 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Halogen- and hydrogen-bonding triazole-functionalised porphyrin-based receptors for anion recognition†
Lydia C. Gilday, Nicholas G. White and Paul D. Beer*
Department of Chemistry, Inorganic Chemistry Laboratory, University of Oxford, South Parks Road, Oxford OX1 3QR, UK. E-mail: paul.beer@chem.ox.ac.uk; Fax: +44 (0)1865 272690; Tel: +44 (0)1865 285142
First published on 10th September 2013
Abstract
Iodotriazole and triazole anion recognition groups have been integrated into a picket-fence zinc(II)-metalloporphyrin scaffold to produce receptors for anion recognition and sensing applications. 1H NMR and UV/visible spectroscopic investigations reveal both host systems exhibit strong anion binding affinities in a range of solvent media. Importantly, the halogen-bonding iodotriazole-containing porphyrin-based host displays halide binding affinities substantially larger than the protic-functionalised analogue concomitant with a reduced strength of oxoanion complexation.
Introduction
Anions are fundamentally important in many biological processes and medical diseases. Moreover, nitrates and phosphates are well-known for their detrimental impact in the aquatic environment. Consequently, the need to selectively complex and detect specific negatively charged species is acute and the field of anion supramolecular chemistry has expanded enormously in recent decades.1 Through the imaginative manipulation of a variety of complementary non-covalent interactions such as electrostatics, hydrogen bonding, Lewis acid–base and anion–π interactions, a plethora of efficient anion receptors have been developed. Constructing receptors which possess anion recognition strengths comparable to that of natural systems, however, is a goal yet to be attained. Halogen bonding (XB), the attractive and highly directional intermolecular interaction between electron-deficient halogen atoms and Lewis bases,2 is beginning to show real promise in solution phase applications such as catalysis, medicinal chemistry and molecular recognition processes.3 Given XB's complementary analogy to ubiquitous hydrogen bonding (HB), it is surprising that there are so few examples of efficient XB anion receptors reported to date.4,5The porphyrin macrocycle is endowed with inherent optical and redox properties that can be exploited for signalling anion recognition via a measurable physical response.6 Indeed, we7 and others8 have reported several porphyrin-based host systems with a variety of integrated hydrogen-bond-donating anion recognition groups, which are able to sense anions through spectroscopic and electrochemical means. To the best of our knowledge, however, porphyrin molecules with pendant XB-donor groups for anion recognition applications are unprecedented. Herein, we describe the synthesis of a XB iodotriazole picket-fence porphyrin receptor which is demonstrated to exhibit superior halide anion binding affinities compared with a protic triazole-functionalised analogue.
Results and discussion
Syntheses
The target iodotriazole- and triazole-containing zinc(II)-metalloporphyrin receptors 5 and 6 were prepared using a copper(I)-catalysed azide–alkyne cycloaddition (CuAAC) reaction9 between a tetra-meso-substituted azide-functionalised zinc(II)-metalloporphyrin and a suitable alkyne species. The required tetra-azide zinc(II)-metalloporphyrin species 3 was prepared as outlined in Scheme 1.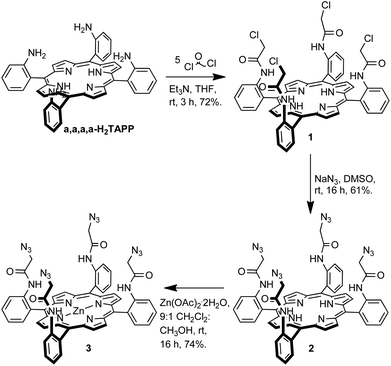 | ||
| Scheme 1 Synthesis of tetra-azide-functionalised porphyrin 3. | ||
H2TAPP was prepared by known literature procedures10 and the proportion of the desired α,α,α,α-isomer was increased using the Lindsey method11 to give α,α,α,α-H2TAPP which was used immediately in the next step. α,α,α,α-Tetrakis(2-(chloroacetamidophenyl)porphyrin was synthesised according to the procedure reported by Collman et al.12 The condensation of α,α,α,α-H2TAPP with five equivalents of chloroacetyl chloride in the presence of base gave porphyrin 1 in 72% isolated yield. A nucleophilic substitution reaction between compound 1 and sodium azide in dimethyl sulfoxide afforded the tetra-azide porphyrin 2 in 61% yield. Metallation with zinc, to give 3 was achieved by stirring porphyrin 2 with a ten-fold excess of zinc(II) acetate dihydrate in dichloromethane–methanol (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v).
