
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe overlooked solvent effects: a reconsideration of the paradigm in semiconductor photocatalysis
Xuejiao
Wu
 *ab,
Jonathan
Van Waeyenberg
b,
Dario
Vangestel
b and
Bert
Sels
*b
*ab,
Jonathan
Van Waeyenberg
b,
Dario
Vangestel
b and
Bert
Sels
*b
aState Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Innovation Laboratory for Sciences and Technologies of Energy Materials of Fujian Province (IKKEM), Xiamen University, Xiamen, China. E-mail: wuxjxm@xmu.edu.cn
bCenter for Sustainable Catalysis and Engineering, Faculty of Bioscience Engineering, KU Leuven, Heverlee 3001, Belgium. E-mail: bert.sels@kuleuven.be
First published on 19th December 2024
Abstract
Semiconductor photocatalysis has seen decades of development, with most attention focused on two key elements: semiconductors and solutes. However, the third “S”—solvents—which play a crucial role in condensed-phase reactions, has been surprisingly largely overlooked in this field. Despite their significant impact on chemical reactions, solvents have not received the attention they deserve in semiconductor photocatalysis. By reviewing the historical development of this area, we argue that the limitations on solvent selection are becoming increasingly impractical. We explore the fundamental effects that solvents have on semiconductor photocatalysis, breaking down their complex influence into three areas: semiconductor properties, interfacial charge transfer, and chemical reactions in the solution. This perspective highlights the urgent need for more comprehensive and systematic research on solvent effects. Although not often the main focus of many studies, several examples are provided to demonstrate the importance of solvent effects. Future research directions are also discussed. Ultimately, this review calls for a rethinking of the current approach to semiconductor photocatalysis, emphasizing the critical role of solvents.
 Xuejiao Wu | Xuejiao Wu is currently a full professor at Xiamen University, China. She received her BS degree (2015) from Xiamen University and obtained her PhD degree (2020) from Xiamen University under the supervision of Prof. Ye Wang. She then carried out postdoctoral research at Prof. Bert F. Sels group at KU Leuven (2020–2024). Her research interest is focused on the valorization of lignocellulose, in particular, by photocatalytic approaches. |
 Jonathan Van Waeyenberg | Jonathan Van Waeyenberg earned his MSc degree in Bioscience Engineering with a specialization in Catalytic Technology from KU Leuven in 2018. He is currently pursuing his PhD at the Center for Sustainable Catalysis and Engineering (CSCE) under the supervision of Prof. Bert F. Sels. His research focuses on the thermal pyrolysis of polyolefin plastics using a conical spouted bed reactor. Additionally, he explores secondary catalytic processes to produce refined lubricants and waxes from the resulting pyrolysis products. |
 Dario Vangestel | Dario Vangestel obtained his MSc degree in Bioscience Engineering from KU Leuven in 2023, specializing in Catalytic Process Technology. During his master's research, he worked at the CSCE under the supervision of Prof. Bert F. Sels. His thesis explored the synthesis of Sustainable Air Fuels from lignin and carbohydrate-derived monomers. Building on this research, Dario began his PhD in the same group, where he investigated the photocatalytic synthesis of renewable alternatives to bisphenol-A. More recently, he transitioned to industry, where he is involved in the mechanical recycling of PET, contributing to the production of sustainable materials. |
 Bert Sels | Bert F. Sels is currently a full professor at KU Leuven. His research is focused on the conversion of renewable feedstock to chemicals, materials, and fuels, and the role of heterogeneous catalysis, with a strong eye at the molecular level. The research covers organic chemistry, fundamental kinetics, reaction network and intermediate product studies, catalytic site identification, and reactor and process designs, including TEA and life cycle assessments. Main topics are biorefineries, biobased chemicals, green chemistry, and catalytic strategies for plastic waste and CO2 conversion. |
Broader contextSemiconductor photocatalysis is a rapidly advancing field with widespread application in various energy and environmental initiatives. While the three “S” components—solutes, semiconductors, and solvents—are all critical to the success of semiconductor photocatalysis, the primary focus has traditionally been on semiconductors and solutes. The solvent, an essential element in condensed-phase reactions, has surprisingly been overlooked for an extended period. This perspective revisits the historical development of semiconductor photocatalysis and identifies that the constraints that once limited solvent selection and the understanding of solvent effects have become progressively outdated. It provides a comprehensive analysis of the fundamentals and summarizes key examples that highlight the influence of solvents on semiconductor photocatalysis. By doing so, this perspective challenges the prevailing view that neglects the role of solvents, advocating for renewed attention to these crucial aspects. In addition to addressing existing gaps in the literature, this perspective emphasizes the challenges and opportunities that arise from gaining a deeper understanding of solvent effects. |
1. Introduction
Semiconductor photocatalysis is a promising strategy to efficiently harness inexpensive, abundant, and renewable solar energy for chemical transformations under mild conditions.1,2 Since the first discovery of semiconductor electrodes, using titanium dioxide (TiO2), for water decomposition by photoillumination in 1972,3 significant progress has occurred in the development of semiconductor-based photocatalytic systems over the past few decades.1,2,4–8 It is worth noting that the majority of these systems are carried out in the liquid phase, constituting a homogeneous continuum with multiple components. Typically, the predominant component, occasionally a mixture, is referred to as the solvent, while the minor components are termed solutes.9 Serving as the principal constituent, solvent molecules engage in mutual interactions with one another and various solution species, including reactants, transition states (surface-bonded and diffused), intermediates, products, and catalyst surfaces. The intricate interplay between solvents and these different species plays a pivotal role in shaping their solubility and energetic states. Consequently, the solvent choice can exert a profound impact on both the thermodynamic equilibrium and the kinetics of solution and surface chemical reactions, and eventually impact the product yield and selectivity.10,11The important roles of solvents were recognized a long time ago, as summarized by the famous ancient Greek philosopher Aristotle (384–322 B.C.): “No Coopora nisi Fluida”, meaning “No reactions in the absence of solvents”.9 Opposed to the knowledge in heterogeneous catalysis, solvent effects in photocatalytic systems are often overlooked and underestimated. Despite a significant surge in academic publications on photocatalysis over the past decade, the proportion of papers dedicated to the study of solvent effects remains low, particularly research on such solvent effects in semiconductor photocatalysis is infrequent. In contrast to the approximately 10% of publications related to solvents in general catalytic and heterogeneous catalytic studies, only around 3% of papers in photocatalysis include the keyword of “solvent”. In the field of semiconductor photocatalysis, this ratio further diminishes, reaching close to 2.5% (Fig. 1). Moreover, the explored solvent influence is predominantly reported on semiconductor preparation processes,12 rather than discusses solvent effects on the catalytic reactions. However, from a fundamental perspective, the influence of interacting solvent molecules on semiconductor photocatalysis should be at least as pronounced, if not more, compared to classic heterogeneous catalysis.
 | ||
| Fig. 1 Numbers of annual publications in Web of Science Core Collection on the topic of “Catalysis”, “Photocatalysis”, “Heterogeneous catalysis”, and “Semiconductor photocatalysis” with and without “Solvent”. Percentages of studies with the keyword “Solvent” to those without the keyword “Solvent” are also illustrated, showing the underestimated solvent effects in “Photocatalysis” compared with “Catalysis”, as well as “Semiconductor photocatalysis” compared with “Heterogenous catalysis”. | ||
Liquid-phase semiconductor photocatalysis is usually initiated by irradiation of light with energy equal to or greater than the band-gap energy (Eg) of the semiconductor to generate charge carriers (electron–hole pairs) in the material, which have reduction (electrons) and oxidation (holes) capabilities to initiate a chemical reaction. The photogenerated charge carriers migrate to the surface, where they activate reactants. Reactant molecules are often pre-oriented in the surface–solvent layer (interfacial area), and activation occurs through interfacial electron transfer or, in some instances, proton-coupled electron transfer.13 The surface activation at the surface–solution interface leads to the generation of primary redox intermediates. These intermediates can either remain on the surface or desorb and diffuse to the solvent region. Subsequent reactions, involving reduction, oxidation, or coupling of redox intermediates, can take place within the interfacial or solution area, ultimately yielding the final products (Fig. 2).
 | ||
| Fig. 2 This perspective analyzes and discusses the effects of solvents on semiconductor photocatalysis. These effects are categorized into three areas: the influence of solvents on semiconductor properties, interfacial charge transfer processes, and chemical reactions in the solution. The highlighted dotted square (on the right) emphasizes non-covalent solvent effects, which have long been overlooked in the field of semiconductor photocatalysis. | ||
Solvent molecules in the semiconductor photocatalytic system, as in any heterogeneous catalytic system, can alter the accessibility and properties of the chemisorption (active) sites of the catalyst. Also, the solvation of the solute molecules, to different degrees along the reaction coordinate, can alter the thermodynamic profile. Additionally, in semiconductor photocatalysis, given the involvement of electron transfer, solvent molecules possess the capability to modulate the redox properties of charge carriers and influence the interfacial charge transfer kinetics.14 Also, the redox species generated through charge transfer, including charged intermediates and open-shell radicals, are highly reactive and can interact strongly with solvent molecules. The solvation of these reactive species, influenced by various molecular interactions—both specific (such as hydrogen bonding and electron–donor/electron–acceptor interactions) and non-specific (like dispersion forces and dipole–dipole interactions)—determines their properties, including their electro- and nucleophilicity, lifetime, and stability. Therefore, these reactive species should not be viewed as isolated solute molecules or radicals, but rather as microclusters of solvent–solute complexes, with their structure and reactivity dependent on the properties of the solvent (as illustrated in Fig. 2). Furthermore, the generation and movement of these reactive species lead to the reorganization of solvent molecules, causing significant changes in energy. Section 3.2 provides a detailed explanation of how solvent molecules influence semiconductor photocatalysis.
