
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of benzofuran-derived sulfamates as dual aromatase-steroid sulfatase inhibitors (DASIs): design, synthesis and biological evaluation†
Ahmed G.
Eissa
 abc,
Francesca
Gozzi
a,
Oqab
Aloqab
a,
Charlotte E.
Parrish
d,
Nadira
Mohamed
d,
Irene
Shiali
a,
Harith
Al-Baldawi
a,
Paul A.
Foster
d and
Claire
Simons
*a
abc,
Francesca
Gozzi
a,
Oqab
Aloqab
a,
Charlotte E.
Parrish
d,
Nadira
Mohamed
d,
Irene
Shiali
a,
Harith
Al-Baldawi
a,
Paul A.
Foster
d and
Claire
Simons
*a
aSchool of Pharmacy and Pharmaceutical Sciences, Cardiff University, King Edward VII Avenue, Cardiff, CF10 3NB, UK. E-mail: simonsc@cardiff.ac.uk
bCollege of Pharmacy, Al Ain University, Abu Dhabi, United Arab Emirates
cDepartment of Medicinal Chemistry, Faculty of Pharmacy, Zagazig University, Zagazig P.C. 44519, Egypt
dDepartment of Metabolism and Systems Science, School of Medical Sciences, College of Medicine and Health, University of Birmingham, Birmingham, B15 2TT, UK
First published on 9th January 2025
Abstract
Resistance of oestrogen receptor-positive (ER+) breast cancer, the most prevalent type of breast cancer accounting for ∼70% of all cases, to current therapies necessitates the study of alternative strategies. One promising strategy is the multi-targeting approach using dual aromatase-steroid sulfatase inhibitors (DASIs). Herein, we describe the development of DASIs using a common benzofuran pharmacophore. Triazole benzofuran sulfamates were found to have low nM aromatase (Arom) inhibitory activity but no steroid sulfatase (STS) inhibitory activity (IC50 > 10 μM); by contrast, benzofuran ketone sulfamates demonstrated low nM STS inhibitory activity but no Arom inhibitory activity (IC50 > 1 μM). The addition of a methyl group at the 3rd position of the benzofuran ring in the benzofuran ketone sulfamate 19 (R1 = CH3) had a notable effect, resulting in dual aromatase and STS inhibitory activities with the 4-chloro derivative 19b (Arom IC50 = 137 nM, STS IC50 = 48 nM) and 4-methoxy derivative 19e (Arom IC50 = 35 nM, STS IC50 = 164 nM) optimal for dual inhibition. Arom/STS inhibition results combined with molecular dynamics studies provided a clear rationale for the activity observed.
Introduction
Considering the treatment of breast cancer, multi-targeting is a very popular research approach owing to the development of drug resistance. Thus, a wide variety of multi-targeted drugs have been developed. Oestrogen receptors (ERs), HER-2, EGFR, VEGFR, PI3K, mTOR, 17β-HSD1, aromatase (CYP19A1) and steroid sulfatase (STS) have all been targets for the development of dual acting inhibitors.1–3 Each of the dual inhibitor target combinations was inspired by the crucial role played by the target in the viability of tumours in different subclasses of breast cancer.In ER-positive (ER+) breast cancer, which accounts for ∼70% of all breast cancers,4 oestrogens promote tumour growth.5 Approximately ten times more oestrone (E1) is derived from oestrone sulfate (E1S) via STS than that from androstenedione via aromatase.6,7 Therefore, the simultaneous dual inhibition of oestrogen synthesis from androgens through aromatase and oestrogen sulfates (E1S and E2S) through STS (Fig. 1) was an interesting approach, leading to the development of several dual aromatase/STS inhibitors (DASIs).8,9
 | ||
| Fig. 1 Biosynthesis of oestrogenic and androgenic steroids. AROM = aromatase (CYP19A1), STS = steroid sulfatase, and 17β-HSD = 17β-hydroxysteroid dehydrogenase (types 1, 2 and 3). | ||
Merging the active pharmacophores for inhibition of the two enzymes was achieved via incorporation of a phenol sulfamate moiety responsible for STS activity into an aromatase inhibitor scaffold depending upon X-ray structures of enzyme–ligand complexes, docking and extensive structure–activity relationship (SAR) studies. Various DASIs were developed over time with activity ranging from a reasonable nanomolar range to outstanding picomolar values for DASIs developed from letrozole and anastrozole aromatase inhibitors and STS inhibitors (Fig. 2).8–14
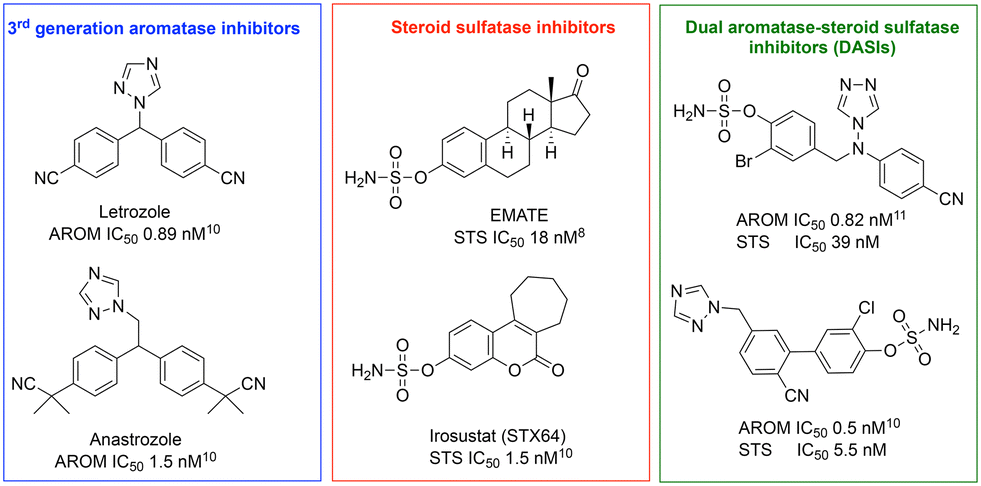 | ||
| Fig. 2 Potent dual aromatase-steroid sulfatase inhibitors (DASIs) based on third-generation aromatase inhibitors, letrozole and anastrozole, and steroid sulfatase inhibitors.8,10,11 | ||
We have previously described potent aromatase inhibitors with a benzofuran/triazole scaffold,15–17 and the research described herein uses the design-in method to build DASIs through incorporation of the sulfamoyl group, required for STS activity, into the phenol scaffold of aromatase inhibitors (Fig. 3). The general idea was to compare three different positions for the sulfamate group: C6 of the benzofuran ring, C5 of the benzofuran ring and C4 of the phenyl ring. Moreover, varying the substituent on the C4 position of the phenyl ring in the 6-benzofuran derivative from chloro to fluoro or nitrile groups has been previously identified as optimal for aromatase inhibitory activity16,17 to help build a clearer SAR (Fig. 3).
 | ||
| Fig. 3 Two sets of comparisons with the structure variations represented in bold; varying position of sulfamate indicated in red and varying phenyl substituent indicated in green. | ||
Results and discussion
Triazole benzofuran sulfamates and carbamates
Starting with the phenol scaffold of the aromatase inhibitors,15–17 preparing the DASI involved two consecutive steps. First sulfamoyl chloride was prepared in situ from the reaction of chlorosulfonyl isocyanate and formic acid13,18 and used without further purification for reaction with phenolic compounds 1–5 (ref. 15–17) to produce the required sulfamate compounds 6–10 (Fig. 4).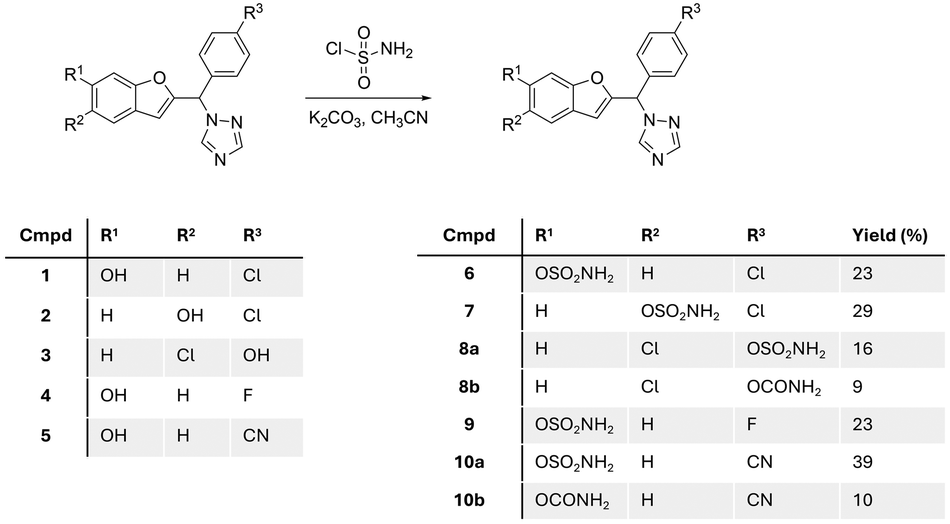 | ||
| Fig. 4 Prepared benzofuran triazole sulfamate and carbamate derivatives 6–10. | ||
Five different sulfamate derivatives were successfully prepared through this synthetic pathway; however, even with the addition of excess base and/or excess sulfamoyl chloride, the reaction did not reach completion with yields ranging from 16% to 39% (Fig. 4). On two occasions, a carbamate compound was also formed as a side product (8b and 10b), in addition to the expected sulfamate (now labelled as 8a and 10a),which may be attributed to the presence of moisture in the in situ preparation of sulfamoyl chloride, resulting in the hydrolysis of the isocyanate starting material. The identity of the sulfamate compounds was confirmed by 1H NMR, which showed the appearance of a broad singlet signal integrating for two protons of the amine group at around 8.0 ppm, with the disappearance of the broad singlet integrating for one proton of the phenolic OH group in the range 9–10 ppm (Fig. S1†). The carbamate side product was identified by 1H NMR, which showed the appearance of two separate broad singlet signals each integrating for one proton between 6.5 and 7.5 ppm (Fig. S1†), related to the tautomerism of the amidic group. 13C NMR supported this assumption by the presence of an extra quaternary carbon in the carbamate product when compared with the 13C NMR spectrum of the sulfamate compound. HRMS of the sulfamate compound and the carbamate side product showed a difference of 36 between the mass ion of the sulfamate and the carbamate. Further verification of the carbamate side product was performed by the intentional synthesis of the carbamate compound using chlorosulfonyl isocyanate/H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 instead of sulfamoyl chloride and comparing the product from the two methods.
