
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of polyoxothiometalates through site-selective post-editing sulfurization of polyoxometalates†
Kentaro
Yonesato
 *,
Kazuya
Yamaguchi
and
Kosuke
Suzuki
*
*,
Kazuya
Yamaguchi
and
Kosuke
Suzuki
*
Department of Applied Chemistry, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: ksuzuki@appchem.t.u-tokyo.ac.jp; k-yonesato@g.ecc.u-tokyo.ac.jp
First published on 14th June 2024
Abstract
Polyoxometalates (POMs) function as platforms for synthesizing structurally well-defined inorganic molecules with diverse structures, metals, compositions, and arrangements. Although post-editing of the oxygen sites of POMs has great potential for development of unprecedented structures, electronic states, properties, and applications, facile methods for site-selective substitution of the oxygen sites with other atoms remain limited. Herein, we report a direct site-selective oxygen–sulfur substitution method that enables transforming POMs [XW12O40]4− (X = Si, Ge) to Keggin-type polyoxothiometalates (POTMs) [XW12O28S12]4− using sulfurizing reagents in an organic solvent. The resulting POTMs retain the original Keggin-type structure, with all 12 surface W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups selectively converted to WS without sulfurization of other oxygen sites. These POTMs show high stability against water and O2 in organic solvents and a drastic change in the electronic states and redox properties. The findings of this study represent a facile method for converting POMs to POTMs, leading to the development of their unique properties and applications in diverse fields, including (photo)catalysis, sensing, optics, electronics, energy conversion, and batteries.
O groups selectively converted to WS without sulfurization of other oxygen sites. These POTMs show high stability against water and O2 in organic solvents and a drastic change in the electronic states and redox properties. The findings of this study represent a facile method for converting POMs to POTMs, leading to the development of their unique properties and applications in diverse fields, including (photo)catalysis, sensing, optics, electronics, energy conversion, and batteries.
Introduction
As a member of group 16, sulfur shares its elemental classification with that of oxygen. Nevertheless, metal sulfides exhibit distinct structures, electronic states, properties, (photo)catalysis, and applications that significantly differ from those of metal oxides because of the strong affinity of sulfur for metal atoms, unique redox properties, and narrow band gaps.1 Therefore, as seen in the photocatalysis of sulfur-doped titania2 and the hydrodesulfurization catalysis of molybdenum oxides activated by hydrogen sulfides,3 the modification of metal oxides with sulfur results in unique physicochemical properties.Polyoxometalates (POMs), which are anionic metal oxide clusters (e.g., W6+, Mo6+, V5+, Nb5+, and Ta5+), exhibit diverse structures and properties, including acidity/basicity, redox properties, and photochemical properties, depending on the structures, constituting atoms, and electronic states.4 These features enable diverse applications that include catalysis, medicine, materials science, sensor, electronics, and batteries. The substitution of the constituting atoms of POMs is an important approach to modify their properties and achieve novel functions: for example, the replacement of metals in POMs has been widely investigated through the direct metal substitution or metal introduction into the vacant sites of lacunary POMs.5,6 In addition, the replacement of the oxygen sites of POMs with various organic ligands, such as phosphonates, silicates, imidos, and pyridines, is also an important approach to synthesize functional materials.4j,7 However, the substitution of the oxygen sites with other atoms is still limited due to the difficulty in controlling the reactivity.8
In the field of synthetic organic chemistry, molecular post-editing has recently become increasingly important to realize late-stage chemical transformations.9 Considering the diverse structures of POMs, site-selective post-editing of POMs has great potential for the development of inorganic molecules with novel properties and applications. This study proposes a selective oxygen–sulfur substitution approach that enables facile and versatile synthesis of polyoxothiometalates (POTMs) from POMs. Several structurally defined POTMs have been synthesized by the self-condensation of mono-, di-, and trinuclear (oxo)thiometalates, or the reaction of these species with organic ligands and/or POMs.10 However, the direct oxygen–sulfur substitution reactions of POMs are limited to the oxygen sites on the substituted metal. For example, the mononiobium and monotantalum-oxo units in Lindqvist-type [(M5+O)W5O18]3− and Keggin-type [(M5+O)PW11O39]4− (M = Nb, Ta) have been converted to mono-sulfurized species [(M5+S)W5O18]3− and [(M5+S)PW11O39]4−, respectively.11 These results showed that sulfurization proceeds only against terminal Nb5+O and Ta5+O, and not on tungstate, which critically hinders the investigation of POTMs. Therefore, the development of site-selective oxygen–sulfur substitution reactions for polyoxotungstates is crucial for establishing a facile and widely applicable method for exchanging the oxygen atoms of various POM precursors.
