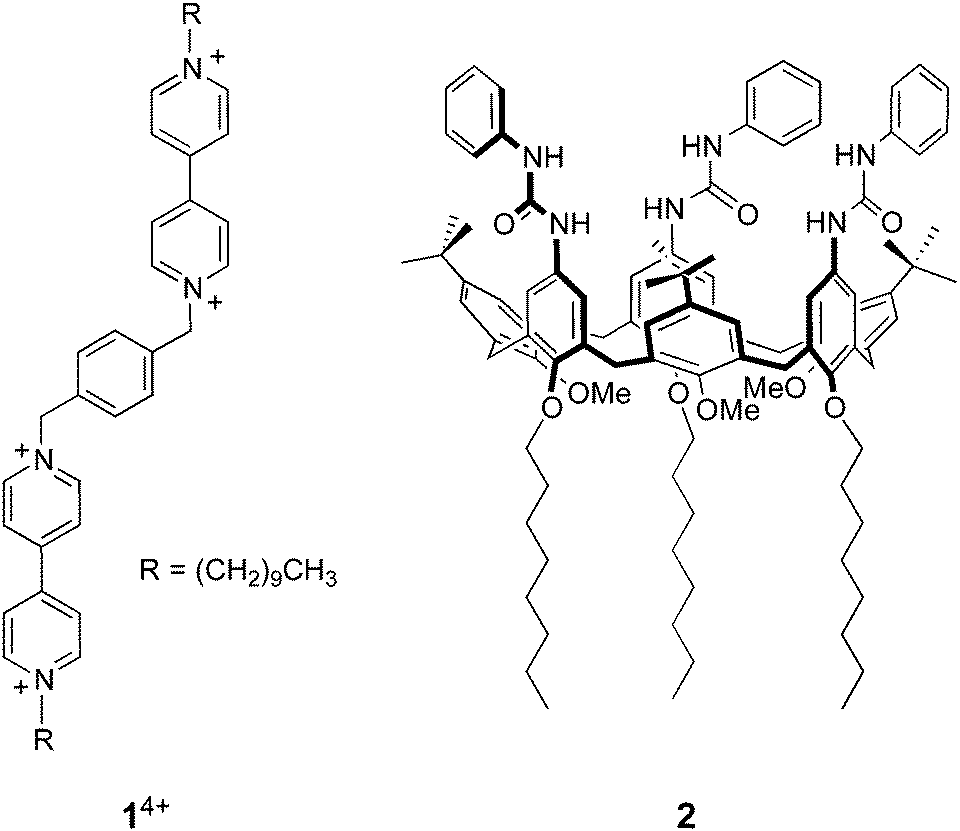
A 1.5 μM solution of CdSe QDs was titrated with 14+ in CHCl3/CH3CN 9:1 (v/v) and the QD emission at 578 nm (λexc = 500 nm) was monitored as a function of the concentration of 14+. As shown in Fig. 1a, the addition of 14+ causes a strong quenching of the QD luminescence. Such an emission decrease cannot be ascribed to dynamic quenching, because of the relatively short lifetime of the nanocrystal emission and the very small concentration of the bipyridinium quencher. Even assuming that dynamic quenching is diffusion controlled (kd = 1.2 × 1010 M−1 s−1),25 the contribution of dynamic quenching to the emission decrease at the highest employed concentration of 14+ would be less than 0.2%. Moreover, the luminescence lifetime of the QDs is just slightly affected by the addition of the quencher (Fig. 1b). All these findings suggest that the quenching is static, that is, the nanocrystals and 14+ are associated in the ground state, as previously observed for related systems.7,9
 | ||
| Fig. 1 (a) Luminescence spectral changes observed upon titration of 1.5 μM CdSe QDs with 14+. (b) Filled circles, left scale: titration plot obtained from the QD luminescence intensity at 578 nm in the presence of increasing amounts of 14+. The dashed and dotted lines show the tangent to the curve at two different slopes and are a guide for the eye, not a fit. Empty triangles, right scale: titration plot obtained from the luminescence lifetime values corresponding to the CdSe emission. Conditions: air equilibrated CHCl3/CH3CN 9:1, room temperature, λexc = 500 nm. | ||
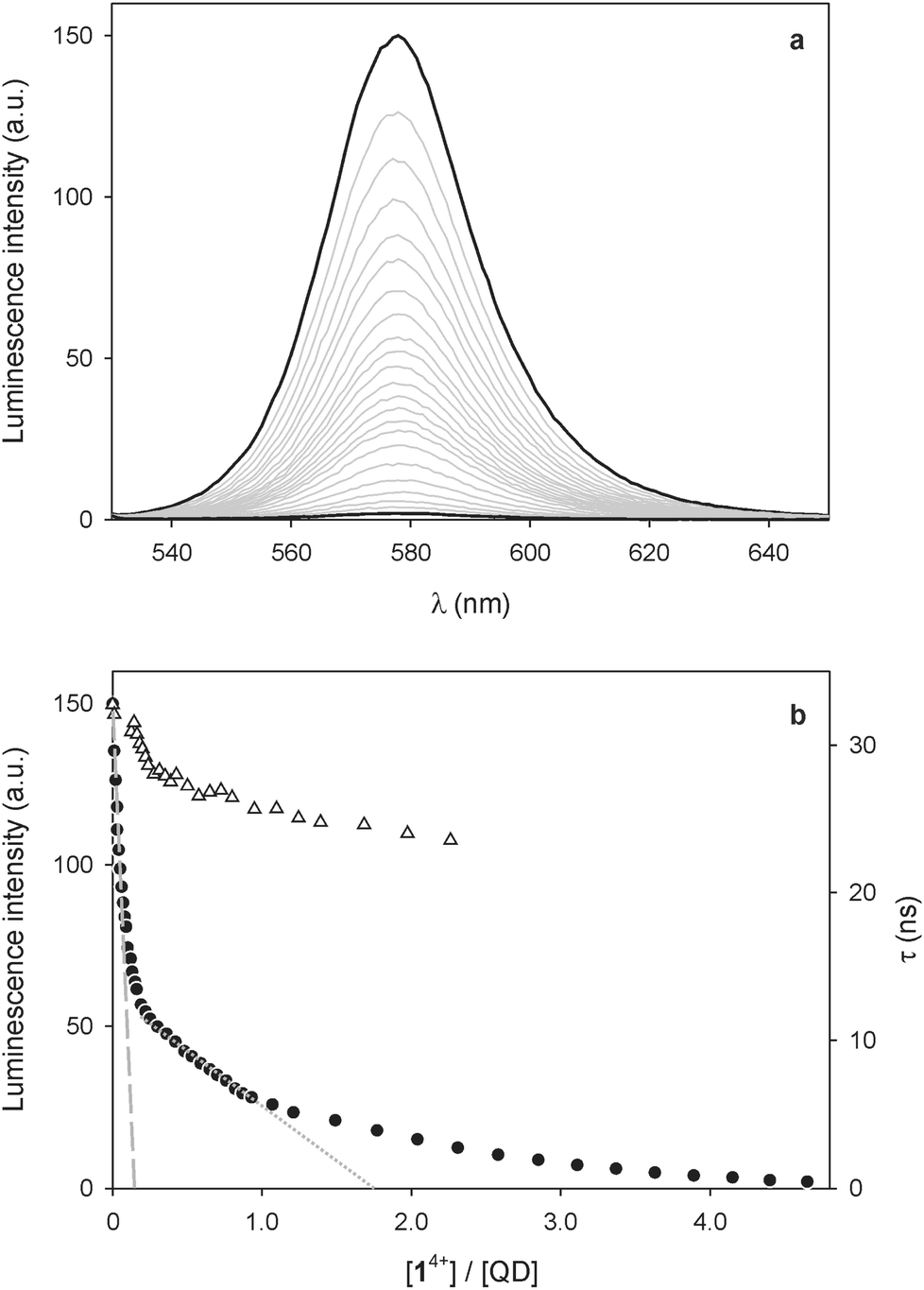
No evidence for ground-state association, however, could be found in the absorption spectra, as the addition of 14+ causes no substantial changes in the absorption features of the QDs (Fig. 2a). Nevertheless, a slight increase in absorbance at around 400 nm and between 500 and 700 nm is observed; the differential spectrum (Fig. 2b) shows the appearance of new bands with λmax = 401 and 612 nm, which can be unequivocally ascribed to the presence of bipyridinium radical cations.24
 | ||
| Fig. 2 (a) Absorption spectrum of a 1.5 μM solution of CdSe QDs before (full line) and after the spectrofluorimetric titration with 4.6 equivalents of 14+ as described in Fig. 1. (b) Difference absorption spectrum of the traces shown in (a), indicating the formation of bipyridinium radical cations. Conditions: air equilibrated CHCl3/CH3CN 9:1, room temperature, λexc = 500 nm. | ||
