Thermally curable fluorinated main chain benzoxazine polyethers via Ullmann coupling†
Kubra Dogan
Demir
a,
Baris
Kiskan
*a,
Sanjay S.
Latthe
c,
A. Levent
Demirel
c and
Yusuf
Yagci
*ab
aIstanbul Technical University, Faculty of Science and Letters, Chemistry Department, Maslak, TR-34469, Istanbul, Turkey. E-mail: yusuf@itu.edu.tr; kiskanb@itu.edu.tr; Fax: +90 212 2856386; Tel: +90 212 2853241
bKing Abdulaziz University, Faculty of Science, Chemistry Department, Jeddah, Saudi Arabia
cKoc University, Chemistry Department, Rumelifeneri Yolu, Sariyer, TR-34450, Istanbul, Turkey
First published on 7th January 2013
Abstract
Fluorinated main chain benzoxazine polyethers were prepared by Ullmann coupling of fluorinated benzoxazines in the presence of a nano-copperoxide catalyst. Various parameters such as the monomer structure, temperature, and the effect of catalyst on the polymerization were studied. The benzoxazine groups present in the polyether structure were shown to readily undergo thermally activated ring-opening polymerization in the absence of an added catalyst forming cross-linked networks. The thermal stability of the cured polymers was investigated and compared to that of classical polybenzoxazines. The lower surface energy of the fluorinated polymers made ultrathin films (∼20 nm thick) stable against dewetting at curing temperatures and resulted in thermally cured smooth coatings on solid substrates.
Introduction
Polybenzoxazines are addition-cure thermosetting polymers exhibiting good mechanical properties, low water sorption, dimensional stability, chemical resistivity, and flame resistance which are associated with the existence of inter- and intra-molecular hydrogen bonds in their structure that make them a superior alternative to phenolics or epoxy resins.1,2 Moreover, these fascinating characteristics make polybenzoxazines suitable for various applications such as electronics, composites, coatings, and adhesives.3–10 The noteworthy dimensional stability feature during polymerization stems from the ring opening polymerization of the corresponding monomers.11,12 And this polymerization is a thermally induced self-polymerization that takes place without any initiator or curative (Scheme 1).13,14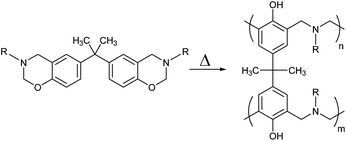 | ||
| Scheme 1 Thermally induced ring opening polymerization of bisbenzoxazine monomers. | ||
Benzoxazines were first synthesized by Cope and Holly in the 1940s.15 However, the polymerization of these molecules was performed much later16 initiating a surge of interest in research on the resulting high performance thermosets.17 Accordingly, there has been a lot of work on new monomers in order to improve the properties of polybenzoxazines for various applications by taking advantage of design flexibility of benzoxazine monomers coming from usage of inexpensive and commercially available phenols, primary amines, and formaldehyde (see Scheme 2).18–25
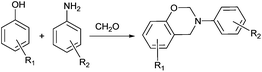 | ||
| Scheme 2 Synthesis of mono-functional 1,3-benzoxazines. | ||
Typically, various new benzoxazine monomers were synthesized with additional polymerizable groups to tune the cross-linking density of the networks.21,23,26–29 However, fabrication of films from monomers is rather difficult especially from mono-functional benzoxazines since the formed films are brittle as a consequence of the low molecular weight and non-flexibility of the network structures.30,31 Linear or side chain benzoxazine polymers as curable precursors were synthesized to overcome these problems.32–42 This way, polymeric benzoxazines behave like a processable and cross-linkable thermoplastic having flexibility, high cross-linking density after cure, reduction in the vapour pressure during processing and fragility of cured end-structures.43–46 A wide range of chemistries can be used for this purpose. Polyesterification,39 Mannich type condensations,32,41 coupling reactions,33 and Huisgen type click reaction40,42,47,48 were shown as successful ways to synthesize new curable precursors. Obviously, each route imparts the characteristics of its chemistry to the resulting polybenzoxazines which can be judiciously chosen for specific applications.
In continuation of our research on the linear polybenzoxazine prepolymers, in this paper, a series of fluorinated and non-fluorinated main chain polybenzoxazine prepolymers have been synthesized by using Ullmann type coupling. These reactive main-chain polymers can be further cross-linked via polymerization of the repeating internal oxazine rings. Combining benzoxazine chemistry with fluorinated aromatic polymers offers additional advantages to improve polybenzoxazines with extended glass transition temperature, thermal stability, low dielectric constant, and low water absorption which could stem from the contribution of fluorinated aromatic polyethers. Particularly, the fluorine content in main-chain polybenzoxazines leads to low surface energy yielding higher water contact angles for both cured and uncured polymers.