Despite its importance, the significant role of solvents in semiconductor photocatalysis has been surprisingly overlooked. In this perspective review, we revisit the early development of semiconductor photocatalysis and thoroughly analyse the reasons behind the neglect of solvent effects. Our examination reveals that the previous justifications for ignoring solvent effects are outdated. To highlight the crucial role of solvents, we explore their fundamental impact at the molecular level. The complex effects of solvents are here divided into three categories: their influence on semiconductor properties, interfacial charge transfer, and chemical reactions in the solution (as shown in Fig. 2). Although not the main focus of most studies, a few examples of solvent effects on semiconductor photocatalysis are presented to demonstrate the emerging opportunities for optimizing these systems by exploring solvent interactions.
This review also provides perspective for future research in this area. We advocate for a reconsideration of the dominant approach in semiconductor photocatalysis, which has primarily focused on semiconductors and solutes—the two “S”s—encompassing semiconductor synthesis, properties, and semiconductor–solute interactions, along with their structure–activity relationships. To our knowledge, this is the first review specifically focused on the solvent effect in semiconductor photocatalysis. It aims to emphasize the importance of the third “S”—solvent—drawing attention to this essential, yet often underappreciated area of research.
2. Why is the solvent effect overlooked?
Reflecting on the factors that have hindered the development of solvent-related semiconductor photocatalysis presents an opportunity to consider the fundamental properties necessary for selecting an appropriate solvent. Moreover, examining the gaps in past research offers valuable insights into future research directions.2.1 Limited solvent choices
Being the first semiconductor discovered for photocatalysis, TiO2 has garnered significant attention from the outset.3 Subsequently, significant efforts have been dedicated to the design and development of metal oxide semiconductors. In the band structure of metal oxides, the atomic orbitals of metal cations and O2− ions undergo hybridization, resulting in numerous closely spaced molecular orbitals that form the conduction band (CB) and the valence band (VB). The band edges, specifically the bottom of the CB and the top of the VB, primarily reflect the characteristics of the outermost orbitals. For metal oxides, the CB edges are mainly derived from the metal's s or p orbitals, while the VB edges are largely contributed by the O 2p orbitals. Notably, O 2p orbitals, which are the major contributors to the VB edges, possess relatively low energy (Fig. 3a). This low energy of O 2p orbitals signifies the high oxidation potential of most metal oxides, but it also results in a wide bandgap, making them primarily responsive to UV light.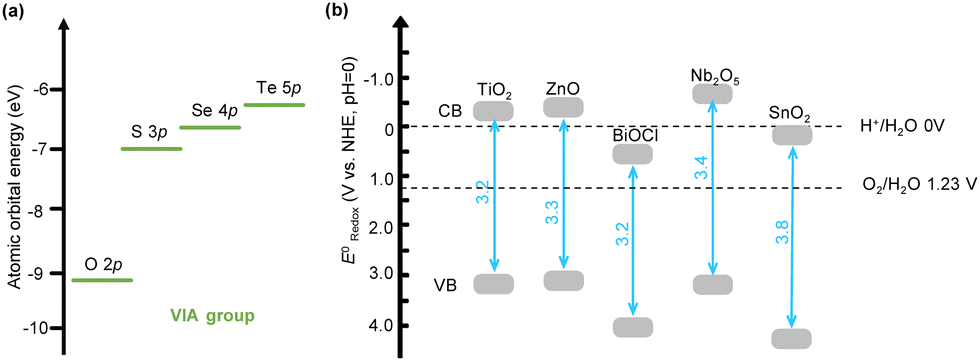 | ||
| Fig. 3 (a) Atomic orbital energies of O, S, Se, and Te in the VIA group, showcasing the low energy of O 2p. The values are from the NIST database. (b) Band position of some typical metal oxides. | ||
Metal oxides that have been widely studied include TiO2, ZnO, BiOCl, Nb2O5, and SnO2, with bandgaps of 3.2 eV, 3.3 eV, 3.2 eV, 3.4 eV, and 3.8 eV, respectively. Consequently, these materials can only absorb UV light with wavelengths below 388 nm, 375 nm, 388 nm, 365 nm, and 326 nm, respectively (Fig. 3b).
As metal oxides dominated the early years of semiconductor photocatalysis research, employed light sources mostly emitted UV light. The most frequently used light sources included low and medium-pressure Hg lamps, which have prominent emissions at 254 nm, 313 nm, 366 nm, 405 nm, and 550 nm (Fig. 4).15 To avoid photon attenuation, i.e., progressive loss of beam energy when travelling through matter, solvents with cut-off wavelengths below 254 nm were commonly chosen for photocatalytic reactions (Fig. 4).
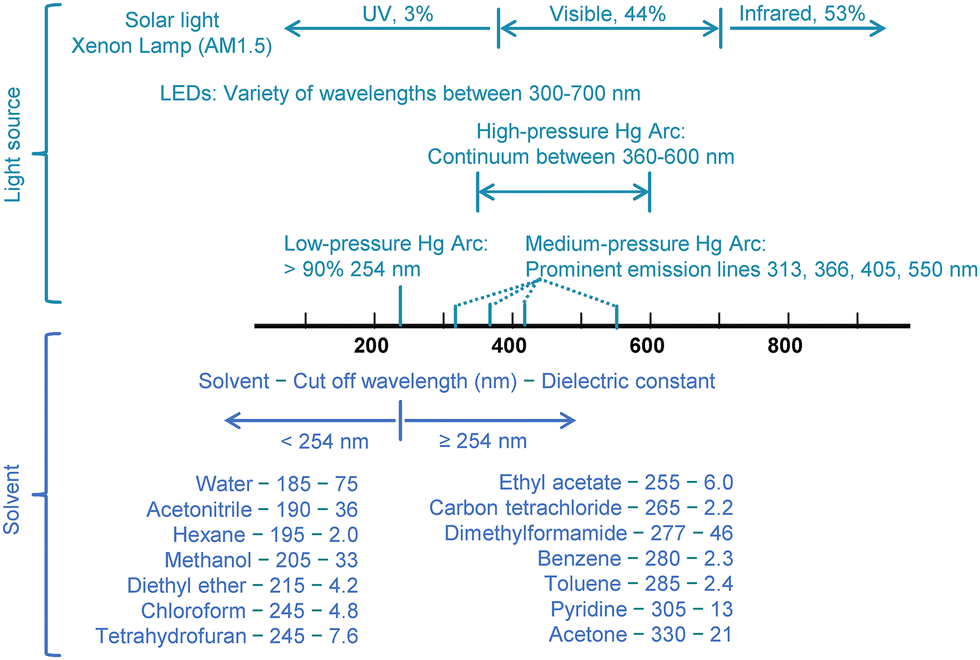 | ||
| Fig. 4 The most commonly used light sources for semiconductor photocatalysis, along with their emission wavelengths, are listed alongside common solvents, with their cut-off wavelengths, and dielectric constants. | ||
In addition to avoiding light absorption, another critical factor in solvent selection is its ability to dissolve substances and facilitate their diffusion, which further narrows the range of suitable solvents. For solid and liquid substrates with low mobility, an effective solvent should be capable of disrupting the lattice structure of solid reactants or breaking the interactions among liquid reactants. This increases the mobility of the reactants once they are dissolved. According to the “like dissolves like” principle, a solvent's ability to dissolve solutes is closely linked to its polarity.16Fig. 4 summarizes some common solvents, along with their cut-off wavelengths and dielectric constants, which indicate their polarity. Water (H2O) is frequently used as a solvent when ionic compounds are involved in a reaction. However, since most chemicals are polar organic compounds that are insoluble in water, acetonitrile (CH3CN) is often used as an alternative, despite its toxicity and associated environmental and health risks,17 as the most popular organic solvent in semiconductor photocatalysis studies.18
Another advantage of acetonitrile is its broad redox potential window, ranging from −3.55 V to 2.35 V when measured with the Pt electrode and the LiCiO4 supporting electrolyte.19 This extensive range allows acetonitrile to be versatile and compatible with semiconductors that have different CB and VB edges.20 It also supports reactions under a broad spectrum of redox potentials.