As these spectral changes are not observed if the QD/14+ solution is kept in the dark, reduction of the dicationic bipyridinium units of 14+ to the corresponding radical cations has to be caused by the light excitation employed in the spectrofluorimetric experiments. Indeed, the photoinduced generation of bipyridinium radical cations was obtained upon visible irradiation of CdSe QD-bipyridinium solutions in the presence of a sacrificial reductant, confirming that the QD luminescence quenching can be ascribed to oxidative electron transfer.11,12 In the present case, it is likely that small amounts of adventitious species (e.g., water) scavenge the photogenerated hole left in the QD valence band, thereby preventing the back electron transfer from the reduced bipyridinium unit to the oxidized nanocrystal. In any instance, the absorption bands of the bipyridinium radical cation disappear after a few minutes in air equilibrated solution, due to slow reoxidation by dioxygen.
In addition to charge transfer, other possible quenching mechanisms in nanocrystal–chromophore complexes are energy transfer (FRET) and formation of non-radiative surface states arising from changes in the capping monolayer.26 FRET quenching can be ruled out in the present case because 14+ cannot act as an energy acceptor for the CdSe QDs. On the other hand, 14+ does not possess functional groups capable of binding to the CdSe surface, and control experiments show that the PF6− counterions have no effect on the QD emission. Moreover, the luminescence quenching occurs immediately after the addition of 14+ and is constant thereafter, whereas ligand exchange typically occurs on the time scale of minutes.26 The absence of kinetic effects and the reversibility of the quenching observed upon addition of the competitive calixarene host (see below) are consistent with the non-covalent nature of the nanocrystal-quencher association.
Insightful information on the interaction between the QDs and 14+ can be gathered from a careful examination of the titration curve (Fig. 1b). At least three different trends for the intensity decrease can be identified, depending on the 14+/QD ratio. It can be noticed that the tangent to the initial part of the curve (dashed line in Fig. 1b) intercepts the x-axis – corresponding to zero emission intensity – at about 0.25 equivalents of 14+. This observation suggests that at low 14+/QD ratios one molecule of 14+ (i) can associate with up to four nanocrystals, and (ii) it is able to completely quench the luminescence of any of them.
