Dressed for success – applying chemistry to modulate aptamer functionality
Fabian
Tolle
and
Günter
Mayer
*
University of Bonn, Gerhard-Domagk-Str.1, 53121, Bonn, Germany. E-mail: gmayer@uni-bonn.de; Tel: +49 (0)228 734808
First published on 23rd October 2012
Abstract
Aptamers entered the stage in the early 1990s. Since then they have proven to be versatile tools for molecular biology and biomedical sciences. The combination of chemical synthesis with aptamer generation and post-SELEX modification now provides means for adapting these nucleic acids to almost every desired application and to tune their properties as required. In this review we discuss chemical approaches towards aptamer variation.
Non-coding nucleic acids and aptamers
For many years, according to the central dogma of molecular biology, it was believed that the sole function of nucleic acids is to code and replicate the information for protein production. Only since the deciphering of the human genome sequence, has it became evident that only a minority of the nucleic acid content in living systems is actually coding for proteins, but that nucleic acids inherit many more regulatory and structural functions. Research efforts during the past two decades revealed that with increasing complexity the percentage of coding nucleic acids decreases, peaking in the human genome, where it is believed that only around 1.5% of genomic DNA encodes for proteins.1 In a very recent estimate 80% of the genome could be associated to a biological function,2 as such indicating that large parts of the genome encode for small non-coding RNA that play crucial roles in the regulation and modulation of biological functions. Nurtured through a variety of structures in which nucleic acids can fold, RNA build stable but dynamic functional molecules.3–5 This is impressively illustrated by riboswitches. Riboswitches are mainly found in the untranslated region of prokaryotic mRNA molecules. These RNA elements regulate the expression of proteins through binding to metabolites and transduction of this information into dynamic structural changes.6Aptamers and their application
Diagnostic and therapeutic regimens rely on tools, which allow a specific interaction with individual (bio-) molecules. This is of even greater importance when dealing with complex mixtures as they are often found in biological systems. One of nature's solutions for the generation of specific protein binders (besides “small molecules”) are antibodies, which themselves are amino acid-based polymers mainly composed of 20 different canonical monomeric building blocks. These amino acids harbour a wide range of chemical entities in their side chains, such as aliphatic- (e.g. leucine) aromatic- (e.g. phenylalanine), positively charged- (e.g. lysine) and negatively charged- (e.g. glutamic acid) groups. The combinatorial assembly of these different chemical entities enables us to build proteins with a wide variety of molecular interaction properties, which form tight non-covalent bonds. This enables the identification of highly specific and selective antibodies, derived naturally or by respective biotechnological approaches, for virtually any target. Although technical advances in the field of chemical biology have enabled the in vitro selection of antibodies, their production still relies on biological, namely cellular, systems. Besides being cost intensive, biological production reveals limitations, for example antibodies can hardly be chemically modified in a site-directed fashion to tune and adapt to specific functionalities.Since 1990, aptamers7,8 have emerged as a potential alternative for protein-based recognition molecules. Aptamers are small single stranded non-coding oligonucleotides (DNA or RNA). These can fold into distinct three-dimensional structures capable of binding to cognate target structures with high affinity and specificity. A remarkably broad range of structurally diverse targets can be addressed by aptamers, ranging from proteins to small molecules such as amino acids9 down to single ions as e.g. fluoride sensing aptamer domains10 found in many bacteria and archaea. Nucleic acids share the advantage of antibodies, that for their amplification and modification, evolution has equipped us with a broad variety of highly optimized enzymatic tools, such as polymerases and nucleases. In turn aptamers are much smaller than antibodies and can be produced efficiently by means of solid phase chemical synthesis. Therefore, aptamers combine the advantages of being “biologics” with the advantage of being “chemicals”.
In this review we want to address some of the modifications that were applied to DNA and RNA aptamers in order to modulate their function, expanding their possible applications in therapy, diagnostics, and as tools in chemical biology.
Where do aptamers come from?
