
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Achieving high-performance OER catalysis with dual-site modulated Fe-based perovskites†
Yixin
Bi‡
a,
Yuhao
Wang‡
a,
Yufei
Song
d,
Qing
Chen
 *a and
Francesco
Ciucci
*abc
*a and
Francesco
Ciucci
*abc
aDepartment of Mechanical and Aerospace Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong S.A.R., China. E-mail: chenqing@ust.hk
bUniversity of Bayreuth, Chair of Electrode Design for Electrochemical Storage, Weiherstraße 26, 95448 Bayreuth, Germany. E-mail: francesco.ciucci@uni-bayreuth.de
cUniversity of Bayreuth, Bavarian Center for Battery Technology (BayBatt), Universitätsstraße 30, 95447 Bayreuth, Germany
dState Key Laboratory of Materials-Oriented Chemical Engineering, College of Chemical Engineering, Nanjing Tech University, Nanjing, 210009, China
First published on 4th June 2025
Abstract
Developing cost-effective, efficient oxygen evolution reaction (OER) catalysts is critical for sustainable hydrogen production through water electrolysis. While noble metal-based catalysts like RuO2 and IrO2 show high activity, their widespread adoption is limited by cost. Fe-based perovskite oxides present a more abundant alternative but typically exhibit inferior OER activities. In this study, we achieved systematic dual-site modulation by incorporating Ba at the A-site and Ni at the B-site of NdFeO3−δ, transforming it into a double perovskite structure. The resulting Nd0.8Ba1.2Fe1.6Ni0.4O6−δ catalyst achieved an overpotential of 320 mV at 10 mA cm−2 in 0.1 M KOH, significantly lower than typical Fe-based perovskites and noble metals. Ab initio simulations revealed that A-site modulation reduces the band gap, which enhances electronic conductivity. Meanwhile, B-site Ni incorporation strengthens metal–oxygen covalency and decreases charge-transfer energy. The synergistic effects between enhanced electronic conductivity and metal–oxygen covalency led to a significantly reduced Tafel slope of 63.23 mV dec−1, compared to 114.85 mV dec−1 for single-site modified Nd0.8Ba1.2Fe2O6−δ and 154.34 mV dec−1 for unmodified NdFeO3−δ. This work provides a framework for understanding and improving performance in Fe-based perovskite OER catalysts through dual-site modulation, paving the way for more cost-effective and sustainable water electrolysis technologies.
1. Introduction
The global transition to renewable energy necessitates efficient energy conversion and storage methods. Water electrolysis is a promising approach for converting renewable energy from solar and wind sources into storable hydrogen fuel.1 However, the oxygen evolution reaction (OER) is still a significant bottleneck for this technology. This four-electron transfer process is inherently sluggish, requiring high activation potentials that limit overall system efficiency.2 While noble metal-based catalysts like RuO2 and IrO2 currently lead the field in OER performance, their widespread adoption is constrained by prohibitive costs.3Transition-metal (TM)-containing perovskite oxides are an alternative to noble metal-based catalysts as they enable strong electrocatalytic activity through flexible compositional adjustments.4,5 Co-based perovskites, particularly Ba0.5Sr0.5Co0.8Fe0.2O3−δ, have demonstrated exceptional catalytic performance.4,6,7 However, growing concerns about Co's toxicity and increasing demand in battery applications have intensified the search for safer, more abundant alternatives. Fe-based perovskites present a promising direction, given Fe's natural abundance, cost-effectiveness, and reduced environmental impact. Despite these advantages, their development as primary catalysts has been hampered by relatively low activity compared to their Co and Ni-based counterparts.8,9 Besides, Fe-based oxides are thermodynamically unstable in alkaline solutions under anodic potentials, resulting in poor OER stability.8 Therefore, Fe has primarily been limited to dopant roles in Co or Ni-based perovskites.
Researchers have pursued various optimization strategies to enhance the OER performance of Fe-based perovskites. The base compound NdFeO3−δ (NF) exhibits relatively weak catalytic activity, requiring an overpotential of 420 mV to achieve a current density of 10 mA cm−2 in 1 M KOH.10 Omari et al.10 demonstrated that introducing A-site deficiency in NF creates oxygen vacancies, reducing the required overpotential to 370 mV for Nd0.9FeO3−δ. Their findings highlight NF's potential for further optimization as an electrochemically active catalyst.
LaFeO3−δ, a material widely investigated for applications in the fields of sensors, photocatalysis, and solid oxide fuel cells,11 showed limited OER activity with a 510 mV overpotential at 10 mA cm−2 in 0.1 M KOH. She et al.11 enhanced its catalytic performance through A-site Sr substitution to make La1−xSrxFeO3−δ (x = 0, 0.2, 0.5, 0.8 and 1). This series of materials underwent a structural transformation from orthorhombic to cubic at x = 0.5, and this transformation was accompanied by a change in surface oxygen content. La0.2Sr0.8FeO3−δ had an optimal performance at a maximum surface O22−/O− content of 31.3%, achieving an overpotential of 370 mV at a current density of 10 mA cm−2.
BaFeO3−δ (BF) is not stable at room temperature, leading to the collapse of the cubic structure.12 Zhu et al.13 demonstrated that B-site Zr doping stabilized a cubic phase that promoted OER activity, achieving a 412 mV overpotential at 10 mA cm−2 in 0.1 M KOH. Zhao et al.14 leveraged the synergy between the perovskite and graphene support to make graphene-supported Ba0.95La0.05FeO3−δ, which achieved 23 mA cm−2 at 0.6 V versus mercury/mercury oxide electrode.
Furthermore, extensive research focused on Fe-based perovskites. Shang et al.15 utilized La0.5Sr0.5FeO3−δ as a foundational material, applying multi-element doping at the A-site to produce high-entropy Ba0.2Sr0.2La0.2Pr0.2Sm0.2FeO3−δ and medium-entropy Ba0.35Sr0.35La0.1Pr0.1Sm0.1FeO3−δ for use as cathode materials in SOFCs. Chen et al.16 doped Pr into the A-site of La0.5Sr0.5FeO3−δ, aiming to improve activity and tolerance to CO2 in SOFC cathodes. Inoue et al.17 incorporated Co as a B-site dopant in both Ba0.5Sr0.5FeO3−δ and LaFeO3 to exploit the synergistic effects of Co and Fe in catalyzing the OER. Omari et al.18 introduced A-site defects into GdFeO3 to enhance its OER catalytic performance. Chu et al.19 doped Pr at the A-site of Bi0.8Ca0.2FeO3−δ to boost activity and CO2 tolerance in SOFC cathodes.