Because of its size and shape in comparison with those of the quantum dots, it is difficult to imagine that 14+ can directly link together more than two nanocrystals. It may be hypothesized, however, that such QD pairs could exhibit a stronger ability to bind other nanoparticles than individual QDs, because of the larger surface available for non-covalent interparticle interactions.14
On increasing the number of 14+ equivalents, non-emissive adducts in which one QD is complexed with two molecules of 14+ are also formed (dotted line in Fig. 1b). Not surprisingly, complexes with higher 14+: QD stoichiometries are obtained as the titration goes on.
CdSe–ZnS core–shell nanocrystals
CdSe–ZnS core–shell QDs are more interesting for technological applications than bare CdSe nanocrystals, because they possess improved stability and photoluminescence efficiency.27 Hence, we studied the effect of the bis(bipyridinium) species 14+ on QDs composed of a CdSe core (d = 3.8 nm) coated with a ZnS shell 1.2 nm thick. These nanocrystal show the typical low-energy absorption (λmax = 579 nm, εmax = 200000 M−1 cm−1)23 and emission (λmax = 608 nm, fwhm = 39 nm) peaks of their CdSe core.
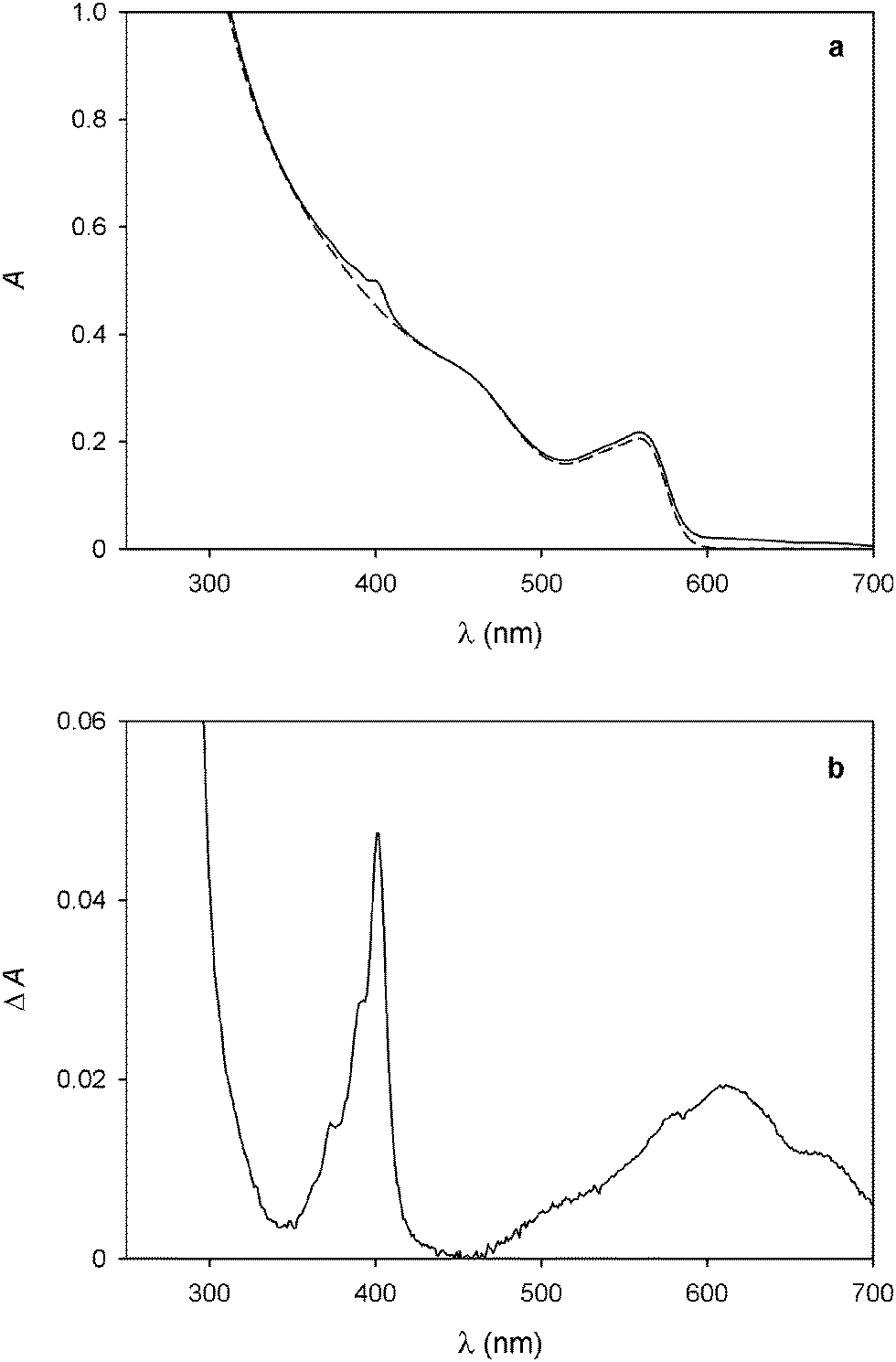
The addition of 14+ to a CHCl3/CH3CN 9:1 solution of the CdSe–ZnS QDs caused a quenching of the emission (Fig. 3a), that is, a qualitatively similar result to that observed for the CdSe nanocrystals. The shape of the titration curve, however (Fig. 3b), is different from that shown in Fig. 1b. In fact, the emission decrease is much less steep than in the case of the CdSe QDs and a full luminescence quenching is observed after addition of a significantly larger number of 14+ equivalents. The initial part of the curve, whose tangent intercepts the x-axis at about 2 equivalents, indicates that at least two 14+ molecules are required to completely quench the luminescence of one QD.