:1, v/v).
The target iodotriazole-containing zinc(II)-metalloporphyrin 5 was prepared in 46% yield by a copper(I)-catalysed cycloaddition reaction between porphyrin 3 and 4.5 eq. of iodoalkyne 4 (prepared from 4-tert-butylphenylacetylene and N-iodosuccinimide)13 in the presence of copper(I) iodide and triethylamine in anhydrous THF (Scheme 2).
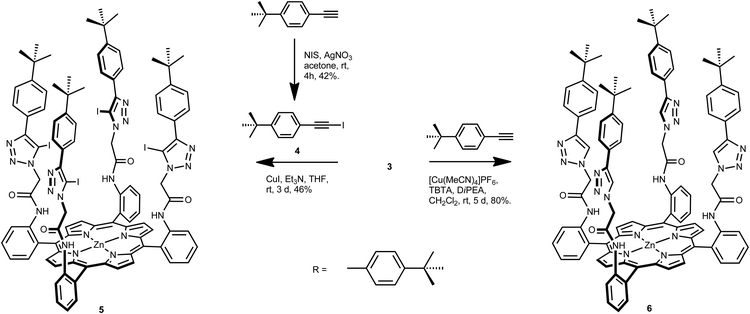 | ||
| Scheme 2 Synthesis of iodotriazole- and triazole-functionalised porphyrins 5 and 6. | ||
An analogous reaction between tetra-azide porphyrin 3 and 4.5 eq. of 4-tert-butylphenylacetylene, tetrakis(acetonitrile)copper(I) hexafluorophosphate as the copper(I) catalyst, TBTA and N,N′-diisopropylethylamine in anhydrous dichloromethane afforded tetrakis-triazole zinc(II)-porphyrin anion receptor 6 in 80% isolated yield (Scheme 2).
Both receptors were characterised by 1H NMR spectroscopy, high-resolution electrospray mass spectrometry, and 13C NMR spectroscopy and UV/visible spectroscopy (see Experimental section and ESI†).
X-ray crystallography
Crystals of iodotriazole-porphyrin 5 of suitable quality for single crystal X-ray structural determination were grown by slow diffusion of diethyl ether into a tetrahydrofuran solution. The structure (Fig. 1, top) reveals a picket-fence porphyrin molecule with all amide and iodotriazole anion recognition motifs on the same side with respect to the porphyrin motif. Intermolecular XB interactions are present between an iodotriazole iodine atom of the receptor and a symmetry-related amide carbonyl group of the adjacent molecule (Fig. 1, bottom). Another iodine atom forms a halogen bond to a THF solvate. In both cases the interactions are close to linear, and the iodine⋯oxygen distance is significantly shorter than the sum of the van der Waals radii [C–I⋯Oamide = 2.920(5) Å, 83% of sum of vdW radii, ∠C–I⋯Oamide = 171.2(2)°; C–I⋯OTHF = 2.961(10) Å, 85% of sum of vdW radii, ∠C–I⋯Oamide = 167.7(3)°].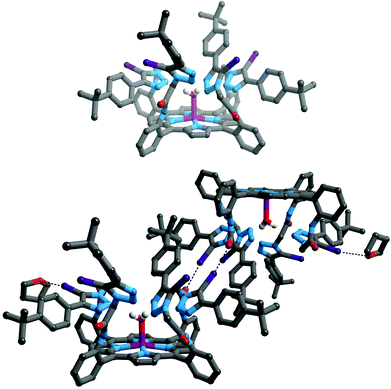 | ||
| Fig. 1 Structure of compound 5 (top); and diagram showing formation of dimers in the solid state. Halogen bonding interactions are shown as black dotted lines. Hydrogen atoms, except those on coordinated water and amide moieties are omitted for clarity, as are both (top) or one (bottom) of the crystallographically independent THF solvates. Key: grey = C, white = H, blue = N, red = O, violet = Zn, purple = I. | ||
Anion binding studies
A preliminary 1H NMR anion binding investigation of 5 and TBA·Cl was undertaken initially in CDCl3 (Fig. 2). Upon addition of one equivalent of chloride, downfield shifts of meso-phenyl proton e, methylene proton g, and phenyl proton h, and relatively smaller perturbation of the amide proton f were observed, which indicate the chloride anion is binding within the pocket formed by the four arms of the picket-fence architecture in the vicinity of the iodotriazole groups by four C–I⋯chloride halogen bonds (Fig. 2).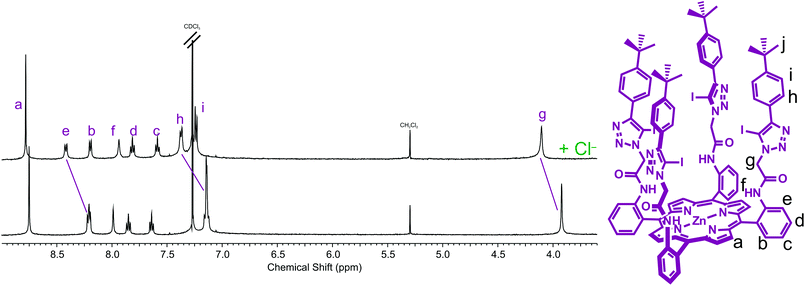 | ||
| Fig. 2 Partial 1H NMR spectra of (a) receptor 5 and (b) receptor 5 and one equivalent of TBA·Cl (500 MHz, CDCl3, 293 K). | ||
An analogous 1H NMR spectroscopic titration experiment with HB triazole receptor 6 revealed the addition of chloride induced downfield shifts in the receptor's triazole h, methylene g and phenyl i protons together with relatively smaller perturbations of the amide proton signal f, which suggests the halide anion is being bound in the vicinity of the triazole groups through C–H⋯anion hydrogen bonds.
The absence of a triazole proton in iodotriazole zinc(II)-metalloporphyrin 5, and relatively modest anion-binding-induced shifts of other protons prevented quantitative assessment of the anion binding properties of XB receptor 5 from being undertaken using 1H NMR spectroscopy. Anion association constant data was determined, however, with HB receptor 6 by monitoring the chemical shift of triazole proton h with increasing concentration of anion (Fig. 3).
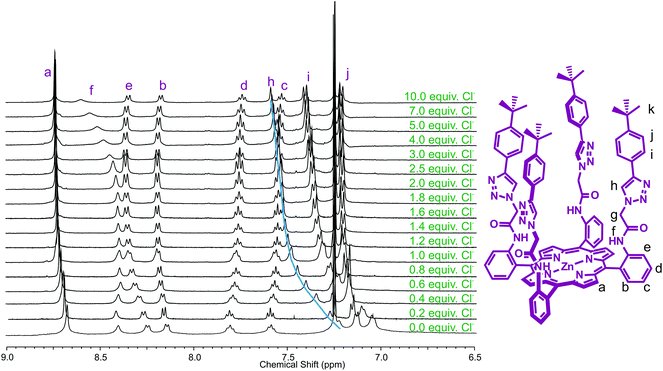 | ||
| Fig. 3 Partial 1H NMR spectra of receptor 6 upon addition of equivalents of TBA·Cl (500 MHz, 293 K, CDCl3). | ||
The observed chemical shift perturbations of the triazole proton h with ten equivalents of anion (Table 1) reveal dihydrogen phosphate causes the largest magnitude of perturbation and the shifts are in general larger for oxoanions than less basic halides. Moreover, no evidence of complex formation was observed with hexafluorophosphate, an anion which typically associates weakly with traditional anion receptors and which is often used as a non-competitive counteranion with positively charged receptors. Monitoring the shift of the porphyrin's triazole proton h as a function of the concentration of anion gave the titration curves reported in Fig. 4. WinEQNMR14 analysis of this titration data gave the 1:1 stoichiometric association constants for anion complexation shown in Table 1.
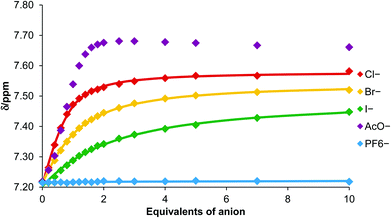 | ||
| Fig. 4 Titration curves for complex formation between porphyrin 6 and various anions obtained by monitoring triazole proton h (CDCl3, 293 K). | ||
:1 complexes of porphyrin host 6 with various anions
| Anion | Δδ (ppm) | Kaa (M−1) |
|---|---|---|
| a All anions added as their TBA salt. Association constants were determined by monitoring the downfield shift of the triazole proton h. Estimated standard errors are given in parentheses. CDCl3, 293 K.b Overlapping peaks in the NMR spectra prevented quantitative binding data to be obtained.c Chemical shift changes too small to allow an accurate association constant to be determined. | ||
| Cl− | 0.37 | 2226 (183) |
| Br− | 0.30 | 662 (23) |
| I− | 0.23 | 224 (23) |
| AcO− | 0.45 | >104 |
| H2PO4− | 0.65 | b |
| PF6− | c | c |
Triazole-zinc(II)-metalloporphyrin host 6 forms the most stable complex with acetate, followed by chloride > bromide > iodide. This anion selectivity trend in general correlates with anion basicity. Hexafluorophosphate binding was extremely weak as anticipated. Addition of dihydrogen phosphate caused changes in the chemical shifts which could not be accurately monitored on account of overlapping signals, so quantitative data for this anion could not be obtained.
UV/visible spectroscopic titration experiments were undertaken to investigate the potential optical sensing capabilities of both receptors and to allow quantitative anion binding data for the iodotriazole-functionalised receptor 5 to be determined.
Anion titration experiments induced significant perturbations of the receptors’ absorbance spectra. In a typical titration experiment (Fig. 5), bathochromic shifts in the Soret band are observed upon addition of anions, which arises from axial ligation to the Lewis acidic zinc metal centre.6 An isosbestic point is observed with increasing anion concentration which corresponds to a 1:1 receptor:anion binding stoichiometry. Association constants for 1:1 stoichiometric complexes were determined using the Specfit® computer program15 to analyse the titration data, and are reported in Table 2.
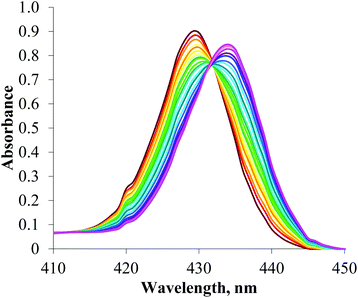 | ||
| Fig. 5 Changes in the Soret band of the UV/visible spectrum of a 2 μm solution of porphyrin 5 upon addition of TBA·Cl (CHCl3, 293 K). | ||
:1 complexes of porphyrin hosts 5 and 6 with various anions
| Anion | Iodotriazole 5Kaa (M−1) | Triazole 6Kaa (M−1) |
|---|---|---|
| a All anions added as their TBA salt. Association constants were determined by monitoring the perturbations of the Soret band. Estimated standard errors are given in parentheses. CHCl3, 293 K.b No evidence of binding. | ||
| Cl− | 3592 (31) | 2587 (128) |
| Br− | 1132 (11) | 685 (16) |
| I− | 305 (23) | 150 (5) |
| AcO− | 258642 (7578) | 335660 (7383) |
| H2PO4− | >106 | >106 |
| SO42− | >106 | >106 |
| PF6− | b | b |
Sulfate is bound strongly by both XB and HB porphyrin receptors, no doubt as a consequence of its higher charge. Of the singly charged anions, a general preference for oxoanions over halides is observed, with both receptors displaying relatively low binding affinities for larger, more diffuse bromide and iodide. It is interesting to note that there is almost an order of magnitude preference for dihydrogen phosphate over acetate, despite acetate being the more basic anion, which suggests the receptors prefer to bind anions of tetrahedral rather than trigonal geometry.
There are some significant differences between the anion binding affinities of the XB and HB receptors. It is noteworthy that the halogen-bonding iodotriazole host 5 has a greater affinity for chloride and bromide—with the strength of bromide association almost double in magnitude that of the protic species (Ka = 1132 M−1versus 685 M−1). There is also a reduction in the acetate binding strength with the XB receptor. Enhanced halide anion recognition together with reduced oxoanion binding affinity is consistent with anion binding trends noted with other XB anion receptors.5
Effect of solvent on anion recognition
The anion binding behaviour of XB receptors in different solvents has not as yet been widely studied. The anion binding properties of XB and HB porphyrin-based receptors 5 and 6, in addition to chloroform, were determined in acetone and acetonitrile to give an insight into solvent effects on anion binding in general, and halogen versus hydrogen bonding in particular. UV/visible spectroscopic titrations were undertaken monitoring the Soret band with increasing anion concentration, producing titration curves (see ESI†) from which Specfit® analysis determined quantitative data shown in Table 3.:1 complexes of porphyrin host 5 and 6 with anions in various solvents
| Kaa (M−1) | Cl− | Br− | I− | AcO− | H2PO4− | SO42− | |
|---|---|---|---|---|---|---|---|
| a All anions added as their TBA salt. Association constants were determined by monitoring the perturbations of the Soret band. Estimated standard errors are given in parentheses. 293 K.b Fitted better to a 1:2 host:guest model. ε = dielectric constants, μ = relative permittivity, AN = Gutmann's acceptor number. | |||||||
| CHCl3 | 5 | 3592 (31) | 1132 (11) | 305 (23) | 258642 (7578) | >106 | >106 |
| ε = 4.81 | |||||||
| μ = 1.15 | 6 | 2587 (128) | 685 (16) | 150 (5) | 335660 (7383) | >106 | >106 |
| AN = 23.1 | |||||||
| MeCN | 5 | 272144 (2613) | 13589 (91) | 656 (11) | 805378 (19409) | >106b | >106 |
| ε = 37.5 | |||||||
| μ = 3.45 | 6 | 34626 (184) | 4494 (369) | 396 (8) | >106 | >106b | >106 |
| AN = 18.9 | |||||||
| Acetone | 5 | >106 | >106 | 60339 (2009) | >106 | >106 | >106 |
| ε = 20.7 | |||||||
| μ = 2.85 | 6 | >106 | 721938 (9818) | 14659 (846) | >106 | >106 | >106 |
| AN = 12.5 | |||||||
As expected there are significant differences in the magnitude of the association constant values obtained in the three solvents. It is important to note, however, that the selectivity trends in anion binding strength remain the same in all cases, where in general, oxoanions are bound more strongly than halides, with sulfate and dihydrogen phosphate displaying the largest association constant values, presumably on account of their tetrahedral shape.
Anion binding strength is maximised by receptors 5 and 6 in acetone, followed by acetonitrile and chloroform. This anion binding behaviour does not correlate with bulk solvent properties such as relative permittivity (ε) or dipole moment (μ). There is however, a correlation between Gutmann's acceptor number (AN) of the solvent,16 which gives a measure of the hydrogen-bond-donor ability of a solvent.
As the hydrogen-bond-donor strength of the solvent increases (larger AN), anion binding affinity of the respective receptor decreases: competing anion–solvent interactions are strongest in chloroform, and weakest in acetone.17
One final observation to note is the magnitude of enhancement of chloride and bromide halide anion recognition that occurs upon changing the solvent from chloroform to acetonitrile is more pronounced with the halogen-bonding receptor 5 than the hydrogen-bonding receptor 6. Indeed chloride anion binding affinity increases 75-fold with XB porphyrin 5 but only 13-fold for HB receptor 6. Future, in-depth thermodynamic investigations will be required to help rationalise these observations, through the determination of entropic and enthalpic contributions of XB anion recognition in a range of solvent media.
Conclusions
The anion binding properties of a XB iodotriazole- and HB triazole-functionalised zinc(II)-metalloporphyrin receptors have been investigated using 1H NMR and UV/visible techniques. Both receptors exhibit strong anion binding affinities forming 1:1 stoichiometric complexes with a range of halide and oxoanions in chloroform, acetone and acetonitrile solution. It is noteworthy that the XB porphyrin-based anion receptor 5 displays enhanced halide anion recognition and reduced acetate oxoanion binding affinities in comparison with the protic-triazole-functionalised analogue 6. The effect of solvent variation on anion binding efficacy reveals the strength of anion recognition correlates with the hydrogen-bond-donor ability of the solvent as measured by the Gutmann acceptor number: the superior halide anion binding affinities of the XB receptor is more pronounced in acetonitrile (AN = 18.9) than chloroform (AN = 23.1).Experimental
General remarks
Unless otherwise stated, commercially available solvents (HPLC grade) and reagents were used without further purification. Triethylamine was distilled from KOH and stored over 3 Å molecular sieves. Pyrrole was distilled over CaH2, under reduced pressure and stored at −25 °C under N2. TBA2·SO4 was azeotroped with toluene and stored in a dessicator containing P2O5. TBA salts of Cl−, Br−, I−, AcO−, H2PO4− were stored in a dessicator. Water was deionised and micro-filtered using a Milli-Q® Millipore machine. Where anhydrous solvents were used, they were degassed with N2, and dried by passing through an MBraun MSPS-800 column.1H, 13C, 19F, 31P NMR spectra were recorded on a Varian Mercury-VX 300, a Varian Unity Plus 500 or a Bruker AVII500 with cryoprobe at 293 K. Mass spectra were obtained using a micromass LCT (ESMS) instrument. Electronic absorption spectra were recorded on a PG instruments T60U spectrometer. Column chromatography was performed on silica gel (particle size: 40–63 μm), preparative TLC was performed on 20 × 20 cm plates, with a silica layer of thickness 1 mm.