Most aptamers are derived from an iterative in vitro selection process, termed “Systematic Evolution of Ligands by EXponential enrichment” (SELEX). Similar to the selection mechanisms working in the evolution of species in nature (survival of the fittest), it is possible to select the best-suited nucleic acid sequence within a given environment. In a typical SELEX experiment a library prepared of 1014 to 1015 different sequences, of up to 100 nucleotides in length, is employed. This library is exposed to a selection pressure, namely binding to a defined target molecule (Fig. 1). After exposure, the nonbinding sequences are partitioned from the bound ones and the latter are subsequently recovered and amplified. After single strand displacement11 the library is reinstated to the next selection cycle. After several rounds of selection and amplification the resulting library is cloned and sequenced. The identified monoclonal aptamers can be produced by solid phase chemistry and thus modified at will.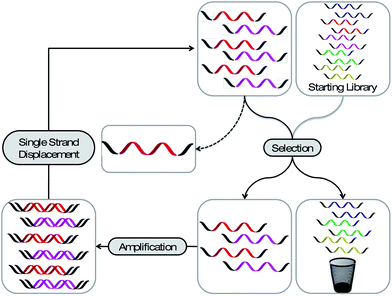 | ||
| Fig. 1 Schematic representation of the SELEX process. A library bearing up to 1015 different sequences is applied to a selection pressure. The strands with the desired property are partitioned from the less desired and amplified. After a single strand displacement step the enriched library can be employed to the next selection cycle. After several rounds the library is analysed for enriched sequences. | ||
Chemistry with aptamers
In principle two strategies are employed to incorporate chemical modifications into aptamers. First, modified (2′-deoxy-) nucleoside triphosphates can be used directly during the selection process. However, the necessity of enzymatic reactions for the amplification during SELEX imposes the limitation that all modifications on the triphosphate-level have got to be compatible with the enzymatic steps of an in vitro selection process. Second, the modification of a distinct aptamer can be done post-selectively, for example by applying modified phosphoramidites during solid phase synthesis. The benefit of the first approach is that the modification is already present during the selection process, thereby assuring that the aptamer once selected is functional despite and/or owing to the modification, a trait that can be compromised when the aptamer is modified post-selectively. Fortunately, many aptamers tolerate modifications, at least when those are restricted to the 5′- or 3′-termini without significant loss of their binding capabilities.Nucleobase modifications for increased chemical diversity of nucleic acids
Nucleic acids are built from four different deoxyribonucleotide – or in the case of RNA ribonucleotide – building blocks and have therefore, compared to antibodies, a more limited chemical diversity. Although several aptamer-target co-structure analyses revealed that nucleic acids employ hydrogen bonds, electrostatic, and pi-stacking interactions, aptamers particularly lack aliphatic, positively charged and extended additional aromatic functional groups that are not involved in hydrogen bond formation and intrahelical stacking interactions. This finding leads to the assumption that the range of targets that can be successfully addressed by aptamers could be extended by increasing the chemical diversity of nucleic acid libraries.12 In this way, nucleobases were equipped with additional chemical groups. Since it is crucial that these modifications do not interfere with the classical base-pairing interface, their possible introduction sites are restricted to non-Watson–Crick sites. In this regard the best-characterized position is the C5 position of pyrimidines, because it is the most permissive site for modifications accepted by DNA polymerases.13–15An early example for the use of modified nucleotides in SELEX was published 1994 by Latham et al., who were able to select an aptamer for thrombin with a DNA library in which they substituted the canonical dT by 5-pentynyl-dU (Fig. 2A).12 The resulting aptamer displayed a similar dissociation constant for thrombin as a previously selected ssDNA aptamer,16 without showing any sequence similarities.
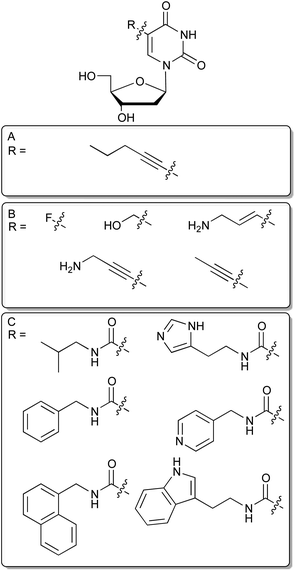 | ||
| Fig. 2 C5 modified deoxyuridines. (A): 5-pentynyl-dU nucleoside. (B): Selection of commercially available modified dU derivatives suitable for SELEX. (C): Modified dU analogs recently employed for in vitro selection. | ||
In a very recent study Vaught et al. were able to select aptamers recognizing the tumour necrosis factor receptor superfamily member 9 (TNFRSF9) that has previously deprived itself from DNA aptamer selection by using six different modifications at C5 of deoxyuridine.17 These were inspired by the side chains found in the proteinogenic amino acids and introduced by an amide linkage at the 5 position of desoxyuridine (Fig. 2C). Benzyl and indole modified nucleobases enabled successful aptamer selection whereas non-modified DNA failed to generate aptamers targeting TNRSF9. This approach has been taken one step further by researchers from Somalogic, significantly increasing the number of addressable targets of the human proteome. When selecting against over 1000 different proteins, they report that the overall success rate for aptamer selection has rose from below 30% using unmodified nucleotides to more than 80% by incorporating C5-modified nucleotides.18 Especially, the 5-tryptaminocarbonyl-dU containing libraries appear to be very successful during the selection of aptamers to otherwise difficult to access targets. These aptamers are coined as SOMAmers, an acronym for “Slow Off-rate Modified Aptamer”, in which additional aromatic side chains provide a molecular basis for generating aptamers with very low off-rates due to enhanced possibilities to form hydrophobic and pi-stacking interactions. These aptamers seem to be especially well suited for diagnostic purposes.19