 | ||
| Fig. 3 (a) Luminescence spectral changes observed upon titration of 0.47 μM CdSe–ZnS QDs with 14+. (b) Titration plot obtained from the QD luminescence intensity at 608 nm in the presence of increasing amounts of 14+. The dashed line shows the tangent to the first part of the curve (it is a guide for the eye, not a fit). Conditions: air equilibrated CHCl3/CH3CN 9:1, room temperature, λexc = 500 nm. | ||
This result contrasts with that obtained for the bare CdSe QDs and can be rationalized on the basis of the different size and structure of the CdSe–ZnS QDs. On the one hand, aggregates in which several QDs are bridged by one 14+ molecule are unlikely to form because of the larger size of the core–shell particles. On the other hand, the presence of the ZnS shell insulates the CdSe core from the external quencher, thus slowing down the photoinduced electron-transfer process at the basis of quenching. This observation is confirmed by the fact that the absorption bands of the bipyridinium radical cations, observed for the CdSe QDs as a consequence of photoexcitation in the spectrofluorimetric titration, were not detected during the same experiments in the case of the CdSe–ZnS QDs. Photoreduction of the bipyridinium units of 14+ was obtained, though, upon illumination of the CdSe–ZnS QDs with high-intensity visible light (λ > 495 nm) in the presence of 24 equivalents of 14+. From the intensity of the absorption band at 401 nm we estimated that, under these conditions, irradiation for 5 min in deoxygenated solution affords ca. 38% reduction of the bipyridinium units.†
QD aggregation triggered by 14+
To investigate the ability of the bis(bipyridinium) compound 14+ to non-covalently link together the nanocrystals we performed dynamic light scattering (DLS) and transmission electron microscopy (TEM) experiments on CdSe QD solutions with and without the presence of 14+. The CdSe QDs described in the previous section could not be utilized in DLS experiments because they absorb and emit light at the wavelength (633 nm) used by the instrument to detect light scattering. Hence, the DLS and TEM measurements were performed on smaller CdSe QDs, exhibiting the first exciton absorption peak at λmax = 517 nm, corresponding to a core diameter of 2.5 nm.23 The hydrodynamic diameter of the nanocrystals (Fig. 4a) resulted to be 4.8 ± 0.1 nm, a figure which is consistent with the core diameter estimated from spectroscopic data and the thickness of the capping ligand shell.28 Indeed, the addition of 0.5 equivalents of 14+ to the QD solution caused a growth of the mean hydrodynamic diameter of the particles to 8.1 ± 0.7 nm as well as an increase in their polydispersity (Fig. 4b). This observation is in full agreement with the formation of aggregates consisting of 2 to 4 nanocrystals, as inferred from luminescence titration data.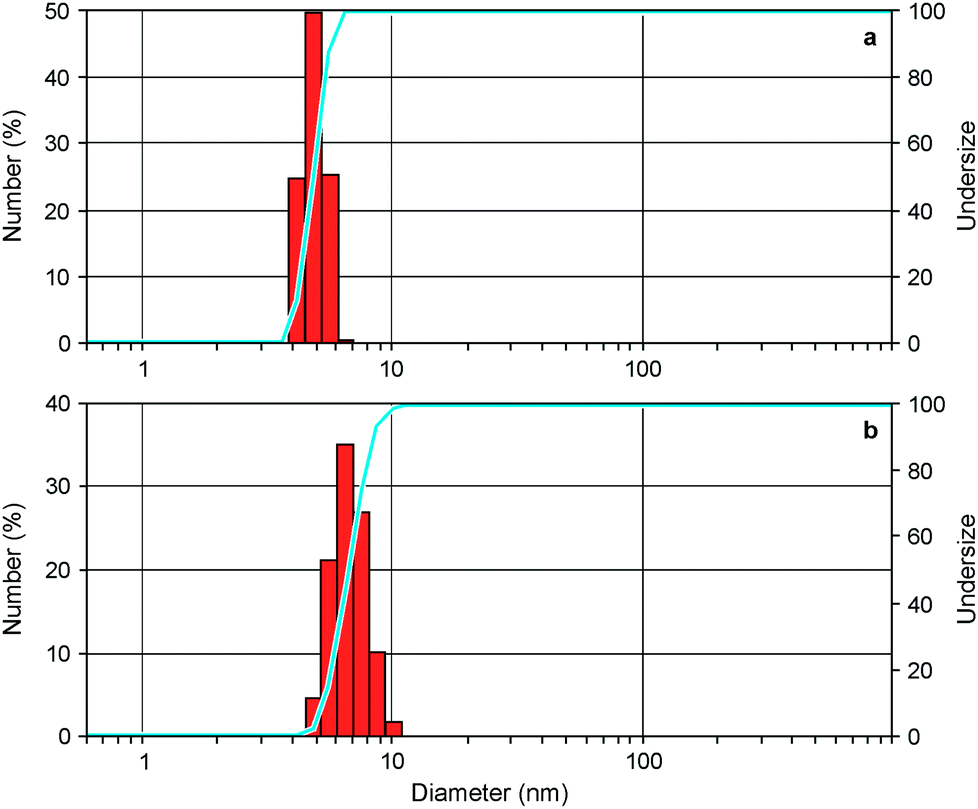 | ||
| Fig. 4 Differential (red bars) and cumulative (blue line) number distributions of particle size obtained from DLS measurements for the CdSe QDs (a) alone and (b) in the presence of 0.5 equivalents of 14+. Conditions: air equilibrated CHCl3/CH3CN 9:1, room temperature. | ||
Further support to the QD aggregation induced by 14+ came from TEM experiments.† The images recorded on substrates obtained from deposition of a dilute solution of the CdSe QDs showed mainly the presence of individual particles in regions with both higher (Fig. 5a) and lower (Fig. 5b) populations of nanocrystals. When 1 equivalent of 14+ was added to the QD solution prior to deposition, single nanocrystals could still be detected (Fig. 5c) but small clusters were also found (Fig. 5d). In the presence of 4 equivalents of 14+ the QDs occurred only in the form of large aggregates and no isolated nanocrystals could be observed (Fig. 5e and f).