2.2. Incorrectly assumed uniformity of solvent effects
During the early development of semiconductor photocatalysis, two of the most investigated applications are environmental remediation and hydrogen evolution.6,21 The aim of these systems is to achieve high activity, while controlling selectivity is not necessary. In many cases, there is strong adsorption of the reactants, making the influence of solvent molecules through non-covalent interactions appear negligible. For instance, in the degradation of pollutants, strong adsorption of the pollutants onto the semiconductor surface is desirable for complete mineralization. Hydrogen bonding is regarded as one of the strongest forms of non-covalent solvent–solute interactions, with typical energy values ranging from 3 to 5 kcal mol−1.22 This value is insignificant compared to the adsorption energy of pollutant molecules on the semiconductor surface. For example, the adsorption energy of phenol on the ZnO surface is ∼50 kcal mol−1 according to density functional theory (DFT) calculations.23Furthermore, to improve the charge transfer process and, consequently, the catalytic activity, these systems are typically highly thermodynamically favourable, indicating a significant reaction driving force, which is the energy difference between the excited reactants and products. Although changing the solvent can still affect the driving force and, in turn, the free activation energy associated with the charge transfer process, the impact is limited due to the already substantial driving force present.
2.3. Barriers to identify solvent contributions
The processes related to charge carriers in photocatalysis occur at extremely rapid rates, typically within the time scale of femtoseconds (fs, 10−15 s) to milliseconds (ms, 10−3 s). For instance, consider TiO2 as an example (Fig. 5).24 Research has shown that the photoexcitation of TiO2, which generates electron–hole pairs (e−/h+), occurs on the femtosecond (fs) time scale. The generated charge carriers can quickly transfer (on the fs scale) and be captured by trap states either in the bulk or on the surface. Trap states are localized energy levels within the band gap that acts as temporary storage sites for charge carriers. Common types of trap states include vacancies and interstitials. Some charge carriers recombine directly within femtoseconds (fs), while the time scale for indirect recombination of photogenerated e−/h+ depends on various trap states, typically ranging from picoseconds (ps, 10−12 s) to nanoseconds (ns, 10−9 s). Trap states can be categorized into two types: shallow traps, which are located near the bottom of the conduction band and the top of the valence band, and deep traps, which have energy levels closer to the center of the band gap (Fig. 5). Generally, recombination processes involving shallow traps occur more rapidly than those involving deep traps. The characteristics of trap states, especially surface trap states, are highly influenced by their surrounding environment, and the presence of adsorbed solvent molecules can significantly modify their properties.25 Interfacial charge transfer processes occur on a time scale of 10−3 to 10−9 seconds. The properties of the semiconductor, such as band position and the accessibility and characteristics of adsorption/activation sites, can be affected by the choice of the solvent. The fundamental principles are elaborated in Section 3.2.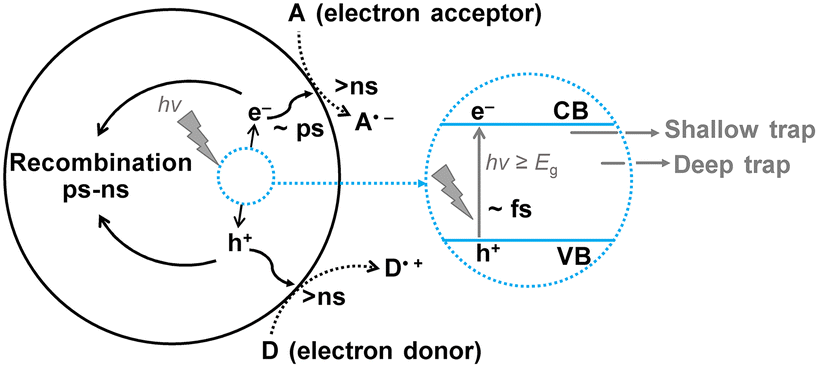 | ||
| Fig. 5 Time scale of typical electron processes in TiO2-based photocatalytic systems. | ||
The charge transfer leads to the formation of radical species under mild conditions. These radical intermediates are typically highly reactive, and their subsequent reactions generally occur rapidly. The rate constants for radical reactions can range from 10−1 to 1010 s−1 for unimolecular reactions or 10−1 to 1010 dm3 mol−3 s−1 for bimolecular reactions.26 The rate constants are generally influenced by the polarity, stereo-electronic properties, and steric structure of the transition states of radical species, all of which can be adjusted by altering the solvent environment.27
These ultrafast processes present a significant challenge in identifying solvent effects, necessitating ultrafast characterization techniques with high time scale sensitivity. Moreover, as previously mentioned, solvents can impact reactions in multiple ways. Altering solvent properties can simultaneously affect semiconductor characteristics, interfacial charge transfer processes, and radical chemistry. These effects are often interrelated, making it difficult to separate them from one another.
3. The critical need to explore solvent effects
The following analysis shows that the historical reasons for disregarding solvent effects are no longer relevant, highlighting the importance of giving greater attention to the often-overlooked influence of solvents. Then, the fundamental importance of solvents in semiconductor photocatalysis is highlighted, emphasizing the critical roles that solvent molecules can play in modulating the system. Additionally, examples of solvent-dependent semiconductor photocatalytic systems have been selected for emphasis, even if solvent effects are not the primary focus of their respective studies. It is important to note that solvent molecules can also directly participate in the reaction as substrates, such as in the case of sacrificial reagents28 and hydrogen sources.29 However, these are not included in the discussion. Instead, our focus is exclusively on the more common but often overlooked non-covalent solvent effects.3.1. Historical constraints on exploring solvent effects are gradually diminishing
Section 2 offers an overview of the historical development of semiconductor photocatalysis and identifies three key reasons why solvent effects have been underexplored. Unfortunately, these constraints, when considered from a historical standpoint, have often been accepted uncritically, resulting in a lack of attention to solvent effects. This section critically reevaluates these past limitations from a modern perspective, illustrating that the restrictions on solvent development have become progressively outdated.Moreover, the growing use of visible light-responsive semiconductors, such as metal sulfides with lower oxidation and reduction potentials, expands the range of solvent options based on the redox potentials. While certain solvents, like methanol and ethanol, remain reactive due to their low oxidation and reduction potentials, many conventional solvents—including sulfolane, tetrahydrofuran (THF), and N,N-dimethylformamide (DMF)—as well as sustainable solvents like γ-valerolactone (GVL), offer wide redox potential windows that are suitable for most visible light-responsive semiconductors.19 The shift from UV to visible light, coupled with the transition from wide-bandgap semiconductors to those with appropriate redox potentials, has rendered the previous limitations on solvent selection obsolete.
Over the past few decades, semiconductor photocatalysis has created new opportunities in fields such as CO2 reduction, organic synthesis, and biomass conversion, where the use of water as a solvent is often less advantageous or even prohibited. For CO2 reduction, alkaline solvents like DMF and dimethyl sulfoxide (DMSO), which have higher CO2 solubility, are more commonly employed because they do not compete for electron consumption as water does. In organic synthesis and biomass valorization, the solubility of organic compounds in water can be limited. Additionally, the highly functionalized nature of these substrates presents a significant challenge for selectivity control, utilizing organic solvents with diverse properties can facilitate the fine-tuning of the reaction system, thereby impacting reaction selectivity.
Recent advancements in time- and spatial-sensitive techniques have significantly enhanced our understanding of semiconductor behaviour during photocatalysis. Transient spectroscopy provides crucial information on the decay dynamics of excited electrons and holes, as well as insights into the lifetime of photogenerated charge carriers and the identification of various exciton types.32 Microscopic techniques have been used to map charge distributions on semiconductor photocatalysts.33 More recently, spatiotemporally resolved surface photovoltage measurements have been introduced to offer a comprehensive mapping of charge transfer processes, encompassing timescales ranging from femtoseconds to seconds.34
A range of in situ techniques have been developed to monitor radicals or reactive intermediates in semiconductor photocatalysis. Time-resolved attenuated total reflection infrared spectroscopy (ATR-IR) is capable of evaluating the behaviour of surface adsorbates. In situ electron spin resonance (ESR) and ultrafast spectroscopy enable the examination of radical intermediates and their lifetimes, shedding light on how solvent properties influence their stability and reactivity.32,35 Nuclear magnetic resonance (NMR) spectroscopy36 allows for the direct observation of the solvation structures of solutes in solution. Sum frequency generation (SFG) spectroscopy is effective in investigating the molecular orientation of adsorbates on semiconductor surfaces.37 The insights gained from these techniques can be further enriched by DFT calculations, which offer a molecular understanding of the solvation structures of key species in semiconductor photocatalysis.