Modifications to improve the pharmacokinetic profile of aptamers
In general, being predestined for therapeutic regimens, the short plasma half-life of aptamers requires improvement to optimize their pharmacokinetic profiles. This can be achieved by chemical modifications (Fig. 3). Unmodified nucleic acids reveal a very short plasma half-life, which is mainly influenced by two factors – enzymatic hydrolysis and renal filtration.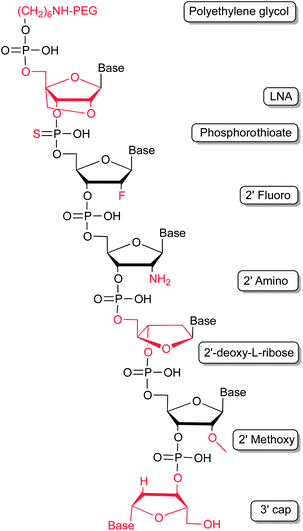 | ||
| Fig. 3 Modifications employed to improve the pharmacokinetic profile of aptamers by stabilizing the nucleic acids against enzymatic hydrolysis and renal filtration. | ||
Nature has evolved a myriad of enzymes in order to synthesize, amplify, modify and hydrolyse nucleic acids. The latter of which, so called nucleases, are important for the turnover of endogenous nucleic acids but also as defence mechanism against pathogenic organisms and viruses. As such these enzymes are found ubiquitously in virtually every organism. Human body fluids contain large amounts of nucleases, thus surging the need to stabilize aptamers against degradation. The most critical position in light of RNA degradation is its 2′-hydroxyl group. Nature makes use of selective protection system by methylation of the 2′-hydroxyl groups in distinct RNA molecules, such as mRNAs. 2′-Methoxy residues can be introduced into aptamers by chemical solid phase synthesis. As for all modifications introduced post-selectively it has to be verified, that the introduction of the modification is not detrimental to the aptamers binding abilities.20 Recent studies also demonstrate that the enzymatic steps of in vitro selection are compatible with 2′-methoxy modifications20,21 which allows the direct selection of modified, and hence, stabilized RNA aptamers. Besides, 2′-deoxy-2′-fluoro and 2′-deoxy-2′-amino modifications of pyrimidines have also been shown to increase the stability of RNA molecules in biological fluids.22 Both modifications are compatible with the enzymatic steps of the in vitro selection processes. In addition, modifications of the ribose–phosphate backbone through introducing phosphorothioates (PS), which are not accepted as a substrate by most nucleases, can lead to further stability.23 A special variant of nucleotides are locked nucleic acids (LNAs).24,25 LNA molecules reveal very interesting properties, which render them attractive for pharmaceutical applications.26 Beside masking the 2′-hydroxyl group, thereby increasing oligonucleotide stability in respect of nuclease degradation, LNA hybrids show an increase in melting temperature, compared to their sole RNA or DNA counterparts. LNA modifications have been introduced in previously selected RNA and DNA aptamers. For example, the TAR element of an HIV-1 binding aptamer was modified with LNAs, stabilizing the stem of a hairpin structure within the aptamer and improving nuclease resistance.27–29 Other examples describe the successful substitution of nucleotides with LNAs in known aptamers, whereas structural features could be stabilized, the sensitivity against nucleases decreased, and, most importantly, the binding to the target molecule retained. These studies include tenascin-C binding aptamers,30 a thrombin binding aptamer31 and an aptamer which recognizes avidin.32 Fortunately, cellular uptake of PS and LNA bearing oligonucleotides is enhanced, what might allow the addressing of even intracellular targets with modified aptamers more rationally.33–35
A very elegant approach to enhance aptamer stability has been described by Klussmann et al., who made use of the chiral nature of nucleic acids and their cognate protein targets.36,37 Aptamers are built of D-ribose monomers whereas proteins, including nucleases, are assembled from L-amino acid enantiomers. First, Klussmann selected aptamers against a peptide, which is identical with regard to primary structure to a natural peptide but composed of the enantiomeric D-amino acids. After successful identification of a D-ribose-based aptamer that recognizes the D-peptide, they synthesized the L-ribose variant of the RNA aptamer, which then is able to recognize the L-peptide, as found in nature. These aptamers are coined “Spiegelmers” (German Spiegel: mirror) and are almost resistant towards degradation by nucleases.38
As well as endonucleases, 3′-and 5′-exonuclease activity is detrimental to aptamer stability. However, capping strategies similar to those found at the 5′-cap of eukaryotic mRNA molecules have been employed. In this way the polarity of the nucleic acid chain is reversed generating nucleic acids that are barely recognized as substrates by exonucleases.39
Besides degradation by nucleases, renal clearance is an important issue when considering aptamers as therapeutics. Renal clearance mainly affects molecules with a molecular weight below 30–50 kDa. Aptamers' half-lives can be easily enhanced through addition of large molecular weight polyethylene glycol (PEG) moieties. These have been introduced either at the 5′- or at the 3′-termini of aptamers and, more importantly, often do not interfere with aptamer function. The circulating half-life of non-pegylated aptamers, which is often only a few minutes, can be extended to up to one day when conjugated with 40 kDa PEG molecules.39
A very prominent example, in which several modifications for the improvement of the pharmacokinetic properties were successfully applied, is Pegaptanib (Macugen®). Pegaptanib is the first and yet sole aptamer approved by the FDA for therapeutic applications. It is a VEGF-specific aptamer used for treatment of the wet form of age-related macular degeneration (AMD).22 This highly modified 28 nucleotide long aptamer is built from 2′-fluoro-pyrimidines and 2′-methoxy-purines and has a capped 3′ end. Additionally, it is conjugated to two 20 kDa PEG groups via 5′-end coupling. All these modifications result in a strong stabilisation so that a terminal apparent plasma half-life of 10 days could be observed.40,41
Linker chemistry for the attachment of “cargo” to aptamers
Small functional chemical entities, for example fluorophores or biotin can be incorporated site-specifically into an aptamer through employing respective phosphoramidite building blocks during synthesis. Alongside the direct incorporation of functional chemical groups it is also desirable to have selective chemical “handles” for the modular introduction of post-synthetic modifications. Therefore, a variety of linker groups have been developed based on bio-orthogonal chemical reactions that leave the aptamer itself intact; thus, displaying no cross-reactivity with the nucleic acid residues. Most of them bear a short alkyl or PEG chain coupled to a reactive group, which can be further covalently modified with the desired cargo (Fig. 4). For example, linker moieties with amine groups enable the straightforward modification with activated carboxyl groups such as N-hydroxy succinimide (NHS)–esters or isothiocyanates, such as the wide-spread use of fluorescein-isothiocyanate (FITC).42,43 Besides these, thiol-linkers are commonly used for a Michael-addition type conjugation with maleimide-derivatives or the functionalization with iodoacetamido bearing molecules. In case of RNA the chemical synthesis is less efficient because of the additional 2′ hydroxyl groups. Therefore the use of so-called initiator nucleotides provides an efficient way to incorporate chemical modifications at the 5′-end of enzymatically synthesized RNA molecules (Fig. 4C). In this way guanosine monophosphorothioate (GMPS) enables the introduction of fluorescent moieties or biotin.44 Alternatively it is possible to introduce an aldehyde group which can be further modified with amino- or hydrazine-modified building blocks.45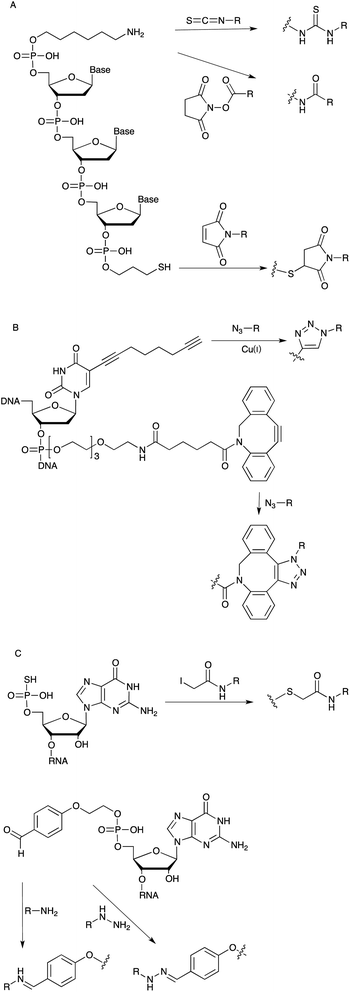 | ||
| Fig. 4 (A): Linker chemistry employed for the post-SELEX modification of DNA aptamers. (B): Linker for DNA functionalization via alkyne–azide cycloaddition. (C): Initiator nucleotides for the chemical modification of the 5′ position of RNA aptamers. | ||
More recently linkers allowing for 1,3 dipolar cycloadditions (“click chemistry”) are widely used especially in aqueous biological systems (Fig. 4B).46,47 The group of Thomas Carell developed and commercialized a “clickable” octadiynyl dU derivative which is available as triphosphate or as phosphoramidites, respectively, and therefore can be introduced into DNA either enzymatically or by solid phase synthesis.48 Beside the Cu(I) catalysed alkyne–azide cycloaddition (CuAAC), copper-free strain-promoted alkyne–azide cycloaddition (SPAAC) reactions are also becoming increasingly popular in order to overcome toxicity issues associated with the use of Cu(I) in cellular or living model systems.49,50
A huge variety of linkers and modifiers are commercially available and can be attached to the 5′-end, the 3′-end but also onto modified nucleobases (Fig. 4). Applying those different linker strategies, a multitude of different combinations of aptamers with other functional molecules have yet been realized. Aptamers recognizing cell surface proteins are frequently internalized into the target cell. Hence, molecular cargos attached to such an aptamer might be co-delivered to the intracellular compartment of distinct cells. This becomes especially important to address the bioavailability of siRNA molecules making them applicable as cell-specific therapeutics.51 For this reason aptamers became an interesting tool to developing targeted delivery approaches of therapeutics. The selective uptake into specific cell types limits off-target effects and is considered to reduce potential unwanted side effects.21,52
Spatiotemporal control of aptamer activity
We have explored ways that allow the exogenous control of aptamer and, thus, target function. In principal this can be achieved by either light-switching approaches or through the introduction of photo-labile groups, so called cages, at strategic positions of an aptamer. These approaches expand the properties of aptamers far beyond what is found in naturally occurring nucleic acids and will allow a precise control over biological functions. Cages are light-sensitive groups, which can be attached on defined positions of nucleobases, thereby interfering for example with their Watson–Crick base pairing capabilities. We started to study caged aptamers using thrombin-binding aptamers that have been well studied in respect to their structure–function relationships. The first example of a caged aptamer was introduced in 2005 where an o-nitrophenylpropyl (NPP) group was introduced into thymidine residues (Fig. 5) of the thrombin binding DNA aptamer HD1 by solid phase synthesis.53 Due to steric hindrance the caged aptamer showed a strongly reduced affinity towards its target. However, upon irradiation and deprotection the binding affinity to the target could be restored. Since thrombin is a key player in the blood-clotting cascade this caged aptamer enabled light-control of thrombin-dependent blood clotting. In a second study we used a similar approach for generating aptamer variants that were inhibited rather than activated upon irradiation. Light-dependent inactivation is achieved by extending the aptamer with a caged antisense region and thereby blocking base pairing properties. When the cage on the complementary antisense region was removed, base pairing was initiated resulting in the formation of a stable hairpin structure and, thus, leading to a loss of aptamer activity.54 The caged aptamer approach was extended to several other aptamers recently. Amongst them light-responsive variants of a different thrombin binding aptamer, namely HD22, which were used for the functional detection of thrombin. In this assay the analyte (thrombin) can be regained allowing for sensitive quantitative measure followed by functional assessment of the analyte.55 Also aptamers that target intracellular signalling proteins have been reported,56 whereas the most sophisticated approach may now be represented by differential regulation of protein sub-domains established through a bivalent aptamer with individually caged aptamer sub-domains. In this approach one of the aptamer domains was caged while the other remained unaffected, thereby allowing the spatiotemporal modulation of a single protein domain.57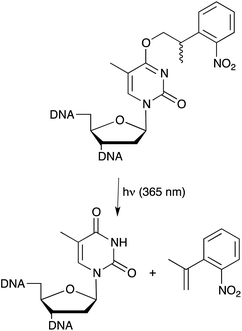 | ||
| Fig. 5 Light induced deprotection of NPP caged thymidine nucleobase for spatiotemporal control of aptamer function. | ||
Beside cages, also photo-reactive switches like the azobenzene have been explored for light dependent aptamer regulation. In this case the HD1 anti thrombin aptamer activity is regulated by cis/trans isomerisation of the azobenzene moiety introduced within a DNA sequence complementary to the aptamer. In the cis-conformation the aptamer remains functional because it cannot hybridize to its complementary strand due to steric hindrance by the azobenzene group. Upon irradiation with visible light the azobenzene switches to the trans-conformation allowing the previously inaccessible complementary strand to form a duplex with the aptamer, thereby inactivating its binding to thrombin.58
Aptamer cross-linking with photo-reactive groups
The use of photo-reactive groups is not only limited to caging applications but can also be used for covalent proximity ligation of the aptamer to its protein target. This is of special interest for the target identification of aptamers that were generated through whole-cell SELEX approaches, in which initially the molecular target is unknown. Replacing some bases post-selectively by photo-reactive nucleobases (Fig. 6) such as 5-iodo-2′-deoxyuridine59 allows the covalent crosslinking of the aptamer with its target through irradiation with UV light. A biotin group conjugated to the aptamer enables the purification of the cross-linked complex and further analysis of the cross-linked protein by mass spectrometry. The downside to this approach being that the positioning of the substitution requires tedious optimisation in order to retain the aptamers’ binding abilities. This can be overcome by photo-SELEX protocols in which libraries with 5-bromo-2′-deoxyuridines are employed thus yielding de novo selected cross-linkable aptamers.60 However, for this approach a complete new selection of an aptamer has to be done. Vinkenborg et al. developed a method termed aptamer-based affinity labelling (ABAL) in which the cross-linking moiety is attached to the 5′-end of a given aptamer.61 Since many aptamers tolerate modifications at the 5′-terminus this approach might be generally applicable and may allow the identification of the molecular target of aptamers or potential secondary targets that otherwise remain unknown.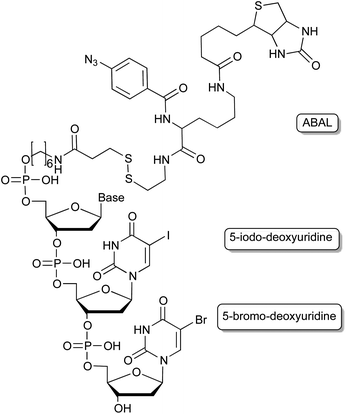 | ||
| Fig. 6 Cross-linking moieties employed in aptamers, enabling e.g. the target identification of cell-SELEX derived aptamers. | ||
Outlook
The general idea to select single polymeric molecules from huge libraries of different polymers has shown to be very powerful and has found various adaptations in nature and chemical biology. Nowadays one of the advantages, namely the sequence specific enzymatic amplification of the enriched libraries, is also one of the major limitations to the employment of strongly modified building blocks, which could bear unprecedented novel functions. Although the active sites of naturally occurring polymerases show a remarkable plasticity in adapting to modified substrates,62 there are clear limitations to the extent to which modified building blocks can be incorporated. Especially, xeno-nucleic acids (XNAs) such as LNAs with altered sugar–phosphate backbones are emerging as a promising new polymer for aptamer selection,63 but are not readily accepted as substrates for the naturally occurring polymerases. Very recent work by Pinheiro et al. tries to overcome this limitation by creating synthetic polymerases capable of handling XNA building blocks.64 Until now an intermediate step using DNA is required, but future work in the field might produce polymerases capable of the direct amplification of XNAs allowing for more efficient XNA selection methods.Until now most modified bases that have been utilized for aptamer selection are based on the canonical Watson–Crick hydrogen bond forming bases. In order to expand the genetic alphabet of DNA by a third base pair, completely new unnatural base pair systems, mimicking the overall shape but not the hydrogen-bonding pattern, are being developed (Fig. 7).65 These bases would allow the introduction of additional chemical interaction possibilities and may be useful in the selection of aptamers with unprecedented structures. Especially, less polar base pairs like the ones that were developed by Kool, Morales66 and Hirao et al.67 could be beneficial to select against targets that are not addressable with traditional SELEX methods. By the employment of a metal-ion complex that forms artificial base pairs68,69 for in vitro selection, aptamers with novel magnetic, conductive or catalytic properties could be generated which could find numerous applications in e.g. nanotechnology.
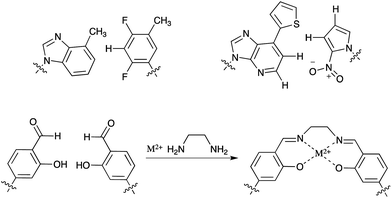 | ||
| Fig. 7 Artificial base pairs that might be employed in SELEX. | ||
Modularity is a factor, which could improve future evolutionary selection methods. Today's selection techniques are often restricted to handling one modification and often have to be re-optimized if a different modification would like to be employed. This creates a need for modular approaches,70 enabling the fast and easy parallel adoption of several modifications, thereby dramatically increasing the chemical space that can be addressed and investigated by aptamers.
Notes and references
- International Human Genome Sequencing Consortium, Nature, 2001, 409, 860–921 CrossRef.
- ENCODE Project Consortium, B. E. Bernstein, E. Birney, I. Dunham, E. D. Green, C. Gunter and M. Snyder, Nature, 2012, 489, 57–74 CrossRef.
- R. Micura and C. Höbartner, ChemBioChem, 2003, 4, 984–990 CrossRef CAS.
- B. Fürtig, C. Richter, J. Wöhnert and H. Schwalbe, ChemBioChem, 2003, 4, 936–962 CrossRef.
- P. Wenter, B. Fürtig, A. Hainard, H. Schwalbe and S. Pitsch, Angew. Chem., Int. Ed., 2005, 44, 2600–2603 CrossRef CAS.
- M. Mandal and R. R. Breaker, Nat. Rev. Mol. Cell Biol., 2004, 5, 451–463 CrossRef CAS.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS.
- M. Famulok, J. Am. Chem. Soc., 1994, 116, 1698–1706 CrossRef CAS.
- J. L. Baker, N. Sudarsan, Z. Weinberg, A. Roth, R. B. Stockbridge and R. R. Breaker, Science, 2012, 335, 233–235 CrossRef CAS.
- M. Svobodová, A. Pinto, P. Nadal and C. K. O'Sullivan, Anal. Bioanal. Chem., 2012, 404, 835–842 CrossRef.
- J. A. Latham, R. Johnson and J. J. Toole, Nucleic Acids Res., 1994, 22, 2817–2822 CrossRef CAS.
- W. Kusser, J. Biotechnol., 2000, 74, 27–38 CAS.
- A. D. Keefe and S. T. Cload, Curr. Opin. Chem. Biol., 2008, 12, 448–456 CrossRef CAS.
- M. Kuwahara and N. Sugimoto, Molecules, 2010, 15, 5423–5444 CrossRef CAS.
- L. C. Bock, L. C. Griffin, J. A. Latham, E. H. Vermaas and J. J. Toole, Nature, 1992, 355, 564–566 CrossRef CAS.
- J. D. Vaught, C. Bock, J. Carter, T. Fitzwater, M. Otis, D. Schneider, J. Rolando, S. Waugh, S. K. Wilcox and B. E. Eaton, J. Am. Chem. Soc., 2010, 132, 4141–4151 CrossRef CAS.
- L. Gold, D. Ayers, J. Bertino, C. Bock, A. Bock, E. N. Brody, J. Carter, A. B. Dalby, B. E. Eaton, T. Fitzwater, D. Flather, A. Forbes, T. Foreman, C. Fowler, B. Gawande, M. Goss, M. Gunn, S. Gupta, D. Halladay, J. Heil, J. Heilig, B. Hicke, G. Husar, N. Janjic, T. Jarvis, S. Jennings, E. Katilius, T. R. Keeney, N. Kim, T. H. Koch, S. Kraemer, L. Kroiss, N. Le, D. Levine, W. Lindsey, B. Lollo, W. Mayfield, M. Mehan, R. Mehler, S. K. Nelson, M. Nelson, D. Nieuwlandt, M. Nikrad, U. Ochsner, R. M. Ostroff, M. Otis, T. Parker, S. Pietrasiewicz, D. I. Resnicow, J. Rohloff, G. Sanders, S. Sattin, D. Schneider, B. Singer, M. Stanton, A. Sterkel, A. Stewart, S. Stratford, J. D. Vaught, M. Vrkljan, J. J. Walker, M. Watrobka, S. Waugh, A. Weiss, S. K. Wilcox, A. Wolfson, S. K. Wolk, C. Zhang and D. Zichi, PLoS One, 2010, 5, e15004 CAS.
- S. Kraemer, J. D. Vaught, C. Bock, L. Gold, E. Katilius, T. R. Keeney, N. Kim, N. A. Saccomano, S. K. Wilcox, D. Zichi and G. M. Sanders, PLoS One, 2011, 6, e26332 CAS.
- P. E. Burmeister, S. D. Lewis, R. F. Silva, J. R. Preiss, L. R. Horwitz, P. S. Pendergrast, T. G. McCauley, J. C. Kurz, D. M. Epstein and C. Wilson, Chem. Biol., 2005, 12, 25–33 CrossRef CAS.
- Z. Xiao, E. Levy-Nissenbaum, F. Alexis, A. Lupták, B. A. Teply, J. M. Chan, J. Shi, E. Digga, J. Cheng, R. Langer and O. C. Farokhzad, ACS Nano, 2012, 6, 696–704 CrossRef CAS.
- J. Ruckman, L. S. Green, J. Beeson, S. Waugh, W. L. Gillette, D. D. Henninger, L. Claesson-Welsh and N. Janjić, J. Biol. Chem., 1998, 273, 20556–20567 CrossRef CAS.
- D. J. King, D. A. Ventura, A. R. Brasier and D. G. Gorenstein, Biochemistry, 1998, 37, 16489–16493 CrossRef CAS.
- A. A. Koshkin, S. K. Singh, P. Nielsen, V. K. Rajwanshi, R. Kumar, M. Meldgaard, C. E. Olsen and J. Wengel, Tetrahedron, 1998, 54, 3607–3630 CrossRef CAS.
- A. A. Koshkin, V. K. Rajwanshi and J. Wengel, Tetrahedron Lett., 1998, 39, 4381–4384 CrossRef CAS.
- S. K. Singh, A. A. Koshkin, J. Wengel and P. Nielsen, Chem. Commun., 1998, 455–456 RSC.
- F. Darfeuille, J. B. Hansen, H. Orum, C. Di Primo and J.-J. Toulme, Nucleic Acids Res., 2004, 32, 3101–3107 CrossRef CAS.
- F. Darfeuille, S. Reigadas, J. B. Hansen, H. Orum, C. Di Primo and J.-J. Toulme, Biochemistry, 2006, 45, 12076–12082 CrossRef CAS.
- I. Lebars, T. Richard, C. Di Primo and J.-J. Toulme, Nucleic Acids Res., 2007, 35, 6103–6114 CrossRef CAS.
- K. S. Schmidt, S. Borkowski, J. Kurreck, A. W. Stephens, R. Bald, M. Hecht, M. Friebe, L. Dinkelborg and V. A. Erdmann, Nucleic Acids Res., 2004, 32, 5757–5765 CrossRef CAS.
- A. Virno, A. Randazzo, C. Giancola, M. Bucci, G. Cirino and L. Mayol, Bioorg. Med. Chem., 2007, 15, 5710–5718 CrossRef CAS.
- F. J. Hernandez, N. Kalra, J. Wengel and B. Vester, Bioorg. Med. Chem. Lett., 2009, 19, 6585–6587 CrossRef CAS.
- C. A. Stein, J. B. Hansen, J. Lai, S. Wu, A. Voskresenskiy, A. Høg, J. Worm, M. Hedtjärn, N. Souleimanian, P. Miller, H. S. Soifer, D. Castanotto, L. Benimetskaya, H. Orum and T. Koch, Nucleic Acids Res., 2010, 38, e3 CrossRef CAS.
- Y. Zhang, Z. Qu, S. Kim, V. Shi, B. Liao, P. Kraft, R. Bandaru, Y. Wu, L. M. Greenberger and I. D. Horak, Gene Ther., 2011, 18, 326–333 CrossRef CAS.
- R. L. Juliano, X. Ming and O. Nakagawa, Bioconjugate Chem., 2012, 23, 147–157 CrossRef CAS.
- A. Nolte, S. Klussmann, R. Bald, V. A. Erdmann and J. P. Fürste, Nat. Biotechnol., 1996, 14, 1116–1119 CrossRef CAS.
- S. Klussmann, A. Nolte, R. Bald, V. A. Erdmann and J. P. Fürste, Nat. Biotechnol., 1996, 14, 1112–1115 CrossRef CAS.
- D. Eulberg and S. Klussmann, ChemBioChem, 2003, 4, 979–983 CrossRef CAS.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537–550 CrossRef CAS.
- E. W. M. Ng, D. T. Shima, P. Calias, E. T. Cunningham, D. R. Guyer and A. P. Adamis, Nat. Rev. Drug Discovery, 2006, 5, 123–132 CrossRef CAS.
- R. S. Apte, Expert Opin. Pharmacother., 2008, 9, 499–508 CrossRef CAS.
- M. Blank, T. Weinschenk, M. Priemer and H. Schluesener, J. Biol. Chem., 2001, 276, 16464–16468 CrossRef CAS.
- H. Ulrich, A. H. B. Martins and J. B. Pesquero, Cytometry, Part B, 2004, 59A, 220–231 CrossRef CAS.
- G. Sengle, A. Jenne, P. S. Arora, B. Seelig, J. S. Nowick, A. Jäschke and M. Famulok, Bioorg. Med. Chem., 2000, 8, 1317–1329 CrossRef CAS.
- S. Pfander, R. Fiammengo, S. I. Kirin, N. Metzler-Nolte and A. Jäschke, Nucleic Acids Res., 2007, 35, e25 CrossRef.
- A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388 RSC.
- E. Paredes and S. R. Das, ChemBioChem, 2011, 12, 125–131 CrossRef CAS.
- J. Gierlich, G. A. Burley, P. M. E. Gramlich, D. M. Hammond and T. Carell, Org. Lett., 2006, 8, 3639–3642 CrossRef CAS.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS.
- I. S. Marks, J. S. Kang, B. T. Jones, K. J. Landmark, A. J. Cleland and T. A. Taton, Bioconjugate Chem., 2011, 22, 1259–1263 CrossRef CAS.
- J. O. McNamara, E. R. Andrechek, Y. Wang, K. D. Viles, R. E. Rempel, E. Gilboa, B. A. Sullenger and P. H. Giangrande, Nat. Biotechnol., 2006, 24, 1005–1015 CrossRef CAS.
- C. Meyer, U. Hahn and A. Rentmeister, J. Nucleic Acids, 2011, 2011, 904750 Search PubMed.
- A. Heckel and G. Mayer, J. Am. Chem. Soc., 2005, 127, 822–823 CrossRef CAS.
- A. Heckel, M. C. R. Buff, M.-S. L. Raddatz, J. Müller, B. Pötzsch and G. Mayer, Angew. Chem., Int. Ed., 2006, 45, 6748–6750 CrossRef CAS.
- A. Pinto, S. Lennarz, A. Rodrigues-Correia, A. Heckel, C. K. O'Sullivan and G. Mayer, ACS Chem. Biol., 2012, 7, 360–366 CrossRef CAS.
- G. Mayer, A. Lohberger, S. Butzen, M. Pofahl, M. Blind and A. Heckel, Bioorg. Med. Chem. Lett., 2009, 19, 6561–6564 CrossRef CAS.
- G. Mayer, J. Müller, T. Mack, D. F. Freitag, T. Höver, B. Pötzsch and A. Heckel, ChemBioChem, 2009, 10, 654–657 CrossRef CAS.
- Y. Kim, J. A. Phillips, H. Liu, H. Kang and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6489–6494 CrossRef CAS.
- P. Mallikaratchy, Z. Tang, S. Kwame, L. Meng, D. Shangguan and W. Tan, Mol. Cell. Proteomics, 2007, 6, 2230–2238 CAS.
- M. Golden, B. Collins, M. Willis and T. Koch, J. Biotechnol., 2000, 81, 167–178 CrossRef CAS.
- J. L. Vinkenborg, G. Mayer and M. Famulok, Angew. Chem., Int. Ed., 2012, 51, 9176–9180 CrossRef CAS.
- S. Obeid, H. Bußkamp, W. Welte, K. Diederichs and A. Marx, Chem. Commun., 2012, 48, 8320–8322 RSC.
- R. N. Veedu and J. Wengel, Mol. BioSyst., 2009, 5, 787–792 RSC.
- V. B. Pinheiro, A. I. Taylor, C. Cozens, M. Abramov, M. Renders, S. Zhang, J. C. Chaput, J. Wengel, S.-Y. Peak-Chew, S. H. McLaughlin, P. Herdewijn and P. Holliger, Science, 2012, 336, 341–344 CrossRef CAS.
- I. Hirao and M. Kimoto, Proc. Jpn. Acad., Ser. B, Phys. Biol. Sci., 2012, 88, 345–367 CrossRef CAS.
- J. C. Morales and E. T. Kool, Nat. Struct. Biol., 1998, 5, 950–954 CrossRef CAS.
- I. Hirao, T. Mitsui, M. Kimoto and S. Yokoyama, J. Am. Chem. Soc., 2007, 129, 15549–15555 CrossRef CAS.
- E. Meggers, P. Holland, W. Tolman, F. Romesberg and P. Schultz, J. Am. Chem. Soc., 2000, 122, 10714–10715 CrossRef CAS.
- G. H. Clever, K. Polborn and T. Carell, Angew. Chem., Int. Ed., 2005, 44, 7204–7208 CrossRef CAS.
- S. Jäger, G. Rasched, H. Kornreich-Leshem, M. Engesser, O. Thum and M. Famulok, J. Am. Chem. Soc., 2005, 127, 15071–15082 CrossRef.
| This journal is © The Royal Society of Chemistry 2013 |