 | ||
| Fig. 5 TEM Images recorded on substrates obtained from deposition of dilute solutions of (a and b) CdSe QDs, (c and d) CdSe QDs and 14+ in a 1:1 ratio, and (e and f) CdSe QDs and 14+ in a 1:4 ratio. Arrows in (d) indicate small QD aggregates. Scale bar is 10 nm for (a, c and e) and 50 nm for (b, d and f). | ||
Luminescence recovery by supramolecular encapsulation of 14+
As pointed out in the introduction, we showed earlier9 that QD-bipyridinium adducts could be disrupted by complexing the bipyridinium species with the calix[6]arene derivative 2, which can efficiently encapsulate bipyridinium-type guests in organic solution.18,29 Hence, we investigated whether the addition of 2 could have an effect on the interaction of 14+ with the luminescent nanocrystals.First of all we performed spectrophotometric titrations to determine the nature and stability of the complex between the bis(bipyridinium) guest and the calixarene host. Upon addition of 2 to 14+ in CHCl3/CH3CN 9:1 an absorption increase in the 450–550 nm region, typical of the inclusion of a bipyridinium unit within the cavity of 2,28 was observed (Fig. 6a). The corresponding titration curve obtained from the absorbance values at 460 nm (Fig. 6b) could be satisfactorily fitted with a binding model implying the formation of two adducts (eqn (1) and (2));30 namely, a 1:1 complex in which one molecule of 2 encircles a bipyridinium unit of 14+ (K1:1 = 1.3 × 105 M−1) and a 2:1 complex in which two calixarene molecules surround both bipyridinium units of one 14+ species (K2:1 = 2.5 × 103 M−1).
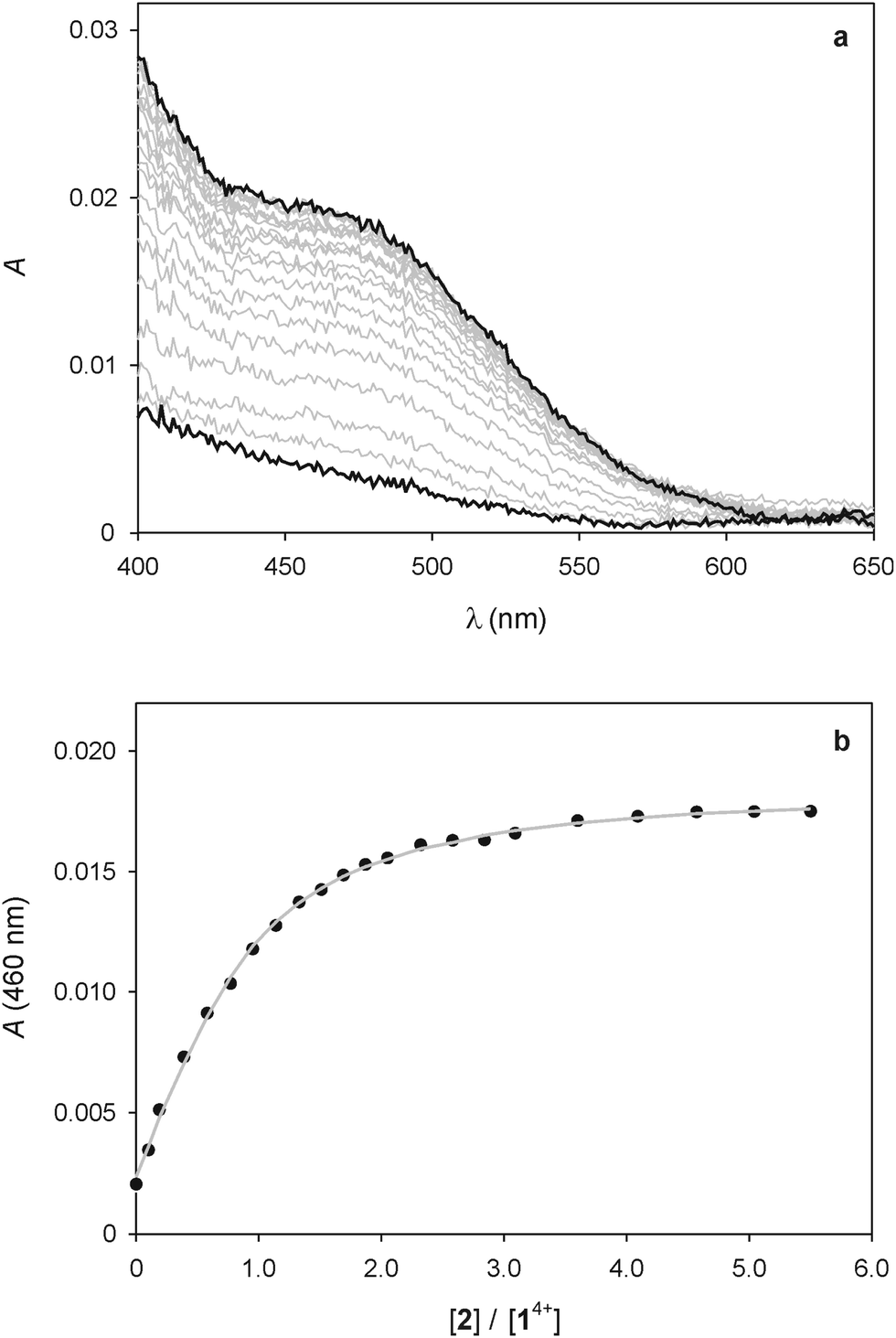 | ||
| Fig. 6 (a) Absorption spectral changes observed upon titration of 25 μM 14+ with 2. (b) Titration plot obtained from the absorption values at 460 nm. The grey curve is the data fitting according to the 1:1 and 2:1 binding model described in the text (eqn (1) and (2)). Conditions: air equilibrated CHCl3/CH3CN 9:1, room temperature. | ||
The value of K1:1 is in line with those found in previous investigations for the 1:1 complex between 2 and 1,1′-dialkyl-4,4′-bipyridinium guests. On the other hand, the significantly lower value of K2:1 indicates that the presence of a first calixarene unit around 14+ hampers the association of a second molecule of 2. This finding is not unexpected if one considers that the distance between the two bipyridinium units of 14+ is short in relation to the length of 2, resulting in a mutual steric hindrance of the two calixarene hosts in the 2:1 complex.