3.2. Fundamentals to emphasize the importance of solvents
Spatially, the effects of solvents on semiconductor photocatalysis can be broadly categorized into three areas: the properties of the semiconductor, interfacial charge transfer, and chemical reactions in the solution phase (Fig. 2). Regarding semiconductor properties, substrate adsorption on the surface typically occurs before charge transfer, competing with solvent adsorption. Additionally, the desorption of intermediates and products is vital for product selectivity. Solvent molecules play a crucial role in influencing the accessibility and characteristics of adsorption and activation sites. Furthermore, the adsorption of solvent molecules on the semiconductor surface can cause a shift in the band edge potentials near the interface, a phenomenon known as semiconductor band bending.38Fig. 6 depicts the upward band bending near the semiconductor surface caused by the adsorption of an acceptor molecule (denoted as “A”). As molecule A approaches the surface, its lowest unoccupied molecular orbital (LUMO) shifts downward and broadens in energy. This lowering of the acceptor's molecular orbital allows electrons to flow from the semiconductor to the molecule, resulting in the formation of a Helmholtz layer. As a result, an electric field is established near the semiconductor surface, causing the band to bend upwards. Conversely, donor molecules would lead to downward band bending.39 | ||
| Fig. 6 Schematic illustration showing the band bending of semiconductors upon adsorption of an acceptor molecule on a semiconductor surface. | ||
Semiconductor flat band potentials (Vfb), which are associated with band bending, exhibit different behaviors in protic and aprotic solvents. A significantly more positive Vfb for TiO2 was observed in water and non-aqueous protic solvents (such as methanol and ethanol) compared to non-aqueous aprotic solvents like acetonitrile, dimethylformamide, and tetrahydrofuran. This discrepancy between protic and aprotic solutions is thought to arise from the presence or absence of proton adsorption–desorption equilibrium.40
The photoelectric properties of semiconductors, such as light absorption capacity and the mobility of photoexcited charge carriers, can also be affected by solvent molecules. A comparison can be made between solvent–semiconductor interactions and ligand–quantum dot (QD) systems, where changes in QD ligands have a substantial impact on light absorption efficiency and exciton mobility.41 Likewise, solvent molecules may exert a comparable influence, albeit lesser, ultimately impacting the photocatalytic quantum yield and charge carrier lifetimes. To the best of our knowledge, explicit studies on such solvent effects have not yet been reported. This is likely due to the perception of semiconductor photoelectric properties as intrinsic features and the current reliance on ex situ characterization, making the development of in situ techniques to examine these properties in the presence of solvent molecules highly desirable.
Substrate activation at the surface–solvent interface through interfacial electron transfer is regarded as a critical step in semiconductor photocatalysis. The influence of solvation effects on the electron transfer process can be explained using the well-established Marcus theory.42,43 To illustrate electron transfer from the semiconductor (denoted as “S”) to the reactant (denoted as “R”), two harmonic free energy curves are employed to represent the system before (S–R) and after (S+–R−) the electron transfer (Fig. 7). It is important to note that the nuclear coordinate encompasses not only the semiconductor and reactant but also the surrounding solvent molecules. The electron transfer between the semiconductor and the reactant leads to the rearrangement of solvent molecules along the reaction coordinate, resulting in significant solvent polarization free energy.44 The rate constant of the electron transfer (kET) process can be predicted by a semi-classical expression:
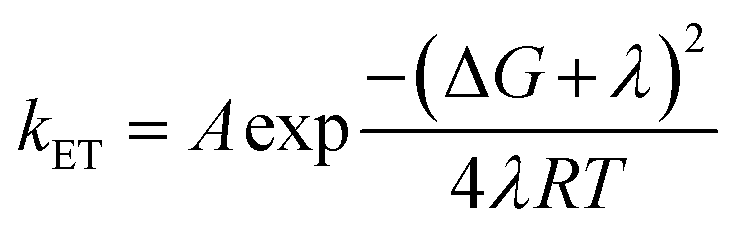
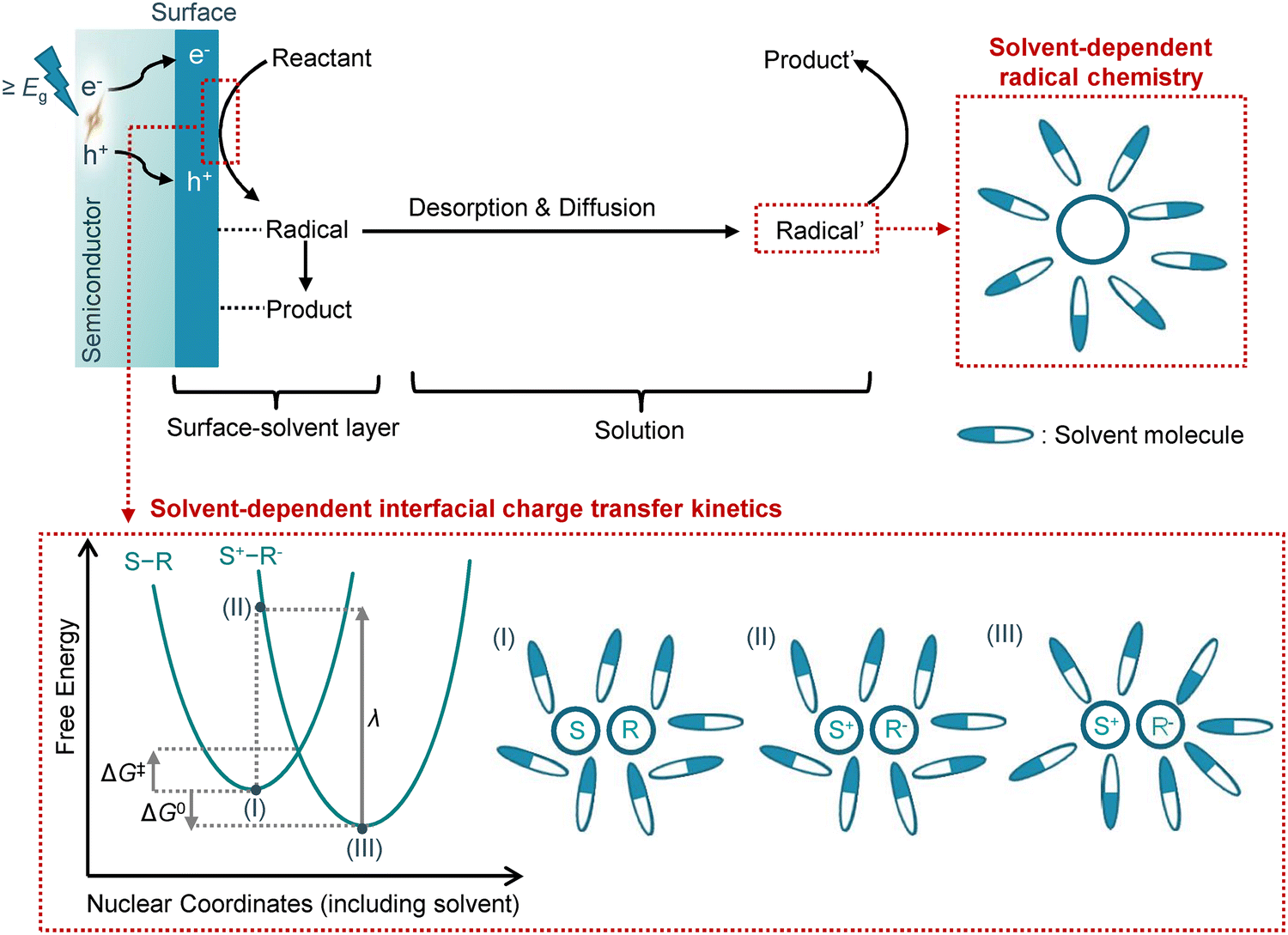 | ||
| Fig. 7 A semiconductor-based photocatalytic system where the solvent effects on the interfacial electron transfer kinetics and the radical chemistry are highlighted. A graphical depiction of solvent-dependent thermodynamics and kinetics for the interfacial charge transfer process according to Marcus theory is illustrated in the lower red-dotted square. S, R, ΔG‡, ΔG0, λ, and kET represent the semiconductor, reactant, activation energy, Gibbs free energy, reorganization energy, and electron transfer rate constant, respectively. | ||
Gibbs free energy (ΔG), a crucial parameter for chemical equilibrium constants, represents the energy difference between states S–R (state (I) in Fig. 7) and S+–R− (state (III) in Fig. 7) at equilibrium coordinates, making it dependent on the solvent. The reorganization energy (λ) is defined as the change in energy when the equilibrium reactant state is distorted to the nuclear coordinates of the product (state (II) in Fig. 7) without any electron transfer. The value of λ primarily arises from the outer shell reorganization energy, which is the energy change resulting from the reorientation of solvent molecules. As the Marcus theory illustrates, the solvation effects can significantly influence the interfacial charge transfer kinetics (kET) by affecting both the ΔG and λ values.