Herein, we report the first synthesis method of Keggin-type POTMs [XW12O28S12]4− (IIX; X = Si, Ge) through direct site-selective oxygen–sulfur substitution of the parent POMs [XW12O40]4− (IX; Fig. 1). By reacting Keggin-type POMs and sulfurizing reagents in organic solvents, the 12 terminal (surface) WO groups are selectively converted to WS groups without undesirable structural changes or over-sulfurization. We also show that the resultant POTMs exhibit high stability and unique electronic states and redox properties, indicating that this sulfurizing method enables the post-editing sulfurization of POMs with various structures, constituent elements, electronic states, and physicochemical properties.
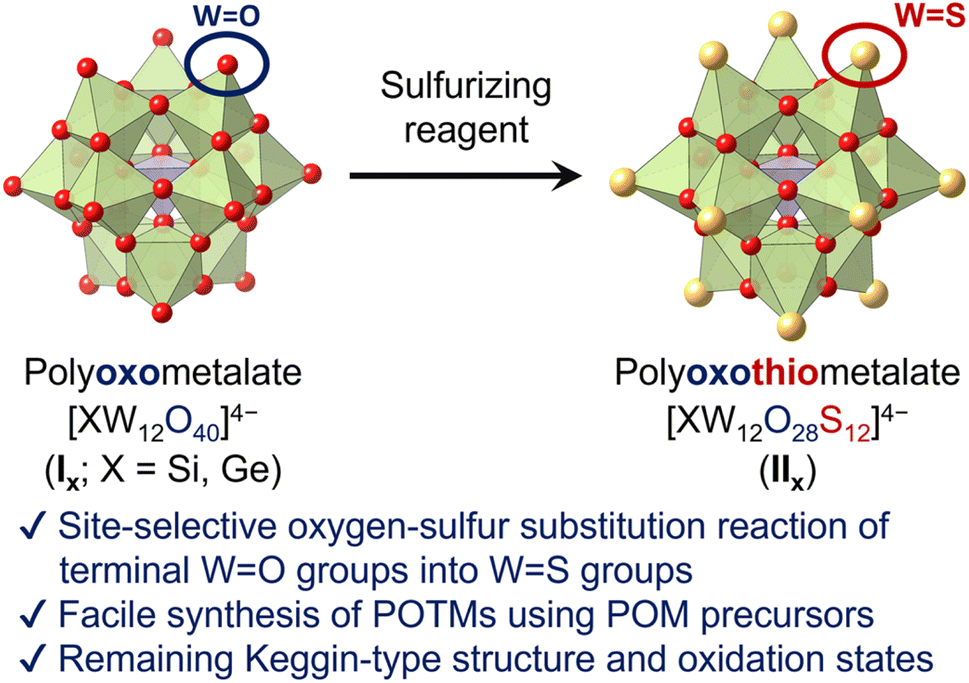 | ||
| Fig. 1 Schematic of the Keggin-type POTMs [XW12O28S12]4− (X = Si, Ge) synthesized via site-selective sulfurization. | ||
Results and discussion
We first examined the oxygen atom substitution of Keggin-type silicotungstate ([SiW12O40]4−; ISi) to sulfur atoms using bis(trimethylsilyl)sulfide (TMS2S), which is a widely employed sulfurizing reagent for d0 metal–oxo species.12 However, the electrospray ionization (ESI)-mass spectrum showed that the oxygen atom substitution of ISi did not proceed using TMS2S (12 equivalents with respect to ISi) in acetonitrile (Fig. S1a†). This result was consistent with that of previous reports showing that the reaction of Nb- or Ta-substituted polyoxotungstates and trialkylsilylsulfides converted the NbO and TaO groups into the NbS and TaS groups, respectively, while the other oxygen atoms remained intact.11a,c
Accordingly, we investigated the reactivity of several sulfurizing reagents toward POMs. When the tetra-n-butylammonium (TBA) salt of ISi (TBA4[SiW12O40]) was reacted with Lawesson's reagent13 (three equivalents with respect to ISi) in acetonitrile at room temperature (∼25 °C), the reaction solution turned colorless to yellow (see ESI† for details). The positive-ion ESI-mass spectrum of the reaction solution revealed a series of signals with the m/z = 16 (z = 1) difference, indicating that the oxygen atoms of ISi were substituted with sulfur atoms (Fig. S2a†).
After further modification of the reaction conditions, the use of six equivalents of Lawesson's reagent provided an ESI-mass spectrum with two prominent signal sets at m/z = 4036.972 and 4278.218 (Fig. 2 and S2†). These signal sets were assigned to [TBA4H(SiW12O28S12)]+ (theoretical m/z = 4037.059) and [TBA5(SiW12O28S12)]+ (theoretical m/z = 4278.336), showing that 12 out of 40 oxygen atoms were substituted with sulfur atoms to form [SiW12O28S12]4− (IISi). In addition, even after the reaction of ISi with excess amounts of Lawesson's reagent (9, 12, and 20 equivalents with respect to ISi), the ESI-mass spectra showed that sulfur atoms were not further introduced into ISi, showing that Lawesson's reagent can selectively convert ISi to IISi (Fig. S2c–e†). Based on the above results and elemental analysis, the formula of IISi was determined as TBA4[SiW12O28S12]. Note that the ESI-mass spectrum of IISi in acetonitrile containing water (ca. 2000 equivalents with respect to IISi) under air showed no significant changes, revealing the high stability of IISi against water and O2 (Fig. S3†). When diphosphorus pentasulfide was used as a sulfuring reagent, ISi was not completely converted to IISi, and several terminal oxygen atoms remained likely due to the very low solubility of diphosphorus pentasulfide in acetonitrile (Fig. S1b†). In contrast, although triphenylphosphine sulfide and dimethyl trisulfide exhibited good solubility, they did not react with ISi under the same conditions (Fig. S1c and d†).
Since X-ray crystallographic analysis of the TBA salt of IISi was unsuccessful likely because of the flexibility of TBA cations, crystallographic analysis was performed using the tetraphenylphosphonium (TPP) salt, which was obtained via cation exchange reaction of IISi with TPPBr (see ESI† for detail). The elemental analysis revealed that the formula of the TPP salt was TPP4[SiW12O28S12], showing that 12 sulfur atoms were retained and all TBA cations were exchanged with TPP cations. X-ray crystallographic analysis of the TPP salt of IISi revealed that the α-Keggin-type {SiW12} structure was retained, and all 12 terminal oxygen atoms of ISi (i.e., the WO groups in [SiW12O40]4−) were substituted with sulfur atoms (Fig. 3a, S4 and Table S1†). Notably, 28 other oxygen atoms remained, that is, four μ4-oxo atoms surrounding the heteroatom (Si) and 24 μ2-oxo atoms bridging the polyatoms (W). These results were consistent with the aforementioned ESI-mass analysis, showing that 12 out of 40 oxygen atoms were substituted with sulfur atoms upon reaction with Lawesson's reagent (Fig. 2). The WS bond lengths in IISi ranged from 2.11 to 2.17 Å, clearly longer than the terminal WO bonds of ISi (1.63–1.74 Å).14 The bond valence sum (BVS) values of the sulfur atoms ranged from 1.80 to 2.13 (Table S2†), indicating that each sulfur atom formed a double bond with a tungsten atom (WS group). The BVS values of the Si (3.89, 3.90) and W (6.12–6.61) atoms also showed that their oxidation states remained at +4 and +6, respectively (Table S3†).
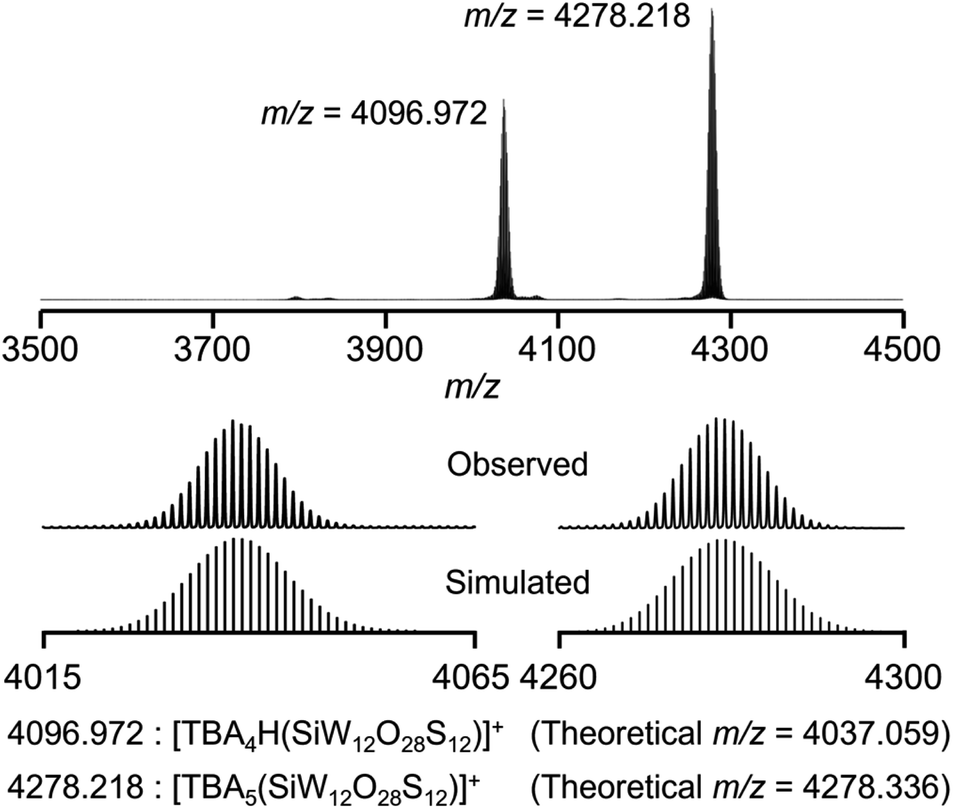 | ||
| Fig. 2 Positive-ion ESI-mass spectrum of IISi in acetonitrile. | ||
The Raman spectrum of ISi showed prominent peaks corresponding to the stretching vibrations of the WO bonds (967 and 988 cm−1; Fig. 3b).15 In contrast, the Raman spectrum of IISi depicted no stretching vibrations of the WO bonds, but clearly illustrated those corresponding to the WS bonds in the 500–600 cm−1 region (Fig. 3b).16 The FT-IR spectrum of IISi also showed the sharp peak at 493 cm−1 assignable to the stretching vibration of WS bonds (Fig. S5†). These results supported the successful synthesis of the Keggin-type POTM [SiW12O28S12]4− (IISi) via the oxygen–sulfur substitution reactions of WO into WS. This is the first report on the synthesis of a structurally defined POTM, in which all the terminal WO groups of parent POM were converted to WS. Previously reported Keggin-type POTMs [γ-XW10O36(M5+2S2O2)]n− (X = Si, P; M = W, Mo) were synthesized by introducing the [M5+2S2O2]2+ moiety into the vacant sites of lacunary POMs [XW10O36]n−, wherein two S atoms bridged two M5+ atoms of the [M5+2S2O2]2+ moiety (i.e., M5+–S2−–M5+).11b,c In contrast, we demonstrated that the direct oxygen–sulfur substitution reaction of POMs enabled the selective incorporation of sulfur atoms at terminal (surface) sites (i.e., W6+ = S).
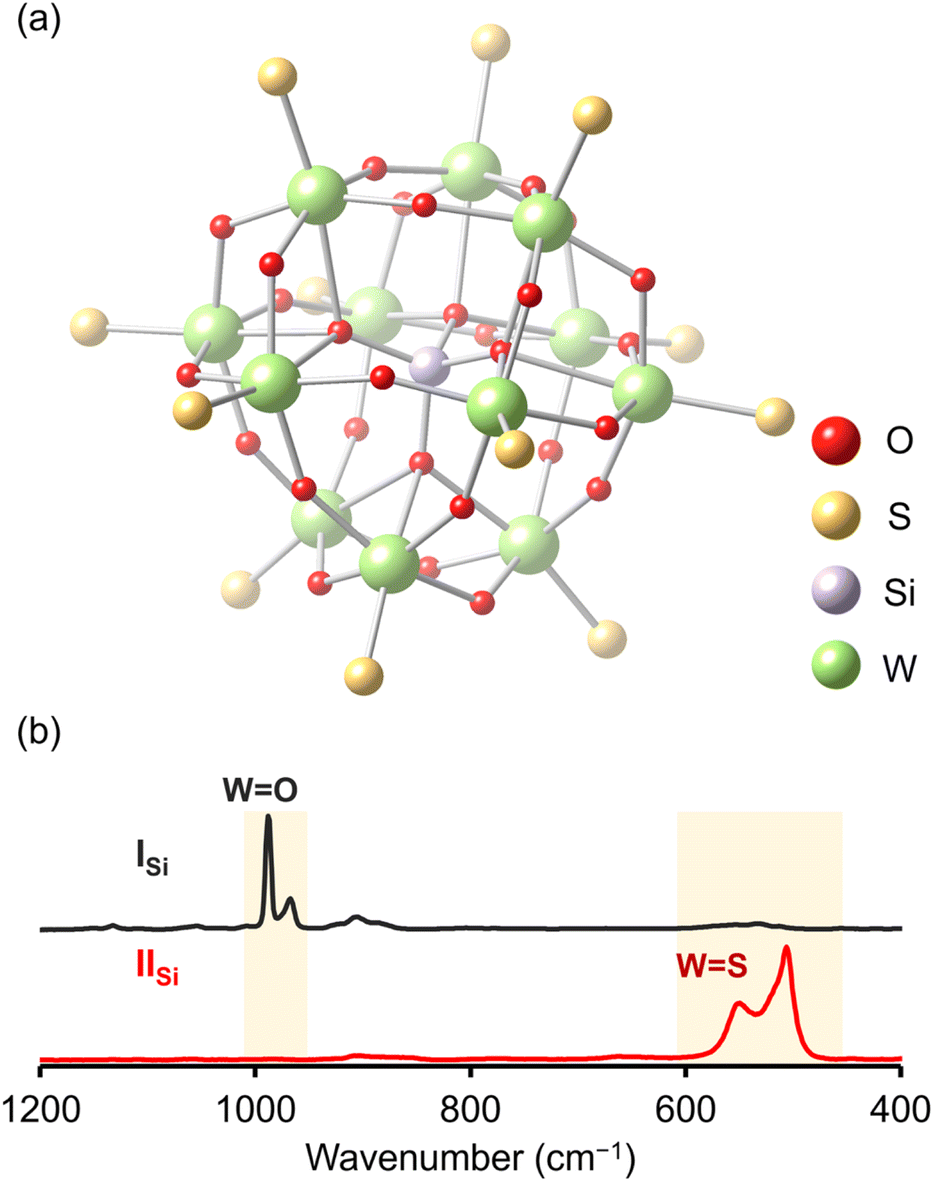 | ||
| Fig. 3 (a) Ball-and-stick representation of the crystal structure of the anion part of IISi. (b) Raman spectra of ISi (black line) and IISi (red line). | ||
The UV-vis spectrum of IISi in acetonitrile exhibited a prominent absorption band at λ = 274 nm (ε = 2.1 × 105 L mol−1 cm−1), which was observed with a higher intensity at a lower wavelength compared with that of ISi (λ = 264 nm; ε = 4.6 × 104 L mol−1 cm−1) (Fig. 4a). The acetonitrile solution of ISi was colorless and showed no absorption band in the visible light region, whereas that of IISi was pale yellow and exhibited weak absorption bands up to approximately 470 nm (Fig. S6†). These results indicated that the introduction of the sulfur atoms led to a drastic change in the electronic state. Thus, to investigate the electronic state, we conducted density functional theory (DFT) calculations on ISi and IISi. In the case of ISi, the highest occupied molecular orbital (HOMO) was mainly derived from the bridging μ2-oxo atoms (Fig. 4b and S7†). In contrast, with the introduced sulfur atoms, the occupied orbitals of IISi (HOMO–HOMO−12) were mainly derived from the terminal S 3p orbitals. The HOMO–LUMO energy gap of IISi became smaller than ISi (6.85 eV for ISi and 5.86 eV for IISi, Fig. 4b and S8†). The lowest unoccupied molecular orbitals (LUMOs) of ISi and IISi were mainly derived from W 5d orbitals. Based on the time-dependent (TD) DFT study, the absorption bands of ISi were assigned to the ligand-to-metal charge transfer from the oxygen atoms to the tungsten atoms. Meanwhile, the absorption bands of IISi in the UV region (λmax = 274 nm) were mainly attributed to the excitation from the S 3p orbitals (HOMO–HOMO−12) and the W–S bonding orbitals (HOMO−13–HOMO−19) to the W–S antibonding orbitals (LUMO+2–LUMO+5). In addition, the broad absorption bands at the longer wavelengths (λ > 350 nm) were mainly attributed to the excitation from the S 3p orbitals and the W–S bonding orbitals to the W 5d orbitals (LUMO, LUMO+1, LUMO+6–LUMO+8) (Fig. S9†). These results revealed the significant contribution of sulfur atoms in the optical properties of IISi.
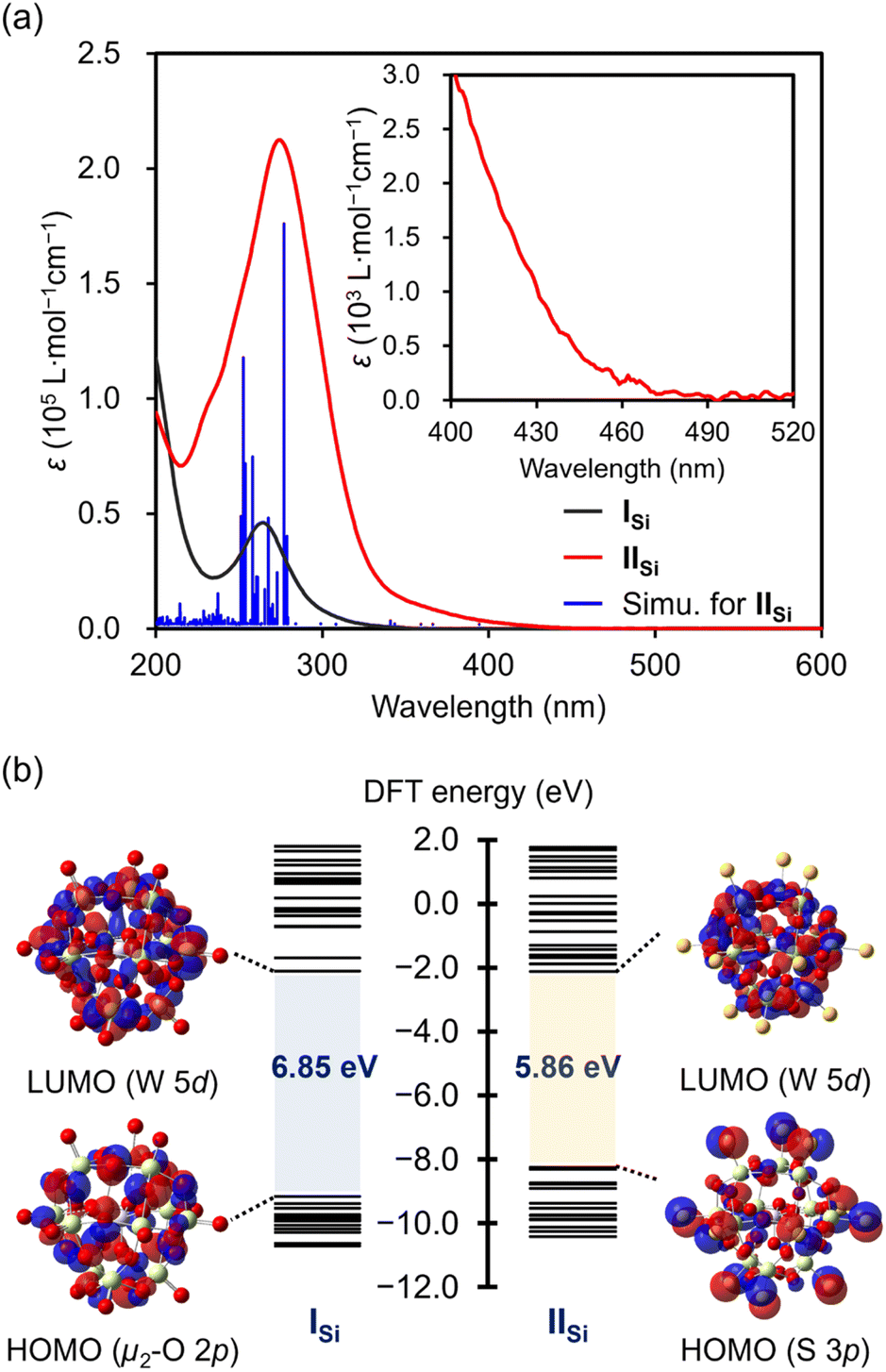 | ||
| Fig. 4 (a) UV-vis spectra of ISi (black line) and IISi (red line), and simulation of IISi based on the TD-DFT study (blue bar). (b) Energy diagrams and visualization of the HOMO and the LUMO of ISi and IISi. | ||
We evaluated the redox behavior of IISi using cyclic voltammetry in acetonitrile. POTM IISi exhibited stable redox cycles, indicating high stability during the reduction/reoxidation reactions (Fig. S10†). Two reduction waves of IISi were observed at −1.08 and −1.48 V (vs. Ag/Ag+ reference electrode), showing that the first redox potential of IISi was similar to that of ISi, whereas the second redox potential shifted from that of ISi (−1.62 V) after the oxygen atom substitution to sulfur atoms.
Finally, in addition to the sulfurization of Si-centered Keggin-type POM (ISi), we investigated the site-selective sulfurization of Ge-centered [GeW12O40]4− (IGe) using Lawesson's reagent. ESI-mass spectrum revealed that site-selective sulfurization of IGe also proceeded to form [GeW12O28S12]4− (IIGe) (Fig. S11†). These results suggest that this method is potentially applicable to sulfurization of various heteropolyoxotungstates.
Conclusions
We have developed a novel synthetic method for polyoxothiotungstates through direct site-selective substitution of terminal oxo ligands (WO groups) of polyoxotungstates into sulfide ligands (WS groups) via reaction with sulfurizing reagents (e.g., Lawesson's reagent). The resulting POTMs (i.e., IIX, X = Si, Ge) retained the parent Keggin-type structure (i.e., IX), while their electronic states and redox properties drastically changed from those of IX. This finding holds the potential to pave the way for the development of the sulfurization method for a broad range of POMs, allowing atomically precise design of molecular metal oxides and sulfides with diverse structures, constituent elements, compositions, and properties. We believe that such advancements will lead to the diverse applications of POMs and related materials, including (photo)catalysis, sensing, optics, energy conversion, and batteries.
Data availability
The data supporting this manuscript is available in the ESI† of and available on request. Crystallographic data for a TPP salt of IISi has been deposited at the CCDC (deposition number 2322226).Author contributions
K. Yo. and K. S. design the project and experiments. K. Yo. performed the major parts of experiments. K. Yo, K. S., and K. Ya cowrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from JSPS KAKENHI (21K20552, 22H04971, 24H02210), JST FOREST (JPMJFR213M), and Mizuho Foundation for the Promotion of Sciences, JSPS Core-to-Core program. Some of the computations were performed using the Research Center for Computational Science, Okazaki, Japan (Project: 23-IMS-C106, 24-IMS-C101). Single-crystal X-ray diffraction measurements at SPring-8 were conducted with the approval of the Japan Synchrotron Radiation Research Institute (proposal numbers 2023A1731, 2023B1842).Notes and references
- (a) H. Beinert, R. H. Holm and E. Münck, Science, 1997, 277, 653 CrossRef CAS PubMed; (b) K. Tanifuji, S. Ohta, Y. Ohki and H. Seino, Coord. Chem. Rev., 2023, 475, 214838 CrossRef CAS; (c) J. Farzana, R. Aqsa, M. Sawaira, H. Junaid, N. Walid, H. Ali, I. Muhammad, N. Ghazanfar, A. Mansur, I. Muhammad, K. Qasim, A. Ghafar, K. Maaz, A. Waqas and M. Muhammad, ACS Appl. Nano Mater., 2023, 6, 7077 CrossRef.
- A. Piątkowska, M. Janus, K. Szymański and S. Mozia, Catalysts, 2021, 11, 144 CrossRef.
- A. Tanimu and K. Alhooshani, Energy Fuels, 2019, 33, 2810 CrossRef CAS.
- (a) M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer, Berlin, 1983 CrossRef; (b) M. Sadakane and E. Steckhan, Chem. Rev., 1998, 98, 219 CrossRef CAS PubMed; (c) H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572 RSC; (d) H. N. Miras, J. Yan, D.-L. Long and L. Cronin, Chem. Soc. Rev., 2012, 41, 7403 RSC; (e) M. Nyman and P. C. Burns, Chem. Soc. Rev., 2012, 41, 7354 RSC; (f) I. A. Weinstock, R. E. Schreiber and R. Neumann, Chem. Rev., 2018, 118, 2680 CrossRef CAS PubMed; (g) M. Lechner, R. Güttel and C. Streb, Dalton Trans., 2016, 45, 16716 RSC; (h) K. Suzuki, N. Mizuno and K. Yamaguchi, ACS Catal., 2018, 8, 10809 CrossRef CAS; (i) S. Uchida, Chem. Sci., 2019, 10, 7670 RSC; (j) J. M. Cameron, G. Guillemot, T. Galambos and S. S. Amin, Chem. Soc. Rev., 2022, 51, 293 RSC.
- (a) S.-T. Zheng and G.-Y. Yang, Chem. Soc. Rev., 2012, 41, 7623 RSC; (b) B. S. Bassil and U. Kortz, Z. Anorg. Allg. Chem., 2010, 636, 2222 CrossRef; (c) K. Suzuki, N. Mizuno and K. Yamaguchi, J. Jpn. Petrol. Inst., 2020, 63, 258 CrossRef CAS.
- (a) T. Minato, D. Salley, N. Mizuno, K. Yamaguchi, L. Cronin and K. Suzuki, J. Am. Chem. Soc., 2021, 143, 12809 CrossRef CAS PubMed; (b) K. Yonesato, D. Yanai, S. Yamazoe, D. Yokogawa, T. Kikuchi, K. Yamaguchi and K. Suzuki, Nat. Chem., 2023, 15, 940 CrossRef CAS PubMed.
- (a) A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009 CrossRef CAS PubMed; (b) A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382 RSC; (c) A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605 RSC; (d) C. Li, N. Mizuno, K. Yamaguchi and K. Suzuki, J. Am. Chem. Soc., 2019, 141, 7687 CrossRef CAS PubMed; (e) M. Yamaguchi, K. Shioya, C. Li, K. Yonesato, K. Murata, K. Ishii, K. Yamaguchi and K. Suzuki, J. Am. Chem. Soc., 2024, 146, 4549 CrossRef CAS PubMed.
- J. Breibeck, N. I. Gumerova and A. Rompel, ACS Org. Inorg. Au, 2022, 2, 477 CrossRef CAS PubMed.
- J. Jurczyk, J. Woo, S. F. Kim, B. D. Dherange, R. Sarpong and M. D. Levin, Nat. Synth., 2022, 1, 352 CrossRef PubMed.
- (a) A. Elliott and H. N. Miras, J. Coord. Chem., 2022, 75, 1467 CrossRef CAS; (b) S. Batool, M. Langer, S. Nagaraju, M. Heiland, D. Eder, C. Streb and A. Cherevan, Adv. Mater., 2024, 36, 2305730 CrossRef CAS PubMed; (c) E. Cadot, M. N. Sokolov, V. P. Fedin, C. Simonnet-Jégat, S. Floquet and F. Sécheresse, Chem. Soc. Rev., 2012, 41, 7335 RSC; (d) E. Cadot, V. Béreau, B. Marg, S. Halut and F. Sécheresse, Inorg. Chem., 1996, 35, 3099 CrossRef CAS PubMed; (e) E. Cadot, V. Béreau, B. Marg, S. Halut and F. Sécheresse, Inorg. Chim. Acta, 1996, 252, 101 CrossRef CAS.
- (a) W. G. Klemperer and C. Schwartz, Inorg. Chem., 1985, 24, 4459 CrossRef CAS; (b) E. Cadot, V. Béreau and F. Sécheresse, Inorg. Chim. Acta, 1995, 239, 39 CrossRef CAS; (c) E. Radkov, Y.-J. Lu and R. H. Beer, Inorg. Chem., 1996, 35, 551 CrossRef CAS.
- J. P. Donahue, Chem. Rev., 2006, 106, 4747 CrossRef CAS PubMed.
- (a) O. Turan, E. Erdal and M. Olcay, Chem. Rev., 2007, 107, 5210 CrossRef PubMed; (b) D. V. Partyka, R. J. Staples and R. H. Holm, Inorg. Chem., 2003, 42, 7877 CrossRef CAS PubMed.
- (a) C. Ritchie, E. Burkholder, P. Kögerler and L. Cronin, Dalton Trans., 2006, 1712 RSC; (b) K. Uehara, H. Nakao, R. Kawamoto, S. Hikichi and N. Mizuno, Inorg. Chem., 2006, 45, 9448 CrossRef CAS PubMed.
- R. Thouvenot, M. Fournier, R. Franck and C. Rocchiccioli-Deltcheff, Inorg. Chem., 1984, 23, 598 CrossRef CAS.
- B. Knies and I. Hartenbach, Z. Anorg. Allg. Chem., 2022, 648, e202200270 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Table S1–S3, and Fig. S1–S11. CCDC 2322226. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02912g |
| This journal is © The Royal Society of Chemistry 2024 |