Variable amounts of calixarene 2 were added to assemblies of 14+ and either CdSe (λmax,abs = 560 nm, d = 3.3 nm) or CdSe–ZnS QDs in CHCl3/CH3CN 9:1 and spectrofluorimetric measurements were carried out on the resulting solutions. Upon addition of 5 equivalents of 2 (with respect to 14+) to a solution containing CdSe QDs (1.5 μM) and 14+ in a 1:4.6 ratio, only less than 1% of the initial emission intensity was restored. Similarly, the addition of 38 equivalents of 2 (with respect to 14+) to a solution containing CdSe–ZnS QDs (0.47 μM) and 14+ in a 1:20 ratio caused <1% recovery of the luminescence. Notably, the emission of CdSe–ZnS QDs (0.43 μM) and 1,1′-dibenzyl-4,4′-bipyridinium in a 1:19 ratio was fully restored after the addition of 20 equivalents of 2 (with respect to the bipyridinium guest).9 The comparison of these results suggests that the binding of the bis(bipyridinium) 14+ species to the nanocrystals is stronger than that of a structurally related compound bearing a single bipyridinium unit. The larger size of 14+, its twice as large electric charge, and its lower affinity for chloroform may be among the reasons for such a behaviour. It may also be considered that complexation of 14+ by 2 could be hampered by aggregation of the nanocrystals triggered by the former.
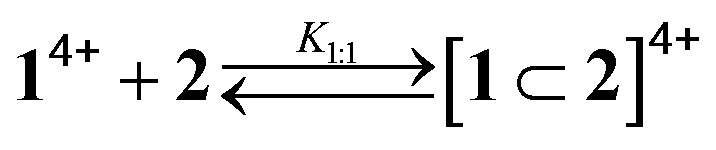 | (1) |
 | (2) |
Nevertheless, the titration of a 1.5 μM 1:1 mixture of CdSe QDs and 14+ (in which ca. 87% of the nanocrystals luminescence is quenched, see Fig. 1b) with 2 shows a recovery of up to 30% of the original emission intensity of the QDs after the addition of a large excess (215 equivalents) of the calixarene host (Fig. 7). Conversely, the addition of the same excess of 2 to the QDs (in the absence of 14+) caused a 8% emission decrease. These observations indicate that complexation of 14+ by 2 causes the disassembly of the non-luminescent adducts consisting of the QDs and the bis(bipyridinium) species.
 | ||
| Fig. 7 Luminescence spectral changes at 578 nm observed upon titration of a 1:1 mixture of CdSe QDs and 14+ (1.5 μM) with the calixarene host 2. Conditions: air equilibrated CHCl3/CH3CN 9:1, room temperature, λexc = 500 nm. | ||
Conclusions
We have investigated the interaction of CdSe and CdSe–ZnS luminescent nanocrystals with the ditopic bis(bipyridinium) species 14+ in organic solution. Our results show that the nanocrystal and molecular components undergo association in the ground state, yielding complexes in which the luminescence of the quantum dots is strongly quenched. Interestingly, 14+ can associate and quench as many as four CdSe nanocrystals, possibly owing to cooperative interparticle binding phenomena. The ability of 14+ to promote aggregation of the quantum dots is confirmed by DLS and TEM experiments.Visible light irradiation of the adducts causes the formation of bipyridinium radical cations, confirming that the luminescence quenching mechanism is an electron-transfer process from the excited electron in the conduction band of the nanocrystal to the LUMO of the bipyridinium quencher. The presence of a ZnS shell around the CdSe core does not prevent the electron-transfer quenching process but it does reduce its efficiency.