Single crystal X-ray diffraction data were collected at 150(2) K using graphite monochromated Cu Kα radiation (λ = 1.54184 Å). Cell parameters and intensity data (including inter-frame scaling) were processed using CrysAlis Pro.18 The structures were solved by charge-flipping methods using SUPERFLIP19 and refined using full-matrix least-squares on F2 within the CRYSTALS suite.20 All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were generally visible in the difference matter, and their positions and displacement parameters were refined using restraints prior to their inclusion in the model using riding constraints.21
:5 CH2Cl2–MeOH) to give compound 1 as purple powder (0.63 g, 0.65 mmol, 72%). λmax(CHCl3)/nm: 418 (ε/dm3 mol−1 cm−1 491 860), 514 (24 580), 546 (5220), 588 (7664), 643 (1634); δH (500 MHz; CDCl3) 8.80 (8 H, s, β-pyrrole-H), 8.73 (4 H, d, 3J = 8.0 Hz, meso-phenyl-H), 8.08 (4 H, d, 3J = 8.0 Hz, meso-phenyl-H), 7.99 (4 H, s, amide-NH), 7.89 (4 H, t, 3J = 8.0 Hz, meso-phenyl-H), 7.61 (4 H, t, 3J = 8.0 Hz, meso-phenyl-H), 3.39 (8 H, s, CH2), −2.66 (2 H, br s, pyrrole-NH); m/z (ES): 1003.2 ([M + Na]+, C52H38Cl4N8NaO4 requires 1003.2).:1 CH2Cl2–MeOH (100 mL), Zn(OAc)2·2H2O (1.10 g, 5.50 mmol) was added and the reaction was stirred for 16 h. The solvents were removed in vacuo and the resulting dark pink residue redissolved in CH2Cl2 (100 mL), washed with H2O (5 × 100 mL) and sat. NaCl(aq) (1 × 100 mL), dried over anhydrous MgSO4, filtered, and the solvent removed in vacuo giving compound 3 as a bright purple solid in (397 mg, 0.37 mmol, 74%). λmax(CHCl3)/nm: 425 (ε/dm3 mol−1 cm−1 726 040), 555 (28 440), 592 (3466); δH (300 Hz; CDCl3) 8.83 (8 H, s, β-pyrrole-H), 8.65 (4 H, d, 3J = 7.7 Hz, meso-phenyl-H), 7.99 (4 H, d, 3J = 7.7 Hz, meso-phenyl-H), 7.84 (4 H, t, 3J = 7.7 Hz, meso-phenyl-H), 7.74 (4 H, s, amide-NH), 7.55 (4 H, t, 3J = 7.7 Hz, meso-phenyl-H), 3.16 (8H, s, CH2); δC (75.5 MHz; CDCl3) 164.1, 150.6, 137.5, 134.5, 132.4, 132.4, 132.2, 129.7, 123.6, 120.5, 115.1, 52.6; m/z (ES): 1091.2408 ([M + Na]+, C52H36N20NaO4Zn requires 1091.2412).:5 CH2Cl2–acetone) to give compound 5 as a purple solid (228 mg, 0.103 mmol, 46%). λmax(CHCl3)/nm: 429 (ε/dm3 mol−1 cm−1 449 000), 559 (13 500), 595 (3530); δH (300 MHz; CDCl3) 8.75 (8 H, s, β-pyrrole-H), 8.21 (4 H, d, 3J = 8.5 Hz, meso-phenyl-H), 8.18 (4 H, d, 3J = 8.5 Hz, meso-phenyl-H), 8.10 (4 H, s, amide-NH), 7.85 (4 H, t, 3J = 7.5 Hz, meso-phenyl-H), 7.63 (4 H, t, 3J = 7.5 Hz, meso-phenyl-H), 7.16 (16 H, s, phenyl-H), 3.99 (8 H, s, CH2), 1.25 (16 H, s, CH3); δC (125.5 MHz; CDCl3) 162.41, 151.89, 150.37, 149.29, 137.65, 135.59, 134.40, 131.78, 128.93, 126.70, 125.74, 125.27, 