Intermediates generated from interfacial charge transfer, primarily consisting of radical species, can desorb from the surface and participate in subsequent reaction steps within the solution phase. These radical intermediates are characterized by open-shell electronic or charged structures, making them highly reactive. While this high reactivity facilitates rapid radical reactions, it also presents challenges in controlling selectivity.45 Notably, kinetic solvent effects have been observed in radical reactions,46 underscoring the potency of solvents in governing desorption, diffusion, activity, persistence, lifetime, and concentration of radicals (Fig. 7). Thus, the selection of solvents can serve as a significant tool for influencing radical selectivity.
In the solution phase, solvent molecules not only influence reactive radicals but also affect the fate of reactions by modulating the diffusion of stable solutes, including reactants, intermediates, and products. When solvents with high viscosity, such as ethylene glycol (18.38 cP), are used instead of commonly employed low-viscosity solvents like water (0.89 cP) and acetonitrile (0.36 cP), solutes can become temporarily confined within a “viscous cage” formed by the solvent molecules. This confinement slows their diffusion, potentially altering reaction rates and pathways. Therefore, the viscosity of the solvent, often overlooked in photocatalysis, should be carefully considered, particularly for reactions where solute diffusion plays a critical role, such as photocatalytic couplings.
3.3. Solvent-driven semiconductor photocatalysis
Recently, a few solvent-sensitive systems have been reported, and we have selected examples where solvents affect both activity and selectivity, emphasizing their crucial role in semiconductor photocatalysis. These examples are categorized according to their spatial regions, as outlined in Section 3.2, specifically focusing on solvent effects on semiconductor properties, the interface where charge transfer occurs, and the surrounding solution.Solvent molecules can influence the accessibility and characteristics of adsorption and activation sites. Through competing with substrates for semiconductor surface adsorption sites, the solvent hinders interfacial electron transfer and thereby reduces the reaction rate. In the photocatalytic oxidation of glycerol to dihydroxyacetone, glyceraldehyde, and hydroxypyruvaldehyde over carbon nitride (C3N4), the reaction was carried out in acetonitrile, DMF, and water. As solvent polarity increased, glycerol conversion decreased, from 43.2% in acetonitrile to 29% in DMF, and just 4.4% in water.47 DFT results revealed that the primary factor contributing to the reduced activity in glycerol oxidation is the competitive adsorption between glycerol and solvent molecules on the surface of C3N4. Solvent molecules, especially in polar solvents, can compete with glycerol for active sites on the photocatalyst. This competition limits the availability of adsorption sites for glycerol, thereby hindering its oxidation and leading to decreased catalytic efficiency. The findings emphasize the critical role of solvent selection in tuning the photocatalytic performance of C3N4 in glycerol oxidation.47
While solvent competition for adsorption sites is typically seen as detrimental to catalytic performance, some cases demonstrate that the presence of the solvent on the semiconductor surface can actually enhance selectivity. For example, in the photocatalytic oxidation of cyclohexane over TiO2, non-polar solvents led to full mineralization, producing CO2 and H2O. However, in polar solvents, there was a high selectivity toward cyclohexanol and cyclohexanone. This increased selectivity is attributed to the competitive adsorption of polar solvents and the products (cyclohexanol and cyclohexanone), which facilitates product desorption and improves selectivity.48 Recently, solvent effects for anaerobic oxidation of benzyl alcohol (BA) to produce benzaldehyde (BAD) over SrTiO3 nanoparticles were investigated.49 The photocatalytic activity was influenced by the concentration of DMF or acetonitrile in water. Control experiments showed that when the solvent competes with the substrate for adsorption sites, it can impede the reaction. However, competition between the solvent and reaction products proved advantageous. Consequently, solvent mixtures with moderate polarity exhibited the highest photocatalytic activity, striking a balance between these competing effects.
The photocatalytic conversion of CO2 into formate (HCOO−) and carbon monoxide over TiO2/SiO2 was studied in different solvents.50 Increasing the dielectric constant of solvents led to higher selectivity for formate production. For example, nearly 100% selectivity for CO was achieved in CCl4, while in water, CO2 was primarily converted to formate (Fig. 8). This solvent effect is attributed to the varying abilities of solvents to stabilize CO2 anion radical intermediates. In low-polarity solvents, the CO2 intermediate strongly adsorbs onto Ti sites on the surface, allowing for the removal of an oxygen atom with a proton, producing CO and H2O. In contrast, high-polarity solvents stabilize the intermediate, reducing its surface interaction and favoring protonation and desorption of formate as the major product (Fig. 8). A similar solvent effect, with CO2 being reduced to CO in chloroform (a low-polarity solvent) and to formate in water (a polar solvent), was also observed on ZnS.51
 | ||
| Fig. 8 Proposed mechanisms of selective conversion of CO2 to CO and formate in low and high polarity solvents, respectively. Reproduced from ref. 50, with the permission from Elsevier, copyright 1997. | ||
The interaction between solvents and catalysts can enhance catalyst dispersion, improving their accessibility to reactants. A notable example is the photocatalytic conversion of native lignin, a key component of lignocellulose that is tightly bound with carbohydrates, using hydrophilic quantum dots (QDs) in polar solvents. This interaction helps facilitate the reaction by increasing the contact between the catalyst and lignin.52–54 The lack of effective contact between the solid catalyst and the solid lignocellulosic biomass, which can hardly be dissolved in any solvent at room temperature,55,56 hurdles the use of semiconductor photocatalysis for its valorization. Control experiments demonstrated that the hydrophilicity of solvents plays a crucial role in maintaining the colloidal stability of hydrophilic quantum dots (QDs), which in turn ensures their accessibility to the targeted C–O linkages.53,54
Changing the pH is expected to induce a shift in the band edge potentials of semiconductors, primarily due to the aforementioned band bending effect. Many semiconductors exhibit Nernstian pH dependence for their band edge potentials, such as TiO2, which has a slope of 59 mV per pH unit, paralleling the behavior of water oxidation and reduction. As a result, the thermodynamics of these semiconductors for hydrogen evolution are typically not significantly impacted by pH changes. In contrast, for CdS, the band edge shift with pH is lower, at 33 mV per pH unit, suggesting that the increasing pH favors the formation of hydroxyl radicals (˙OH) during water oxidation. This reduced pH dependence of the CdS band edge, compared to water oxidation, leads to increased ˙OH production at higher pH levels, significantly enhancing the rates of ethanol photoreforming.57
The coupling of radical intermediates generated from the photocatalytic C–H activation of alcohols, such as methanol, ethanol, and butanol, into diols is thought to occur primarily in the solution phase.58,59 Competing reactions involve the further oxidation of radical species, leading to the formation of aldehydes, and back electron transfer to the original alcohol reactant, both of which occur on the semiconductor surface. To enhance selectivity for diol synthesis, it is crucial to effectively remove radical intermediates from the semiconductor surface. Recently, Wang and colleagues demonstrated the photocatalytic coupling of ethanol to produce 2,3-butanediol (BDO), with acetaldehyde (AA) identified as a major side product.60 Compared with the reaction results using pure ethanol, with the addition of 5% water, the productivity of BDO was 2.4 times higher, and the selectivity of BDO increased from 37% to 57%.60 Combined experimental and computational findings demonstrated that hydrogen bonding between radical intermediates, influenced by the addition of water, enhanced selectivity. The α-hydroxyethyl radicals (αHRs), generated from the cleavage of the Cα–H bond in ethanol during photoirradiation, form hydrogen bonds with water molecules adsorbed on the surface, facilitating the desorption of αHRs. Additionally, the presence of water aids in stabilizing the desorbed αHRs in the bulk solution, thereby reducing the likelihood of their re-adsorption onto the surface (Fig. 9).60 Consequently, the addition of water inhibited side reactions, such as oxidation to acetaldehyde (AA) and reverse reactions back to ethanol, resulting in enhanced activity and selectivity for the coupling of ethanol to 2,3-butanediol (BDO).
 | ||
| Fig. 9 Schematic illustration of the proposed mechanism for the presence of water in steering the reaction paths, changing the reaction of αHRs (α-hydroxyethyl radicals) from oxidation for the acetaldehyde generation toward C–C bond coupling by forming hydrogen bonds. Reproduced from ref. 60, with the permission of American Chemical Society, copyright 2022. | ||
Photocatalytic activation of substrates in semiconductor-based systems can also occur in the solution phase, initiated by additives that generate active species. A common example is the hydroxyl radical (˙OH), which is frequently utilized in reactions requiring highly reactive species, such as methane oxidation. However, the highly reactive nature of ˙OH can limit reaction selectivity, necessitating careful control of its concentration. Regulation of the radical concentration can be achieved by adjusting the solvent composition. The nitrite ion serves as both a source and a scavenger of ˙OH radicals, effectively acting as an “˙OH buffer” to manage the ˙OH concentration. By adding a small amount of nitrite, the formation of undesired CO2 can be completely suppressed, thereby enhancing the selectivity for the oxidation of methane to methanol (CH3OH) over BiVO4.61 Enhanced generation of hydroxyl radicals (˙OH) was achieved by introducing Fenton reagents (Fe2+ and H2O2) into the solution. The Fenton reaction (Fe2+ + H2O2 → Fe3+ + ˙OH + OH−) facilitated the production of ˙OH, which in turn increased the yield of methanol (CH3OH) from methane (CH4) oxidation. Notably, the photogenerated electrons over BiVO4 reduced Fe3+ ions back to Fe2+ ions.62
The diffusion behavior of radical species is significantly influenced by the properties of the solvent. In the context of photocatalytic C–N coupling, specifically the addition of 2,5-dihydrofuran (2,5-DHF) to azobenzene, radical mobility in the diffusion phase was studied by varying the pressure and utilizing different alcohol solvents.63 The rates of the C–N addition reaction decreased as pressure increased from 0.1 to 120 MPa. This suggests that the higher viscosity and, consequently, lower radical mobility at elevated pressures are responsible for the reduced activity. However, this conclusion is not definitive, as the increased pressure also raises the dielectric constant of the solvent.63 These two possibilities were differentiated by measuring reaction rates in a series of alcohols, where the viscosity and the dielectric constant changed in opposite directions. As expected, the viscosity continued to reduce the reaction rates, even with the increase in dielectric constants.63 The combined results emphasize the impact of solvents on the diffusion behavior of radical species in solution.