19 instead of sulfamoyl chloride and comparing the product from the two methods.
Seven compounds including the two carbamate side products were investigated for their aromatase inhibitory activity at 10 nM using the previously reported modified tritiated water assay.20 All compounds, except compound 8a, showed more than 50% aromatase inhibition and were further investigated for IC50 confirming retention of aromatase inhibitory activity in the low nanomolar range (Table 1). The results provided some preliminary SARs in terms of position and nature of substituents and showed that the carbamate side products 8b and 10b were more active against the aromatase enzyme. There was clear superiority for the 6-position of benzofuran for the sulfamate group over the 5-benzofuran or the 4-phenyl positions. Moreover, the chloro and cyano derivatives 6 and 10 displayed improved aromatase inhibitory activity compared with the fluoro derivative 9 (Table 1).
Although low nanomolar aromatase activity was observed for the benzofuran triazole derivatives, no inhibitory activity was observed against STS (IC50 ≥ 10 μM) using the previously described JEG-3 lysate assay.21 A similar profile of nanomolar aromatase inhibition and >10 μM STS inhibitory activity has been reported for vorozole-derived sulfamates having a similar geometry to the benzofuran derivatives described herein,13,14 which was suggested to arise from the triazole ring causing steric hindrance within the STS active site. To investigate the relationship between the lack of STS inhibitory activity and the geometric orientation of the compounds 6–10, a series of truncated sulfamate and carbamate compounds 18–21 (Scheme 1), lacking the triazole group but retaining the benzofuran scaffold, were designed based on STX64 (irosustat) (Fig. 1) structural similarity. These truncated compounds had the triazole group replaced by a carbonyl group to offer some planarity and rigidity to the structure.
 | ||
| Scheme 1 Reagents and conditions: (i) K2CO3, CH3CN, 70 °C, 3 h, (ii) HCl/dioxane, 1 h, (iii) method 1: sulfamoyl chloride, K2CO3, CH3CN or DMF, 16 h. Method 2: sulfamoyl chloride, DMA, 3 h, (iv) chlorosulfonyl isocyanate, CH3CN, 1.5–2 h, then H2O, 1 h [R2: a = 4-F, b = 4-Cl, c = 4-Br, d = 2,4-diCl, e = 4-OCH3, f = 4-CN]. | ||
Ketone benzofuran sulfamates and carbamates
The ketone sulfamates 18/19 and two carbamate compounds 20/21 were prepared via a three-step synthetic pathway. The first step employed a Rap–Stoermer condensation reaction22,23 between tetrahydropyran (THP)-protected 2-hydroxybenzaldehyde (11) or THP-protected 2-hydroxyacetophenone (12)24,25 and 2-bromo-acetophenone (13) to form the THP-protected benzofuran ketones 14/15. Anomalous behaviour was observed for the 3-methylbenzofuran 2,4-dichloro derivative 15d with the THP deprotected compound 17d in a yield of 45%. The THP protecting in all other derivatives (14/15) was then removed using HCl/dioxane26 to give the phenolic derivatives 16/17. Using the in situ formation of sulfamoyl chloride described for the preparation of the triazole compounds 6–10 resulted in very low yields (3–7%) of the benzofuran ketone sulfamates 18/19; however, a commercial source was available with improved yields obtained (Table 2) using K2CO3 as the base and DMF solvent,18 although it should be noted that the sulfamoylation reaction did not go to completion even using three molecular equivalents of sulfamoyl chloride.| Cmpd | R1 | R2 | Yield (%) | m.p. (°C) | |
|---|---|---|---|---|---|
| Method 1 | Method 2 | ||||
| NR no reaction observed. | |||||
| Sulfamates | |||||
| 18a | H | 4-F | NR | 76 | 200–202 |
| 18b | H | 4-Cl | 18 | — | 159–161 |
| 18c | H | 4-Br | 46 | — | 166–168 |
| 18d | H | 2,4-diCl | 33 | — | 198–200 |
| 18e | H | 4-OCH3 | 52 | — | 182–184 |
| 18f | H | 4-CN | NR | 74 | 200–204 |
| 19a | CH3 | 4-F | 60 | — | 181–183 |
| 19b | CH3 | 4-Cl | 32 | — | 181–182 |
| 19c | CH3 | 4-Br | 25 | — | 178–181 |
| 19d | CH3 | 2,4-diCl | NR | 22 | 138–140 |
| 19e | CH3 | 4-OCH3 | 20 | 56 | 172–176 |
| 19f | CH3 | CN | NR | NR | — |
| Carbamates | |||||
| 20 | H | 4-Cl | 58 | 190–192 | |
| 21 | CH3 | 4-Cl | 63 | 210–212 | |
For derivatives 18a, 18f and 19d, no sulfamate product was obtained using this method (method 1, Table 2); however, using the method of Okada et al.,27 which employed two equivalents of sulfamoyl chloride, DMA as solvent and no base (method 2, Table 2), the sulfamates were successfully obtained, and in the case of 19e, an improved yield was obtained with method 2 compared with method 1 (56% vs. 20%). The preparation of the nitrile derivative 19f using either method 1 or 2 was unsuccessful with no product observed. The carbamates were obtained in good yields on reaction of the phenolic derivatives 16/17 with chlorosulfonyl isocyanate followed by hydrolysis of the (chlorosulfonyl)carbamate intermediate.19
The sulfamates 18/19 and carbamates 20/21 were evaluated for inhibitory activity against aromatase20 and STS21 (Table 3). The sulfamates 18a–f displayed low aromatase inhibitory activity (Arom IC50 > 1 μM); however, all displayed sub micromolar STS inhibitory activity (STS IC50 0.115–0.74 μM), with the 4-methoxy derivative 18e (STS IC50 0.115 μM) and 4-nitrile derivative 18f (STS IC50 0.28 μM) optimal.
| Cmpd | Arom IC50 (μM) | 95% CIa (μM) | STS IC50 (μM) | 95% CIa (μM) |
|---|---|---|---|---|
| a Each data point measured in triplicate and the error in the IC50 calculation represented as 95% confidence interval (CI). | ||||
| 18a | >1 | — | 0.74 | 0.657–0.828 |
| 18b | >1 | — | 0.41 | 0.372–0.447 |
| 18c | >1 | — | 0.51 | 0.385–0.673 |
| 18d | >1 | — | 0.31 | 0.240–0.399 |
| 18e | >1 | — | 0.115 | 0.083–0.159 |
| 18f | >1 | — | 0.28 | 0.198–0.627 |
| 19a | 0.050 | 0.037–0.089 | 0.654 | 0.471–0.778 |
| 19b | 0.137 | 0.114–0.151 | 0.048 | 0.023–0.058 |
| 19c | 0.022 | 0.012–0.027 | 0.83 | 0.516–2.816 |
| 19d | 0.020 | 0.013–0.030 | 1.503 | 0.937–2.412 |
| 19e | 0.035 | 0.018–0.067 | 0.164 | 0.089–0.299 |
| 20 | >1 | — | 1.22 | — |
| 21 | >1 | — | 0.98 | — |
| Letrozole | 0.007 | — | >1 | — |
| STX64 | >1 | — | 0.006 | — |
The addition of a methyl group at the 3-position of the benzofuran ring in sulfamates 19a–e had a notable effect, resulting in dual aromatase and STS inhibitory activity with the 4-chloro derivative 19b (Arom IC50 137 nM, STS IC50 48 nM) and 4-methoxy derivative 19e (Arom IC50 35 nM, STS IC50 164 nM) optimal for dual inhibition. The two carbamates 20/21 did not show promising inhibitory activity (Arom/STS IC50 ≥ 1 μM).
Computational studies
 | ||
| Fig. 5 Exemplar triazoles 6 and 10b binding within the CYP19A1 active site after MD simulation (150 ns). Haem is shown in orange with the central iron as a brown sphere, H2O molecules shown as red spheres, H-bonding cyan lines/barrels, hydrophobic interactions (van der Waals, π–π stacking) shown as green lines/barrels, and key binding amino acids in light grey. | ||
 | ||
| Fig. 6 2D LigPlot figures of exemplar ketone sulfamates 18e and 19e within the CYP19A1 active site after MD simulation (150 ns), illustrating all amino acids within the CYP19A1 active site surrounding the sulfamate ligands. | ||
The R-enantiomer of the triazole derivatives formed a Fe–N interaction via N2 of the triazole, while the S-enantiomers formed a preferable (perpendicular) Fe–N interaction via N4 of the triazole. The sulphonamide and carbamate moieties formed H-bonding interactions with two or three key amino acids, namely Asp309, Ser478 and His480 with van der Waals or π–π stacking interactions observed between the benzofuran ring/phenyl group and Trp224 (R-enantiomers) or Thr310 (S-enantiomers) and Phe221 (R-6 and S-10b). The nitrile group of the triazole derivative R/S-10b also formed a H-bonding interaction with Met374 (Fig. 5).
Generally, the ketone sulfamates 18 interacted indirectly (type I binding) with haem via a water molecule (e.g.18e, Fig. 6), while the 3-methyl derivatives 19 either formed a direct (type II) binding Fe–O interaction with the haem through the carbonyl group (e.g.19e, Fig. 6) or was positioned close to the haem. However, no haem interaction, direct or indirect, was observed for the carbamates 20 and 21. For all the ketone derivatives 18–21, the sulfamate and carbamate moieties formed the same H-bonding interactions observed with the triazole derivative 6–10, most commonly with Asp309, Ser478 and Leu479.
 | ||
| Fig. 7 Steroid sulfatase (STS) catalytic cycle and point of STS inhibition. | ||
Computational studies for STS first involved modification of the FGS75 amino acid in the crystal structure of human placental oestrone sulfatase (pdb: 8EG3)37 to the diol (FGH) using the Builder tool in MOE. Waters and additional ligands were removed leaving the protein and Ca2+ and the resulting FGH-sulfatase protein prepared for docking to generate protein–ligand complexes. The protein–ligand complexes were then subject to 150 ns molecular dynamics simulations. The exemplar sulfamates 18f and 19b are used for the illustration of binding interactions (Fig. 8).
 | ||
| Fig. 8 Exemplar ketone sulfamates 18f and 19b binding in the STS active site after MD simulation (150 ns). The Ca2+ cation is shown as an orange sphere, H2O molecules shown as red spheres, H-bonding cyan lines/barrels, and key binding amino acids in light grey. Pocket grid colours: green for lipophilic and pink for hydrophilic. | ||
The benzofuran sulfamates 18 and 19 sit within the binding pocket, with the benzofuran moiety in the lipophilic domain, while the sulfamate moiety is positioned in the catalytic hydrophilic domain. In the catalytic domain, there are ten important residues: Asp35, Asp36, FGS75, Arg79, Lys134, His136, His290, Asp342, Lys368 and Gln343, with Asp35, Asp36, FGS75, Asp342 and Gln343 side chains forming a H-bonding network with the Ca2+ cation.38 In the protein–ligand complexes generated here, FGH75 has replaced FGS75 and using the exemplars 18f and 19b, the sulfamate moiety forms H-bonding interactions either directly or indirectly through H2O molecules with this network (Fig. 8), placing the sulfamate group in close proximity to the FGH diol. The ketone group is positioned to form a H2O-mediated interaction with the backbone NH of either Gly100 or Val101, and for 18f, the nitrile group is positioned to form a H2O-mediated interaction with the backbone NH of Thr493.
Conclusions
The study with the triazole sulfamates accidently led to the preparation of additional carbamates, which showed potent Arom inhibitory activity (8b, IC50 4.1 nM; 10b, IC50 0.65 nM); however, neither the triazole sulfamates (6, 7, 8a, 9, 10a) nor the triazole carbamates (8b, 10b) displayed STS inhibitory activity; this can be explained by the reduced size of the STS active site compared with the Arom active site, which cannot accommodate the triazole ring. The superiority for the 6-position of the benzofuran for the sulfamate group over the 5-benzofuran or the 4-phenyl positions was also consistent with our previous findings for the alkylated 4th-generation Arom inhibitors.17 Replacing the CH-triazole with the ketone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O resulted in the benzofuran ketone sulfamates 18 with good STS inhibitory activity but loss of the potent Arom inhibitory activity. A computational molecular dynamics study showed that generally, the ketone sulfamates 18 formed a type I (indirect) binding with haem (e.g.18e, Fig. 6), which would explain the reduction in Arom inhibitory activity. Introducing a methyl group in the 3-position of the furan in the benzofuran ring to give the methyl benzofuran ketone sulfamate derivatives 19 had a significant impact with dual Arom/STS inhibitory activity observed. Computational analysis of these DASIs would suggest that the introduction of the methyl group was sterically sufficient to move the compounds close enough to the haem to form a type II direct binding interaction (e.g.19e, Fig. 6), which would strengthen the binding translating to Arom inhibition, while the small steric increase was still accommodated within the STS active site retaining STS inhibitory activity. Further research is required to investigate optimal steric requirements with respect to DASI activity and to obtain a better understanding of structure–activity relationships.
O resulted in the benzofuran ketone sulfamates 18 with good STS inhibitory activity but loss of the potent Arom inhibitory activity. A computational molecular dynamics study showed that generally, the ketone sulfamates 18 formed a type I (indirect) binding with haem (e.g.18e, Fig. 6), which would explain the reduction in Arom inhibitory activity. Introducing a methyl group in the 3-position of the furan in the benzofuran ring to give the methyl benzofuran ketone sulfamate derivatives 19 had a significant impact with dual Arom/STS inhibitory activity observed. Computational analysis of these DASIs would suggest that the introduction of the methyl group was sterically sufficient to move the compounds close enough to the haem to form a type II direct binding interaction (e.g.19e, Fig. 6), which would strengthen the binding translating to Arom inhibition, while the small steric increase was still accommodated within the STS active site retaining STS inhibitory activity. Further research is required to investigate optimal steric requirements with respect to DASI activity and to obtain a better understanding of structure–activity relationships.
Experimental
General
All commercially available starting materials and solvents were of general purpose or analytical grade and used without further purification. Solvents were appropriately dried over activated molecular sieves (4 Å). Melting points were determined using a Gallenkamp melting point apparatus and are uncorrected. 1H, 19F and 13C NMR (APT) spectra were recorded using a Bruker Advance DP500 spectrometer operating at 500, 400 and 125 MHz, respectively. Chemical shifts are given in parts per million (ppm) relative to the internal standard tetramethylsilane (Me4Si). Analytical thin-layer chromatography (TLC) was carried out on pre-coated silica plates (ALUGRAM® SIL G/UV254) with visualisation via UV light (254 nm). HPLC was performed either by the Department of Pharmacy & Pharmacology, University of Bath, Bath, UK using a Zorbax Eclipse plus C18 Rapid at a resolution of 2.1 × 50 mm a particle size of 1.8 μm using gradient (methanol:H2O) with 0.1% formic acid (method A) or in house using a Shimadzu LC-2030C Plus C18 Rapid at a resolution of 250 × 4.6 mm and a particle size of 5 μm using isocratic 80:20 (methanol:H2O) (method B). All biologically evaluated compounds are ≥95% pure by HPLC analysis or within 0.5% by elemental analysis. Phenolic compounds (1–5)15–17 and THP protected and phenolic derivatives (14 and 16)16,17,22,23 were prepared as described previously.
Chemistry
:1 v/v, Rf = 0.13. 1H NMR (DMSO-d6) δ: 8.77 (s, 1H, CH-triazole), 8.11 (s, 1H, CH-triazole), 7.99 (bs, 2H, NH2), 7.69 (d, J = 8.5 Hz, 1H, Ar), 7.54 (d, J = 8.6 Hz, 2H, Ar), 7.51 (d, J = 1.6 Hz, 1H, Ar), 7.49 (d, J = 8.6 Hz, 2H, Ar), 7.42 (s, 1H, CH), 7.21 (dd, J = 2.1, 8.5 Hz, 1H, Ar), 6.74 (s, 1H, Ar). 13C NMR (DMSO-d6) δ: 155.25 (C), 154.64 (C), 152.63 (CH), 148.17 (C), 144.94 (CH), 135.59 (C), 134.09 (C), 130.29 (2 × CH), 129.40 (2 × CH), 126.33 (C), 122.45 (CH), 118.79 (CH), 107.50 (CH), 106.35 (CH), 59.62(CH). HPLC (method B): 97.7% at R.T. = 3.99 min.
:1 v/v, Rf = 0.13. 1H NMR (DMSO-d6) δ: 8.76 (s, 1H, CH-triazole), 8.11 (s, 1H, CH-triazole), 7.92 (bs, 2H, NH2), 7.65 (d, J = 8.9 Hz, 1H, Ar), 7.56 (d, J = 2.5 Hz, 1H, Ar), 7.54 (d, J = 8.6 Hz, 2H, Ar), 7.49 (d, J = 8.6 Hz, 2H, Ar), 7.43 (s, 1H, CH), 7.23 (dd, J = 2.5, 8.9 Hz, 1H, Ar), 6.76 (s, 1H, Ar). 13C NMR (DMSO-d6) δ: 155.62 (C), 153.04 (C), 152.65 (CH), 146.52 (C), 144.93 (CH), 135.55 (C), 134.09 (C), 130.31 (2 × CH), 129.40 (2 × CH), 128.58 (C), 120.25 (CH), 115.69 (CH), 112.53 (CH), 107.90 (CH), 59.65(CH). HPLC (method B): 97.5% at R.T. = 3.92 min.
:3 v/v, Rf = 0.52 (carbamate) 0.35 (sulfamate). NMR 8a1H NMR (DMSO-d6) δ: 8.76 (s, 1H, CH-triazole), 8.10 (s, 1H, CH-triazole), 8.03 (bs, 2H, NH2), 7.76 (d, J = 2.3 Hz, 1H, Ar), 7.61 (d, J = 8.9 Hz, 1H, Ar), 7.57 (d, J = 8.6 Hz, 2H, Ar), 7.45 (s, 1H, CH), 7.34 (m, 3H, Ar), 6.71 (t, J = 0.9 Hz, 1H, Ar). 13C NMR (DMSO-d6) δ: 155.85 (C), 153.56 (C), 152.65 (CH), 150.81 (C), 144.90 (CH), 134.76 (C), 130.08 (2 × CH), 129.55 (C), 128.09 (C), 125.45 (CH), 123.17 (2 × CH), 121.56 (CH), 113.41 (CH), 107.22 (CH), 59.75(CH). NMR 8b1H NMR (DMSO-d6) δ: 8.70 (s, 1H, CH-triazole), 8.13 (s, 1H, CH-triazole), 7.95 (d, J = 2.1 Hz, 1H, Ar), 7.78 (d, J = 8.8 Hz, 1H, Ar), 7.65 (d, J = 8.7 Hz, 2H, Ar), 7.49 (bs, 1H, NH2), 7.01 (dd, J = 2.1, 8.8 Hz, 1H, CH), 6.97 (bs, 1H, NH2), 6.70 (d, J = 8.7 Hz, 2H, Ar), 6.45 (s, 1H), 6.67 (s, 1H, Ar). 13C NMR (DMSO-d6) δ: 154.32 (C), 153.94 (C), 153.68 (C), 152.54 (CH), 151.43 (C), 144.82 (CH), 132.38 (C), 129.05 (C), 128.92 (2 × CH), 128.77 (C), 125.62 (CH), 122.42 (2 × CH), 121.14 (CH), 112.59 (CH), 107.37 (CH), 61.55 (CH). HPLC 8a (method A) 100% at R.T. = 4.35 min. HPLC 8b (method A) 100% at R.T. = 4.33 min. HRMS 8a (EIC): calculated 404.0346 [M]+, found 404.0352 [M]+. HRMS 8b (EIC): calculated 368.0676 [M]+, found 368.0680 [M]+.
:1 v/v, Rf = 0.15. 1H NMR (DMSO-d6) δ: 8.81 (s, 1H, CH-triazole), 8.16 (s, 1H, CH-triazole), 8.03 (bs, 2H, NH2), 7.75 (d, J = 8.5 Hz, 1H, Ar), 7.61 (dd, J = 5.4, 8.8 Hz, 2H, Ar), 7.56 (d, J = 1.8 Hz, 1H, Ar), 7.46 (s, 1H), 7.37 (t, J = 8.9 Hz, 2H, Ar), 7.26 (dd, J = 2.1, 8.5 Hz, 1H, Ar), 6.77 (t, J = 0.9 Hz, 1H, Ar). 13C NMR (DMSO-d6) δ: 163.64 (d, 1JC,F = 243.75 Hz, C), 155.57 (C), 154.62 (C), 152.59 (CH), 148.13 (C), 144.85 (CH), 132.88 (d, 4JC,F = 2.5 Hz, C), 130.74 (d, 3JC,F = 7.5 Hz, 2 × CH), 126.35 (C), 122.42 (CH), 118.78 (CH), 116.35 (d, 2JC,F = 22.5 Hz, 2 × CH), 107.33(CH), 106.34 (CH), 59.63 (CH). HPLC (method B): 100% at R.T. = 3.69 min.
:1 v/v, Rf = 0.22 (carbamate) 0.20 (sulfamate). NMR 10a1H NMR (DMSO-d6) δ: 8.79 (s, 1H, CH-triazole), 8.14 (s, 1H, CH-triazole), 7.99 (bs, 2H, NH2), 7.95 (d, J = 8.5 Hz, 2H, Ar), 7.70 (d, J = 8.5 Hz, 1H, Ar), 7.63 (d, J = 8.2 Hz, 2H, Ar), 7.56 (s, 1H, CH), 7.52 (d, J = 1.7 Hz, 1H, Ar), 7.22 (dd, J = 2.1, 8.5 Hz, 1H, Ar), 6.78 (t, J = 0.9 Hz, 1H, Ar). 13C NMR (DMSO-d6) δ: 154.68 (C), 154.52 (C), 152.79 (CH), 148.25 (C), 145.13 (CH), 141.73 (C), 133.38 (2 × CH), 129.29 (2 × CH), 126.25 (C), 122.54 (CH), 118.87 (CH), 112.18 (C), 107.89 (CH), 106.38 (CH), 60.22 (C), 59.75(CH). NMR 10b1H NMR (DMSO-d6) δ: 8.79 (s, 1H, CH-triazole), 8.13 (s, 1H, CH-triazole), 7.95 (d, J = 8.5 Hz, 2H, Ar), 7.62 (d, J = 8.2 Hz, 2H, Ar), 7.60 (d, J = 8.7 Hz, 1H, Ar), 7.52 (s, 1H, CH), 7.38 (d, J = 1.6 1H, Ar), 7.23 (bs, 1H, NH2), 7.03 (dd, J = 2.1, 8.5 Hz, 1H, Ar), 6.93 (bs, 1H, NH2), 6.71 (t, J = 1.0, 1H, Ar). 13C NMR (DMSO-d6) δ: 155.32 (C), 155.01 (C), 153.67 (C), 152.75 (CH), 149.45 (C), 145.13 (CH), 141.88 (C), 133.35 (2 × CH), 129.28 (2 × CH), 124.78 (C), 121.89 (CH), 118.88 (C), 118.70 (CH), 112.11 (C), 107.91 (CH), 106.09 (CH), 59.85 (CH). HPLC 10a (method A) 100% at R.T. = 3.94 min. HPLC 10b (method A) 100% at R.T. = 3.85 min. HRMS 10a (EI): calculated 396.0766 [M + H]+, found 396.0761 [M + H]+. HRMS 10b (EI): calculated 382.0916 [M + Na]+, found 382.0908 [M + Na]+.
:1 v/v, Rf = 0.65. 1H NMR (DMSO-d6) δ: 8.11 (m, 2H, Ar), 7.77 (d, J = 8.5 Hz, 1H, Ar), 7.43 (m, 2H, Ar), 7.32 (d, J = 2.0 Hz, 1H, Ar), 7.10 (dd, J = 2.0, 8.5 Hz, 1H, Ar), 5.62 (t, J = 3.5 Hz, 1H, CH-pyran), 3.79 (m, 1H, CH2-pyran), 3.61 (m, 1H, CH2-pyran), 2.56 (s, 3H, CH3), 1.93 (m, 3H, CH2-pyran), 1.68 (m, 3H, CH2-pyran). 13C NMR (DMSO-d6) δ: 183.43 (C), 166.05 (1JC,F = 250 Hz, CF), 158.46 (C), 155.30 (C), 147.82 (C), 134.61 (4JC,F = 3.13 Hz, CF), 132.70 (2JC,F = 9.625 Hz, 2 × CH), 127.83 (C), 123.19 (C), 122.79 (CH), 116.11 (3JC,F = 21.75 Hz 2 × CH), 115.52 (CH), 99.30 (CH), 96.57 (CH), 62.10 (CH2), 30.15 (CH2), 25.08 (CH2), 18.96 (CH2), 10.34 (CH3). 19F NMR (DMSO-d6) δ: −106.52.
:1 v/v, Rf = 0.63. 1H NMR (DMSO-d6) δ: 8.03 (d, J = 9.0 Hz, 2H, Ar), 7.78 (d, J = 9.0 Hz, 1H, Ar), 7.67 (d, J = 8.4 Hz, 2H, Ar), 7.32 (d, J = 2.4 Hz, 1H, Ar), 7.11 (dd, J = 1.8, 8.4 Hz, 1H, Ar), 5.62 (t, J = 3.0 Hz, 1H, CH-pyran), 3.79 (m, 1H, CH2-pyran), 3.62 (m, 1H, CH2-pyran), 2.56 (s, 3H, CH3), 1.92 (m, 3H, CH2-pyran), 1.67 (m, 3H, CH2-pyran).13C NMR (DMSO-d6) δ: 183.68 (C), 158.55 (C), 155.35 (C), 147.74 (C), 137.92 (C), 136.70 (C), 131.57 (2 × CH), 129.08 (2 × CH), 128.12 (C), 123.17 (C), 122.86 (CH), 115.57 (CH), 99.27 (CH), 96.56 (CH), 62.10 (CH2), 30.14 (CH2), 25.07 (CH2), 18.95 (CH2), 10.36 (CH3).
:2 v/v to give the product as a yellow solid. Yield: 0.54 g (51%), m.p.: 96–98 °C, TLC: petroleum ether–EtOAc 3:1 v/v, Rf = 0.68. 1H NMR (DMSO-d6) δ: 7.94 (d, J = 9.0 Hz, 2H, Ar), 7.81 (d, J = 9.0 Hz, 2H, Ar), 7.78 (d, J = 8.4 Hz, 1H, Ar), 7.32 (d, J = 1.8 Hz, 1H, Ar), 7.11 (dd, J = 2.4, 9.0 Hz, 1H, Ar), 5.62 (t, J = 3.6 Hz, 1H, CH-pyran), 3.78 (m, 1H, CH2-pyran), 3.61 (m, 1H, CH2-pyran), 2.56 (s, 3H, CH3), 1.90 (m, 3H, CH2-pyran), 1.66 (m, 3H, CH2-pyran). 13C NMR (DMSO-d6) δ: 183.88 (C), 158.55 (C), 155.36 (C), 147.73 (C), 137.05 (C), 132.02 (2 × CH), 131.67 (2 × CH), 128.14 (C), 127.02 (C), 123.17 (C), 122.87 (CH), 115.58 (CH), 99.26 (CH), 96.57 (CH), 62.104 (CH2), 30.14 (CH2), 25.06 (CH2), 18.95 (CH2), 10.37 (CH3).
:2 v/v and then recrystallisation from CH3CN to obtain the product as a pale yellow solid. Yield: 0.56 g (45%), m.p.: 88–90 °C, TLC: petroleum ether–EtOAc 3:1 v/v, Rf = 0.69. 1H NMR (DMSO-d6) δ: 8.05 (d, J = 9.0 Hz, 2H, Ar), 7.75 (d, J = 9.0 Hz, 1H, Ar), 7.33 (d, J = 2.4 Hz, 1H, Ar), 7.13 (d, J = 9.0 Hz, 2H, Ar), 7.10 (dd, J = 2.4, 9.0 Hz, 1H, Ar), 5.61 (t, J = 3.0 Hz, 1H, CH-pyran), 3.88 (s, 3H, O CH3), 3.80 (m, 1H, CH2-pyran), 3.62 (m, 1H, CH2-pyran), 2.54 (s, 3H, CH3), 1.92 (m, 2H, CH2-pyran), 1.79 (m, 1H, CH2-pyran), 1.66 (m, 3H, CH2-pyran). 13C NMR (DMSO-d6) δ: 183.39 (C), 163.33 (C), 158.16 (C), 155.07 (C), 148.26 (C), 132.22 (2 × CH), 130.50 (C), 126.70 (C), 122.60 (CH), 155.32 (CH), 114.30 (2 × CH), 99.32 (CH), 96.57 (CH), 62.10 (CH2), 56.01 (CH2), 30.17 (CH2), 25.08 (CH2), 19.0 (CH2), 10.30 (CH3).
:2 v/v and two hot washes in CH3CN to give the product as a yellow solid. Yield: 0.36 g (23%), m.p.: 198–200 °C, TLC: petroleum ether–EtOAc 3:1 v/v, Rf = 0.70. 1H NMR (CDCl3) δ: 8.20 (d, J = 8.4 Hz, 2H, Ar), 7.84 (d, J = 9.0 Hz, 2H, Ar), 7.62 (d, J = 7.6, 1H, Ar), 7.26 (d, J = 1.8 Hz, 1H, Ar), 7.11 (dd, J = 1.8, 8.4 Hz, 1H, Ar), 5.52 (t, J = 3.0 Hz, 1H, CH-pyran), 3.49 (m, 1H, CH2-pyran), 3.69 (m, 1H, CH2-pyran), 2.69 (s, 3H, CH3), 2.07 (m, 1H, Ar), 1.95 (m, 2H, CH2-pyran), 1.78 (m, 1H, CH2-pyran), 1.67 (m, 2H, CH2-pyran). 13C NMR (CDCl3) δ: 183.39 (C), 158.94 (C), 155.78 (C), 147.61 (C), 141.50 (2 × C), 132.06 (2 × CH), 130.06 (2 × CH), 129.55 (C), 123.20 (C), 121.98 (CH), 118.28 (C), 115.37 (CH), 98.82 (CH), 96.82 (CH), 62.14 (CH2), 30.22 (CH2), 25.08 (CH2), 18.61 (CH2), 10.28 (CH3).
:2 v/v and two washes with CH2Cl2 to obtain (2,4-dichlorophenyl)(6-hydroxy-3-methylbenzofuran-2-yl)methanone 17d as a white solid. Yield: 0.70 g (45%), TLC: petroleum ether–EtOAc 2:1 v/v, Rf = 0.57. 1H NMR (DMSO-d6) δ: 10.32 (s, 1H, OH), 7.82 (d, J = 1.5 Hz, 1H, Ar), 7.67 (dd, J = 1.5, 7.5 Hz, 1H, Ar), 7.62 (s, 1H, Ar), 7.62 (d, J = 1.5, 1H, Ar), 6.9 (m, 2H, Ar), 2.37 (s, 3H, CH3).
:1 v/v, Rf = 0.31. 1H NMR (DMSO-d6) δ: 10.19 (s, 1H, OH), 8.08 (m, 2H, Ar), 7.66 (d, J = 8.5 Hz, 1H, Ar), 7.42 (m, 2H, Ar), 6.95 (d, J = 2.0 Hz, 1H, Ar), 6.91 (dd, J = 2.0, 8.5 Hz, 1H, Ar), 2.53 (s, 3H, CH3). 13C NMR (DMSO-d6) δ: 183.19 (C), 165.94 (1JC,F = 249.3 Hz, CF), 160.0 (C), 155.92 (C), 147.19 (C), 134.78 (4JC,F = 2.63 Hz, CF), 132.59 (3JC,F = 9.12 Hz, 2 × CH), 128.30 (C), 122.92 (CH), 121.40 (C), 116.06 (2JC,F = 21.6 Hz, 2 × CH), 114.55 (CH), 97.86 (CH), 10.42 (CH3). 19F NMR (DMSO-d6) δ: −106.80.
:1 v/v, Rf = 0.32. 1H NMR (DMSO-d6) δ: 10.21 (s, 1H, OH), 7.99 (d, J = 8.8 Hz, 2H, Ar), 7.64 (d, J = 8.8 Hz, 2H, Ar), overlapping 7.66 (d, J = 8.6 Hz, 1H, Ar), 6.94 (d, J = 2.0, Hz, 1H, Ar), 6.90 (dd, J = 2.0, 8.6 Hz, 1H, Ar), 2.53 (s, 3H, CH3). 13C NMR (DMSO-d6) δ: 183.42 (C), 160.11 (C), 155.98 (C), 147.13 (C), 137.73 (C), 136.91 (C), 131.50 (2 × CH), 129. 05 (2 × CH), 128.59 (C), 123.00 (CH), 121.40 (C), 114.62 (CH), 97.85 (CH), 10.43 (CH3).
:1 v/v, Rf = 0.29. 1H NMR (DMSO-d6) δ: 10.21 (s, 1H, OH), 7.99 (d, J = 8.5 Hz, 2H, Ar), 7.67 (m, 3H, Ar), 6.95 (d, J = 2.0 Hz, 1H, Ar), 6.91 (dd, J = 2.0, 8.5 Hz, 1H, Ar), 2.53 (s, 3H, CH3). 13C NMR (CDCl3) δ: 183.62 (C), 160.14 (C), 156.00 (C), 147.12 (C), 137.28 (C), 132.00 (2 × CH), 131.62 (2 × CH), 128.62 (C), 126.83 (C), 123.02 (CH), 121.41 (C), 114.64 (CH), 97.86 (CH), 10.44 (CH3).
:1 v/v, Rf = 0.32. 1H NMR (DMSO-d6) δ: 10.15 (s, 1H, OH), 8.03 (d, J = 9.0 Hz, 2H, Ar), 7.64 (d, J = 8.4 Hz, 1H, Ar), 7.12 (d, J = 9.0 Hz, 2H, Ar), 6.96 (d, J = 2.4 Hz, 1H, Ar), 6.90 (dd, J = 1.8, 8.4 Hz, 1H, Ar), 3.84 (s, 3H, OCH3), 2.52 (s, 3H, CH3). 13C NMR (DMSO-d6) δ: 183.22 (C), 163.20 (C), 159.64 (C), 155.67 (C), 147.61 (C), 132.11 (2 × CH), 130.69 (C), 127.16 (C), 122.71 (CH), 121.44 (C), 114.33 (CH), 114.26 (2 × CH), 97.86 (CH), 55.98 (O CH3), 10.36 (CH3).
:1 v/v, Rf = 0.45. 1H NMR (DMSO-d6) δ: 10.29 (s, 1H, OH), 8.09 (m, 2H, Ar), 8.05 (m, 2H, Ar), 7.69 (d, J = 8.5 Hz, 1H, Ar), 6.93 (d, J = 2.0 Hz, 1H, Ar), 6.91 (dd, J = 2.0, 8.5 Hz, 1H, Ar), 2.53 (s, 3H, CH3). 13C NMR (DMSO-d6) δ: 183.50 (C), 160.40 (C), 156.21 (C), 146.88 (C), 142.00 (C), 132.91 (2 × CH), 130.09 (2 × CH), 129.47 (C), 133.20 (CH), 121.38 (C), 118.75 (C), 114.81 (CH), 114.72 (C), 97.83 (CH), 10.46 (CH3).
:4 v/v). Yield: 0.22 g (76%), m.p.: 200–202 °C, TLC: petroleum ether–EtOAc 2:1 v/v, Rf = 0.37. 1H NMR (DMSO-d6) δ: 8 16 (s, 2H, NH2), 8.13 (m, 2H, Ar), 7.95 (dd, J = 8.5 Hz, 1H, Ar), 7.88 (dd, J = 1.0 Hz, 1H, Ar), 7.73 (s, 1H, CH-furan), 7.48 (m, 2H, Ar), 7.352 (dd, J = 2.0, 8.5 Hz, 1H, Ar). 13C NMR (DMSO-d6) δ: 182.23 (C), 166.51 (d, 1JC,F = 251.25 Hz, C), 155.59 (C), 152.90 (C), 150.80 (C), 133.63 (d, 4JC = 3.75 Hz, C), 132.75 (d, 3JC = 8.75 Hz, 2 × CH), 125.67 (C), 125.02 (CH), 119.90 (CH), 117.30 (CH), 116.48 (d, 2JC = 21.25 Hz, 2 × CH), 106.93 (CH). 19F NMR (DMSO-d6) δ: −105.60. Anal. calculated for C15H10FNO5S (335.3054): C, 53.73%; H, 3.01%; N, 4.18. Found: C, 53.63%; H, 3.07%; N, 3.96.
:3 v/v). Yield: 61 mg (18%), m.p.: 159–161 °C, TLC: petroleum ether–EtOAc 2:1 v/v, Rf = 0.23. 1H NMR (CDCl3) δ: 8.15 (s, 2H, NH2), 8.04 (d, J = 8.5 Hz, 2H, H-2′ and H-6′), 7.94 (d, J = 8.6 Hz, 1H, H-4),7.89 (s, 1H, H-7), 7.73 (s, 1H, H-3), 7.70 (d, J = 8.5 Hz, 2H, H-3′ and H-5′), 7.34 (dd, J = 2.0, 8.6 Hz, 1H, H-5). 13C NMR (CDCl3) δ: 182.48 (CO), 155.60 (C), 152.82 (C), 150.87 (C), 138.60 (C), 135.68 (C), 131.59 (CH-2′ and CH-6′), 129.38 (CH-3′ and CH-5′), 125.64 (C), 125.06 (CH), 119.92 (CH), 117.52 (CH), 106.93 (CH). Anal. calculated for C15H10ClNO5S (351.7570) C, 51.22%; H, 2.87%; N, 3.98%. Found: C, 51.70%; H, 3.01%; N, 3.72%.
:3 v/v). Yield: 0.125 g (46%), m.p.: 166–168 °C, TLC: petroleum ether–EtOAc 2:1 v/v, Rf = 0.33. 1H NMR (DMSO-d6) δ: 8. 15 (s, 2H, NH2), 7.97 (d, J = 8.4 Hz, 2H, Ar), 7.95 (d, J = 8.4 Hz, 1H, Ar), 7.90 (d, J = 1.2 Hz, 1H, Ar), 7.85 (d, J = 9.0 Hz, 2H, Ar), 7.73 (s, 1H, CH-furan), 7.36 (dd, J = 2.4, 9.0 Hz, 1H, Ar). 13C NMR (DMSO-d6) δ: 182.176 (C), 160.01 (C), 157.77 (C), 150.68 (C), 136.65 (C), 136.65 (2 × CH), 131.42 (2 × CH), 127.07 (C), 124.98 (CH), 119.49 (C), 119.04 (CH), 115.24 (CH), 97.93 (CH). Anal. calculated for C15H10BrNO5S·0.3 H2O (401.6156): C, 44.86%; H, 2.66%; N, 3.49%. Found: C, 44.92%; H, 2.59%; N, 3.09%. HPLC (method A) 100% at R.T. = 4.20 min. HRMS (EIC): calculated 397.9522 [M + H]+, found 397.9515 [M + H]+.
:3 v/v). Yield: 0.10 g (33%), m.p.: 198–200 °C, TLC: petroleum ether–EtOAc 2:1 v/v, Rf = 0.64. 1H NMR (DMSO-d6) δ: 8.16 (s, 2H, NH2), 7.91 (d, J = 8.5 Hz, 1H, Ar), 7.88 (s, 1H, Ar), 7.79 (d, J = 8.5 Hz, 1H, Ar), 7.73 (s, 1H, Ar), 7.71 (s, 1H, CH-furan), 7.67 (dd, J = 8.5 Hz, 1H, Ar), 7.34 (d, J = 9.0 Hz, 1H, Ar). 13C NMR (DMSO-d6) δ: 182.35 (C), 156.07 (C), 152.71 (C), 151.31 (C), 136.82 (C), 136.04 (C), 131.99 (C), 131.48 (CH), 130.23 (CH), 128.16 (CH), 125.59 (C), 125.35 (CH), 120.14 (CH), 119.07 (CH), 107.00 (CH). Anal. calculated for C15H9Cl2NO5S (386.199): C, 46.65%; H, 2.35%; N, 3.63. Found: C, 46.80%; H, 2.41%; N, 3.37.
:3 v/v). Yield: 0.17 g (52%), m.p.: 182–184 °C, TLC: petroleum ether–EtOAc 2:1 v/v, Rf = 0.36. 1H NMR (DMSO-d6) δ: 8.14 (s, 2H, NH2), 8.08 (d, J = 9.0 Hz, 2H, Ar), 7.94 (d, J = 8.4 Hz, 1H, Ar), 7.82 (d, J = 1.2 Hz, 1H, Ar), 7.72 (s, 1H, CH-furan), 7.35 (dd, J = 2.4, 9.0 Hz, 1H, Ar), 7.17 (d, J = 9.0 Hz, 2H, Ar), 3.90 (s, 3H, OCH3). 13C NMR (DMSO-d6) δ: 181.74 (C), 163.42 (C), 159.54 (C), 157.41 (C), 151.26 (C), 131.84 (2 x CH), 130.04 (C), 124.67 (CH), 119.49 (C), 117.53 (CH), 114.94 (CH), 114.47 (2 x CH), 97.94 (CH), 56.04 (CH3). Anal. calculated for C16H13NO6S·0.2 H2O (350.9441): C, 54.76%; H, 3.84%; N, 3.99%. Found: C, 54.93%; H, 3.79%; N, 3.59%. HPLC (method A) 100% at R.T. = 4.39 min. HRMS (EIC): calculated 348.0543 [M + H]+, found 348.0534 [M + H]+.
:6 v/v). Yield: 0.27 g (74%), m.p.: 200–204 °C, TLC: petroleum ether–EtOAc 2:1 v/v, Rf = 0.38. 1H NMR (DMSO-d6): δ 8.18 (s, 2H, NH2), 8.14 (d, J = 8.6 Hz, 2H, Ar), 8.10 (d, J = 8.6 Hz, 2H, Ar), 7.95 (d, J = 8.6 Hz, 1H, Ar), 7.91 (d, J = 1.0 Hz, 1H, CH-furan), 7.73 (d, J = 1.6 Hz, 1H, Ar), 7.35 (dd, J = 2.0, 8.6 Hz, 1H, Ar). 13C NMR (DMSO-d6) δ: 182.69 (C), 155.77 (C), 152.53 (C), 151.07 (C), 140.67 (C), 133.20 (2 × CH), 130.26 (2 × CH), 125.62 (C), 125.25 (CH), 120.03 (CH), 118.61 (C), 118.36 (CH), 115.50 (C), 106.95 (CH). Anal. calculated for C16H10N2O5S·0.5 H2O (351.3326): C, 54.69%; H, 3.16%; N, 7.97%. Found: C, 54.73%; H, 3.21%; N, 7.53. HPLC (method A) 100% at R.T. = 4.06 min.
:3 v/v). Yield: 0.202 g (60%), m.p.: 181–183 °C, TLC: petroleum ether–EtOAc 2:1 v/v, Rf = 0.33. 1H NMR (DMSO-d6): δ 8.12 (m, 4H, H-2′, H-4′ and NH2), 7.97 (d, J = 8.6 Hz, 1H, H-4), 7.64 (d, J = 2.0 Hz, 1H, H-7), 7.44 (t, J = 8.9 Hz, 2H, H-3′ and H-5′), 7.34 (dd, J = 2.0, 8.6 Hz, 1H, H-5), 2.60 (s, 3H, CH3). 13C NMR (DMSO-d6): δ 183.80 (CO), 166.29 (C, 1JCF = 252 Hz), 153.86 (C), 150.99 (C), 148.95 (C), 134.23 (C, 4JCF = 3.7 Hz), 132.87 (2 × CH, 3JCF = 9.8 Hz), 127.40 (C), 127.00 (C), 123.15 (CH), 119.34 (CH-5), 116.19 (2 × CH, 2JCF = 22.0 Hz), 106.77 (CH), 10.20 (CH3). 19F NMR (DMSO-d6): δ −105.9. Anal. calculated for C16H12FNO5S (349.3324) C, 55.01%; H, 3.46%; N, 4.01%. Found: C, 55.12%; H, 3.66%; N, 3.94%.
:3 v/v). Yield: 0.106 g (32%); m.p. 181–182 °C; TLC (petroleum ether–EtOAc 2:1 v/v) Rf 0.40. 1H NMR (DMSO-d6): δ 8.13 (s, 2H, NH2), 8.04 (d, J = 8.8 Hz, 2H, H-2′, and H-4′), 7.97 (dd, J = 0.5, 8.6 Hz, 1H, H-4), 7.68 (d, J = 8.8 Hz, 2H, H-3′ and H-5′), 7.64 (dd, J = 0.5, 2.0 Hz, 1H, H-5), 7.34 (dd, J = 2.0, 8.6 Hz, 1H, H-7), 2.60 (s, 3H, CH3). 13C NMR (DMSO-d6): δ 184.07 (CO), 153.90 (C), 151.07 (C), 148.87 (C), 138.35 (C), 136.32 (C), 131.72 (2 × CH), 129.22 (2 × CH), 127.38 (C), 127.27 (C), 123.21 (CH), 119.38 (CH), 106.77 (CH), 10.22 (CH3). Anal. calculated for C16H12ClO5NS (365.7840) C, 52.54%; H, 3.31%; N, 3.83%. Found: C, 52.75%; H, 3.32%; N, 3.64%.
:3 v/v). Yield: 0.093 g (25%); m.p. 178–181 °C; TLC (petroleum ether–EtOAc 2:1 v/v) Rf 0.30. 1H NMR (CDCl3) δ: 8.13 (s, 2H, NH2), 7.96 (m, 3H, H-2′, H-6′ and H-4), 7.83 (d, J = 8.6 Hz, 2H, H-3′ and H-5′), 7.64 (d, J = 2.0 Hz, 1H, H-7), 7.34 (dd, J = 2.1, 8.6 Hz, 1H, H-5), 2.51 (s, 3H, CH3). 13C NMR (CDCl3) δ: 184.27 (CO), 153.90 (C), 151.07 (C), 148.86 (C), 136.66 (C), 132.16 (2 × CH), 131.80 (2 × CH), 127.48 (C), 127.38 (C), 127.28 (C), 123.22 (CH), 119.38 (CH), 106.77 (CH), 10.23 (CH3). Anal. calculated for C16H12BrO5NS (410.2380) C, 46.84%; H, 2.95%; N, 3.41%. Found: C, 47.13%; H, 3.25%; N, 3.31%.
:4 v/v). Yield: 0.107 g (22%); m.p. 138–140 °C; TLC (petroleum ether–EtOAc 2:1 v/v) Rf 0.42. 1H NMR (DMSO-d6): δ 8.13 (s, 2H, NH2), 7.98 (d, J = 8.6 Hz, 1H, Ar), 7.86 (d, J = 1.9 Hz, 1H, Ar), 7.69 (d, J = 8.3 Hz, 1H, Ar), 7.65 (dd, J = 2.0, 8.3 Hz, 1H, Ar), 7.58 (d, J = 2.0 Hz, 1H, Ar), 7.33 (dd, J = 2.0, 8.6 Hz, 1H, Ar), 2.49 (s, 3H, CH3). 13C NMR (DMSO-d6) δ: 184.21 (CO), 154.34 (C), 151.51 (C), 148.35 (C), 137.20 (C), 136.58 (C), 131.62 (C), 131.05 (CH), 130.00 (CH), 128.41 (CH), 127.84 (C), 127.45 (C), 123.69 (CH), 119.54 (CH), 106.74 (CH), 9.85 (CH3). Anal. calculated for C16H11Cl2O5NS (400.2260) C, 48.02%; H, 2.77%; N, 3.50%. Found: C, 47.91%; H, 2.82%; N, 3.31%.
:4 v/v). Yield: 0.138 g (56%); m.p. 172–176 °C; TLC (petroleum ether–EtOAc 2:1 v/v) Rf 0.22. 1H NMR (DMSO-d6): δ 8.13 (s, 2H, NH2), 8.06 (d, J = 8.9 Hz, 2H, Ar), 7.94 (d, J = 8.6 Hz, 1H, Ar), 7.64 (d, J = 2.0 Hz, 1H, Ar), 7.33 (dd, J = 2.0, 8.6 Hz, 1H, Ar), 7.14 (d, J = 8.9 Hz, 2H, Ar), 3.89 (s, 3H, OCH3), 2.49 (s, 3H, CH3). 13C NMR (DMSO-d6): δ 183.64 (CO), 163.65 (C), 153.66 (C), 150.68 (C), 149.42 (C), 132.40 (2 × CH), 130.12 (C), 127.49 (C), 125.87 (C), 122.93 (CH), 119.24 (CH), 114.45 (2 × CH), 106.76 (CH), 56.08 (OCH3), 10.14 (CH3). Anal. calculated for C17H15O6NS·0.1 H2O (363.1695) C, 56.22%; H, 4.22%; N, 3.86%. Found: C, 56.07%; H, 4.20%; N, 3.75%. HPLC (method A) 100% at R.T. = 4.35 min. HRMS (EIC): calculated 362.0699 [M + H]+, found 362.0688 [M + H]+.
:1 v/v) Rf 0.26. 1H NMR (DMSO-d6) δ: 8.02 (d, J = 8.3 Hz, 2H, H-2′ and H-6′), 7.84 (m, 2H, H-4 and H-7), 7.68 (d, J = 8.6 Hz, 2H, H-3′ and H-5′), 7.60 (s, 1H, H-3), 7.36 (br s, 1H of NH2), 7.17 (dd, J = 2.0, 8.6 Hz, 1H, H-5), 7.05 (br s, 1H of NH2). 13C NMR (DMSO-d6) δ: 182.51 (CO, C-1), 155.99 (C), 154.95 (C), 152.28 (C), 152.26 (C), 138.43 (C), 135.87 (C), 131.53 (2 × CH, Ar), 129.34 (2 × CH, Ar), 127.37 (CH), 124.26 (C), 120.01 (CH), 117.88 (CH), 106.53 (CH). Anal. calculated for C16H10ClNO4 (315.7090): C, 60.87%; H, 3.19%; N, 4.43. Found: C, 61.03%; H, 3.27%; N, 4.01.
:1 v/v) Rf 0.12. 1H NMR (DMSO-d6) δ: 8.02 (d, J = 8.7 Hz, 2H, H-2′ and H-6′), 7.86 (d, J = 8.6 Hz, 1H, H-4), 7.66 (d, J = 8.7 Hz, 2H, H-3′ and H-5′), 7.51 (d, J = 1.9 Hz, 1H, H-7), 7.35 (br s, 1H of NH2), 7.17 (dd, J = 2.0, 8.6 Hz, 1H, H-5), 7.05 (br s, 1H of NH2), 2.58 (s, 3H, CH3). 13C NMR (DMSO-d6) δ: 184.10 (CO, C-1), 154.96 (C), 154.24 (C), 152.43 (C), 148.41 (C), 138.17 (C), 136.49 (C), 131.68 (2 × CH, Ar), 129.15 (2 × CH, Ar), 127.47 (C), 126.04 (C), 122.49 (CH), 119.41 (CH), 106.44 (CH), 10.28 (CH3). Anal. calculated for C17H12ClNO4 (329.7360): C, 61.92%; H, 3.67%; N, 4.25%. Found: C, 61.57%; H, 3.79%; N, 3.96%.
Cell culture
JEG-3 cells (ATCC) were cultured in Eagle's minimal essential medium (EMEM) supplemented with 10% fetal calf serum (FCS). The cells were maintained at 37 °C in a humidified incubator with 5% CO2. Then, the cells were cultured to approximately 80% confluence before any treatments were applied.Aromatase activity assay
The aromatase activity was measured using a modified tritiated water assay. The JEG-3 cells were cultured in six-well plates in EMEM until reaching 80% confluence. The substrate androst-4-ene-3,17-dione [1β-3H] was added to each well in a serum-free medium. In the presence and absence of dual inhibitors (at concentrations ranging from 0.001 to 10 nM), the cells were incubated at 37 °C for 1 hour. The reaction was stopped by placing the plates on ice for 5 minutes. The medium (500 μL) was obtained, vortexed with 2% dextran-treated charcoal in PBS, and centrifuged at 4000 rpm. [3H]H2O in the supernatant was quantified using scintillation counting. The aromatase activity was normalized to protein concentration, determined by the Pierce BCA assay kit (Thermo Fisher Scientific), and expressed as pmol of product per mg of protein per hour.Steroid sulfatase activity assay
STS activity was measured using an in vitro, cell-free assay. Briefly, lysates from JEG-3 cells, known to have high endogenous STS activity, were prepared by lysing the cells with a buffer (50 mM Tris-HCl, pH 7.4, and 1 mM EDTA). After centrifugation, the supernatant was collected and protein concentrations were determined using a BCA assay kit.For the steroid sulfatase assay, 125 μg of JEG-3 lysate was incubated with the substrate [3H]oestrone sulfate (E1S, 4 × 105 dpm) diluted to a final concentration of 20 μM using non-radioactive E1S. The mixture was incubated in the presence of different concentrations of inhibitors (ranging from 10−11 M to 10−5 M) at 37 °C for 1 hour. After incubation, the reaction was stopped by placing the tubes on ice, and the estrone product formed was separated from the substrate by extraction with toluene. [14C]oestrone was added to the organic phase as an internal standard to monitor recovery and procedural losses.
The organic phase was subjected to liquid scintillation counting to measure both 3H and 14C. The amount of E1S hydrolysed was calculated from the 3H counts, corrected for recovery using the 14C standard, and converted into molar concentrations of hydrolysed product. The inhibitory activity of test compounds was expressed as the IC50 value, representing the concentration required to inhibit 50% of the STS activity.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
AGE, FG, OA, IS and HA-B performed the chemical synthesis supervised by CS. CEP and ND performed the enzyme assays supervised by PAF. CS performed the computational studies. The manuscript was prepared by CS and PAF and reviewed by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Egyptian Ministry of Higher Education-Mission Sector and Zagazig University, Egypt and the Saudi Arabia Cultural Bureau and Jouf University, Saudi Arabia for funding this research in Cardiff University through PhD scholarships to AEG and OA respectively. Molecular dynamics simulations were undertaken using the supercomputing facilities at Cardiff University operated by Advanced Research Computing at Cardiff (ARCCA) on behalf of the Cardiff Supercomputing Facility and the HPC Wales and Supercomputing Wales (SCW) projects. We acknowledge support of the latter, which is part-funded by the European Regional Development Fund (ERDF) via the Welsh Government.Notes and references
- E. Proschak, H. Stark and D. Merk, J. Med. Chem., 2019, 62, 420–444 CrossRef CAS PubMed
.
- J. Zhou, X. Jiang, S. He, H. Jiang, F. Feng, W. Liu, W. Qu and H. Sun, J. Med. Chem., 2019, 62, 8881–8914 CrossRef CAS PubMed
- M. Salah, A. S. Abdelsamie and M. Frotscher, J. Med. Chem., 2017, 60, 4086–4092 CrossRef CAS PubMed
- N. Harbeck, F. Penault-Llorca, J. Cortes, M. Gnant, N. Houssami, P. Poortmans, K. Ruddy, J. Tsang and F. Cardoso, Nat. Rev. Dis. Primers, 2019, 5, 66 CrossRef PubMed
- R. C. Travis and T. J. Key, Breast Cancer Res., 2003, 5, 239–247 CrossRef CAS PubMed
- S. J. Santner, P. D. Deil and R. J. Santen, J. Clin. Endocrinol. Metab., 1984, 59, 29–33 CrossRef CAS PubMed
- R. J. Santen, D. Leszczynski, N. Tilson-Mallet, P. D. Feil, C. Wright, A. Manni and S. J. Santner, Ann. N. Y. Acad. Sci., 1986, 464, 126–137 CrossRef CAS PubMed
- L. W. L. Woo, A. Purohit and B. V. L. Potter, Mol. Cell. Endocrinol., 2011, 340, 175–185 CrossRef CAS PubMed
- P. A. Foster, Molecules, 2021, 26, 2852 CrossRef CAS PubMed
- L. W. L. Woo, T. Jackson, A. Putey, G. Cozier, P. Leonard, K. R. Acharya, S. K. Chander, A. Purohit, M. J. Reed and B. V. L. Potter, J. Med. Chem., 2010, 53, 2155–2170 CrossRef CAS PubMed
- L. W. L. Woo, C. Bubert, O. B. Sutcliffe, A. Smith, S. K. Chander, M. F. Mahon, A. Purohit, M. J. Reed and B. V. L. Potter, J. Med. Chem., 2007, 50, 3540–3560 CrossRef CAS PubMed
- L. W. L. Woo, P. M. Wood, C. Bubert, M. P. Thomas, A. Purohit and B. V. L. Potter, ChemMedChem, 2013, 8, 779–799 CrossRef CAS PubMed
- P. M. Wood, L. W. L. Woo, M. P. Thomas, M. F. Mahon, A. Purohit and B. V. L. Potter, ChemMedChem, 2011, 6, 1423–1438 CrossRef CAS PubMed
- T. Jackson, L. W. L. Woo, M. N. Trusselle, S. K. Chander, A. Purohit, M. J. Reed and B. V. L. Potter, Org. Biomol. Chem., 2007, 5, 2940–2952 RSC
- T. K. Vinh, M. Ahmadi, P. O. L. Delgado, S. F. Perez, H. M. Walters, H. J. Smith, P. J. Nicholls and C. Simons, Bioorg. Med. Chem. Lett., 1999, 9, 2105–2108 CrossRef CAS PubMed
- M. R. Saberi, T. K. Vinh, S. W. Yee, B. J. N. Griffiths, P. J. Evans and C. Simons, J. Med. Chem., 2006, 49, 1016–1022 CrossRef CAS PubMed
- A. G. Eissa, D. Barrow, J. Gee, L. E. Powell, P. A. Foster and C. Simons, Eur. J. Med. Chem., 2022, 240, 114569 CrossRef CAS PubMed
- H. A. M. Hejaz, L. W. L. Woo, A. Purohit, M. J. Reed and B. V. L. Potter, Bioorg. Med. Chem., 2004, 12, 2759–2772 CrossRef CAS PubMed
- R. Singh, K. Nagesh and M. Parameshwar, ACS Catal., 2016, 6, 6520–6524 CrossRef CAS
- P. A. Foster, S. K. Chander, S. P. Newman, L. W. L. Woo, O. B. Sutcliffe, C. Bubert, D. Zhou, S. Chen, B. V. L. Potter, M. J. Reed and A. Purohit, Clin. Cancer Res., 2008, 14, 6469–6477 CrossRef CAS PubMed
- A. Purohit, G. J. Williams, N. M. Howarth, B. V. L. Potter and M. J. Reed, Biochemistry, 1995, 24, 11508–11514 CrossRef PubMed
- V. Pestellini, A. Giolitti, F. Pasqui, L. Abelli, C. Cutrufo, G. De Salvia, S. Evangelista and A. Meli, Eur. J. Med. Chem., 1988, 23, 203–206 CrossRef CAS
- S. Mahboobi, A. Sellmer, H. Höcher, C. Garhammer, H. Pongratz, T. Maier, T. Ciossek and T. Beckers, J. Med. Chem., 2007, 50, 4405–4418 CrossRef CAS PubMed
- R. M. Moriarty, S. Grubjesic, B. C. Surve, S. N. Chandersekera, O. Prakash and R. Naithani, Eur. J. Med. Chem., 2006, 41, 263–267 CrossRef CAS PubMed
- S. Kawashita, K. Aoyagi, H. Yamanaka, R. Hantani, S. Naruoka, A. Tanimoto, Y. Hori, Y. Toyonaga, K. Fukushima, S. Miyazaki and Y. Hantani, Bioorg. Med. Chem. Lett., 2019, 29, 2464–2467 CrossRef CAS PubMed
- T. H. Jepsen, M. Larsen, M. Jørgensen, K. A. Solanko, A. D. Bond, A. Kadziola and M. B. Nielsen, Eur. J. Org. Chem., 2011, 2011, 53–57 CrossRef
- M. Okada, S. Iwashita and N. Koizumi, Tetrahedron Lett., 2000, 41, 7047–7051 CrossRef CAS
- D. Ghosh, C. Egbuta and J. Lo, J. Steroid Biochem. Mol. Biol., 2018, 181, 11–19 CrossRef CAS PubMed
-
Molecular Operating Environment (MOE), 2024.06, Chemical Computing Group ULC, 910-1010 Sherbrooke St. W., Montreal, QC H3A 2R7, 2024 Search PubMed
-
Schrödinger Release 2020–1: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2020Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2020, [https://www.schrodinger.com/products/desmond] Search PubMed
- K. J. Bowers, E. Chow, H. Xu, R. O. Dror, M. P. Eastwood, B. A. Gregersen, J. L. Klepeis, I. Kolossvary, M. A. Moraes, F. D. Sacerdoti, K. K. Salmon, Y. Shan and D. E. Shaw, Molecular dynamics---Scalable algorithms for molecular dynamics simulations on commodity clusters, Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, Florida, November 11–17, 2006.
- B. L. Carlson, E. R. Ballister, E. Skordalakes, D. S. King, M. A. Breidenbach, S. A. Gilmore, J. M. Berger and C. R. Bertozzi, J. Biol. Chem., 2008, 283, 20117–20125 CrossRef CAS PubMed
- T. Dierks, M. R. Lecca, P. Schlotterhose, B. Schmidt and K. von Figura, EMBO J., 1999, 18, 2084–2091 CrossRef CAS PubMed
- B. Schmidt, T. Selmer, A. Ingendoh and K. von Figura, Cell, 1995, 82, 271–278 CrossRef CAS PubMed
- M. Recksiek, T. Selmer, T. Dierks, B. Schmidt and K. von Figura, J. Biol. Chem., 1998, 273, 6096–6103, DOI:10.1074/jbc.273.11.6096
- G. Lukatela, N. Krauss, K. Theis, T. Selma, V. Gieseimann, K. Von Figura and W. Saenger, Biochemistry, 1998, 37, 3654–3664, DOI:10.1021/bi9714924
- D. Ghosh, J. Steroid Biochem. Mol. Biol., 2023, 227, 106228, DOI:10.1016/j.jsbmb.2022.106228
- D. Ghosh, Methods Enzymol., 2005, 400, 273–293, DOI:10.1016/S0076-6879(05)00016-9
- J. B. D. Mackenzie, A. Robertson, A. Bushra and R. Towers, J. Chem. Soc., 1949, 2057–2061 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00795f |
| This journal is © The Royal Society of Chemistry 2025 |