124.40, 123.51, 114.89, 78.16, 53.75, 53.11, 34.61, 31.27, 31.19, 29.24, 27.77, m/z (ES): 2227.2660 ([M + Na]+, C100H88I4N20NaO4Zn requires 2227.2661).:3 CH2Cl2–MeOH) to give compound 6 as a purple crystalline solid (0.13 g, 72.0 mmol, 80%). λmax(CHCl3)/nm: 427 (ε/dm3 mol−1 cm−1 511000), 557 (22500) and 597 (4500); δH (500 MHz; CDCl3) 8.70 (8 H, s, β-pyrrole-H), 8.42 (4 H, s, amide-NH), 8.27 (4 H, d, 3J = 7.8 Hz, meso-phenyl-H), 8.17 (4 H, d, 3J = 8.2 Hz, meso-phenyl-H), 7.83 (4 H, t, 3J = 7.8 Hz, meso-phenyl-H), 7.61 (4 H, t, 3J = 7.8 Hz, meso-phenyl-H), 7.25 (4 H, s, triazole-H), 7.17 (8 H, d, 3J = 8.6 Hz, phenyl-H), 7.07 (8 H, d, 3J = 8.6 Hz, phenyl-H), 3.93 (8 H, s, CH2), 1.21 (36 H, s, CH3); δC (125.5 MHz; CDCl3) 163.1, 151.6, 150.4, 147.4, 137.8, 134.4, 131.7, 128.9, 128.7, 126.3, 125.5, 125.4, 124.3, 121.1, 114.8, 52.7, 34.6, 31.2; m/z (ES): 1725.6824 ([M + Na]+, C100H92N20NaO4Zn requires 1725.6853).Acknowledgements
We thank the EPSRC (L.C.G.), Trinity College and the Clarendon Fund (N.G.W.) for funding, and Prof. S. Faulkner for use of UV/visible equipment.Notes and references
- Anion Receptor Chemistry, ed. J. L. Sessler, P. A. Gale and W.-S. Cho, RSC, Cambridge, 2006 Search PubMed; Supramolecular Chemistry of Anions, ed. A. Bianchi, K. Bowman-Jones and E. Garcia-Espana, Wiley-VCH, New York, 1997 Search PubMed.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed.
- S. Derossi, L. Brammer, C. A. Hunter and M. D. Ward, Inorg. Chem., 2009, 48, 1666–1677 CrossRef CAS PubMed; T. Beweries, L. Brammer, N. A. Jasim, J. E. McGrady, R. N. Perutz and A. C. Whitwood, J. Am. Chem. Soc., 2011, 133, 14338–14348 CrossRef PubMed; M. Erdelyi, Chem. Soc. Rev., 2012, 41, 3547–3557 RSC; T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC.
- A. Caballero, F. Zapata, N. G. White, P. J. Costa, V. Felix and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 1876–1880 CrossRef CAS PubMed; M. G. Sarwar, B. Dragisic, L. J. Salsberg, C. Gouliaras and M. S. Taylor, J. Am. Chem. Soc., 2010, 132, 1646–1653 CrossRef PubMed; M. G. Sarwar, B. Dragisic, S. Sagoo and M. S. Taylor, Angew. Chem., Int. Ed., 2010, 49, 1674–1677 CrossRef PubMed; A. Mele, P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, J. Am. Chem. Soc., 2005, 127, 14972–14973 CrossRef PubMed; M. Cametti, K. Raatikainen, P. Metrangolo, T. Pilati, G. Terraneo and G. Resnati, Org. Biomol. Chem., 2012, 10, 1329–1333 Search PubMed; A. Caballero, N. G. White and P. D. Beer, Angew. Chem., Int. Ed., 2011, 50, 1845–1848 CrossRef PubMed; F. Zapata, A. Caballero, N. G. White, T. D. W. Claridge, P. J. Costa, V. Felix and P. D. Beer, J. Am. Chem. Soc., 2012, 134, 11533–11541 CrossRef PubMed.
- M. G. Chudzinski, C. A. McClary and M. S. Taylor, J. Am. Chem. Soc., 2011, 133, 10559–10567 CrossRef CAS PubMed.
- M. Nappa and J. S. Valentine, J. Am. Chem. Soc., 1978, 100, 5075–5080 CrossRef CAS.
- P. D. Beer, M. G. B. Drew and R. Jagessar, J. Chem. Soc., Dalton Trans., 1997, 881–886 RSC; P. D. Beer, D. P. Cormode and J. J. Davis, Chem. Commun., 2004, 414–415 RSC; D. P. Cormode, M. G. B. Drew, R. Jagessar and P. D. Beer, Dalton Trans., 2008, 6732–6741 RSC; D. P. Cormode, J. J. Davis and P. D. Beer, J. Inorg. Organomet. Polym. Mater., 2008, 18, 32–40 CrossRef CAS; A. Brown and P. D. Beer, Dalton Trans., 2012, 41, 118–129 RSC.
- R. C. Jagessar, M. Y. Shang, W. R. Scheidt and D. H. Burns, J. Am. Chem. Soc., 1998, 120, 11684–11692 CrossRef CAS; M. J. Gunter, S. M. Farquhar and K. M. Mullen, New J. Chem., 2004, 28, 1443–1449 Search PubMed; C. Lee, D. H. Lee and J. I. Hong, Tetrahedron Lett., 2001, 42, 8665–8668 CrossRef; M. Takeuchi, T. Shioya and T. M. Swager, Angew. Chem., Int. Ed., 2001, 40, 3372–3376 CrossRef; M. Dudic, P. Lhotak, I. Stibor, K. Lang and P. Proskova, Org. Lett., 2003, 5, 149–152 CrossRef PubMed; P. K. Panda and C. H. Lee, J. Org. Chem., 2005, 70, 3148–3156 CrossRef PubMed; C. Bucher, C. H. Devillers, J. C. Moutet, G. Royal and E. Saint-Aman, New J. Chem., 2004, 28, 1584–1589 RSC; S. D. Starnes, S. Arungundram and C. H. Saunders, Tetrahedron Lett., 2002, 43, 7785–7788 CrossRef; C.-H. Lee, S. Lee, H. Yoon and W.-D. Jang, Chem.–Eur. J., 2011, 17, 13898–13903 CrossRef PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; V. D. Bock, H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2006, 51–68 CrossRef.
- T. N. Sorrell, Inorg. Synth., 1980, 20, 161 CrossRef CAS; J. P. Collman, R. R. Gagne, C. Reed, T. R. Halbert, G. Lang and W. T. Robinson, J. Am. Chem. Soc., 1975, 97, 1427–1439 CrossRef.
- J. Lindsey, J. Org. Chem., 1980, 45, 5215–5215 CrossRef CAS.
- J. P. Collman, B. Boitrel, L. Fu, J. Galanter, A. Straumanis and M. Rapta, J. Org. Chem., 1997, 62, 2308–2309 CrossRef CAS.
- J. E. Hein, J. C. Tripp, L. B. Krasnova, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2009, 48, 8018–8021 CrossRef CAS PubMed.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
- Specfit v. 2.02 ed, Spectrum Software Associates, Chapel Hill, NC, USA Search PubMed.
- U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem. Chem. Mon., 1975, 106, 1235–1257 CrossRef CAS.
- P. D. Beer and M. Shade, Chem. Commun., 1997, 2377–2378 RSC.
- CrysAlisPro, Agilent Technologies, 2011 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487–1487 CrossRef CAS.
- R. I. Cooper, A. L. Thompson and D. J. Watkin, J. Appl. Crystallogr., 2010, 43, 1100–1107 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra and ESI mass spectra or porphyrins 5 and 6; titration protocols and UV/visible titration curves. CCDC 953393. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt52093e |
| This journal is © The Royal Society of Chemistry 2013 |