High ortho selectivity for the photoelectrocatalytic C–H/N–H coupling of anisole with pyrazole was accomplished using a co-solvent system of hexafluoroisopropanol (HFIP) and methanol (CH3OH). The yields and product selectivity changed with different ratios of HFIP and CH3OH, with optimal conditions producing a 77% yield of the coupling product and an ortho/para ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in an HFIP/MeOH mixture (4:1) (Fig. 10).64 It is important to note that the para product was observed in other photoredox and electrochemical systems that utilized 1,2-dichloroethane or CH3CN as the solvent. One possible explanation for this difference is that amination at the ortho position is favored due to the hydrogen-bonding network formed between anisole, HFIP, and pyrazole (Fig. 10, within the dotted square).64 Control experiments with various substrates and solvents were conducted to validate the theory. In the absence of hydrogen bonding, such as when using diphenyl and 9,9-dimethyl-9H-fluorene as substrates, only the para product was formed. When CF3CH2OH/MeOH (4:1) was used as the solvent, the yield of the ortho product was 61%, with a selectivity ratio of 2 (ortho):1 (para); the ortho product was still favored, although its selectivity was lower than that observed in HFIP/MeOH. In contrast, when non-fluorinated CH2ClCH2Cl was employed as the solvent, no selectivity was observed, resulting in a ratio of 1:1 for ortho to para products.64 These results support the hypothesis that hydrogen bonding from fluorinated alcohols enhances ortho selectivity.
:1 in an HFIP/MeOH mixture (4:1) (Fig. 10).64 It is important to note that the para product was observed in other photoredox and electrochemical systems that utilized 1,2-dichloroethane or CH3CN as the solvent. One possible explanation for this difference is that amination at the ortho position is favored due to the hydrogen-bonding network formed between anisole, HFIP, and pyrazole (Fig. 10, within the dotted square).64 Control experiments with various substrates and solvents were conducted to validate the theory. In the absence of hydrogen bonding, such as when using diphenyl and 9,9-dimethyl-9H-fluorene as substrates, only the para product was formed. When CF3CH2OH/MeOH (4:1) was used as the solvent, the yield of the ortho product was 61%, with a selectivity ratio of 2 (ortho):1 (para); the ortho product was still favored, although its selectivity was lower than that observed in HFIP/MeOH. In contrast, when non-fluorinated CH2ClCH2Cl was employed as the solvent, no selectivity was observed, resulting in a ratio of 1:1 for ortho to para products.64 These results support the hypothesis that hydrogen bonding from fluorinated alcohols enhances ortho selectivity.
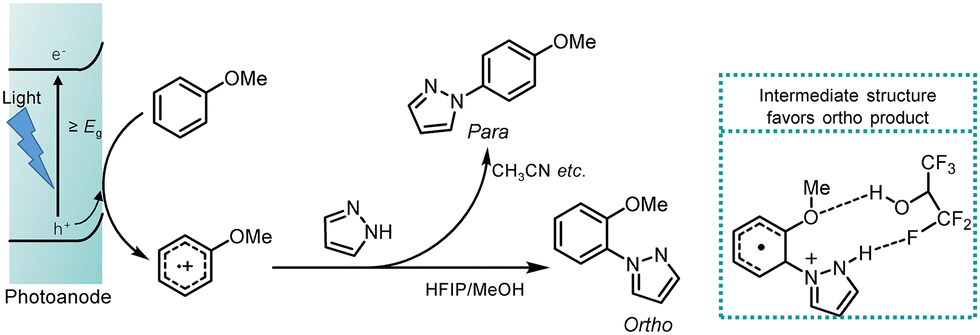 | ||
| Fig. 10 Proposed mechanism for photoelectrocatalytic C–N coupling of anisole with pyrazole, and the green dotted square includes the proposed hydrogen bonding among anisole, pyrazole, and hexafluoroisopropanol (solvent), which may contribute to the favorable formation of the ortho product. Reproduced from ref. 64, with permission from Springer-Nature, copyright 2019. | ||
Recently, high activity-solvent dependency was observed in the photocatalytic coupling of toluene by Zn2In2S5. A comprehensive investigation of the intricate solvent effects, including the influence of solvent molecules on the semiconductor surface, the charge transfer process, and the solution phase, has been conducted. Solvent molecules with a high Gutmann donor number (DN), such as dimethyl acetamide and dimethyl formamide, can retard the reaction by competitive adsorption on the hole-delocalized active sites. For the hole-transfer-induced C–H bond activation, it has been illustrated that solvent molecules with a large dielectric constant (ε) and a refractive index (n) are favorable. This can be rationalized by the dielectric continuum theory, where a large ε can increase the driving force, likely through higher solvation stabilization of the benzyl radical compared to the reactant; while a higher n of the solvent indicates greater polarizability, which facilitates easier reorganization and helps in decreasing reorganization energy. After C–H activation, the generated benzylic radical can undergo a quick coupling reaction in the solution phase to form coupling products. Solvents with high viscosity, including Cyrene and ethylene glycol, hinder fast coupling and thus result in low coupling product yields (Fig. 11).65
 | ||
| Fig. 11 Solvent effects on the photocatalytic coupling of toluene by Zn2In2S5. “1. Competitive adsorption”, “2. Solvent-dependent hole-transfer-induced C–H activation kinetics”, and “Solvent-dependent coupling” indicate the influence of solvent molecules on the semiconductor surface, the charge transfer process, and the solution phase, respectively. These are summarized from ref. 65. | ||
It is noteworthy that the photocatalytic coupling of toluene by Zn2In2S5 demonstrated the highest efficiency when performed in the environmentally friendly solvent γ-valerolactone (GVL). GVL offers several advantageous properties that promote the photocatalytic coupling process. Its relatively low donor number (DN) and viscosity minimize competitive adsorption on the active sites, preventing the reaction from being slowed down during the coupling step. Additionally, GVL's combination of a large dielectric constant (ε) and a high refractive index (n) contributes to a lower energy barrier for the hole-transfer-induced activation of the C–H bond. These synergistic factors enhance the overall photocatalytic efficiency, establishing GVL as the optimal solvent for toluene coupling reactions.65
4. Conclusions and outlook
As the dominant component of liquid-phase systems, solvent molecules play a pivotal and omnipresent role in shaping chemical reactions. However, in the realm of semiconductor photocatalysis, the focus has largely been confined to the semiconductor and the solute—the two “S”s—while the equally significant impact of the third “S”, the solvent, has often been overlooked. Revisiting the early history of semiconductor photocatalysis reveals that the choice of solvents was initially constrained by the compatibility requirements of both the catalyst and the light source. Additionally, there seems to have been an early misconception that solvent effects were uniform, particularly in the study of widely researched systems like environmental remediation and hydrogen evolution. This neglect of solvent influence has made it challenging to characterize the solvation structures of reactive, short-lived species and to unravel the complex network of solvent effects at play.A deeper analysis of solvent effects, particularly from a molecular perspective, reveals the crucial role solvents play in guiding both activity and selectivity in semiconductor photocatalysis. As this field experiences rapid advancements—marked by the discovery of visible-light-sensitive photocatalysts and novel materials such as quantum dots, and expanding applications in areas like CO2 reduction, organic synthesis, and biomass valorization—new solvent-sensitive systems have begun to emerge. Recent breakthroughs in techniques such as DFT calculations and in situ/operando characterization have further enabled a more nuanced understanding of these effects.
Despite these advancements, research specifically dedicated to investigating solvent effects in semiconductor photocatalytic systems remains limited. Even in cases where solvents demonstrably influence the regulation of activity and selectivity, their role often remains secondary to the primary focus of research. This lack of attention is problematic, as fully understanding and controlling solvent effects is essential for optimizing the performance of semiconductor photocatalysts. Therefore, future research must prioritize this often-neglected aspect. We propose several key areas for future exploration, emphasizing the need for a more deliberate focus on the influence of solvents in these systems.