Experimental part
Materials
Phenol (Carlo Erba, 99%), 4,4-isopropylidenediphenol (bisphenol A) (Aldrich, 97%), 4,4′-(hexafluoroisopropylidene)diphenol (bisphenol AF) (Alfa Aesar, 98%), 4-fluoroaniline (Aldrich, 99%), 2,3,4,5,6-pentafluoroaniline (Aldrich, 99%), paraformaldehyde (Acros, 96%), nano CuO (Aldrich, <50 nm), Cs2CO3 (Fluka, ≥98%), sodium hydroxide (Acros, 97+%), 1,4-dioxane (Aldrich, ≥99%), xylene (Merck, extra pure), toluene (Carlo Erba, 99.5%), ethylacetate (Merck, 99.5%), and chloroform (Acros, 99+%) were used as received. Dimethylsulfoxide (DMSO) (Riedel-de Haen, 99.5%) was distilled under vacuum prior to use.Characterization
1H NMR spectra were recorded in CDCl3 with Si(CH3)4 as an internal standard, using a Bruker AC250 instrument at a proton frequency of 250 MHz. The FTIR spectra were recorded using Perkin Elmer Spectrum One with an ATR Accessory (ZnSe, Pike Miracle Accessory) and a cadmium telluride (MCT) detector. The resolution was 4 cm−1 and 24 scans with 0.2 cm s−1 scan speed. Differential scanning calorimetry (DSC) was performed on a Perkin-Elmer Diamond DSC with a heating rate of 20 °C min−1 under nitrogen flow (20 mL min−1). Thermogravimetric analysis (TGA) was performed on a Perkin-Elmer Diamond TA/TGA with a heating rate of 10 °C min−1 under nitrogen flow (200 mL min−1).For surface characterization, thin films of polymeric benzoxazine precursors were spin coated (Specialty Coating Systems, P67080 Spin Coater) on silicon wafers from 3 to 10 mg mL−1 solutions in chloroform at 2000 rpm for 1 min. The water contact angles (WCAs) were measured using the sessile drop method with a water drop volume of 8 μl on a contact angle system (Dataphysics, Contact Angle System OCA 20) at ambient temperature. More than five different positions were measured on a given sample and the average values were plotted. The surface morphology of the coatings was characterized by Atomic Force Microscopy (AFM) (NT-MDT, Solver P47) in tapping mode using ultra-sharp silicon cantilevers (force constant ∼48 N m−1). The thickness of the coatings was measured by ellipsometry (Microphotonics, EL X-01R).
Synthesis
BisF-Benz: 1H-NMR (CDCl3): δ = 1.59 (s, 6H, CH3), 4.50 (s, 4H, O–CH2–N), 5.10 (s, 4H, Ar–CH2–N), 6.77–7.25 (m, 14H, aromatics).
DecaF-Benz: 1H-NMR (CDCl3): δ = 1.61 (s, 6H, CH3), 4.49 (s, 4H, O–CH2–N), 5.09 (s, 4H, Ar–CH2–N), 6.79–7.27 (m, 6H, aromatics).
OctaF-PolyBenzPre: yield: 55%. 1H-NMR (CDCl3): δ = 1.61 (s, 12H, CH3), 4.52 (s, 4H, O–CH2–N), 5.12 (s, 4H, Ar–CH2–N), 6.73–7.11 (m, 14H, aromatics).
TetradecaF-PolyBenzPre: yield: 70%. 1H-NMR (CDCl3): δ = 1.61 (s, 6H, CH3), 4.53 (s, 4H, O–CH2–N), 5.14 (s, 4H, Ar–CH2–N), 6.79–6.99 (m, 14H, aromatics).
NonF-PolyBenzPre: yield: 34%. 1H-NMR (CDCl3): δ = 1.51 (s, 12H, CH3), 4.47 (s, 4H, O–CH2–N), 5.23 (s, 4H, Ar–CH2–N), 6.68–7.05 (m, 22H, aromatics).
HexaF-PolyBenzPre: yield: 25%. 1H-NMR (CDCl3): δ = 1.49 (s, 6H, CH3), 4.49 (s, 4H, O–CH2–N), 5.24 (s, 4H, Ar–CH2–N), 6.60–7.14 (m, 22H, aromatics).
Results and discussion
A successful synthesis of fluoro containing benzoxazine monomers has been achieved by typical benzoxazine synthesis. Bisphenol A was reacted with paraformaldehyde and 4-fluoroaniline to yield bisfluorobenzoxazine (BisF-Benz) as depicted in Scheme 3. This monomer was obtained without any purification step. | ||
| Scheme 3 Synthesis of the bisfluorobenzoxazine monomer (BisF-Benz). | ||
However, the synthesis of octafluorophenylbenzoxazine (OctaF-Benz) from 2,3,4,5,6-pentafluoroaniline gave the benzoxazine monomer with a low yield as several by-products were formed. Thus, the synthetic strategy was adapted and firstly, triazines were formed in the reaction medium, using paraformaldehyde and 2,3,4,5,6-pentafluoroaniline prior to addition of the bisphenol derivative and additional paraformaldehyde (Scheme 4). This way, a pure product namely DecaF-Benz was obtained with a higher yield and better purity.