Calix[6]arene 2, which is able to encapsulate both bipyridinium units of 14+ by virtue of supramolecular interactions, was used to promote the decomplexation of 14+ from the quantum dots. Indeed, the addition of an excess of 2 to a solution containing the nanocrystal/14+ complexes causes a recovery of the luminescence of the quantum dots, indicating the partial disassembly of the complexes.
These results demonstrate that supramolecular interactions are a convenient way to control the aggregation of semiconductor nanocrystals. The combination of molecular recognition and self-assembly concepts with luminescent quantum dots is a promising route for the generation of nanostructured arrays with controlled morphologies and properties. Such a task is important for improving the performances of quantum dot-based optoelectronic devices.
Acknowledgements
This work was supported by the European Union (EU) (Hysens Project, grant no. 263091, ATOMIN project, grant no. POIG.02.01.00-12-023/08), the Ministero dell'Università e della Ricerca (PRIN 2010CX2TLM) and the University of Bologna. M. O. thanks the National Science Centre (grant no. UMO 2011/01/N/ST5/02550) and the Foundation for Polish Science for a MPD programme fellowship co-financed by the EU European Regional Development Fund. K. S. acknowledges financial support from the National Science Centre (grant no. UMO-2011/03/B/ST5/01495).Notes and references
- Semiconductor nanocrystal quantum dots, ed. A. L. Rogach, Springer-Verlag, Wien, Austria, 2008 Search PubMed.
- A. M. Smith and S. Nie, Acc. Chem. Res., 2010, 43, 190 CrossRef CAS PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763 CrossRef CAS PubMed.
- Quantum dot sensors: technology and commercial applications, ed. J. F. Callan and F. M. Raymo, Pan Stanford Publishing, Singapore, 2013 Search PubMed.
- I. Yildiz, E. Deniz and F. M. Raymo, Chem. Soc. Rev., 2009, 38, 1859 RSC; I. L. Medintz and H. Mattoussi, Phys. Chem. Chem. Phys., 2009, 11, 17 RSC; N. Hildebrandt, ACS Nano, 2011, 5, 5286 CrossRef CAS PubMed; T. L. Doane and C. Burda, Chem. Soc. Rev., 2012, 41, 2885 RSC; R. Freeman and I. Willner, Chem. Soc. Rev., 2012, 41, 4067 RSC; R. Freeman, J. Girsh and I. Willner, ACS Appl. Mater. Interface, 2013, 5, 2815 CrossRef PubMed; T. Avellini, C. Lincheneau, F. Vera, S. Silvi and A. Credi, Coord. Chem. Rev., 2014, 263–264, 151 CrossRef PubMed.
- See, e.g.: Y. Chen, R. Thakar and P. T. Snee, J. Am. Chem. Soc., 2008, 130, 3744 CrossRef CAS PubMed; I. L. Medintz, M. H. Stewart, S. A. Trammell, K. Susumu, J. B. Delehanty, B. C. Mei, J. S. Melinger, J. B. Blanco-Canosa, P. E. Dawson and H. Mattoussi, Nat. Mater., 2010, 9, 676 CrossRef PubMed; Z. Erno, I. Yildiz, B. Gorodetsky, F. M. Raymo and N. R. Branda, Photochem. Photobiol. Sci., 2010, 9, 249 Search PubMed; M. Amelia, A. Lavie-Cambot, N. D. McClenaghan and A. Credi, Chem. Commun., 2011, 47, 325 RSC; W. R. Algar, D. Wegner, A. L. Huston, J. B. Blanco-Canosa, M. H. Stewart, A. Armstrong, P. E. Dawson, N. Hildebrandt and I. L. Medintz, J. Am. Chem. Soc., 2012, 134, 1876 CrossRef PubMed; S. Impellizzeri, B. McCaughan, J. F. Callan and F. M. Raymo, J. Am. Chem. Soc., 2012, 134, 2276 CrossRef PubMed; R. Freeman, T. Finder, L. Bahshi, R. Gill and I. Willner, Adv. Mater., 2012, 24, 6416 CrossRef PubMed; J. Aguilera-Sigalat, V. F. Pais, A. Domenech-Carbo, U. Pischel, R. E. Galian and J. Perez-Prieto, J. Phys. Chem. C, 2013, 117, 7365 Search PubMed.
- M. Amelia, M. Font and A. Credi, Dalton Trans., 2011, 40, 12083 RSC; M. Amelia and A. Credi, Inorg. Chim. Acta, 2012, 381, 247 CrossRef CAS PubMed.
- A. Boulesbaa, A. Issac, D. Stockwell, Z. Huang, J. Huang, J. Guo and T. Lian, J. Am. Chem. Soc., 2007, 129, 15132 CrossRef CAS PubMed; S.-C. Cui, T. Tachikawa, M. Fujitsuka and T. Majima, J. Phys. Chem. C, 2011, 115, 1824 Search PubMed; J. Aguilera-Sigalat, J. M. Casas-Solvas, M. C. Morant-Minana, A. Vargas-Berenguel, R. E. Galian and J. Perez-Prieto, Chem. Commun., 2012, 48, 2573 RSC; A. Iagatti, R. Flamini, M. Nocchetti and L. Latterini, J. Phys. Chem. C, 2013, 117, 23996 Search PubMed.