To deepen our understanding of solvent effects, it is crucial to explore a wider variety of solvents. As mentioned earlier, acetonitrile is currently the most widely used organic solvent in semiconductor photocatalytic systems due to its favorable properties—low cut-off wavelength, high polarity, and photostability. However, acetonitrile's toxicity poses a challenge to the “green” nature of semiconductor photocatalysis. Given that most solvents have cut-off wavelengths between 200 nm and 400 nm, many are suitable for use under visible light or simulated solar light conditions. With the increasing focus on visible-light-driven systems, a range of organic solvents offers promising alternatives to acetonitrile. This is especially important in modern photocatalytic applications, which are gaining traction in fine chemical and pharmaceutical industries that often require large volumes of solvents. The shift toward more sustainable and environmentally friendly solvent options is therefore not only desirable but imperative for advancing green photocatalysis.66
It is worth noting that despite the introduction of numerous sustainable solvents over the past few decades, the use of eco-friendly or bio-renewable solvents in semiconductor-based photocatalytic systems remains uncommon. While efforts have been made to promote greener alternatives in other chemical processes, their integration into semiconductor photocatalysis has been relatively limited. This highlights an area in need of further exploration and development as the field continues to evolve.67 A recent study has demonstrated that the sustainable solvent γ-valerolactone (GVL) can serve as a viable alternative to acetonitrile in several key semiconductor photocatalytic systems. This finding highlights the potential of GVL as an eco-friendly option in advancing more sustainable photocatalytic processes.65 We believe that the vast potential of sustainable solvents in semiconductor photocatalysis remains largely untapped, and exploring other eco-friendly solvent alternatives is strongly encouraged. The limited range of solvent options not only hinders the future application of photocatalysis across diverse fields but also constrains the study of solvent effects. To advance semiconductor photocatalysis, it is essential to investigate novel solvents, particularly those that are environmentally friendly.
While there are few examples demonstrating their use in semiconductor photocatalysis, we believe that ionic liquids68 and deep eutectic solvents (DES)69 represent a new class of sustainable solvents with numerous advantages, including low toxicity, widespread availability, reduced flammability, recyclability, low volatility, and cost-effectiveness. These properties could pave the way for new advancements in semiconductor photocatalysis. In addition to their safety and environmental advantages, ionic liquids and DES offer the ability to precisely tune various solvent parameters through compositional modifications, potentially taking semiconductor photocatalysis in synthetic chemistry to new heights.
Furthermore, there is an urgent need to broaden the toolkit for investigating solvent effects in photocatalytic systems. These systems often generate highly reactive species, such as radicals, which exhibit stronger interactions with solvent molecules than their ground-state counterparts. However, due to their typically short lifetimes, capturing these species ex situ is a significant challenge. In this regard, the application of in situ and operando characterization techniques becomes invaluable. Although new techniques with time and spatial sensitivity, such as transient spectroscopy32 and spatiotemporally resolved surface photovoltage measurements,34 have been developed to provide information about reactive species in semiconductor photocatalysis, their application in real environments containing solvents remains challenging. First, the presence of solvent molecules can significantly reduce signal intensity. Additionally, non-covalent solvent interactions are typically weak and difficult to detect. Therefore, it is essential to develop in situ techniques and measurement modes that can accommodate solvent molecules, and the sensitivity of these tools needs to be enhanced.
Another important aspect of developing in situ techniques is the integration of various characterization methods to distinguish multiple interfering solvent effects. For instance, when a high-polarity solvent is used, it can reduce activity through competitive adsorption on the surface while simultaneously enhancing activity by increasing interfacial charge transfer. This interplay may lead to volcano-shaped dependency of activity on solvent polarity. By combining an adsorption-based imaging technique to detect molecule adsorption33 and ultrafast spectroscopy to assess electron decay dynamics,32 it may be possible to identify the contribution of each factor to overall activity. As a result, the combined technique can identify the optimal solvent that yields the highest activity.
Computational methods can offer a molecular-level illustration of the structural details of solvent–solute clusters in their excited states, providing deeper insights into their behavior. However, incorporating the complexities of solvent molecules into computational models remains a significant challenge. Achieving the accurate system representation is crucial for aligning computational results with experimental data, but increasing the number of molecules involved in simulations significantly raises computational demands. A comprehensive understanding of solvation structures at the molecular level, obtained through a combination of experimental characterization and theoretical calculations, not only aids in rationalizing experimental outcomes but also helps guide the design of optimized systems.
Furthermore, employing appropriate solvent parameters to semi-quantify solvent effects can enhance the understanding of solvent functions and aid in selecting suitable solvent systems. For instance, while many studies highlight the importance of solvent “polarity” for activity and selectivity, this is often represented by the dielectric constant, a macroscopic property resulting from multiple interactions. The concept of “polarity” oversimplifies the solvent's role. Instead, empirical solvent parameters, such as Kamlet–Taft parameters, Catalán parameters, and Hansen solubility parameters, offer a more nuanced description of the solvent's ability to engage in both specific and non-specific interactions, providing a clearer understanding of its influence on photocatalytic processes. For example, the hydrogen-bond donor (α) parameter in Kamlet–Taft16 can be used to quantify solvent capability in forming hydrogen bonds. Advanced statistical analyses can be utilized to correlate solvent empirical parameters with photocatalytic activity, helping to disentangle various solvent effects and offering deeper insights into the dominant influences. This approach provides a more structured understanding of how solvents impact reaction outcomes.
Screening solvent systems experimentally is particularly challenging due to the vast range of possible solvent types and compositions for most reactions. Machine learning presents a powerful tool for interpreting and simulating solvent effects, allowing for the prediction of optimal solvent choices without the need for extensive experimental work.70 While computational chemistry has been employed with some success to guide solvent selection, the complexity and large number of interacting species involved make achieving precise experimental predictions a non-trivial task. Machine learning has the potential to streamline this process, offering a more efficient path to identifying the best solvent systems. Machine learning can help analyze complex environments and accelerate the prediction of experimental outcomes by leveraging physics-based simulation data, as demonstrated by the success of machine learning for solvent effects in several general heterogeneous catalytic processes.71,72 In the realm of semiconductor photocatalysis, it is imperative that further efforts focus on incorporating solvent effects into our understanding of the properties of active species. This includes a detailed examination of the solvation structures of radical–solvent molecule clusters, which play a crucial role in influencing reaction mechanisms and efficiencies. By understanding how solvent interactions affect these active species, researchers can gain valuable insights into optimizing photocatalytic processes for various applications.
The collection of data necessary for machine learning applications in this context is heavily reliant on advancements in both computational techniques and experimental characterization methods. As our computational capabilities evolve, the accuracy and reliability of the data generated will improve, creating a robust foundation for machine learning models. Conversely, the insights gained from machine learning outcomes are expected to feed back into the development of computational methods and characterization techniques, leading to a synergistic relationship that enhances both fields.
Furthermore, with the establishment of a sufficiently comprehensive database that includes a variety of solvent interactions and their effects on photocatalytic activity, machine learning algorithms will be equipped to predict solvent effects with greater precision. This predictive power could revolutionize the way researchers approach the selection of optimal combinations of reactants, solvents, and catalysts. Ultimately, such advancements would not only streamline the design of more effective photocatalytic systems but also pave the way for innovative applications across a range of chemical processes. By leveraging the interplay between machine learning, computational advancements, and experimental data, the field of semiconductor photocatalysis can significantly advance toward more efficient and sustainable solutions.
Author contributions
X. W. contributed to the conceptualization of this review, design and drawing of figures, and writing of the original draft, J. V. W. and D. V. contributed to the editing and reviewing of the draft, and B. S. contributed to conceptualization, editing, writing, reviewing, and supervising. All authors discussed and commented on the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (W2412025 and 22472135), the Research Foundation-Flanders-the FWO (12S1822N), and the Xiamen Natural Science Foundation (3502Z202471025).Notes and references
- H. Kisch, Angew. Chem., Int. Ed., 2013, 52, 812–847 CrossRef CAS PubMed.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- H. Kisch, Acc. Chem. Res., 2017, 50, 1002–1010 CrossRef CAS PubMed.
- X. Wu, S. Xie, H. Zhang, Q. Zhang, B. F. Sels and Y. Wang, Adv. Mater., 2021, 33, 2007129 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- X. Wu, N. Luo, S. Xie, H. Zhang, Q. Zhang, F. Wang and Y. Wang, Chem. Soc. Rev., 2020, 49, 6198–6223 RSC.
- C. Reichardt, Org. Process Res. Dev., 2007, 11, 105–113 CrossRef CAS.
- D. S. Potts, D. T. Bregante, J. S. Adams, C. Torres and D. W. Flaherty, Chem. Soc. Rev., 2021, 50, 12308–12337 RSC.