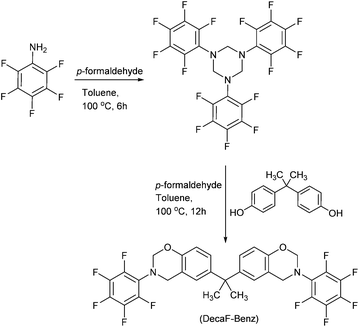 | ||
| Scheme 4 Synthesis of the decafluorobisbenzoxazine (DecaF-Benz) monomer. | ||
The structures of the benzoxazine monomers were confirmed by spectral analysis. The 1H NMR spectrum of DecaF-Benz presented in Fig. 1a exhibits the typical proton signals attributed to the oxazine structure at 5.09 (Ar–CH2–N) and 4.49 ppm (O–CH2–N). Moreover, the aromatic protons were observed between 7.27 and 6.79 ppm. Similarly, the oxazine structure of BisF-Benz was proved by the peaks at 5.10 (Ar–CH2–N) and 4.50 ppm (O–CH2–N). Aromatic protons were also detected between 7.25 and 6.77 ppm (Fig. 2a). 13C NMR spectra of the monomers also reveal signals for Ar–CH2–N and O–CH2–N carbons at 82.5, 54 ppm and 82.8, 53.3 ppm belonging to BisF-Benz and DecaF-Benz, respectively (Fig. S1 and S2†). The infrared absorption peaks of the monomers (Fig. S3–S6†) also confirm the formation of oxazine rings in each monomer. The out-of-plane absorption bands of oxazine functional benzene emerge between 967 and 985 cm−1 for the DecaF-Benz monomer and 964 and 993 cm−1 for the BisF-Benz monomer, respectively. Furthermore, the presence of the aromatic ether of the benzoxazine ring could be observed as the C–O–C symmetric stretching band, centered in the range of 1120–1241 cm−1. Additionally, the asymmetric stretching bands between 1172 and 1243 for DecaF-Benz and 1192 and 1238 cm−1 for BisF-Benz confirm the presence of the oxazine ring. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–C aromatic ring stretch of both monomers could also be detected at around 1502 cm−1.
C–C aromatic ring stretch of both monomers could also be detected at around 1502 cm−1.
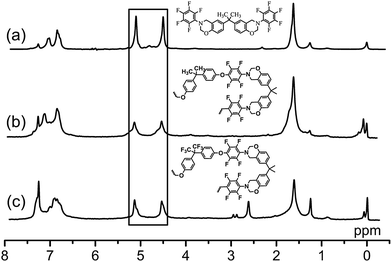 | ||
| Fig. 1 1H NMR spectra of (a) DecaF-Benz, (b) OctaF-PolyBenzPre and (c) TetradecaF-PolyBenzPre in CDCl3. | ||
 | ||
| Fig. 2 1H NMR spectra of (a) BisF-Benz, (b) nonF-PolyBenzPre and (c) HexaF-PolyBenzPre in CDCl3. | ||
In the subsequent step, Ullmann coupling was utilized to obtain main-chain polybenzoxazine precursors from the corresponding fluoro containing bisbenzoxazine monomers (Scheme 5). It is known that Ullmann chemistry along with related methods has served successfully for C–N, C–S, C–O bond formation reactions in organic chemistry for decades.49,50 Moreover, Ullmann coupling of aryl halides with phenols is popular for the synthesis of polyethers on laboratory and industrial scales.51 However, the practicality of traditional Ullmann coupling has been greatly limited by its harsh reaction conditions like prolonged heating at 200 °C in polar high-boiling solvents, the abundant use of copper compounds and the low conversion of non-activated aryl halides.
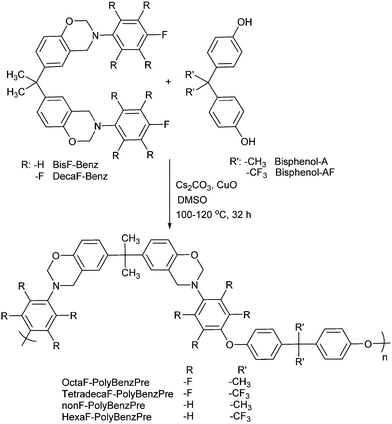 | ||
| Scheme 5 Syntheses of polymeric benzoxazine precursors. | ||
Accordingly, nanoparticles with reactive high surface areas have been widely studied in catalyst chemistry. Very recently, the employment of nanocrystalline metal oxides as catalysts in organic syntheses has attracted much attention, due to the ease of recycling of the catalysts and minimization of metal residues in the products and decrease in the reaction temperatures.52 Hence, performing Ullmann cross-coupling of phenols with aryl halides in the presence of the nano-CuO catalyst under ligand-free conditions could eliminate the harsh reaction conditions, which is particularly important to prevent ring opening reactions of benzoxazine moieties during the synthesis. Polymerizations conducted at around 100–120 °C with the nano-CuO catalyst gave corresponding polybenzoxazine precursors. Notably, the molecular weights of the obtained polyethers, TetradecaF-PolyBenzPre and OctaF-PolyBenzPre, were affected by the applied temperature. The molecular weights (Mn) were around 1.4 kDa and 3.5 kDa for the polymerization at 100 °C and 120 °C, respectively. It was observed that higher temperatures than 120 °C increased by-product formation. The syntheses of nonF-PolyBenzPre and HexaF-PolyBenzPre yielded reasonable molecular weights even when lower temperatures (>100 °C) were applied. The molecular weight characteristics of all polymeric precursors are tabulated in Table 1. In this connection, it should be pointed out that ‘Ullmann type condensation’ of the ‘dibromo monomers’ with bisphenol-A and bisphenol-AF also yields aromatic polyethers with similar low molecular weights.53,54

Moreover, both TetradecaF-PolyBenzPre and OctaF-PolyBenzPre have higher yields and higher molecular weights compared to nonF-PolyBenzPre and HexaF-PolyBenzPre. The main reason for these results is the activity of fluorine groups on the highly fluorinated monomers compared to lower F containing ones. Additional F atoms attached to aromatic benzene withdraw electrons from the ring which makes the C–F bond more polar and reactive against aryl nucleophilic substitution.55,56
The spectral analysis confirms the structures of fluorinated polybenzoxazine prepolymers. As shown in Fig. 1, 1H NMR spectra of OctaF-PolyBenzPre and TetradecaF-PolyBenzPre showed the characteristic benzoxazine peaks at 5.12, 4.52 and 5.14, 4.53 ppm, respectively. It is clear from the spectra that oxazine rings of the highly fluorinated benzoxazines remained under coupling conditions. Moreover, the 1H NMR spectra of the prepolymers, nonF-PolyBenzPre and HexaF-PolyBenzPre, revealed peaks for oxazine protons at 5.23, 4.47 and 5.24, 4.49 ppm, respectively (Fig. 2). Additional peaks at 3.7 and 4.2 ppm indicate the presence of some ring opened oxazine structures in the main chain for both prepolymers. The peaks of ring opened oxazines disappeared when the polyetherification temperature was set below 85 °C; however, in this case a drastic decrease in the molecular weights was observed.
The IR studies also evidenced the expected chemical structures of prepolymers. The transmittance spectra revealed that out-of-plane absorption bands of oxazine functional benzene emerge at 980 and 970 cm−1 for OctaF-PolybenzPre and TetradecaF-PolyBenzPre, respectively. The CC–C aromatic ring stretch of both polymers is detectable at 1494 cm−1. Moreover, C–O–C symmetric stretching mode centered at ∼1260 cm−1 indicates the aromatic ether bond formation by Ullmann coupling (see Fig. S3 and S4†). Similar FT-IR spectra were recorded for HexaF-PolyBenzPre and nonF-PolyBenzPre (Fig. S5 and S6†).
In order to get more insight into the polyetherification reaction of benzoxazines, several model reactions were conducted (see Scheme 6). The preservation of the oxazine ring under the reaction conditions was evidenced by tracking the reactions with 1H NMR (Fig. S7†). It was also verified that nano-CuO is indispensable for a successful polyetherification process. In the absence of CuO, a low molecular weight product (Mn = 650) corresponding to a dimer between BisF-Benz and bisphenol A was obtained.
 | ||
| Scheme 6 Model reactions of benzoxazine monomers. | ||
It is well known that thermally activated ring opening polymerization of 1,3-benzoxazines is an exothermic process which exhibits a peak at around 180–270 °C depending on the functionalities and concentration of the benzoxazines. Fig. 3 illustrates DSC profiles of the highly fluorinated difunctional benzoxazine, DecaF-Benz, and its polyether prepolymers. The ring opening polymerization (ROP) of the DecaF-Benz monomer starts at 194 °C and has a peak at 244 °C with a total 30 cal g−1 exotherm. Moreover, the DSC thermograms exhibit the ROP temperatures for TetradecaF-PolyBenzPre and OctadecaF-PolyBenzPre at 182 °C and 244 °C, respectively. The DSC thermogram also reveals another exothermic reaction at 297 °C for OctadecaF-PolyBenzPre, starting just after the maximum curing temperature at 244 °C, which can be attributed to the initiation of degradation. It is noteworthy that a similar exotherm is also detectable for the monomer TetradecaF-PolyBenzPre.
 | ||
| Fig. 3 DSC thermograms of (a) DecaF-Benz, (b) OctaF-PolyBenzPre and (c) TetradecaF-PolyBenzPre. | ||
The curing behaviours of the BisF-Benz monomer and its prepolymers are depicted in Fig. 4. The maximum curing temperatures for BisF-Benz, nonF-PolyBenzPre and HexaF-PolyBenzPre are 253 °C, 243 °C, and 227 °C, respectively. High ROP temperature for the BisF-Benz monomer can be attributed to the purity of the monomer compared to its prepolymers since it is known that pure benzoxazines have high ROP temperatures. During the synthesis of prepolymers some of the oxazine rings can be opened and the formed free phenols can catalyze the ring opening reaction leading to a decrease in ROP temperature. The ring opened structures in the main chains of nonF-PolyBenzPre and HexaF-PolyBenzPre could be detected in 1H NMR spectra in Fig. 2b and c. Besides, for all samples the disappearance of the exotherms was observed in the second run indicating the completion of the ring-opening process in the first run. Hence, all the precursors became insoluble after the first thermal treatment. For better clarity, a comparison of curing characteristics of monomers and their polymers is tabulated in Table 2.
 | ||
| Fig. 4 DSC thermograms of (a) BisF-Benz, (b) nonF-PolyBenzPre and (c) HexaF-PolyBenzPre. | ||
| Benzoxazine precursor | R | Onset curing temperature (°C) | Maximum curing temperature (°C) | Amount of exotherm (cal g−1) |
|---|---|---|---|---|

|
CF3 | 236 | 244 | 7 |
| CH3 | 160 | 182 | 6 | |
|
CF3 | 182 | 227 | 45 |
| CH3 | 223 | 243 | 19 |
Thermal stability of the polybenzoxazines was investigated by thermogravimetric analysis (TGA) under nitrogen atmosphere. The TGA profiles and derivatives of cured TetradecaF-PolyBenzPre, OctadecaF-PolyBenzPre, nonF-PolyBenzPre and HexaF-PolyBenzPre are shown in Fig. 5 and 6 and the results are summarized in Table 3.
 | ||
| Fig. 5 TGA thermograms of cured (at 250 °C for 0.5 h) prepolymers (a) OctaF-PolyBenzPre, (b) TetradecaF-PolyBenzPre, (c) nonF-PolyBenzPre and (d) HexaF-PolyBenzPre. | ||
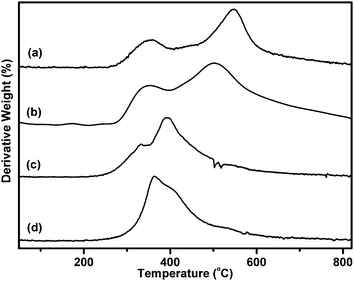 | ||
| Fig. 6 Rate of weight loss of cured (at 250 °C for 0.5 h) prepolymers (a) TetradecaF-PolyBenzPre, (b) OctaF-PolyBenzPre, (c) HexaF-PolyBenzPre and (d) nonF-PolyBenzPre. | ||
| Benzoxazine precursor | R | T 5% (°C) | T 10% (°C) | T max (°C) | Char yield (%) at 800 °C |
|---|---|---|---|---|---|
| a T 5%: temperature at which the weight loss is 5%; T10%: temperature at which the weight loss is 10%; char %: char yields at 800 °C under a nitrogen atmosphere; Tmax: temperature for maximum weight loss. | |||||

|
CF3 | 346 | 400 | 353, 545 | 57 |
| CH3 | 346 | 391 | 354, 505 | 54 | |

|
CF3 | 318 | 353 | 333, 392 | 57 |
| CH3 | 330 | 353 | 361 | 50 | |
The degradation temperatures T5%, T%10, Tmax of cured TetradecaF-PolyBenzPre and OctadecaF-PolyBenzPre are higher than prepolymers nonF-PolyBenzPre and HexaF-PolyBenzPre which can be possibly attributed to the delayed Mannich base cleavage and phenol degradation of the benzoxazine units due to the thermal stability of fluoro groups. Accordingly, the weight loss becomes more significant for the non-fluorous prepolymer nonF-PolyBenzPre at ∼800 °C.
The pronounced effect of the fluoro groups is detectable for both initial degradation temperatures and char yields for all polymers. Additionally, all the cured polymers have comparable char yields to classical polybenzoxazines derived from, for example, bisphenol-A and allyl amine, which have a char yield around 25% at 800 °C.
The low surface energy of the synthesized fluorine containing polymeric benzoxazine precursors allowed formation of stable ultrathin coatings (film thickness ∼20 nm) after curing on solid surfaces. Thin films of polymeric benzoxazine precursors having film thicknesses between 20 and 80 nm were prepared by spin coating on silicon wafers. Water contact angle (WCA), θ, values immediately after spin coating were measured to be between 85 and 100° as shown in Fig. 7. A slight increase in the number of F atoms in the precursor was observed for nonF-PolyBenzPre, OctaF-PolyBenzPre and TetradecaF-PolyBenzPre. The solid line is a linear fit to these three data points (squares). HexaF-PolyBenzPre deviated from this trend and showed a larger WCA (θ = 97°). The origin of this deviation will be discussed below.
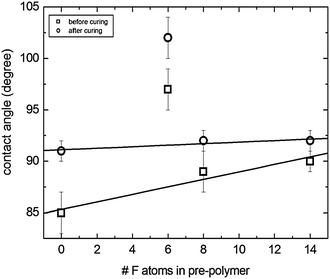 | ||
| Fig. 7 The change of water contact angles on thin films of polymeric benzoxazine precursors as a function of the number of F atoms in the precursor. Before curing (□), after curing (○), and error bars (I). | ||
The increase in the WCA from 85° to 90° before curing indicates the contribution of the F atoms in the precursors. The fraction of F atoms per precursor, f#F = #F/(#F + #H), goes from 0 to 0.33 with #F going from 0 to 14. These values are consistent with WCA values of previously reported partially fluorinated polymers. The WCA of poly(vinylidene fluoride) (f#F = 0.50) was reported to be 80°, of the ethylene-chlorotrifluoroethylene copolymer (f#F = 0.43) 99° and of the ethylene-tetrafluoroethylene copolymer (f#F = 0.50) 108°.57 WCA values are seen to depend not only on f#F, but also on the chemical structure of the polymers. In the case of poly(butyl methacrylate-co-perfluoroalkyl acrylate) copolymers WCA values varied between 85 and 115° depending on the length of the perfluoroalkyl acrylate block.58 Together with f#F, molecular architecture is also important. The WCA was 68° for fluorodecyl M2 (straight chain where 2 fluorodecyl groups (–(CH2)2–(CF2)7–CF3) were attached to two Si atoms, each bonded to a single oxygen atom), 116° for fluorodecyl Q4 (8 fluorodecyl groups attached to the ring with four Si atoms, each bonded to four oxygen atoms) and 122° for fluorodecyl T8 (8 fluorodecyl groups attached to the cage containing eight silicon atoms each bonded to three oxygen atoms).59
After curing the films at 250 °C for 30 min, the WCA increased ∼2 to 6° for all precursors. For nonF-PolyBenzPre, OctaF-PolyBenzPre and TetradecaF-PolyBenzPre, the WCA was nearly constant at ∼91 to 92° after curing. The solid line is a linear fit to these three data points (circles). The HexaF-PolyBenzPre WCA value was 102° which deviated from this trend similar to the WCA values before curing.
Fig. 8 shows the AFM height pictures of ∼20 nm thick precursor films before and after curing. nonF-PolyBenzPre (Fig. 8a), OctaF-PolyBenzPre (Fig. 8c) and TetradecaF-PolyBenzPre (Fig. 8d) films before curing all showed similar smooth morphologies with some holes on the top surface. These holes originate from evaporation of the solvent at the top surface during the spin coating process. For spin coating thin films, the precursor solutions were prepared in chloroform and such surface dips and holes are especially typical for chloroform due to its relatively low boiling temperature (∼61 °C).
 | ||
| Fig. 8 AFM height pictures of the precursor films before (a–d) and after curing (e–h). Before curing: (a) nonF-PolyBenzPre, (b) HexaF-PolyBenzPre, (c) OctaF-PolyBenzPre, (d) TetradecaF-PolyBenzPre. After curing: (e) nonF-PolyBenzPre, (f) HexaF-PolyBenzPre, (g) OctaF-PolyBenzPre, (h) TetradecaF-PolyBenzPre. | ||
The morphology of HexaF-PolyBenzPre right after spin coating (Fig. 8b) was significantly different than the other three precursors. It showed a ∼10 nm thick smooth layer in touch with the substrate and a ∼8 nm thick incomplete top layer. We attribute this morphology to the phase separation in the film between hydrogenated and fluorinated parts of the HexaF-PolyBenzPre precursor (see Fig. S8 in ESI for phase pictures†). HexaF-PolyBenzPre consists of two –CF3 groups located at one end of the monomer. In ultrathin films, the much longer hydrogenated part of the molecule prefers to be in touch with the underlying substrate while the lower surface energy fluorinated parts prefer to be at the top surface (film–air interface). The phase separation and the orientation of the hydrogenated and fluorinated parts of the molecules (hydrogenated at the substrate, fluorinated at the top surface) increase the density of –CF3 groups at the top surface. This, together with the resulting surface roughness, leads to a larger WCA compared to other three precursors (Fig. 7).
The surface morphology of all precursors after curing was much smoother compared to that before curing. The dips and holes smoothened out or became smaller after 30 min at 250 °C. The smoothening of the top surface was especially significant for HexaF-PolyBenzPre. The rough surface disappeared almost completely, probably as a result of spreading of the fluorinated parts of the molecules at the film–air interface. The WCA of HexaF-PolyBenzPre after curing (θ = 102°) was larger than those of the other three precursors (θ = 91–92°). This indicates that the orientation of the hydrogenated and fluorinated parts of the molecules after curing was still maintained such that the –CF3 groups were predominantly at the top surface.
Conclusions
In conclusion, we have demonstrated that aromatic ether containing benzoxazine moieties in the main chain can be prepared by Ulmann coupling of aryl halide functional benzoxazines in the presence of a nano-copperoxide catalyst. It is clear that the process is selective and it is possible to prepare polybenzoxazine precursors without affecting the benzoxazine moieties under polymerization conditions. The benzoxazine groups were shown to readily undergo thermally activated ring-opening reaction in the absence of an added catalyst and formed cross-linked networks. The polymers cured in this way exhibited comparable thermal stability to classical polybenzoxazines. Moreover, the fluoro content in the main chains led to higher WCAs for both cured and uncured polymers. The low surface energy of the fluorinated polymers allowed ultrathin films to be stable against dewetting at curing temperatures and resulted in thermally cured smooth coatings, even in the case of phase separation of hydrogenated and fluorinated parts of the molecules.Notes and references
- N. N. Ghosh, B. Kiskan and Y. Yagci, Prog. Polym. Sci., 2007, 32, 1344–1391 CrossRef CAS.
- X. Ning and H. Ishida, J. Polym. Sci., Part B: Polym. Phys., 1994, 32, 921–927 CrossRef CAS.
- B. Kiskan, N. N. Ghosh and Y. Yagci, Polym. Int., 2011, 60, 167–177 CrossRef CAS.
- C. Sawaryn, K. Landfester and A. Taden, Polymer, 2011, 52, 3277–3287 Search PubMed.
- C. Sawaryn, K. Landfester and A. Taden, Macromolecules, 2011, 44, 5650–5658 Search PubMed.
- S.-W. Kuo, Y.-C. Wu, C.-F. Wang and K.-U. Jeong, J. Phys. Chem. C, 2009, 113, 20666–20673 CrossRef CAS.
- K. D. Demir, M. A. Tasdelen, T. Uyar, A. W. Kawaguchi, A. Sudo, T. Endo and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4213–4220 Search PubMed.
- B. Kiskan, A. L. Demirel, O. Kamer and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6780–6788 CrossRef CAS.
- X. Li, Y. Xia, W. Xu, Q. Ran and Y. Gu, Polym. Chem., 2012, 3, 1629–1633 RSC.
- H. C. Chang, H. T. Lin and C. H. Lin, Polym. Chem., 2012, 3, 970–978 RSC.
- H. D. Kim and H. Ishida, Macromolecules, 2003, 36, 8320–8329 Search PubMed.
- H. Ishida and D. J. Allen, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 1019–1030 CrossRef CAS.
- V. M. Russell, J. L. Koenig, H. Y. Low and H. Ishida, J. Appl. Polym. Sci., 1998, 70, 1413–1425 Search PubMed.
- V. M. Russell, J. L. Koenig, H. Y. Low and H. Ishida, J. Appl. Polym. Sci., 1998, 70, 1401–1411 Search PubMed.
- F. W. Holly and A. C. Cope, J. Am. Chem. Soc., 1944, 66, 1875–1879 CrossRef CAS.
- H. Schreiber, Ger. Pat., 2 255 504, 1973.
- X. Ning and H. Ishida, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1121–1129 CrossRef CAS.
- B. Kiskan, B. Koz and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6955–6961 CrossRef CAS.
- B. Kiskan and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1670–1676 CrossRef CAS.
- H. Ishida and S. Ohba, Polymer, 2005, 46, 5588–5595 CrossRef CAS.
- T. Takeichi, K. Nakamura, T. Agag and H. Muto, Des. Monomers Polym., 2004, 7, 727–740 Search PubMed.
- M. A. Espinosa, M. Galia and V. Cadiz, Polymer, 2004, 45, 6103–6109 CrossRef CAS.
- T. Agag and T. Takeichi, Macromolecules, 2003, 36, 6010–6017 CrossRef CAS.
- J. P. Liu and H. Ishida, Polym. Polym. Compos., 2002, 10, 191–203 Search PubMed.
- T. Agag and T. Takeichi, Macromolecules, 2001, 34, 7257–7263 CrossRef CAS.
- B. Koz, B. Kiskan and Y. Yagci, Polym. Bull., 2011, 66, 165–174 Search PubMed.
- M. Sponton, D. Estenoz, G. Lligadas, J. C. Ronda, M. Galia and V. Cadiz, J. Appl. Polym. Sci., 2012, 126, 1369–1376 Search PubMed.
- K. S. S. Kumar, C. P. R. Nair, T. S. Radhakrishnan and K. N. Ninan, Eur. Polym. J., 2007, 43, 2504–2514 CrossRef.
- Y.-C. Yen, C.-C. Cheng, Y.-L. Chu and F.-C. Chang, Polym. Chem., 2011, 2, 1648–1653 RSC.
- Y. Yagci, B. Kiskan and N. N. Ghosh, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5565–5576 CrossRef CAS.
- K. D. Demir, B. Kiskan, B. Aydogan and Y. Yagci, React. Funct. Polym., DOI:10.1016/j.reactfunctpolym.2012.1004.1016.
- T. Takeichi, T. Kano and T. Agag, Polymer, 2005, 46, 12172–12180 CrossRef CAS.
- K. D. Demir, B. Kiskan and Y. Yagci, Macromolecules, 2011, 44, 1801–1807 CrossRef.
- S. Ates, C. Dizman, B. Aydogan, B. Kiskan, L. Torun and Y. Yagci, Polymer, 2011, 52, 1504–1509 Search PubMed.
- C. Altinkok, B. Kiskan and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2445–2450 CrossRef CAS.
- A. Tuzun, B. Kiskan, N. Alemdar, A. T. Erciyes and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4279–4284 CrossRef CAS.
- B. Aydogan, D. Sureka, B. Kiskan and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5156–5162 CrossRef CAS.
- B. Kiskan, B. Aydogan and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 804–811 CrossRef CAS.
- B. Kiskan, Y. Yagci and H. Ishida, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 414–420 CrossRef CAS.
- A. Chernykh, T. Agag and H. Ishida, Polymer, 2009, 50, 382–390 CrossRef CAS.
- A. Chernykh, J. P. Liu and H. Ishida, Polymer, 2006, 47, 7664–7669 CrossRef CAS.
- A. Nagai, Y. Kamei, X. S. Wang, M. Omura, A. Sudo, H. Nishida, E. Kawamoto and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2316–2325 CrossRef CAS.
- C. H. Lin, S. L. Chang, T. Y. Shen, Y. S. Shih, H. T. Lin and C. F. Wang, Polym. Chem., 2012, 3, 935–945 RSC.
- W.-H. Hu, K.-W. Huang and S.-W. Kuo, Polym. Chem., 2012, 3, 1546–1554 RSC.
- A. D. Baranek, L. L. Kendrick, J. Narayanan, G. E. Tyson, S. Wand and D. L. Patton, Polym. Chem., 2012, 3, 2892–2900 RSC.
- B. Kiskan and Y. Yagci, Polymer, 2008, 49, 2455–2460 CrossRef CAS.
- M. Kukut, B. Kiskan and Y. Yagci, Des. Monomers Polym., 2009, 12, 167–176 Search PubMed.
- B. Kiskan, G. Demiray and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3512–3518 CrossRef CAS.
- I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337–2364 CrossRef CAS.
- J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1470 CrossRef CAS.
- P. J. Fagan, E. Hauptman, R. Shapiro and A. Casalnuovo, J. Am. Chem. Soc., 2000, 122, 5043–5051 CrossRef CAS.
- J. Zhang, Z. Zhang, Y. Wang, X. Zheng and Z. Wang, Eur. J. Org. Chem., 2008, 5112–5116 CrossRef CAS.
- J. L. Yang and H. W. Gibson, Macromolecules, 1997, 30, 5629–5633 CrossRef CAS.
- J. I. Lee, L. Y. Kwon, J. H. Kim, K. Y. Choi and D. H. Suh, Angew. Makromol. Chem., 1998, 254, 27–32 Search PubMed.
- S. Masaki, N. Sato, A. Nishichi, S. Yamazaki and K. Kimura, J. Appl. Polym. Sci., 2008, 108, 498–503 Search PubMed.
- Y. Song, J. Wang, G. Li, Q. Sun, X. Jian, J. Teng and H. Zhang, Polymer, 2008, 49, 4995–5001 Search PubMed.
- S. Lee, J.-S. Park and T. R. Lee, Bull. Korean Chem. Soc., 2011, 32, 41–48 Search PubMed.
- K. Li, P. Wu and Z. Han, Polymer, 2002, 43, 4079–4086.
- S. S. Chhatre, J. O. Guardado, B. M. Moore, T. S. Haddad, J. M. Mabry, G. H. McKinley and R. E. Cohen, ACS Appl. Mater. Interfaces, 2010, 2, 3544–3554 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 13C NMR, FT-IR spectra of monomers and polymers, 1H NMR spectra of model reactions and AFM phase pictures. See DOI: 10.1039/c2py21029k |
| This journal is © The Royal Society of Chemistry 2013 |