- B. Gadenne, I. Yildiz, M. Amelia, F. Ciesa, A. Secchi, A. Arduini, A. Credi and F. M. Raymo, J. Mater. Chem., 2008, 17, 2022 RSC.
- J. Bebie, M. A. A. Schoonen, M. Fuhrmann and D. R. Strongin, Geochim. Cosmochim. Acta, 1998, 62, 633 CrossRef CAS; M. Tiemann, F. Marlow, F. Brieler and M. Lindén, J. Phys. Chem. B, 2006, 110, 23142 CrossRef PubMed; M. Wang, Q. Zhang, W. Hao and Z.-X. Sun, Chem. Cent. J., 2011, 5, 73 CrossRef PubMed; J. Saikiaa, B. Sahab and G. Das, J. Colloid Interface Sci., 2014, 416, 235 CrossRef PubMed.
- I. Yildiz and F. M. Raymo, J. Mater. Chem., 2006, 16, 1118 RSC; I. Yildiz, M. Tomasulo and F. M. Raymo, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11457 CrossRef CAS PubMed.
- S. Logunov, T. Green, S. Marguet and M. A. El-Sayed, J. Phys. Chem. A, 1998, 102, 5652 CrossRef CAS; C. Burda, T. C. Green, S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 1783 CrossRef.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591 CrossRef CAS PubMed; R. Schreiber, J. Do, E.-M. Roller, T. Zhang, V. J. Schüller, P. C. Nickels, J. Feldmann and T. Liedl, Nat. Nanotechnol., 2014, 9, 74 CrossRef PubMed.
- T. Wang, D. LaMontagne, J. Lynch, J. Zhuang and Y. C. Cao, Chem. Soc. Rev., 2013, 42, 2804 RSC.
- H. Zhang, E. W. Edwards, D. Y. Wang and H. Möhwald, Phys. Chem. Chem. Phys., 2006, 8, 3288 RSC; Y. Gao and Z. Tang, Small, 2011, 7, 2133 CrossRef CAS PubMed; C. Zhang, R. J. Macfarlane, K. L. Young, C. H. J. Choi, L. Hao, E. Auyeung, G. Liu, X. Zhou and C. A. Mirkin, Nat. Mater., 2013, 12, 741 CrossRef PubMed.
- D. Vanmaekelbergh and P. Liljeroth, Chem. Soc. Rev., 2005, 34, 299 RSC; D. V. Talapin, J. S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389 CrossRef CAS PubMed.
- R. J. Alvarado, J. Mukherjee, E. J. Pacsial, D. Alexander and F. M. Raymo, J. Phys. Chem. B, 2005, 109, 6164 CrossRef CAS PubMed.
- A. Credi, S. Dumas, S. Silvi, M. Venturi, A. Arduini, A. Pochini and A. Secchi, J. Org. Chem., 2004, 69, 5881 CrossRef CAS PubMed.
- A. Credi and L. Prodi, J. Mol. Struct., 2014 DOI:10.1016/j.molstruc.2014.03.028 , in press.
- Z. A. Peng and X. Peng, J. Am. Chem. Soc., 2001, 123, 183 CrossRef CAS; J. Li, Y. A. Wang, W. Guo, J. C. Keay, T. D. Mishima, M. B. Johnson and X. Peng, J. Am. Chem. Soc., 2003, 125, 12567 CrossRef PubMed.
- Z. A. Peng and X. Peng, J. Am. Chem. Soc., 2002, 124, 3343 CrossRef CAS PubMed; C. R. Bullen and P. Mulvaney, Nano Lett., 2004, 4, 2303 CrossRef.
- D. Chen, F. Zhao, H. Qi, M. Rutherford and X. Peng, Chem. Mater., 2010, 22, 1437 CrossRef CAS.
- W. W. Yu, L. Qu, W. Guo and X. Peng, Chem. Mater., 2003, 15, 2854 CrossRef CAS.
- R. Ballardini, A. Credi, M. T. Gandolfi, C. Giansante, G. Marconi, S. Silvi and M. Venturi, Inorg. Chim. Acta, 2007, 360, 1072 CrossRef CAS PubMed.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of photochemistry, CRC Press, Boca Raton, 3rd edn, 2006 Search PubMed.
- T. Blaudeck, E. I. Zenkevich, M. Abdel-Mottaleb, K. Szwaykowska, D. Kowerko, F. Cichos and C. von Borczyskowski, ChemPhysChem, 2012, 13, 959 CrossRef CAS PubMed.
- M. A. Hines and P. Guyot-Sionnest, J. Phys. Chem., 1996, 100, 468 CrossRef CAS.
- J. M. Tsay, S. Doose and S. Weiss, J. Am. Chem. Soc., 2006, 128, 1639 CrossRef CAS PubMed.
- A. Arduini, R. Bussolati, A. Credi, A. Secchi, S. Silvi, M. Semeraro and M. Venturi, J. Am. Chem. Soc., 2013, 135, 9924 CrossRef CAS PubMed; M. Semeraro, A. Secchi, S. Silvi, M. Venturi, A. Arduini and A. Credi, Inorg. Chim. Acta, 2014, 417, 258 CrossRef PubMed.
- R. A. Binstead, SPECFIT, Spectrum Software Associates, Chapel Hill, NC, 1996 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV-visible absorption spectra, luminescence spectra and lifetimes, and TEM images. See DOI: 10.1039/c4ra03259d |
| This journal is © The Royal Society of Chemistry 2014 |