Previous studies have primarily focused on enhancing performance through modifications of either the A-site or the B-site individually; however, the improvements from these single-site adjustments have been limited, resulting in suboptimal performance. This situation highlights the need for a more comprehensive and effective approach to materials optimization. Zhao et al.20 addressed this by exploring the potential of simultaneously adjusting both the A-site and B-site PrCoO3, which appeared to be a more promising strategy than single-site modulation. They implemented dual-site substitution in PrCoO3 by substituting Sr and Ru at the A and B sites, respectively. Their experimental results showed that Pr0.7Sr0.3Co1−xRuxO3 exhibited superior catalytic activity compared to both Pr0.7Sr0.3CoO3 and PrCoO3. However, a critical aspect missing from their analysis is a direct comparison between Pr0.7Sr0.3Co1−xRuxO3 and PrCo1−xRuxO3. This lack of direct comparison means that while the effectiveness of Ru substitution at the B site is evident, the individual contributions of A-site and B-site modifications remain unclear. Therefore, a comprehensive investigation into the effects of dual-site substitution is essential to quantify the individual and combined impacts on performance enhancement and to elucidate the underlying mechanisms.
In this study, we selected NF as the base compound due to its capability for compositional tuning and the specific properties of Nd atoms at the A-site, which allows for the adjustment of the double perovskite structure. We aimed to investigate dual-site modulation on NF to overcome the limitations of single-site modulation for OER catalysis. A site substitution of large diameter Ba in NF allows structure evolution from single perovskite to double perovskite. Furthermore, substituting low-valence Ni at the B site is expected to optimize the surface oxygen content and the electronic structure, further benefiting OER activity.
First-principles calculations validated the effectiveness of dual-site modification through electronic structure and bonding characteristics analysis. A-site Ba substitution reduced the band gap, enhancing electronic conductivity, while B-site Ni incorporation strengthened metal–oxygen covalency and reduced charge-transfer energy, creating optimal pathways for oxygen evolution. The optimized Nd0.8Ba1.2Fe1.6Ni0.4O6−δ (NBFN) catalyst demonstrated superior performance with a Tafel slope of 63.23 mV dec−1, significantly outperforming both single-site modified Nd0.8Ba1.2Fe2O6−δ (NBF) (114.85 mV dec−1) and unmodified NF (154.34 mV dec−1).
These findings establish dual-site modulation as an effective method for enhancing double perovskite catalysts, positioning NBFN among the most efficient Fe-based perovskite OER catalysts. Our work provides a rational framework for designing high-performance perovskite catalysts through precise electronic structure engineering.
2. Methods
2.1 Experimental methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CA = 1:1:2, with ammonia added to adjust the pH to 7. The solution was heated and stirred to form the gel; then, drying at 250 °C led to the formation of the solid precursor. The precursor was calcined at 1000 °C for 10 hours in air to obtain the perovskite powders.
:CA = 1:1:2, with ammonia added to adjust the pH to 7. The solution was heated and stirred to form the gel; then, drying at 250 °C led to the formation of the solid precursor. The precursor was calcined at 1000 °C for 10 hours in air to obtain the perovskite powders.
To evaluate catalytic activity, we performed linear sweep voltammetry (LSV) by scanning from 0.2 to 1 V versus Ag/AgCl at 5 mV s−1 while maintaining the RDE rotation at 1600 rpm. All LSV curves were IR-corrected to account for solution resistance. Double-layer capacitance (Cdl) measurements employed cyclic voltammetry within a non-faradaic window of 0.2–0.3 V versus Ag/AgCl, using scan rates from 40 to 100 mV s−1. We calculated Cdl from the linear relationship between current density and scan rate at 0.25 V. Catalyst stability was assessed through chronopotentiometry at a constant current density of 10 mA cm−2.
High-resolution imaging and elemental mapping were conducted using a JEOL JEM 2010F transmission electron microscope (TEM) equipped with energy-dispersive X-ray spectroscopy (EDS). Surface morphology was examined using a JSM-6390 scanning electron microscope (SEM). We probed the electronic structure using soft X-ray absorption spectroscopy (sXAS) at the MCD-A and MCD-B beamlines of the National Synchrotron Radiation Laboratory (NSRL).
2.2 Computational methods
We used density functional theory (DFT) to study the electronic structure and catalytic properties of NBF and NBFN systems. To study the bulk of these materials, we constructed various 2 × 2 × 2 supercells, which were initialized using the supercell program.21 Following electrostatic energy minimization of 1568 randomly generated configurations of NBFN, we selected the ten lowest-energy structures for detailed optimization using the Vienna Ab initio Simulation Package (VASP).22 These electronic structure calculations employed the projector-augmented wave method with spin polarization, utilizing the Perdew–Burke–Ernzerhof function within the generalized gradient approximation.23 To represent the localization of the Fe-3d and Ni-3d electrons, we used the DFT+U approach with Ueff values of 5.3 eV and 6.2 eV, respectively. The Ueff values were obtained from the Materials Project.24 Furthermore, the DFT calculation employed a plane-wave basis energy cutoff of 450 eV and a 3 × 3 × 1 Monkhorst-Pack k-point mesh for Brillouin zone sampling. Structure optimization utilized the Broyden method with convergence criteria of 0.02 eV Å−1 for forces and 10−5 eV for energy.For OER pathway analysis, we constructed symmetric slab models based on experimentally observed (100) surfaces on NBF and NBFN, incorporating a 15 Å vacuum layer along the c direction to prevent periodic image interactions. The energetics of OER intermediates were evaluated using the Rossmeisl–Nørskov free energy framework,25 with Gibbs free energy changes calculated as follows:
| ΔG = ΔE + ΔZPE − TΔS | (1) |
3. Results and discussion
3.1 Electronic structure and catalytic activity correlation
DFT calculations were used to simulate how dual-site substitution impacts the electronic structure and catalytic properties. Projected density of states (PDOS) analysis of Fe, Ni, and O revealed electronic structure evolution across our materials series (Fig. 1a). Shifting from single to double perovskite structures can lower the band gap, and substituting with Ni also contributes to this reduction. In turn, a decreased band gap leads to enhanced electronic conductivity. It is worth mentioning that the pristine NF had a substantial band gap of 2.36 eV, whereas the A-site modified NBF, B-site modified NFN (NdFe0.8Ni0.2O3−δ), and A/B-site co-modified NBFN all exhibited metallic character with a high density of states near the Fermi level. This electronic structure suggested increased electronic conductivity and improved electrocatalytic performance.26 | ||
| Fig. 1 Electronic structure and reaction mechanism analysis. (a) Projected density of states (PDOS) for NF, NBF, NFN, and NBFN, showing O 2p band center and transition metal 3d band center positions relative to the Fermi level. (b) Integrated PDOS of Fe and Ni d-orbitals in NBFN demonstrating spin-dependent orbital occupancy. (c) Schematic illustration of band alignment changes due to dual-site modification. (d) Four-step adsorbate evolution mechanism for OER on perovskite surfaces in alkaline condition. (e) Free energy diagrams showing reduced RDS for NBFN (1.01 eV) compared to NBF (1.10 eV). | ||
The O 2p band center energy has been used as a descriptor for OER activity in perovskites.27 Prior research by Zhao et al.28 established a direct correlation between the position of the O 2p band center and catalytic performance; the closer this band center is to the Fermi level, the higher the OER activity. This occurs because the closer the O 2p band center is to the Fermi level, the easier it becomes for lattice oxygen to participate in the OER process by undergoing oxidation. Our calculations show that modifying both A-site and B-site can move the O 2p band center closer to the Fermi level, with simultaneous modifications at both sites resulting in a higher O 2p band center compared to modifications at individual sites. The original NF displayed an O 2p band center of −3.74 eV, while modifications at the A-site raised it to −1.84 eV, and B-site modifications raised it to −2.14 eV. In contrast, simultaneous modifications at both the A-site and B-site increased the O 2p band center to −1.6 eV, highlighting the synergistic benefits of dual-site modifications. This indicates that the O 2p band center in NBFN is closest to the Fermi level, suggesting that NBFN is likely to exhibit superior catalytic performance.
In addition to analyzing the O 2p band center, we examined charge-transfer energy (Δ) as a complementary descriptor of OER activity. This parameter, defined as the energy difference between the O 2p band center and unoccupied transition metal 3d orbital, has also been shown to correlate with catalytic performance.29 A lower Δ facilitates easier electron transfer from oxygen to the transition metal centers during the OER process, thus enhancing activity. Our analysis of the Fe 3d–O 2p orbital interactions showed that the original NF had the highest ΔFe–O at 4.83 eV. In contrast, A-site adjustment (NBF) reduced ΔFe–O to 4.48 eV, and B-site adjustment (NFN) decreased it to 4.76 eV. Notably, simultaneous A/B-site adjustments resulted in an even lower ΔFe–O of 4.28 eV, making NBFN the most active catalyst for OER, which underscores the synergistic effect of A/B-site modifications. Furthermore, the investigation of Ni 3d–O 2p orbital interactions in NBFN showed a significantly lower charge-transfer energy (ΔNi–O = 2.87 eV), indicating that the Ni sites possess a more favorable electronic structure for OER in comparison to the Fe sites. For NFN, the ΔNi–O (3.36 eV) is also less than ΔFe–O (4.76 eV), suggesting that the electronic structure of Ni is optimized compared to that of Fe.
To investigate metal–oxygen bonding in NBFN, we analyzed the integrated projected density of states (IPDOS) for Fe and Ni d-orbitals. In the spin-up channel (Fig. 1a), Fe d-orbitals exhibited pronounced PDOS peaks below the Fermi level (0 eV), indicating complete orbital filling. IPDOS analysis confirmed this observation quantitatively (Fig. 1b), with electronic state density exceeding 0.5 at the Fermi level.30 In contrast, the spin-down (spin-dw) channel exhibited Fe d-orbital PDOS peaks above the Fermi level, with IPDOS occupancy below 0.5, demonstrating the presence of unoccupied states. This electronic configuration, characterized by five d-electrons in the spin-up channel and none in the spin-down channel, suggested that the valence state in NBFN is 3+. Conversely, Ni displayed different electronic occupation in NBFN (Fig. 1b). The Ni d-orbitals showed full occupancy in the spin-up channel, while the spin-down channel exhibited partial filling, with dxy, dyz, and dxz orbitals showing occupancy rates near 1. This electronic distribution, with five d-electrons in the spin-up channel and three in the spin-down channel, indicated that the valence state of Ni is 2+.
Analysis of TM–O covalency follows the methodology established by Jiang et al.,31 which quantifies orbital interactions through the energy difference between TM 3d and oxygen 2p band centers. In NBFN, this analysis revealed distinct bonding characteristics: the Fe–O interaction showed a band center difference of 4.587 eV, while the Ni–O interaction exhibited a smaller difference of 2.901 eV. The reduced energy separation for Ni–O indicated a stronger covalent character, enabling more efficient electron transfer between Ni sites and oxygen adsorbates.32 This enhanced Ni–O covalency identified Ni sites as more active for OER, which is consistent with our charge transfer energy calculations. These simulations suggested that by combining double perovskite formation with Ni substitution, we can reduce the band gap, shift the O 2p band center towards the Fermi energy, enhance metal–oxygen covalency, and minimize charge transfer energy (Fig. 1c). These electronic structure modifications work synergistically to enhance catalytic performance.
To understand the mechanistic basis of catalyst performance, we investigated the oxygen evolution reaction (OER) pathway using the adsorbate evolution mechanism (AEM) framework for perovskite oxides in alkaline conditions (Fig. 1d and e). The reaction proceeded through four elementary steps, each involving the formation and transformation of key oxygen-containing intermediates (*O, OH, and OOH, where * represents the active site). An analysis of all possible reaction pathways across Fe and Ni sites (structures detailed in Fig. S1 and S2†) identified the deprotonation of OH to O as the rate-determining step (RDS) for both NBF and NBFN catalysts. Notably, the energy barrier for this critical step was lower at the Ni sites in NBFN (1.01 eV) compared to the Fe sites in NBF (1.10 eV), suggesting optimized reaction pathways on the Ni sites. This observation regarding optimized reaction pathways on the Ni sites is consistent with the study of charge transfer energy and TM–O covalency. The lower RDS energy barrier at Ni sites in NBFN, as predicted by DFT, directly accounts for the superior OER activity of NBFN compared to NBF.
3.2 Structure and surface analysis
The crystal structures of the synthesized perovskite oxides were characterized using X-ray diffraction (XRD) (Fig. 2a). The analysis revealed distinct crystal structures: NF is orthorhombic, while NBF is cubic. Upon substituting Ni, the crystal symmetry was preserved, with NBFN maintaining the cubic structure of NBF, whereas NFN (NdFe0.8Ni0.2O3−δ) retained the orthorhombic structure of NF. The incorporation of smaller Ni atoms (0.124 nm) in place of larger Fe atoms (0.126 nm) at the B-site caused the diffraction peaks of NBFN and NFN to shift towards higher angles compared to their parent compounds NBF and NF, respectively (see Fig. S3†), indicating lattice shrinkage. Notably, XRD patterns showed no detectable secondary phases.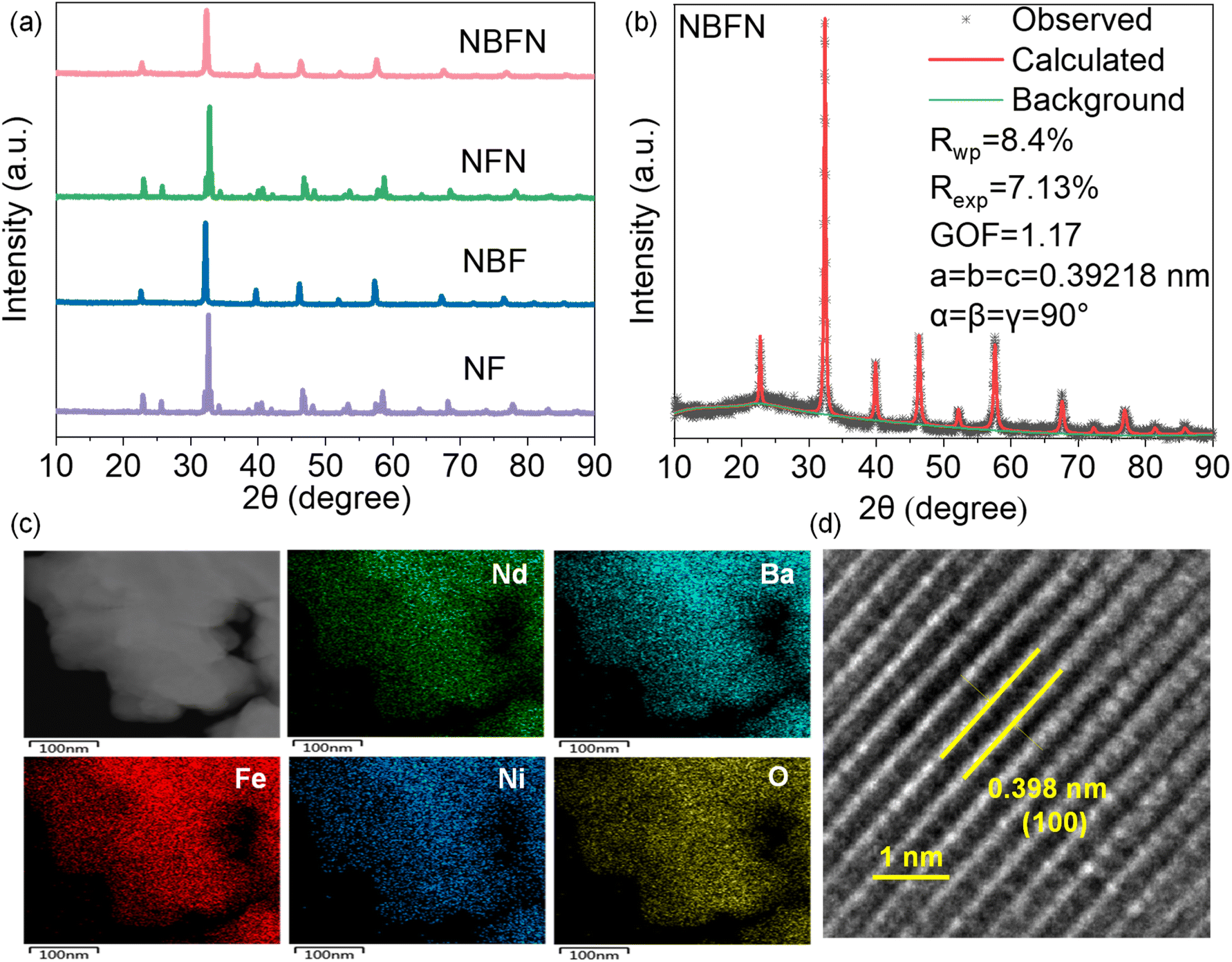 | ||
| Fig. 2 (a) X-ray diffraction (XRD) patterns of the synthesized NF, NBF, NFN, and NBFN perovskite powders. (b) Rietveld refinement of NBFN XRD profile. (c) Scanning transmission electron microscopy (STEM) image with corresponding energy-dispersive X-ray (EDX) elemental mapping of NBFN, showing the elemental distribution. (d) High-resolution transmission electron microscopy (HR-TEM) image showing the lattice spacing of NBFN. | ||
Rietveld refinement analysis (Fig. 2b) confirmed that NBFN crystallized in the cubic space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m with lattice parameters a = b = c = 3.92 Å. The XRD pattern is similar to double perovskite NdBaFe2−xMnxO5+δ reported by Mao et al.33 Scanning transmission electron microscopy (STEM) and energy-dispersive X-ray (EDX) mapping (Fig. 2c) revealed uniform element distribution within individual NBFN particles, suggesting the successful dual-site substitution and homogeneity of the NBFN catalyst. High-resolution transmission electron microscopy (HR-TEM, Fig. 2d) showed a crystalline cubic structure with clear lattice fringes, with a lattice spacing of 0.398 nm, corresponding to (100) lattice planes from Rietveld refinement results. The combination of SEM-EDS and ICP results (Fig. S4 and Table S1†) confirmed that the composition of NBFN elements aligned with the intended stoichiometry. SEM images (Fig. S5†) showed that NF had a smaller grain size compared to BF, with NBF and NBFN having smaller grain size than BF.
m with lattice parameters a = b = c = 3.92 Å. The XRD pattern is similar to double perovskite NdBaFe2−xMnxO5+δ reported by Mao et al.33 Scanning transmission electron microscopy (STEM) and energy-dispersive X-ray (EDX) mapping (Fig. 2c) revealed uniform element distribution within individual NBFN particles, suggesting the successful dual-site substitution and homogeneity of the NBFN catalyst. High-resolution transmission electron microscopy (HR-TEM, Fig. 2d) showed a crystalline cubic structure with clear lattice fringes, with a lattice spacing of 0.398 nm, corresponding to (100) lattice planes from Rietveld refinement results. The combination of SEM-EDS and ICP results (Fig. S4 and Table S1†) confirmed that the composition of NBFN elements aligned with the intended stoichiometry. SEM images (Fig. S5†) showed that NF had a smaller grain size compared to BF, with NBF and NBFN having smaller grain size than BF.
We measured surface oxygen vacancies and transition metal valence state using X-ray photoelectron spectroscopy (XPS) and soft X-ray absorption spectroscopy (sXAS). Analysis of the O 1s XPS spectra (Fig. 3a and Table S2†) reveals that pristine NF had the highest lattice oxygen content. In contrast, both A-site modified NBF and B-site modified NFN exhibited decreased lattice oxygen levels, suggesting that adjustments at both sites promote the creation of oxygen vacancies. Interestingly, the NBFN with simultaneous A/B-site adjustments displayed the lowest lattice oxygen content, indicating that this combined modification is more effective for forming oxygen vacancies than modifications at individual sites. Oxygen vacancies significantly change how electrocatalysts interact with reactants (both in absorption and desorption) and also alter the bulk properties like electronic structure and conductivity, benefiting OER.34
 | ||
| Fig. 3 (a) O 1s X-ray photoelectron spectroscopy (XPS) spectra of NF, NBF, NFN and NBFN, demonstrating different surface oxygen contents. (b) Fe 2p XPS spectra of NF, NBF, NFN and NBFN, revealing oxidation states of Fe. (c) Fe L3-edge soft X-ray absorption (sXAS) spectra of NF, NBF, and NBFN. (d) Ni L3-edge sXAS spectra comparing NBFN with reference NiO, and showing the valence state of Ni. | ||
A detailed analysis of the Fe 2p and Ni 2p XPS spectra (Fig. 3b and S6,† respectively), along with the corresponding peak deconvolution results (Tables S3†), provided insights into the oxidation states of the transition metals. The Fe ions in NBFN exhibited a valence state of +3, while the Ni ions displayed a valence state of +2.36, which aligns broadly with the trends predicted by our DFT calculations concerning electron occupancy in the Fe and Ni d-orbitals. It is significant that Ni demonstrated a lower oxidation state compared to Fe, aligning with previous findings.2,8 The addition of low valence Ni to Fe-based perovskites encourages the development of oxygen vacancies, which subsequently reduces the oxidation state of Fe. This occurrence is illustrated by the shifts from NF to NFN and from NBF to NBFN (Table S3†). Moreover, the presence of Ba has minimal impact on the oxidation states of B-site transition metals, mainly affecting the control of the crystal structure.
We employed soft X-ray absorption spectroscopy (sXAS) at the Fe and Ni L-edges to measure the electronic structures of NF, NBF, and NBFN catalysts and to confirm the XPS findings (Fig. 3c and d). The Fe L3-edge sXAS spectra (Fig. 3c) indicate that NBFN exhibited a characteristic peak shift toward lower energies compared to NBF, suggesting a reduction in the valence state of Fe, consistent with the XPS results. The similar peak positions of NF and NBF suggest that A-site modification had minimal effect on the oxidation state of Fe, which aligns with the XPS results. Analysis of the Ni L3 edge sXAS spectra (Fig. 3d) revealed a peak shift toward higher energies for NBFN in comparison to the NiO reference, suggesting that the Ni in NBFN had a valence state higher than +2. Furthermore, the higher peak intensity of NBFN compared to NiO at around 856 eV indicates the presence of Ni3+ (Fig. 3d).35 This observation aligns with XPS characterization and provides additional evidence for the mixed-valence character of Ni in NBFN (Fig. S6†).
3.3 Oxygen evolution reaction (OER) performance evaluation
A systematic comparison of single/dual site modifications (Fig. 4a) revealed clear performance differences among the catalysts. Using NF as the baseline, we compared NBF (A-site modification), NFN (B-site modification), and NBFN (dual-site modification). Current densities at 1.7 V were NF (0.71 mA cm−2), NBF (1.84 mA cm−2, 2.6-fold increase), NFN (4.7 mA cm−2, 6.6-fold increase), and NBFN (20.55 mA cm−2, 28.9-fold increase). Single-site modulated NBF and NFN showed a slight improvement compared to the unmodified NF, while dual-site modulated NBFN showed great improvement. The large improvement with NBFN compared to single-site-modified NBF and NFN demonstrated the synergistic effect of dual-site modulation. Furthermore, NBFN had a lower Tafel slope (63.23 mV dec−1) compared to NF (154.34 mV dec−1), NBF (114.85 mV dec−1), and NFN (82.92 mV dec−1), indicating faster reaction kinetics and a more efficient OER process (Fig. 4b). The experimentally observed lower Tafel slope for NBFN is consistent with the DFT predicted lower RDS energy barrier, suggesting that the dual-site modulation indeed facilitates faster OER kinetics as theoretically predicted. | ||
| Fig. 4 Electrochemical performance evaluation of perovskite catalysts. (a) LSV curves comparing OER activity of NF, NBF, NFN, and NBFN. (b) Tafel plots derived from LSV curves used to evaluate reaction kinetics. (c) Current density versus scan rate plots to determine electrochemical double-layer capacitance (Cdl) and ECSA. (d) Overpotential comparison of NBFN at 10 mA cm−2 against: LaNiO3, La0.95FeO3−δ, BSCF, SrNb0.1Co0.7Fe0.2O3−δ, LaCr0.5Fe0.5O3, Sr2Fe1.5Mo0.5O6−δ, PrBa0.85Ca0.15FeMnO5+δ, PrBaCo2O5+δ, RuO2, IrO2. (e) Assessment of the stability of NF, NBF, NFN and NBFN via chronopotentiometry conducted at 10 mA cm−2. | ||
We also examined the effects of varying the substitution levels of Ba at the A site and Ni at the B site. As shown in Fig. S7 and S8,† changes in A-site Ba concentration did not affect the double perovskite structure, and all series of double perovskites exhibited higher performance compared to the single perovskite NF and BF (0.81 mA cm−2 at 1.7 V). Additionally, our analysis of Ni concentration indicated that when the Ni:Fe ratio is greater than 2:8, it results in the degradation of both the perovskite structure and catalytic performance. We also investigated other potential dopant levels such as NdxBa2−xFe1.6Ni0.4O6−δ (Fig. S9†) or NdxBa2−xFe1.2Ni0.8O6−δ (Fig. S10†) to see if NBFN is the optimized composition. The results show that there are no double perovskites with higher performance than the NBFN.
The electrochemical active surface area (ECSA) was quantified through double-layer capacitance (Cdl) analysis (Fig. 4c and S11†).36 Compared to the pristine NF, the NBF and NFN showed modest ECSA enhancements of 1.03 and 1.16 times, respectively. In contrast, NBFN displayed a substantial 1.74-fold increase in ECSA. The larger ECSA for NBFN suggested a greater number of electrochemically active sites available for OER, contributing to its improved performance.37,38
We assessed the stability through chronopotentiometry at a constant current density of 10 mA cm−2 (Fig. 4e). NF exhibited rapid potential increase after only 500 s at a current density of 10 mA cm−2, which agrees with the argument in ref. 8 about the thermodynamically unstable characteristics of Fe based oxides in alkaline OER conditions. In contrast, NBF demonstrated improved stability, functioning effectively for approximately 5 hours before failing, while NFN operated steadily for about 7.8 hours before ceasing to work. Notably, following an initial activation period, NBFN maintained a stable operating voltage of 1.55 V for over 20 hours. To confirm the robustness of NBFN, XRD pattern (Fig. S12†) and HR-TEM image (Fig. S13†) were obtained after stability testing, which showed no evidence of phase transformation. The XRD pattern did not detect any secondary phases, and the HR-TEM image indicated that the lattice structure remained stable, with no collapse or distortion, thereby demonstrating the robustness of NBFN. The performance of NBFN was benchmarked against state-of-the-art catalysts, including both transition metal perovskites and precious metal-based materials2,36,39–46 (Fig. 4d and Table S4†). This comparison showed that the overpotential of NBFN is significantly lower than that of other perovskite catalysts and even surpasses the precious metal benchmarks IrO2 and RuO2.
4. Conclusions
This study demonstrates the effectiveness of dual-site modulation for enhancing OER catalysis in Fe-based perovskites. By simultaneously incorporating Ba into the A-site and Ni into the B-site of NdFeO3−δ, we successfully engineered a double perovskite NBFN catalyst that exhibits remarkable electrochemical performance. The catalyst achieves a low onset potential of 237 mV, maintains stable operation at a 320 mV overpotential (10 mA cm−2) for 20 hours, and demonstrates rapid kinetics with a Tafel slope of 63.23 mV dec−1, outperforming conventional noble metal catalysts IrO2 and RuO2. Mechanistic investigations reveal that this enhanced performance stems from two complementary effects: (1) A-site Ba incorporation induces a double perovskite structure with a reduced band gap, improving electronic conductivity, and (2) B-site Ni substitution optimizes the electronic structure by reducing charge-transfer energy and strengthening transition metal–oxygen covalency. These findings not only establish dual-site modulation as a powerful approach for developing high-performance Fe-based catalysts but also provide fundamental insights into structure–property relationships in perovskite catalysts. This earth-abundant, high-performance catalyst offers a promising alternative to noble metal-based catalysts, potentially reducing costs and improving the sustainability of hydrogen production via water electrolysis.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the support of the Hong Kong Research Grant Council (16201820, 16201622) and Green Tech Fund (GTF202020131). F. C. thanks the University of Bayreuth and the Bavarian Center for Battery Technology (BayBatt) for providing start-up funds. Y. Wang acknowledges the support from the RGC Junior Research Fellow Scheme (JRFS2526-6S08). During the editing of the manuscript, Gemini Advanced 2.0 (Google) was employed to enhance text clarity and style. Subsequent to AI assistance, the authors reviewed and edited the manuscript. A final editorial review was conducted by Dr Mark Ellwood of HKUST. The authors retain full responsibility for the publication content.References
- K. Zhu, X. Zhu and W. Yang, Application of In Situ Techniques for the Characterization of NiFe-Based Oxygen Evolution Reaction (OER) Electrocatalysts, Angew. Chem., Int. Ed., 2019, 58, 1252–1265 CrossRef CAS PubMed
.
- H. Sun, X. Xu, Z. Hu, L. H. Tjeng, J. Zhao, Q. Zhang, H.-J. Lin, C.-T. Chen, T.-S. Chan, W. Zhou and Z. Shao, Boosting the oxygen evolution reaction activity of a perovskite through introducing multi-element synergy and building an ordered structure, J. Mater. Chem. A, 2019, 7, 9924–9932 RSC
- T. Reier, M. Oezaslan and P. Strasser, Electrocatalytic Oxygen Evolution Reaction (OER) on Ru, Ir, and Pt Catalysts: A Comparative Study of Nanoparticles and Bulk Materials, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, A Perovskite Oxide Optimized for Oxygen Evolution Catalysis from Molecular Orbital Principles, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed
- F. Abdelghafar, X. Xu, D. Guan, Z. Lin, Z. Hu, M. Ni, H. Huang, T. Bhatelia, S. P. Jiang and Z. Shao, New Nanocomposites Derived from Cation-Nonstoichiometric Bax(Co, Fe, Zr, Y)O3−δ as Efficient Electrocatalysts for Water Oxidation in Alkaline Solution, ACS Mater. Lett., 2024, 6, 2985–2994 CrossRef CAS
- X. Xu, Y. Pan, W. Zhou, Y. Chen, Z. Zhang and Z. Shao, Toward Enhanced Oxygen Evolution on Perovskite Oxides Synthesized from Different Approaches: A Case Study of Ba0.5Sr0.5Co0.8Fe0.2O3−δ, Electrochim. Acta, 2016, 219, 553–559 Search PubMed
- X. Xu, C. Su and Z. Shao, Fundamental Understanding and Application of Ba0.5Sr0.5Co0.8Fe0.2O3−δ Perovskite in Energy Storage and Conversion: Past, Present, and Future, Energy Fuels, 2021, 35, 13585–13609 CrossRef CAS
- S. She, Y. Zhu, X. Wu, Z. Hu, A. Shelke, W.-F. Pong, Y. Chen, Y. Song, M. Liang, C.-T. Chen, H. Wang, W. Zhou and Z. Shao, Realizing High and Stable Electrocatalytic Oxygen Evolution for Iron-Based Perovskites by Co-Doping-Induced Structural and Electronic Modulation, Adv. Funct. Mater., 2022, 32, 2111091 Search PubMed
- J. Wang, S. Choi, J. Kim, S. W. Cha and J. Lim, Recent Advances of First d-Block Metal-Based Perovskite Oxide Electrocatalysts for Alkaline Water Splitting, Catalysts, 2020, 10, 770 CrossRef CAS
- E. Omari and M. Omari, Enhancing catalytic activity of NdFeO3 perovskite by tuning A-site cation deficiency for oxygen evolution reaction, Int. J. Hydrogen Energy, 2022, 47, 14542–14551 CrossRef CAS
- S. She, J. Yu, W. Tang, Y. Zhu, Y. Chen, J. Sunarso, W. Zhou and Z. Shao, Systematic Study of Oxygen Evolution Activity and Stability on La1−xSrxFeO3−δ Perovskite Electrocatalysts in Alkaline Media, ACS Appl. Mater. Interfaces, 2018, 10, 11715–11721 CrossRef CAS PubMed
- Y. Lu, H. Zhao, K. Li, X. Du, Y. Ma, X. Chang, N. Chen, K. Zheng and K. Świerczek, Effective calcium doping at the B-site of BaFeO3−δ perovskite: towards low-cost and high-performance oxygen permeation membranes, J. Mater. Chem. A, 2017, 5, 7999–8009 RSC
- K. Zhu, H. Liu, X. Li, Q. Li, J. Wang, X. Zhu and W. Yang, Oxygen evolution reaction over Fe site of BaZrxFe1−xO3−δ perovskite oxides, Electrochim. Acta, 2017, 241, 433–439 CrossRef CAS
- H. Zhao, C. Chen, D. Chen, M. Saccoccio, J. Wang, Y. Gao, T. H. Wan and F. Ciucci, Ba0.95La0.05FeO3−δ–multi-layer graphene as a low-cost and synergistic catalyst for oxygen evolution reaction, Carbon, 2015, 90, 122–129 CrossRef CAS
- D. Shang, B. Zhang, L. Zhang, J. Li, Y. Zhao, Y. Huang, K. Tang and C. Xia, Improving electrocatalytic activity through multi-element doping to A-site of Fe-based perovskite cathode for solid oxide fuel cells, Chem. Eng. J., 2025, 506, 160067 CrossRef CAS
- Y. Chen, J. Zhu, T. Xia, Y. Feng, J. Chang, X. Zhu, J. Bai, J. Wang, W. Yan and D. Zhou, Improved electrocatalytic activity and CO2 tolerance of iron-based perovskite as an intermediate temperature SOFC cathode, Fuel, 2024, 375, 132546 CrossRef CAS
- Y. Inoue, Y. Miyahara, K. Miyazaki, C. Lee, R. Sakamoto and T. Abe, Synergistic Interplay between Fe-Based Perovskite Oxides and Co in Electrolyte for Efficient Oxygen Evolution Reaction, ChemSusChem, 2025, 18, e202401982 CrossRef CAS PubMed
- E. Omari and M. Omari, Gd1−xFeO3 perovskite oxides for catalyzing oxygen evolution reaction via A-site cation deficiency in alkaline media, Ceram. Int., 2024, 50, 25509–25517 CrossRef CAS
- Z. Chu, J. Gao, Q. Li, T. Xia, L. Sun, H. Zhao, I. V. Kovalev, R. D. Guskov, M. P. Popov and A. P. Nemudry, Highly oxygen reduction activity and CO2 resistance of Fe-based cathode electrocatalysts for solid oxide fuel cells, J. Mater. Sci. Technol., 2025, 212, 303–311 CrossRef CAS
- Y.-N. Zhao, C. Liu, S. Xu, S. Min, W. Wang, N. Mitsuzaki and Z. Chen, A/B-Site Management Strategy to Boost Electrocatalytic Overall Water Splitting on Perovskite Oxides in an Alkaline Medium, Inorg. Chem., 2023, 62, 12590–12599 CrossRef CAS PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, Commentary: The Materials Project: A materials genome approach to accelerating materials innovation, APL Mater., 2013, 1, 011002 CrossRef
- Z. M. Baiyee, C. Chen and F. Ciucci, A DFT+U study of A-site and B-site substitution in BaFeO3−δ, Phys. Chem. Chem. Phys., 2015, 17, 23511–23520 RSC
- A. B. Muñoz-García and M. Pavone, First-Principles Design of New Electrodes for Proton-Conducting Solid-Oxide Electrochemical Cells: A-Site Doped Sr2Fe1.5Mo0.5O6−δ Perovskite, Chem. Mater., 2016, 28, 490–500 CrossRef
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Electrolysis of water on (oxidized) metal surfaces, Chem. Phys., 2005, 319, 178–184 CrossRef CAS
- Y. Zhou, Z. Zhou, L. Hu, R. Tian, Y. Wang, H. Arandiyan, F. Chen, M. Li, T. Wan, Z. Han, Z. Ma, X. Lu, C. Cazorla, T. Wu and D. Chu, A facile approach to tailor electrocatalytic properties of MnO2 through tuning phase transition, surface morphology and band structure, Chem. Eng. J., 2022, 438, 135561 CrossRef CAS
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Double perovskites as a family of highly active catalysts for oxygen evolution in alkaline solution, Nat. Commun., 2013, 4, 2439 CrossRef PubMed
- J.-W. Zhao, Y. Li, D. Luan and X. W. Lou, Structural evolution and catalytic mechanisms of perovskite oxides in electrocatalysis, Sci. Adv., 2024, 10, eadq4696 Search PubMed
- W. T. Hong, K. A. Stoerzinger, Y.-L. Lee, L. Giordano, A. Grimaud, A. M. Johnson, J. Hwang, E. J. Crumlin, W. Yang and Y. Shao-Horn, Charge-transfer-energy-dependent oxygen evolution reaction mechanisms for perovskite oxides, Energy Environ. Sci., 2017, 10, 2190–2200 RSC
- Y. Wang, Z. Wang, K. Yang, J. Liu, Y. Song, J. Li, Z. Hu, M. J. Robson, Z. Zhang, Y. Tian, S. Xu, Y. Lu, H. M. Law, F. Liu, Q. Chen, Z. Yang and F. Ciucci, Self-Recoverable Symmetric Protonic Ceramic Fuel Cell with Smart Reversible Exsolution/Dissolution Electrode, Adv. Funct. Mater., 2024, 34, 2404846 CrossRef CAS
- C. Jiang, H. He, H. Guo, X. Zhang, Q. Han, Y. Weng, X. Fu, Y. Zhu, N. Yan, X. Tu and Y. Sun, Transfer learning guided discovery of efficient perovskite oxide for alkaline water oxidation, Nat. Commun., 2024, 15, 6301 CrossRef CAS PubMed
- S. Li, T. Xia, Y. Dou, Y. Xie, J. Wang, H. Zhao and L. Huo, Phosphatizing Engineering of Perovskite Oxide Nanofibers for Hydrogen Evolution Reaction to Achieve Extraordinary Electrocatalytic Performance, Adv. Funct. Mater., 2022, 32, 2112164 Search PubMed
- X. Mao, T. Yu and G. Ma, Performance of cobalt-free double-perovskite NdBaFe2−xMnxO5+δ cathode materials for proton-conducting IT-SOFC, J. Alloys Compd., 2015, 637, 286–290 CrossRef CAS
- K. Zhu, F. Shi, X. Zhu and W. Yang, The roles of oxygen vacancies in electrocatalytic oxygen evolution reaction, Nano Energy, 2020, 73, 104761 CrossRef CAS
- W. H. Lee, M. H. Han, U. Lee, K. H. Chae, H. Kim, Y. J. Hwang, B. K. Min, C. H. Choi and H.-S. Oh, Oxygen Vacancies Induced NiFe-Hydroxide as a Scalable, Efficient, and Stable Electrode for Alkaline Overall Water Splitting, ACS Sustain. Chem. Eng., 2020, 8, 14071–14081 Search PubMed
- J. Wang, Y. Gao, D. Chen, J. Liu, Z. Zhang, Z. Shao and F. Ciucci, Water Splitting with an Enhanced Bifunctional Double Perovskite, ACS Catal., 2018, 8, 364–371 CrossRef CAS
- J. Yu, X. Wu, D. Guan, Z. Hu, S.-C. Weng, H. Sun, Y. Song, R. Ran, W. Zhou, M. Ni and Z. Shao, Monoclinic SrIrO3: An Easily Synthesized Conductive Perovskite Oxide with Outstanding Performance for Overall Water Splitting in Alkaline Solution, Chem. Mater., 2020, 32, 4509–4517 CrossRef CAS
- H. Sun, L. Li, Y. Chen, H. Kim, X. Xu, D. Guan, Z. Hu, L. Zhang, Z. Shao and W. Jung, Boosting ethanol oxidation by NiOOH-CuO nano-heterostructure for energy-saving hydrogen production and biomass upgrading, Appl. Catal., B, 2023, 325, 122388 CrossRef CAS
- C. Liu, D. Ji, H. Shi, Z. Wu, H. Huang, Z. Kang and Z. Chen, An A-site management and oxygen-deficient regulation strategy with a perovskite oxide electrocatalyst for the oxygen evolution reaction, J. Mater. Chem. A, 2022, 10, 1336–1342 RSC
- Y. Zhu, W. Zhou, J. Yu, Y. Chen, M. Liu and Z. Shao, Enhancing Electrocatalytic Activity of Perovskite Oxides by Tuning Cation Deficiency for Oxygen Reduction and Evolution
Reactions, Chem. Mater., 2016, 28, 1691–1697 CrossRef CAS
- L. Tang, Y. Rao, L. Wei, H. Zheng, H. Liu, W. Zhang and K. Tang, A-site Cation Defects (Ba0.5Sr0.5)1−xCo0.8Fe0.2O3−δ Perovskites as Active Oxygen Evolution Reaction Catalyst in Alkaline Electrolyte, Chin. J. Chem., 2021, 39, 2692–2698 CrossRef CAS
- Y. Zhu, W. Zhou, Z.-G. Chen, Y. Chen, C. Su, M. O. Tadé and Z. Shao, SrNb0.1Co0.7Fe0.2O3−δ Perovskite as a Next-Generation Electrocatalyst for Oxygen Evolution in Alkaline Solution, Angew. Chem., Int. Ed., 2015, 54, 3897–3901 CrossRef CAS PubMed
- X. Gao, Z. Sun, J. Ran, J. Li, J. Zhang and D. Gao, High efficiency electrocatalyst of LaCr0.5Fe0.5O3 nanoparticles on oxygen-evolution reaction, Sci. Rep., 2020, 10, 13395 CrossRef CAS PubMed
- J. Zhou, T. Liu, J. Zhang, L. Zhao, W. He and Y. Wang, Rational design of ultrafine cobalt free electrospun nanofibers as efficient and durable binfunctional oxygen electrocatalysts for rechargeable zinc-air battery, Sep. Purif. Technol., 2023, 304, 122316 CrossRef CAS
- H. Sun, G. Chen, Y. Zhu, B. Liu, W. Zhou and Z. Shao, B-Site Cation Ordered Double Perovskites as Efficient and Stable Electrocatalysts for Oxygen Evolution Reaction, Chem.–Eur. J., 2017, 23, 5722–5728 CrossRef CAS PubMed
- B. Hua, Y.-F. Sun, M. Li, N. Yan, J. Chen, Y.-Q. Zhang, Y. Zeng, B. Shalchi Amirkhiz and J.-L. Luo, Stabilizing Double Perovskite for Effective Bifunctional Oxygen Electrocatalysis in Alkaline Conditions, Chem. Mater., 2017, 29, 6228–6237 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta01754h |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2025 |