- P. J. Dyson and P. G. Jessop, Catal. Sci. Technol., 2016, 6, 3302–3316 RSC.
- J. Lee, S. A. Park, S. U. Ryu, D. Chung, T. Park and S. Y. Son, J. Mater. Chem. A, 2020, 8, 21455–21473 RSC.
- X. Wu, X. Fan, S. Xie, I. Scodeller, X. Wen, D. Vangestel, J. Cheng and B. Sels, Nat. Commun., 2024, 15, 4967 CrossRef CAS PubMed.
- M. J. Weaver, Chem. Rev., 1992, 92, 463–480 CrossRef CAS.
- Y. Su, N. J. W. Straathof, V. Hessel and T. Noël, Chem. – Eur. J., 2014, 20, 10562–10589 CrossRef CAS.
- A. R. Katritzky, D. C. Fara, H. Yang, K. Tämm, T. Tamm and M. Karelson, Chem. Rev., 2004, 104, 175–198 CrossRef CAS.
- N. A. Dirgha Raj Joshi, J. Pharm. Res. Int., 2019, 28, 1–18 Search PubMed.
- L. Chen, J. Tang, L.-N. Song, P. Chen, J. He, C.-T. Au and S.-F. Yin, Appl. Catal., B, 2019, 242, 379–388 CrossRef CAS.
- O. R. Luca, J. L. Gustafson, S. M. Maddox, A. Q. Fenwick and D. C. Smith, Org. Chem. Front., 2015, 2, 823–848 RSC.
- Q. Lu, Y. Yu, Q. Ma, B. Chen and H. Zhang, Adv. Mater., 2016, 28, 1917–1933 CrossRef CAS.
- D. Zhu and Q. Zhou, Environ. Nanotechnol., Monit. Manage., 2019, 12, 100255 Search PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- F. Maldonado, L. Villamagua and R. Rivera, J. Phys. Chem. C, 2019, 123, 12296–12304 CrossRef CAS.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS.
- A. A. Cordones and S. R. Leone, Chem. Soc. Rev., 2013, 42, 3209–3221 RSC.
- M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- X. Wu, J. Li, S. Xie, P. Duan, H. Zhang, J. Feng, Q. Zhang, J. Cheng and Y. Wang, Chem, 2020, 6, 3038–3053 CAS.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef.
- V. Esen, Ş. Sağlam and B. Oral, Renewable Sustainable Energy Rev., 2017, 77, 1240–1250 CrossRef CAS.
- M. L. Bols, J. Ma, F. Rammal, D. Plessers, X. Wu, S. Navarro-Jaén, A. J. Heyer, B. F. Sels, E. I. Solomon and R. A. Schoonheydt, Chem. Rev., 2024, 124, 2352–2418 CrossRef CAS.
- X. Mao, C. Liu, M. Hesari, N. Zou and P. Chen, Nat. Chem., 2019, 11, 687–694 CrossRef CAS PubMed.
- R. Chen, Z. Ren, Y. Liang, G. Zhang, T. Dittrich, R. Liu, Y. Liu, Y. Zhao, S. Pang, H. An, C. Ni, P. Zhou, K. Han, F. Fan and C. Li, Nature, 2022, 610, 296–301 CrossRef CAS PubMed.
- M. Maiuri, M. Garavelli and G. Cerullo, J. Am. Chem. Soc., 2020, 142, 3–15 CrossRef CAS.
- M. Hunger and J. Weitkamp, Angew. Chem., Int. Ed., 2001, 40, 2954–2971 CrossRef CAS.
- A. Ge, B. Rudshteyn, P. E. Videla, C. J. Miller, C. P. Kubiak, V. S. Batista and T. Lian, Acc. Chem. Res., 2019, 52, 1289–1300 CrossRef CAS.
- Z. Zhang and J. T. Yates, Chem. Rev., 2012, 112, 5520–5551 CrossRef CAS.
- Z. Zhang and J. T. Yates, Jr., J. Phys. Chem. Lett., 2010, 1, 2185–2188 CrossRef CAS.
- G. Redmond and D. Fitzmaurice, J. Phys. Chem., 1993, 97, 1426–1430 CrossRef CAS.
- M. D. Peterson, L. C. Cass, R. D. Harris, K. Edme, K. Sung and E. A. Weiss, Annu. Rev. Phys. Chem., 2014, 65, 317–339 CrossRef CAS.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811, 265–322 CrossRef CAS.
- E. R. Barthel, I. B. Martini and B. J. Schwartz, J. Phys. Chem. B, 2001, 105, 12230–12241 CrossRef CAS.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed.
- G. Litwinienko, A. L. J. Beckwith and K. U. Ingold, Chem. Soc. Rev., 2011, 40, 2157–2163 RSC.
- P. Zhang, C. Yue, M. Fan, A. Haryonob, Y. Leng and P. Jiang, Catal. Sci. Technol., 2021, 11, 3385–3392 RSC.
- C. B. Almquist and P. Biswas, Appl. Catal., A, 2001, 214, 259–271 CrossRef CAS.
- Y. Hu, Z. Shen, B. Li, S. Li, J. Yue, G. Zhao, M. Muhler and X. Wang, ACS Appl. Nano Mater., 2021, 4, 9254–9264 CrossRef CAS.
- B.-J. Liu, T. Torimoto, H. Matsumoto and H. Yoneyama, J. Photochem. Photobiol., A, 1997, 108, 187–192 CrossRef CAS.
- T. Baran, S. Wojtyła, A. Dibenedetto, M. Aresta and W. Macyk, Appl. Catal., B, 2015, 178, 170–176 CrossRef CAS.
- D. W. Wakerley, M. F. Kuehnel, K. L. Orchard, K. H. Ly, T. E. Rosser and E. Reisner, Nat. Energy, 2017, 2, 17021 CrossRef CAS.
- X. Wu, S. Xie, C. Liu, C. Zhou, J. Lin, J. Kang, Q. Zhang, Z. Wang and Y. Wang, ACS Catal., 2019, 9, 8443–8451 CrossRef CAS.
- X. Wu, X. Fan, S. Xie, J. Lin, J. Cheng, Q. Zhang, L. Chen and Y. Wang, Nat. Catal., 2018, 1, 772–780 CrossRef CAS.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- T. Renders, S. Van den Bosch, S. F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- T. Simon, N. Bouchonville, M. J. Berr, A. Vaneski, A. Adrović, D. Volbers, R. Wyrwich, M. Döblinger, A. S. Susha, A. L. Rogach, F. Jäckel, J. K. Stolarczyk and J. Feldmann, Nat. Mater., 2014, 13, 1013–1018 CrossRef CAS.
- H. Zhang, S. Xie, J. Hu, X. Wu, Q. Zhang, J. Cheng and Y. Wang, Chem. Commun., 2020, 56, 1776–1779 RSC.
- S. Xie, Z. Shen, J. Deng, P. Guo, Q. Zhang, H. Zhang, C. Ma, Z. Jiang, J. Cheng, D. Deng and Y. Wang, Nat. Commun., 2018, 9, 1181 CrossRef.
- Z. Gao, J. Mu, J. Zhang, Z. Huang, X. Lin, N. Luo and F. Wang, J. Am. Chem. Soc., 2022, 144, 18986–18994 CrossRef CAS.
- S. Murcia-López, K. Villa, T. Andreu and J. R. Morante, Chem. Commun., 2015, 51, 7249–7252 RSC.
- Y. Zeng, H. C. Liu, J. S. Wang, X. Y. Wu and S. L. Wang, Catal. Sci. Technol., 2020, 10, 2329–2332 RSC.
- A. Reinheimer, R. van Eldik and H. Kisch, J. Phys. Chem. B, 2000, 104, 1014–1024 CrossRef CAS.
- L. Zhang, L. Liardet, J. Luo, D. Ren, M. Grätzel and X. Hu, Nat. Catal., 2019, 2, 366–373 CrossRef CAS.
- X. Wu, S. Xie, D. Vangestel, H. Zhao and B. F. Sels, Angew. Chem., Int. Ed., 2024, e202409826 CAS.
- D. Friedmann, A. Hakki, H. Kim, W. Choi and D. Bahnemann, Green Chem., 2016, 18, 5391–5411 RSC.
- C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- B. Wang, L. Qin, T. Mu, Z. Xue and G. Gao, Chem. Rev., 2017, 117, 7113–7131 CrossRef CAS.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- H. Wen, S. Nan, D. Wu, Q. Sun, Y. Tong, J. Zhang, S. Jin and W. Shen, Ind. Eng. Chem. Res., 2023, 62, 20473–20491 CrossRef CAS.
- M. Gastegger, K. T. Schütt and K.-R. Müller, Chem. Sci., 2021, 12, 11473–11483 RSC.
- L. Je, G. W. Huber, R. C. Van Lehn and V. M. Zavala, Curr. Opin. Chem. Eng., 2022, 36, 100796 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |