
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of the dual roles of Al2O3 coatings on NMC811-cathodes via theory and experiment†
Richard L. B.
Chen
 ab,
Farheen N.
Sayed
ab,
Hrishit
Banerjee
abc,
Israel
Temprano
abd,
Jing
Wan
ae,
Andrew J.
Morris
bf and
Clare P.
Grey
*ab
ab,
Farheen N.
Sayed
ab,
Hrishit
Banerjee
abc,
Israel
Temprano
abd,
Jing
Wan
ae,
Andrew J.
Morris
bf and
Clare P.
Grey
*ab
aYusuf Hamied Department of Chemistry, The University of Cambridge, Cambridge CB2 1EW, UK. E-mail: cpg27@cam.ac.uk
bThe Faraday Institution, Didcot OX11 0RA, UK
cSchool of Science and Engineering, University of Dundee, Dundee, DD1 4HN, Scotland, UK
dCICA – Interdisciplinary Center for Chemistry and Biology, University of A Coruña, A Coruña, 15071, Spain
eDepartment of Engineering, The University of Cambridge, CB3 0FA, UK
fSchool of Metallurgy and Materials, University of Birmingham, Birmingham, B15 2TT, UK
First published on 20th January 2025
Abstract
Metal-oxide coatings are a favoured strategy for mitigating surface degradation problems in state-of-the-art lithium-ion battery Ni-rich layered positive electrode materials. Despite their extensive use, a full, fundamental understanding of the role of coatings in reducing degradation and extending cycling lifetimes is currently lacking. In this work, the interactions between an atomic layer deposited (ALD) alumina coating on polycrystalline LiNi0.8Mn0.1Co0.1O2 (NMC811) and a carbonate-based battery electrolyte are studied. Solid-state nuclear magnetic resonance (ssNMR) heteronuclear experiments show that the Al2O3 coating transforms by reacting with electrolyte species present before and during electrochemical cycling, scavenging protic and acidic species. Density-functional theory calculations highlight the additional chemical effect of the coating in locally stabilising the structure of the NMC811, limiting oxidation of the oxygen atoms coordinated to both Al and Ni, thereby limiting the surface reconstruction process and improving the electrochemical performance. Improved NMC811 surface stability is confirmed by monitoring gaseous degradation species by online electrochemical mass-spectrometry and via X-ray spectroscopic analysis of the electrochemically aged samples to examine changes in Ni and O oxidation state and local structure. The combination of this experimental and theoretical analysis suggests that Al2O3 coatings have a dual role: as a protective barrier against attack from chemical species in the electrolyte, and as an artificial passivating layer hindering oxygen loss and surface phase transformations. This holistic approach, which provides a fundamental understanding of how the surface stability is improved by the coating, will aid the design of the state-of-the-art and future positive electrode materials.
Broader contextRechargeable lithium-ion batteries (LIBs) are crucial for transitioning towards a carbon neutral society. One barrier towards widespread utilisation of LIBs in electric vehicles is the limited capacity of the positive electrode (cathode) materials. Nickel-rich layered oxides (e.g. LiNi0.8Mn0.1Co0.1O2 (NMC811)) are the state-of-the-art material of choice for cathodes, offering high practical capacities. However, accessing this high capacity induces structural degradation and parasitic electrolyte reactions at the surface. Degradation of these cathodes via the reaction of electrolyte components is a well-established and major mode of overall battery failure. One strategy to mitigate this problem is surface modification with coating materials like Al2O3 – cycling life is extended but the fundamental reasons behind the protection are not well understood. The comprehensive experimental and theoretical work described here provides a significant advance in the field in that it clearly shows that atomic layer deposition (ALD) Al2O3 coatings (i) scavenge electrolyte degradation species, which would otherwise corrode the surface of the transition metal oxide materials, and (ii) stabilise the surface oxygen of these materials at high potentials to prevent losses in capacity. In this study we address the challenge in characterising the evolution of a nanometre thin coating layer by using extremely high field MAS NMR (magic angle spinning nuclear magnetic resonance) spectroscopy and double-resonance NMR experiments that are selective to specific degradation and reaction products. Online electrochemical mass spectrometry (OEMS) along with solution NMR spectroscopy allowed the effect of the coatings on electrolyte degradation reactions to be determined. Additionally, first principles studies of model AlOx coatings explained how the wide bandgap nature of Al–O bonding helps to stabilise hybridised Ni–O environments at high potentials, thereby reducing surface degradation and the evolution of highly oxidising surface oxygen species. These results provide fundamental insight into how, why and when these coatings will extend the lifetime of Ni-rich layered oxides. |
Introduction
Ni-rich layered oxide positive electrode materials such as LiNixMnyCozO2 (x > 0.6, y < 0.2 and z = 1 − x − y; NMCxyz) are the state-of-the-art choice for the positive electrode in high-energy density rechargeable Li-ion batteries and used in the electrification of vehicles.1–3 However, accessing the higher energy density comes with challenges, affecting the lifetime and safety of these materials; this includes cracking of the secondary particles (the agglomerates of primary nanoparticles that are present in polycrystalline NMC samples),4–6 increased reactivity with electrolytes, which leads to cross-talk of transition metals (TM)7–9 and electrolyte degradation species,8,10 and surface and sub-surface phase changes (e.g., reconstruction of the surface layered structure to rock salt), following reactions that in general increase impedance.11–13 One proposed strategy for addressing these surface degradation phenomena in Ni-rich layered materials is to modify the NMC surfaces with a thin layer of metal oxide such as Al2O3.14–16Al2O3 coatings improve the capacity retention during electrochemical cycling and improve the rate performance of high Ni NMC materials.17–19 However, a fundamental understanding of the mechanism is lacking – in particular, the structural evolution of Al2O3 on electrochemical cycling and its impact on the NMC particle surface and sub-surface is unclear. Moreover, the variety of coating methods adds uncertainty to the analysis of the coating's function. Coating methods include wet-chemistry based processes like solution deposition20–22 or sol–gel,23–25 dry coating processes26–29 and deposition processes such as atomic layer deposition (ALD).30–32 Compared with wet-chemistry based methods, in which the subsequent annealing temperature determines the structure and uniformity of the coating and doping into the subsurface,33 ALD offers a route to synthesise nanometre thin, highly conformal, continuous and dense coatings even when the underlying host material has a complex surface morphology.16,30,34–36 A carefully controlled coating limited to the surface is advantageous when deconvoluting the mechanisms behind the protective effect of the coating.
Al2O3 coatings on NMC811 (by ALD) extend the lifetime and reduce the growth in charge-transfer resistance impedance.14,16 This improvement in electrochemical cycling performance is concomitant with mitigated layered-to-rock salt transformation (surface reconstruction) of the NMC811 as seen in the Ni L-edge and O K-edge electron energy loss spectroscopy (EELS) of electrodes in the discharged state after extended cycling.14 That is, the layered structure of NMC811 is well-preserved by the coating, resulting in little chemical difference in the Ni–O bonding environment between Ni on the surface and sub-surface of a particle.14 However, different findings were seen in Ni K-edge X-ray absorption spectroscopy (XAS) of layered oxides charged to high states-of-charge (SoC)37 – the Ni edge energy of Ni ions close to the surface (approximately ∼20 nm) was not seen to shift to the higher energies seen for the Ni in the sub-surface (∼150 nm), even when an Al2O3 coating was present. The authors concluded that the surface Ni, susceptible to surface reconstruction, was not oxidised to the same extent as bulk Ni and this instability was not improved by coating with Al2O3.
Whilst these previous X-ray spectroscopy studies have characterised the surface transition metal chemistry of layered oxides to understand the effect of coatings on surface degradation, changes to the chemical environment of the coating were not clearly identified. Analysing the evolution of the coating with solid-state nuclear magnetic resonance (ssNMR) could reveal unique insights into its function. ssNMR is sensitive to the local chemical and structural environments around an observed spin (i.e.27Al), allowing structural characterisation of the different Al coordination environments found in Al2O3 coatings38 to be linked to the electrochemical performance of the coated Ni-rich layered oxide.39–42 Post-mortem analysis of Al2O3 coated NMC after electrochemical cycling with ssNMR to determine the role of the coating has been more scarce in the literature. The 27Al spectra in past studies40,43 suggest that little structural and chemical evolution of the Al2O3 coating have occurred, in contrast to reported HF scavenging reactions that might be expected to occur given the degradation mechanisms proposed in the literature.44–48 It should be noted that these 27Al spectra were measured on coatings deposited using wet-chemistry methods where the “bulk” alumina signals likely dominate the signal rather than the signals from alumina surface species. The use of double-resonance NMR experiments, not widely employed to study these systems, provides a strategy to obtain more detailed local chemical information. For example, the proximity of 7Li to 27Al spins in Al–F based coatings deposited by ALD, as found in 27Al{7Li} and 7Li{19F} rotational echo double resonance (REDOR) NMR experiments, suggest the formation of a lithiated coating.42
In this study we characterise the ALD Al2O3 coatings formed on polycrystalline NMC811 and how the coatings evolve with cycling. This is achieved by using a combination of single and double-resonance ssNMR experiments that monitor the local coordination environment of Al, measuring gaseous and solution electrolyte degradation species arising from NMC811–electrolyte interactions at high potentials, and density functional theory using a Hubbard correction (DFT+U) to examine the surface Ni/O electronic states to explore improved capacity retention and surface stability. The coating is shown to have a dual protective role: namely, the coating plays a key role in scavenging electrolyte decomposition species (from the commonly used LiPF6-based carbonates) and in stabilising the surface oxygen of NMC811.
Experimental methods
Materials
Commercial polycrystalline NMC811 (Targray) stored in an Ar-filled glovebox (GB, MBraun, Germany; O2 and H2O < 0.1 ppm) was used as a baseline material. The polycrystalline NMC811 powders were coated by Forge Nano via a proprietary ALD process after keeping the powders at 120 °C overnight in a fluidised reactor bed. The ALD reaction proceeded at 120 °C in the same reactor bed with cycles of trimethyl aluminium (TMA) precursor gas followed by water vapour and then purging with N2 – the resulting concentration of Al after coating the NMC811 was 223 ppm (baseline powder, uncoated, had 34 ppm of Al) as determined by inductively coupled plasma mass spectrometer (ICP-MS). Powders were transported under N2 before taking them into the Ar-filled glovebox (GB). The polycrystalline Al2O3-coated NMC811 is referred to as ALD NMC811 herein.Characterisation of the pristine powders
ALD Al2O3 coated and uncoated NMC811 powders were pressed onto carbon tape for scanning electron microscopy (SEM) or onto carbon-coated Cu grids for transmission electron microscopy (TEM) imaging inside the Ar-filled glovebox but samples were briefly exposed to air during loading into the microscopes. SEM (TESCAN MIRA3 FEG-SEM) coupled with energy-dispersive X-ray spectroscopy (EDX) mapping (5.0 kV operating voltage) was used for elemental analysis – at least 8 area scans for 3 different secondary particles were averaged to obtain a representative value. TEM (Thermo Scientific Talos F200X G2 TEM) coupled with EDX mapping was also performed at 200 kV for elemental comparisons (averaged across 3 secondary particles) between the edge and bulk (100–200 nm further in) regions of the secondary particle.Gas adsorption experiments on pristine and Al-coated NMC811 powders were conducted using an automatic gas sorption analyser (Quantachrome Corp., NOVA 4200, USA) with high-purity nitrogen (99.999%, BOC) as the adsorptive gas. The specific surface area, total pore volume, and pore size distribution were determined via multipoint Brunauer–Emmett–Teller (BET) analysis. Samples were initially degassed under vacuum at 100 °C for 12 h to remove adsorbed impurities. Adsorption and desorption measurements were conducted at 77 K using liquid nitrogen, and full adsorption/desorption isotherms were obtained over 75 relative pressure (P/P0) data points. The sample mass of 1.50 ± 0.01 g was analysed throughout, with free space calibration performed using ultra-high-purity helium (99.9995%, BOC). An equilibration time of 120 s was applied at each pressure step, ensuring sufficient time for pressure stabilisation and attainment of equilibrium during the adsorption and desorption processes.
X-ray powder diffraction patterns collected at RT on a PANalytical Empyrean diffractometer (Cu Kα irradiation, λ = 1.5406 Å with a Ni filter) for the uncoated NMC811 and the ALD Al2O3 coated NMC811 (material just referred to as ALD NMC811 henceforth). Rietveld refinements49 were performed with Academic TOPAS (v.6.1) software50 to obtain structural parameters and determine bulk structural changes, if any, after the coating process. A R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group with transition metals (TM) at the 3a Wyckoff positions, Li on the 3b sites and O on the 6c sites was used with the oxygen positions, lattice parameters and Li/TM anti-site occupancies being allowed to iterate (details in ES1). Displacement parameters were assumed isotropic and given values of 1.
m space group with transition metals (TM) at the 3a Wyckoff positions, Li on the 3b sites and O on the 6c sites was used with the oxygen positions, lattice parameters and Li/TM anti-site occupancies being allowed to iterate (details in ES1). Displacement parameters were assumed isotropic and given values of 1.
Soaking experiments of the pristine material
For soaking experiments of the pristine material, ethylene carbonate (EC) and ethyl methyl carbonate (EMC) (EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EMC = 3:7 (v/v), SoulBrain MI) solvents and lithium hexafluorophosphate (LiPF6) salt (Solvionic, 99.99%) were used. Polypropylene vials were initially dried at 60 °C before transferring into the glovebox to reduce introduced moisture. 30 mg of active material (ALD NMC811 or uncoated NMC811) were soaked for 24 h in 600 μL of either the EC/EMC solvent or freshly mixed 1 M LiPF6 in EC/EMC electrolyte. After soaking, 400 μL of the solvent (electrolyte) was pipetted out for analysis by 1H and 19F solution NMR using a C6D6 capillary for shift referencing to avoid any contamination from trace H2O in deuterated solvents. The remaining solvent (electrolyte) was pipetted out and the residual slurry was rinsed with excess dimethyl carbonate (DMC, Sigma-Aldrich, anhydrous, ≥99%) before drying in vacuo. After drying, the NMC811 powders were packed for ssNMR measurements in 1.3 mm rotors. All sample handling was done inside the Ar GB.
:EMC = 3:7 (v/v), SoulBrain MI) solvents and lithium hexafluorophosphate (LiPF6) salt (Solvionic, 99.99%) were used. Polypropylene vials were initially dried at 60 °C before transferring into the glovebox to reduce introduced moisture. 30 mg of active material (ALD NMC811 or uncoated NMC811) were soaked for 24 h in 600 μL of either the EC/EMC solvent or freshly mixed 1 M LiPF6 in EC/EMC electrolyte. After soaking, 400 μL of the solvent (electrolyte) was pipetted out for analysis by 1H and 19F solution NMR using a C6D6 capillary for shift referencing to avoid any contamination from trace H2O in deuterated solvents. The remaining solvent (electrolyte) was pipetted out and the residual slurry was rinsed with excess dimethyl carbonate (DMC, Sigma-Aldrich, anhydrous, ≥99%) before drying in vacuo. After drying, the NMC811 powders were packed for ssNMR measurements in 1.3 mm rotors. All sample handling was done inside the Ar GB.
Assembly of Swagelok cells for solid-state NMR
Ex situ ssNMR studies were performed after electrochemical cycling on materials (without binder and current collector to minimise background signals) in oven-dried Swagelok cells. ALD NMC811 and the Super P conductive carbon were dried at 120 °C under vacuum before being mixed (80:20 wt%) using an agate mortar and pestle (dried at 100 °C in a vacuum oven prior to use) inside the Ar GB. This mixture was transferred into a ½-inch swagelok cell with the cell stack and completed with borosilicate glass fibre separators (Whatmann, GF/B, 0.68 mm thick, 1 μm pore size, dried at 120 °C for 12 h under vacuum) flooded with freshly mixed 300 μL of 1 M LiPF6 in EC/EMC electrolyte and a Li metal disk (12 mm diameter; MTI Corporation, 99.9%). The inside of the cell was lined with Kapton polyimide film (DuPont) film to ensure that no loose positive electrode powder would be displaced and come into direct contact with the Li metal leading to a short circuit. The entire Swagelok cell was (dis)assembled inside the Ar GB. After electrochemical cycling, the positive electrode powder mix was soaked in 1 mL DMC for 5 min before in vacuo drying. Analogous cells with the uncoated NMC811 were also (dis)assembled for electrochemical cycling comparisons. Details of coin cell preparation for electrochemistry and ex situ experiments are in provided in ESI 2:† Fabrication of coin cells.
Electrochemical measurements
Electrochemical cycling of (ALD) NMC811|Li Swagelok cells was performed on a MPG2 potentiostat (Biologic) using the EC-lab software at room temperature (RT, 20 ± 2 °C). The cell was rested for 4 h at the open-circuit voltage (OCV) before galvanostatic (dis)charging at C/20 to different cut-off voltages (assuming a practical capacity of 200 mA h g−1). A potentiostatic hold was applied at the upper cut-off voltage (UCV = 4.4 VLi) until the current decayed to the C/40 value to account for Li-ion concentration gradients in the NMC811 particles.Electrochemical cycling of LFP|Li and (ALD) NMC811|delithiated LFP coin cells was performed on a BT-2000 potentiostat (Arbin) running the MITS Pro software at RT. LFP|Li cells were rested for 4 h before galvanostatic charging at a C/20 rate (practical capacity of 127 mA h g−1) to 4.0 VLi with a hold until the current decays to the C/40 value followed by galvanostatic discharge until 3.8 VLi and were then disassembled for the delithiated LFP. (ALD) NMC811|delithiated LFP cells were rested for 4 h before C/10 charging (assuming a practical capacity of 200 mA h g−1) up to 1.16 V (∼4.6 VLi). The cells were held at this potential for 60 h before C/10 galvanostatic discharge −0.46 V (∼3.0 VLi) with this protocol being repeated for 5 cycles.
Electrochemical cycling of NMC811|graphite coin cells were performed on a BT-2000 potentiostat at RT. Cells were first charged to 1.5 V at C/20 (assuming 200 mA h g−1 practical capacity) and held for 15 min before resting at OCV for 6 h to passivate the Cu current collector of the graphite. Cells underwent formation cycles (C/20) between 2.5–4.3 V for 2 cycles to form a stable SEI on the graphite and diagnose the available capacity. Extended electrochemical cycling was conducted over 1040 cycles in sets with each set consisting of 48 ageing C/2 cycles (UCV holds until the current decays to the C/20 value) followed by 3 diagnostic C/20 cycles (UCV holds until the current decays to C/40). All NMC811|Li cells (with pristine or aged electrodes) were charged to 4.4 VLi with a UCV hold until the current value decayed to the C/100.
Solid state NMR characterisation
1H rotor-synchronised Hahn-echo NMR experiments under magic-angle spinning (MAS, 50 kHz) conditions were acquired at 16.4 T using a Bruker 1.3 mm double-resonance (DR) probe. Excitation was achieved with 125 kHz applied radio frequency (rf) field strength i.e. π pulse of 4 μs. Evolution and refocusing times, τ, were set to five rotor periods to filter out broad background signals (with fast spin–spin T2 relaxation), and a recycle delay of 1 s was used. 1H chemical shifts were referenced externally to the lower frequency peak of adamantane (1.85 ppm).
NMR experiments under magic-angle spinning (MAS, 50 kHz) conditions were acquired at 16.4 T using a Bruker 1.3 mm double-resonance (DR) probe. Excitation was achieved with 125 kHz applied radio frequency (rf) field strength i.e. π pulse of 4 μs. Evolution and refocusing times, τ, were set to five rotor periods to filter out broad background signals (with fast spin–spin T2 relaxation), and a recycle delay of 1 s was used. 1H chemical shifts were referenced externally to the lower frequency peak of adamantane (1.85 ppm).
19F Hahn-echo NMR experiments were acquired at 4.7 T with a 1.3 mm DR probe whilst spinning with a MAS frequency of 50 kHz (1.3 mm rotor). 19F pulses of 263 kHz rf field strength, τ was one rotor period and 250 ms recycle delays were used. 19F chemical shifts were referenced externally to AlF3 (−172.0 ppm).
27Al Hahn-echo NMR experiments were acquired at either 23.5 T with a 1.9 mm triple resonance (TR) probe at a 40 kHz MAS frequency and 156 kHz rf field strength or at 16.4 T with a 1.3 mm DR probe at a 50 kHz MAS frequency and 179 kHz rf field strength. A  pulse length is normally used to excite all environments with different quadrupolar couplings uniformly and be quantitative.51 However, the short pulse length results in lower excitation compared to
pulse length is normally used to excite all environments with different quadrupolar couplings uniformly and be quantitative.51 However, the short pulse length results in lower excitation compared to 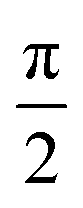 pulses so to achieve sufficient signal-to-noise ratio (SNR) in these very low Al concentration samples, longer
pulses so to achieve sufficient signal-to-noise ratio (SNR) in these very low Al concentration samples, longer  pulse lengths at the cost of being fully quantitative. 75 ms recycle delays were used (see ESI 3:† Details about NMR parameters for further details about additional recycle delay times). 27Al chemical shifts were referenced externally to AlF3 (−17 ppm).
pulse lengths at the cost of being fully quantitative. 75 ms recycle delays were used (see ESI 3:† Details about NMR parameters for further details about additional recycle delay times). 27Al chemical shifts were referenced externally to AlF3 (−17 ppm).
Double resonance REDOR and TRAPDOR experiments52,53 were also used to obtain detailed chemical information – signal attenuation indicates that two spins (different elements) are in close proximity. Calibration curves were first performed on Gibbsite Al(OH)3, α-AlF3 and γ-LiAlO2 to determine appropriate dipolar evolution times – experimental times on the samples take ∼3 days per spectrum, ruling out obtaining a full REDOR curve for the sample. Further experimental details on the double resonance experiments can be found in ESI 4:† Experimental details for double resonance experiments.
Solution state NMR characterisation
Cycled (ALD) NMC811|LFP coin cells were disassembled in the Ar GB and the glass fibre soaked in 1 mL of DMSO-d6 (Sigma-Aldrich, 99.9 atom% D, 99% CP) for 5 min. 0.7 mL of solution was then transferred to an NMR tube fitted with a J-Young tap.One-dimensional 1H and 19F{1H} NMR spectra and two-dimensional 1H–1H correlation spectroscopy (COSY) were collected at 11.7 T (500 GHz 1H Larmor frequency) on a Bruker AVANCE III HD spectrometer with a BBO probe. 1H was referenced internally to the DMSO-d6 solvent at 2.50 ppm whilst 19F was referenced internally to LiPF6 at −74.5 ppm.
Online electrochemical mass spectrometry (OEMS)
Gas evolution from (ALD) NMC811|Li cells with 1 M LiPF6 in EC:EMC (3:7, v/v) electrolyte were investigated using OEMS. The experimental setup has previously been described elsewhere.54,55 The cell is cycled using an Ivium Vertex potentiostat with the following protocol: 6 h rest at OCV, C/10 galvanostatic charge to 4.6 VLi coupled with a potential hold at 4.6 VLi for 1 h, C/10 galvanostatic discharge until 3.0 VLi, an OCV rest for 2 h and then the cycle is repeated at C/5.
X-ray absorption spectroscopy (XAS)
XAS measurements on the NMC811|Li samples charged to 4.4 VLi were conducted at the I09 beamline, Diamond Light Source using the total electron yield (TEY) mode. The Ni metal L-edge spectra were background corrected with a straight-line baseline and normalised to the peak intensity of the L3 peak. O K-edge spectra, on the other hand, were only baseline corrected with a straight line through the pre-peak region. For the cycled electrodes of pristine and ALD coated NMC811, O K-edge spectra were normalised with the O K-edge of pristine electrodes.First-principles calculations
First-principles calculations were carried out on the plane-wave basis as implemented in the Vienna ab initio simulation package (VASP)56,57 version 5.4 with projector-augmented wave (PAW)58 potential. The exchange–correlation functional was chosen to be that of generalised gradient approximation (GGA) implemented following the Perdew–Burke–Ernzerhof59 prescription for solids (PBESol).60 For ionic relaxations, the internal positions of the atoms were allowed to relax until the forces became less than 0.005 eV Å−1. An energy cutoff of 550 eV, and 8 × 8 × 1 Gamma centred k-point mesh (for the slab calculations with vacuum on top) were found to provide a good convergence of the total energy in self-consistent field calculations.The NMC811 cell was constructed based on a 5 × 4 × 1 supercell of the rhombohedral LiNiO2 (LNO) unit cell. In each layer 2 Ni ions were replaced by Mn and Co respectively, with all dopant atoms surrounded only by Ni atoms, and the maximum possible separation between dopant ions in each layer possible within the supercell was considered, to prevent unphysical clustering of dopant ions. 75% of the Li was removed from the pristine NMC811 structure to achieve an experimentally relevant state of 75% delithiation, and both pristine as well as 75% delithiated structures were studied to decouple the effects of coating from the effects of delithiation. Three different structures were created, in each case, each with a vacuum of ∼20 Å along the b direction to mimic the effect of a coated/uncoated surface. One structure was uncoated, and the surface was left exposed to vacuum, while in the other two structures, surface TM atoms were partially (56.25%) and fully replaced by Al atoms to mimic the effect of a coating on the surface. These structures were fully relaxed using the PBESol functional,60 as it is considered to reproduce the experimental lattice constants of solids accurately. The choice of the surface was motivated by the active surface chosen in a previous study by Genreith Schriever et al.,61 who showed that the O loss was most profound in the (012) facet of LNO. In this case, the plane perpendicular to the b-direction is chosen as it is analogous to the (012) plane in this rhombohedral unit cell.
To account for the correlation effects at Ni, Mn, and Co sites beyond GGA, a Hubbard U correction was incorporated (following the DFT+U as implemented by Liechtenstein et al.),62 where we use a Hubbard interaction parameter of UNi = 6 eV, UMn = 4.5 eV, and UCo = 5 eV and a uniform Hund's coupling parameter J = 0.75 eV. Averaged Crystal Orbital Overlap Population (COOP)63 were calculated based on the LOBSTER64 package to estimate the Ni–O hybridisation in each case. O p charges were calculated following the Bader charge partitioning method.65
Results
Characterisation of the coating
 | ||
| Fig. 1 SEM images of the as-synthesised (a) NMC811 and (b) the ALD NMC811 secondary particles (c) bright field image of an ALD NMC811 particle with (d) a corresponding EDX map of Ni (blue) and Al (red). The X-ray (λ = 1.5406 Å) diffraction patterns (e) of the NMC811 (black) and ALD NMC811 (red) are compared, showing the experimental data (solid line), the Rietveld refinement (open circles), the difference between the data and fit (grey line) and the position of the expected reflections of the Rm space group (vertical ticks). | ||
Closer inspection of the particles with HR-TEM (Fig. 1(c)), coupled with EDX mapping (Fig. 1(d)), shows that the coating (∼1 nm thick) has a disordered structure that is conformal to the secondary particles. Crystalline fringes in the NMC811 particle indicate the ordered, repeating transition metal layers of the layered oxide structure. Overlaying the Al elemental map with the Ni shows that the Al is concentrated at the edge of the particle, supporting the assignment of the Al2O3 coating being surface-limited. The microscopy observation agrees with ICP-MS measurements, whereby the ALD NMC811 has an Al concentration (by weight) of 223 ppm which indicates successful coating (34 ppm for the baseline NMC811 with no coating). A more detailed structural analysis of the disordered coating is performed below with NMR (Fig. 2).
 | ||
| Fig. 2
27Al MAS NMR spectra of the ALD NMC811 in the (a) as-synthesised/pristine state, (b) the material after soaking in the EC/EMC (3:7 (v/v)) solvent for 24 h (then dried under vacuum) and (c) the material after soaking in 1 M LiPF6 in the EC/EMC (3:7 (v/v)) electrolyte for 24 h (then dried under vacuum). The spectrum from a reference AlF3 with a chemical shift at −17 ppm is included. The spectrum obtained in (a) was acquired at 23.5 T whilst spectra in (b) and (c) were acquired at 16.4 T. Spectra were fitted using Czjzek models and the Al(IV), Al(V) and Al(VI) components are plotted in blue, red and black respectively (fits found in ESI 5:† Supporting NMR experiments). The grey dashed lines mark the average isotropic chemical shift extracted from the fits; these are offset from the centre of mass due to the second order quadrupolar interaction. Mass-normalised 1H MAS NMR spectra of ALD NMC811 in the (d) pristine state, (e) the ALD NMC811 after soaking in the EC/EMC solvent for 24 h and (f) the ALD NMC811 after soaking in 1 M LiPF6 in the EC/EMC electrolyte for 24 h. Spectra are fitted with Gaussian/Lorentzian line shapes and incorporate chemical shift anisotropy to model spinning sidebands (see ESI 5:† Supporting NMR experiments for further fitting details). Isotropic chemical shifts of each fitted component are marked with a dashed line. Spinning sidebands are marked with asterisks (*). | ||
Destructive focussed ion beam (FIB) cross-sectioning of the NMC811 particles was not performed, because the primary and secondary structures of these agglomerated polycrystalline particles have been well studied.66–68 We have instead evaluated the effect of ALD coating on the particle surface area and accessible pore structure by recording N2 adsorption–desorption isotherms on pristine and ALD-coated NMC811 samples, Fig. S0 in the ESI.† The BET surface area of NMC811 was 0.85 m2 g−1, which decreases for ALD coated alumina samples to 0.64 m2 g−1. The reduction in surface area upon ALD coating is ascribed to the blocking of some pore entrances by the thin alumina layer, some internal porosity now being unavailable for N2 adsorption. The pore size distribution plots showed a negligible change in the actual distribution of pore size between pristine and coated samples, but we note that a more detailed analysis of porosity would require the use of Kr adsorption–desorption isotherms.
XRD analysis (Fig. 1(e) and Table S1, ESI†) of the powders confirms that the bulk crystal structure is unaffected by the coating process, where the a- and c-lattice parameters from Rietveld refinements of the ALD NMC811 and uncoated NMC811 were 2.8715(3) Å, 14.205(2) Å and 2.8716(3) Å, 14.205(2) Å respectively. Therefore, any changes to the NMC811 electrochemical performance must be due to the presence of the coating and the reduced surface area. The disordered Al-based coating (seen in TEM (Fig. 1(c))) and its interactions with the electrolyte are first characterised with NMR. Revealing structural changes in the absence of electrochemical cycling provides a foundation for understanding the effect of the coating on cycling performance.
27Al NMR of the pristine ALD NMC811 in Fig. 2(a) shows that the coating consists of tetrahedral (Al(IV)), penta-coordinated (Al(V)) and octahedral (Al(VI)) local environments for the Al3+ ions. The presence of the Al(V) environments and broad peaks indicate an amorphous or disordered phase69 (in agreement with the TEM in Fig. 1(c)) meaning each type of environment will have a distribution of second order quadrupolar interactions and isotropic chemical shifts, resulting in the asymmetric lineshape with a tail at lower chemical shifts. The size of the quadrupolar interaction, CQ, measures the interaction between the local electric field gradient and the quadrupole and is therefore indicative of local distortions around the Al3+ ions – this interaction is reduced at higher magnetic field strengths. Quadrupolar nuclei in disordered solids can be fit with a Czjzek model; where the distribution of quadrupolar interactions and isotropic chemical shifts are given by averaged values (CQ and δiso respectively).70,71 Fitting the spectrum with a Czjzek model (see ESI 5:† Supporting NMR experiments for the fit parameters) gives approximate proportions for Al(IV):Al(V):Al(VI) of 47:37:16 whilst the grey dashed lines in the spectrum indicate the extracted average isotropic chemical shifts of each Al environment (δiso,IV = 83 ppm, δiso,V = 47 ppm and δiso,VI = 17 ppm). The fit in the Al(IV) region in particular is poor and there are likely further species at lower frequencies (but still in the chemical shift range of Al(IV)). A range of possible Al(IV) sites (with chemical shifts ranging from 50–100 ppm) was observed in our previous work investigating amorphous ALD alumina deposited on a silicon wafer – the Al(IV) region was dominated by signals at a lower frequency of ∼70 ppm38 and could justify the inclusion of other Al(IV) components in the fitting in Fig. 2(a). In this previous work,38 the Al(IV):Al(V):Al(VI) proportion in the ALD alumina deposited on a silicon wafer was approximately 50:38:12 which gives similar populations of Al environments to the ALD alumina-coated NMC811 studied in this work. No Al signal was observed at negative frequencies (∼−900 ppm)72,73 in the region expected for Al substitution in the bulk72,73 indicating that the ALD process does not result in any Al doping into the NMC811 crystal structure.
Exposing ALD NMC811 to the EC/EMC carbonate-based solvent (Fig. 2(b)) shows that the saturated Al(VI) coordination environments increase at the expense of the undercoordinated Al(IV) sites (i.e. Al(IV):Al(V):Al(VI) changes to 32:36:32). However, the chemical shifts remain similar to the pristine case with δiso,IV = 81 ppm, δiso,V = 46 ppm and δiso,VI = 14 ppm. When the material is exposed to the electrolyte (LiPF6 in EC/EMC), Al environments show a similar proportion of environments (Fig. 2(c)) to the EC/EMC soaked sample (Al(IV):Al(V):Al(VI) now 31:30:39). However, the 27Al NMR shows on average lower chemical shifts of δiso,IV = 63 ppm, δiso,V = 38 ppm and δiso,VI = 4 ppm. These changes indicate that the coating is affected by both, the solvent (salt-free soaking in Fig. 2(b)) and salt components of the electrolyte (Fig. 2(c)), but in different ways.
The relation between Al and H at the surface, before and after the material is soaked in EC/EMC solvent (or the LiPF6 in EC/EMC) is probed with 1H NMR. To separate out the species (and respective resonances) not associated with the coating, the 1H spectrum of the uncoated NMC811 (Fig. S7, ESI†) was first fit. Three diamagnetic resonances were seen at 1.2 (most likely grease from the glovebox or tentatively terminal TM–OH groups with hydrogen bonding), 3.8 (tentatively organic carbonates, 3.6–4.5 ppm8,10 or physisorbed water) and 6.1 ppm (tentatively lithium bicarbonate). Additional broad features centred around −5.0 to −6.7 ppm (tentatively ascribed to OH groups nearby a TM ion)74 and −93.2 ppm (Ni2M–OH groups where M = TM or Al ions, based on the shifts seen for layered nickel metal hydroxides)75,76 are observed, with further discussion on their assignment in ESI 5:† Supporting NMR experiments. On the other hand, three different diamagnetic resonances related to the Al2O3 coating are observed at 4.5, 7.5 and 9.5 ppm in pristine ALD NMC811 (Fig. 2(d)). Previous NMR characterisation of aluminium oxide hydroxide and aluminium hydroxide phases77–79 has shown that protic species nearby Al can have a range of chemical shifts from approximately 2.3 ppm for terminal OH groups and in the range 5.5–10.8 ppm for protons on bridging oxygens (Al–![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) –Al/Al2– or Al2––Al/Al3–) with hydrogen bonding (Scheme 1). Therefore, the 1H resonances at 7.5 and 9.5 ppm of the amorphous Al2O3 coating are tentatively assigned to OH groups chemisorbed to Al (Al2–OH and Al3–OH respectively). 1H resonances at around 4.8 ppm are normally assigned to the bulk water80,81 but based on the 1H{27Al} TRAPDOR experiment below, the resonance at 4.5 ppm for the coating appears to contain a contribution from an Al-bonded OH. In amorphous aluminium hydroxides, bound OH groups have previously been reported at 4.5 ppm whilst physisorbed water on these aluminium hydroxides had a resonance around 5 ppm.79 A broad paramagnetic resonance is also observed at −50.0 ppm which is tentatively assigned to Al2Ni–OH environments.75,82 The 27Al{1H} REDOR spectra for the pristine ALD NMC811 (Fig. S9(a), ESI†) shows an overall attenuation in signal intensity of the Al(IV), Al(V) and Al(VI) environments. This indicates that at least a subset of Al3+ ions in all three Al coordination environments are experiencing dipolar interactions with 1H and are thus nearby protons. The REDOR experiments are supported by 1H{27Al} TRAPDOR (Fig. S9(b), ESI†) experiments showing the influence of 1H-27Al dipolar coupling on the 1H spectrum: two dominant peaks (4.9 and 9.5 ppm) are seen in the TRAPDOR difference spectrum along with a broad peak at −7.0 ppm. Therefore, these resonances belong to protic species that are located within a few Å to Al3+ cations. Furthermore, these experiments suggest that the broad peaks at −5 to −7 ppm are likely due to alumina environments nearby both paramagnetic and Al ions, possibly in configurations involving H-bonding to an oxygen nearby a TM or in environments such as (NiMnAl)–OH (see ESI 5: Supporting NMR experiments†); the difference in chemical shift between the peak at 4.5 ppm of the Hahn-echo experiment (Fig. 2(d)) and the peak in the difference spectrum of the 1H{27Al} TRAPDOR at 4.9 ppm (Fig. S9(b), ESI†) suggests that there may be more components (configurations of Al2–OH) than fit but see further discussion about other proposed resonances in ESI 5: Supporting NMR experiments.†
–Al/Al2– or Al2––Al/Al3–) with hydrogen bonding (Scheme 1). Therefore, the 1H resonances at 7.5 and 9.5 ppm of the amorphous Al2O3 coating are tentatively assigned to OH groups chemisorbed to Al (Al2–OH and Al3–OH respectively). 1H resonances at around 4.8 ppm are normally assigned to the bulk water80,81 but based on the 1H{27Al} TRAPDOR experiment below, the resonance at 4.5 ppm for the coating appears to contain a contribution from an Al-bonded OH. In amorphous aluminium hydroxides, bound OH groups have previously been reported at 4.5 ppm whilst physisorbed water on these aluminium hydroxides had a resonance around 5 ppm.79 A broad paramagnetic resonance is also observed at −50.0 ppm which is tentatively assigned to Al2Ni–OH environments.75,82 The 27Al{1H} REDOR spectra for the pristine ALD NMC811 (Fig. S9(a), ESI†) shows an overall attenuation in signal intensity of the Al(IV), Al(V) and Al(VI) environments. This indicates that at least a subset of Al3+ ions in all three Al coordination environments are experiencing dipolar interactions with 1H and are thus nearby protons. The REDOR experiments are supported by 1H{27Al} TRAPDOR (Fig. S9(b), ESI†) experiments showing the influence of 1H-27Al dipolar coupling on the 1H spectrum: two dominant peaks (4.9 and 9.5 ppm) are seen in the TRAPDOR difference spectrum along with a broad peak at −7.0 ppm. Therefore, these resonances belong to protic species that are located within a few Å to Al3+ cations. Furthermore, these experiments suggest that the broad peaks at −5 to −7 ppm are likely due to alumina environments nearby both paramagnetic and Al ions, possibly in configurations involving H-bonding to an oxygen nearby a TM or in environments such as (NiMnAl)–OH (see ESI 5: Supporting NMR experiments†); the difference in chemical shift between the peak at 4.5 ppm of the Hahn-echo experiment (Fig. 2(d)) and the peak in the difference spectrum of the 1H{27Al} TRAPDOR at 4.9 ppm (Fig. S9(b), ESI†) suggests that there may be more components (configurations of Al2–OH) than fit but see further discussion about other proposed resonances in ESI 5: Supporting NMR experiments.†
 | ||
| Scheme 1 The five different types of hydroxylated Al environments on the surface of γ-Al2O3 proposed by Peri.83 The references mentioned have assigned chemical shifts for these environments based on experimental data: Piedra et al.77 and Fitzgerald et al.78 AlIv denotes Al with tetrahedral coordination whilst AlVI denotes octahedral coordination. | ||
EC/EMC solvent-soaked ALD NMC811 shows an evolution of 1H signals likely resulting from reactions between trace water in the solution and the undercoordinated Al(IV) and Al(V) sites in the Al2O3 coating, increasing the proportion of Al(VI) sites without significantly changing the chemical shift (Fig. 2(b)). This is correlated with the increase in overall 1H signal intensity of the spectrum (Fig. 2(e)), three new resonances appearing at 1.5, 3.6 and 6.0 ppm. Probing the 27Al–1H dipolar interactions with 27Al{1H} REDOR (Fig. S9(c), ESI†) shows that (at least some of) the species giving rise to the Al(VI) signal are closer to protons than the Al(IV) and Al(V) environments. Thus the growth of the Al(VI) sites, with adsorption of protons, is concomitant with a decrease in the undercoordinated Al environments. The difference spectrum of the 1H{27Al} TRAPDOR experiment (Fig. S9(d), ESI†) shows two distinct (5.8 and 1.4 ppm) resonances that can be matched to those found in the Hahn-echo experiment (Fig. 2(e)). Therefore, the resonance at 5.8 ppm is tentatively assigned to Al2OH like clusters with hydrogen bonding between Al2OH groups whilst the 1.4 ppm is assigned to terminal Al–OH groups based on the chemical shifts discussed above. The resonance at 3.6 ppm in the Hahn-echo spectrum (Fig. 2(e)) was not clearly observed in the TRAPDOR difference experiment (Fig. S9(d), ESI†) so is assigned to the organic carbonates as for the uncoated NMC811 samples (Fig. S7, ESI†). Analysis of the EC/EMC solvent with 1H solution NMR (Fig. S10, ESI†), after soaking the (un)coated NMC811 powder for 24 h, shows a reduction in normalised signal intensity ascribed to water in the solvent. This supports the assertion that chemisorption of trace water occurs on soaking and results in an increase in 1H signal intensity of the ALD NMC sample.
The diamagnetic 1H environment (Fig. 2(f)) of the ALD NMC811 material continues to evolve after soaking in the electrolyte, with an increase in the 1H signal intensity of the resonances at 3.1, 5.8 and 7.7 ppm. 27Al{1H} REDOR (Fig. S9(a), ESI†) show that (some of the) Al(VI) sites are still protonated whilst 1H{27Al} TRAPDOR (Fig. S9(b), ESI†) verifies the persistence of Al2O![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) species. The same three resonances are observed in the difference spectrum with the range in chemical shifts indicating that Al2O types of protons can be structurally different. However, these 1H–27Al heteronuclear experiments do not explain the origin of the decrease in the 27Al chemical shifts observed for the electrolyte-soaked ALD NMC811 material. This indicates that the Al2O3 coating also interacts with the LiPF6 salt (or its products) in the electrolyte. LiPF6 readily undergoes hydrolysis to form HF84,85 and this acid can react with the Al2O3 or the NMC811.44–46,48 Unsurprisingly, the 19F NMR spectra (Fig. S15, ESI†) of the pristine ALD NMC811 and EC/EMC-soaked material show no resonances; i.e. no fluorine-containing surface species are observed. In the electrolyte soaked ALD NMC811 (point I in Fig. 3(a)), the resonances are assigned to residual LiPF6 salt (−76 to −80 ppm),8,10,84 difluorophosphate species from hydrolysis of the salt (−88 ppm),10 an aluminium oxyfluoride species (−184 ppm),86 and LiF (−206 ppm).86 Fluorination of the Al2O3 coating could explain the decrease in 27Al chemical shifts of the electrolyte-soaked ALD NMC811 (Fig. 2(c), e.g. δiso,VI = 4 ppm) but the extent of fluorination does not reach AlF3 (δVI = −17 ppm). 27Al{19F} REDOR (Fig. S13(c), ESI†) shows a decrease in the signal of the Al(VI) intensity when the 19F irradiation is applied during the dipolar evolution. This supports the proposed fluorination of the coating; further investigation of the 19F–27Al dipolar interactions with 19F{27Al} TRAPDOR (Fig. S13(d), ESI†) was not possible due to the poor signal-to-noise.
species. The same three resonances are observed in the difference spectrum with the range in chemical shifts indicating that Al2O types of protons can be structurally different. However, these 1H–27Al heteronuclear experiments do not explain the origin of the decrease in the 27Al chemical shifts observed for the electrolyte-soaked ALD NMC811 material. This indicates that the Al2O3 coating also interacts with the LiPF6 salt (or its products) in the electrolyte. LiPF6 readily undergoes hydrolysis to form HF84,85 and this acid can react with the Al2O3 or the NMC811.44–46,48 Unsurprisingly, the 19F NMR spectra (Fig. S15, ESI†) of the pristine ALD NMC811 and EC/EMC-soaked material show no resonances; i.e. no fluorine-containing surface species are observed. In the electrolyte soaked ALD NMC811 (point I in Fig. 3(a)), the resonances are assigned to residual LiPF6 salt (−76 to −80 ppm),8,10,84 difluorophosphate species from hydrolysis of the salt (−88 ppm),10 an aluminium oxyfluoride species (−184 ppm),86 and LiF (−206 ppm).86 Fluorination of the Al2O3 coating could explain the decrease in 27Al chemical shifts of the electrolyte-soaked ALD NMC811 (Fig. 2(c), e.g. δiso,VI = 4 ppm) but the extent of fluorination does not reach AlF3 (δVI = −17 ppm). 27Al{19F} REDOR (Fig. S13(c), ESI†) shows a decrease in the signal of the Al(VI) intensity when the 19F irradiation is applied during the dipolar evolution. This supports the proposed fluorination of the coating; further investigation of the 19F–27Al dipolar interactions with 19F{27Al} TRAPDOR (Fig. S13(d), ESI†) was not possible due to the poor signal-to-noise.
 | ||
| Fig. 3 (a) Voltage profile of the first charge (C/20 rate assuming 200 mA h g−1 practical capacity) for NMC811 (black) and ALD NMC811 (red) where crosses indicate sample states analysed with post-mortem NMR: pink cross (I) indicates the ALD NMC811 soaked in 1 M LiPF6 in EC/EMC, the red cross (II) indicates the ALD NMC811 charged to 4.4 VLi (rinsed with DMC), and the black cross (III) is the NMC811 charged to 4.4 VLi (rinsed with DMC). Discharge capacities over long-term cycling in sets of 49× ageing cycles at C/2 rate and 2× diagnostic cycles at C/20 rate are presented in (b) for the NMC811 and ALD NMC811. | ||
In conclusion, exposure of the ALD-coated NMC811 surface to the electrolyte results in a reduction of the concentration of lower-coordinated (and more reactive) Al sites, along with partial fluorination of the coating. Fluorination is ascribed to reactions of the hydroxyl/water groups in the coating and trace water in the electrolyte with LiPF6 generating species (such as HF) that can react with the undercoordinated Al sites.
Electrochemical cycling of ALD NMC811
Further binder-free (i.e., initially fluorine-free) samples were then prepared to assess the evolution of coating structure as a function of state-of-charge (SoC) using NMR spectroscopy. One sample was discharged until 3.0 VLi after reaching an upper cut-off voltage of 4.4 VLi (point IV in Fig. S17, ESI†) whilst a second sample was charged back to 4.4 VLi after one cycle (3.0–4.4 VLi, point V in Fig. S17, ESI†). This subsequent discharge and charge continue to show the typical voltage-capacity profile during (de-)lithiation of NMC811 and indicates unchanged solid-solution behaviour of the bulk structure as seen with other Al2O3 coated Ni-rich NMCs.14,33
Analysis of cycled samples – theory and experiment
Observing the average O electronic density of states (DOS) (Fig. S22, ESI†) in the supercell shows a large contribution from O p orbitals near the Fermi energy due to hybridisation with Ni d orbitals. No significant difference in the average O p orbital populations, which are dominated by O in the bulk structure, for these (de-)lithiated structures are observed between the uncoated, partially coated and fully coated cases. This highlights that Al present at the surface does not influence the electronic structure of bulk oxygen. This is further demonstrated by calculated averaged crystal orbital overlap population (COOP) values, in which lower values indicate relatively higher covalency in the average Ni–O bonds of the bulk of the (de-)lithiated structures. Both the pristine as well as the 75% delithiated case were analysed using averaged COOP values for NiO6 octahedra in the bulk of the structures. Average COOP values for Ni–O bonds in the uncoated, 56.25% partly coated, and fully coated NMC811 structures with full Li inventories are 1.93, 1.95, and 1.92 respectively. Average COOP values of 2.87, 2.86, and 2.86 are obtained for Ni–O bonds after 75% delithiation in the uncoated, 56.25% coated, and 100% coated NMC811 structures respectively. The comparisons show that coating the surface has a negligible effect on the average Ni–O bonds within the bulk structure (∼1.93) but there is a significant increase in covalency on charging (∼2.87). Similar conclusions can be drawn from charge analysis of oxygen in the NMC811 structure. For example, Wannier projections of LNO (end member of Ni-rich NMC)61 have indicated that the charge on O is closer to 2s2 2p5.5 (i.e. 7.5e−) due to covalency and charge transfer (from O p orbitals to Ni d orbitals) as opposed to 8e− for an idealised O2− (2s2 2p6). Given that the VASP software only considers the valence shell and Bader charge partitioning is more simplistic than Wannier projections, the estimated Bader charges are 7e− for the bulk oxygen (<5 Å below the surface) in the fully lithiated NMC811 (irrespective of any coating, Fig. 4 right). Delithiating the structures results in a decrease in average Bader charge (6.75e−) that can be explained by oxidation/ligand hole formation on the O 2p orbitals of the hybridised Ni 3d–O 2p bond at high SoC as calculated in LNO.61,88 Bader charge densities on the surface O p orbitals are discussed below.
 | ||
| Fig. 4 Electronic density of states and Bader charges: (a) the Projected DOS (PDOS) for surface oxygens in both pristine and 75% delithiated (charged) NMC811 structure with either no Al present (green), Al partially (56.25%) substituting the surface transition metals (blue) or fully substituting the surface transition metals (purple). A vacuum (extending up to 20 Å) above the surface in the b-direction is applied to the surface. The black dashed line indicates the Fermi energy. On the other hand, (b) show the Bader charges as a function of distance in the b-direction going from the bulk (0 Å) to the surface (10.8 Å, marked by the brown dashed line). The charges of the valence electrons around an idealised O2− would give 8 electrons but is underestimated in Bader partitioning (ranging from ∼7 in the bulk for the fully lithiated structure to ∼6.75 in the bulk of the delithiated case). | ||
The effect of the coating is seen in the projected DOS (obtained by projection of DOS onto scaled spherical harmonics in VASP) of the surface monolayer of oxygen atoms bonded to the coating layer (Fig. 4(a)). For the uncoated NMC811, a large population of O p orbitals is filled near the Fermi energy – indicative of the hybridisation with Ni d orbitals. However, in both the fully lithiated and 75% delithiated cases of the 56.25% coated and the 100% coated structure, there is a significant decrease in energy states near the Fermi energy, and a shift of surface O DOS to lower energies is observed. Increasingly filled lower energy states of O p orbitals, as Al substitutes Ni, imply a lower degree of hybridisation with the Ni d orbitals, and a higher and more stable oxidation state of O. Incidentally, computed electronic DOS of amorphous Al2O3 in our previous work38 indicated that the material is a wide bandgap insulator. The Al-bonding results in the O p orbitals of the valence and conduction bands being well separated (∼2.6 eV) and this trend is mirrored in the change in surface O DOS for these Al2O3 coated NMC. Looking at the Bader charge of O near the surface (from 6–10.8 Å at the very surface, Fig. 4(b)) shows an increase in charge density, compared to the bulk oxygen anions, when the Al is present in either partially or fully coated structures. This confirms the highly local effect that Al has on decreasing Ni d–O p orbital hybridisation in both the fully lithiated and 75% delithiated structures.
The formation of ligand holes in LNO89 and NMC90 has previously been proposed as being responsible for the redox of the oxygen into an unstable peroxide state that leads to O gas loss at high SoC and concomitant surface reconstruction from the layered to rock salt structure.61,88 Therefore, the action of Al in locally increasing the charge around O (reduced Ni 3d–O 2p hybridisation) suggests that coatings using Al have an additional effect of improving the resilience of NMC811 against oxidation of surface O to reactive oxygen species.
 | ||
| Fig. 5 The (a) 19F MAS NMR (4.7 T, 50 kHz MAS, 250 ms recycle delay) of the three samples I, II and III presented in the voltage profile shown in Fig. 3(a). The 19F{27Al} TRAPDOR in (b) is for sample II (ALD NMC811 at 4.4 VLi): a spectrum with no 27Al adiabatic irradiation (S0, cyan) over 25 μs dipolar evolution period and with 27Al irradiation (S, magenta with 122 kHz 27Al rf) for the same period are compared to yield a difference spectrum (Δ, black). Experiments with other dephasing times can be found in ESI 5:† Supporting NMR experiments. In (c) is the corresponding 27Al MAS NMR spectra of sample II acquired at 23.5 T. This spectrum is fitted using a Czjzek model with Al(IV), Al(V) and Al(VI) components plotted in blue (and cyan), red and black respectively. Isotropic chemical shifts of each fitted component are marked with a dashed line. The 27Al{19F} REDOR (23.5 T) in (d) shows the experiment without the 19F irradiation (S0, cyan) during the total dipolar evolution period of 269 μs and with 19F π pulses (S, magenta) during the total dipolar evolution period. Spinning sideband are marked with asterisks (*). | ||
The 27Al NMR features in the charged sample (II, Fig. 5(c)) show resonances at lower chemical shifts than the samples without exposure to LiPF6 related products. 27Al{19F} REDOR (Fig. 5(d)) shows a clearer attenuation in the signal of the Al(VI) site after the ALD NMC811 material has been charged to 4.4 VLi (compared to the electrolyte-soaked sample in Fig. S13(c), ESI†). These results are consistent with the 19F{27Al} TRAPDOR spectrum, confirming fluorination of the Al2O3 coating to form aluminium oxyfluoride species as the LiPF6 salt undergoes hydrolysis to generate HF. The rate of hydrolysis is proposed to increase at high potentials due to the chemical oxidation of EC that produces water.8–10,87
Interestingly, fitting the 27Al spectrum (Fig. 5(c)) reveals a shoulder peak to the main Al(IV) environment at higher chemical shifts (δiso = 75 ppm). This resonance contributes 17% to the total signal (Al(IV):Al(V):Al(VI) ratios from this fit yield 41:27:32). The additional Al(IV) component was not clearly observed in the spectra of the uncycled materials acquired at lower fields amongst the three other major components. Fitting the 27Al spectra of further electrochemically cycled samples indicates irreversibility of the structural evolution as the original Al(IV):Al(V):Al(VI) ratios (47:37:16) are never recovered and resonances remain at lower chemical shifts. Analysing the spectra (acquired at 16.4 T) of the ALD NMC811 in the discharged state (IV, Fig. S18(a), ESI†), the Al(IV):Al(V):Al(VI) ratio of 48:18:34 whilst the sample charged to 4.4 VLi on the second cycle (V, Fig. S18(b), ESI†) had an Al(IV):Al(V):Al(VI) ratio of 44:14:42. Interestingly, secondary Al components (δiso = 15 ppm and δiso > 70 ppm) were only required when fitting spectra of samples charged to 4.4 VLi (II, Fig. 5(c) and V, Fig. S18(b), ESI†), indicating these components may be SoC dependent and are not observed in the fully lithiated states (Fig. 2(a)–(c) and IV, Fig. S18(a), ESI†).
27Al–7Li double-resonance experiments were performed on the materials at high SoC to identify any lithiated alumina phases that could be attributed to the additional fitted components. The delithiated state also offers the additional benefit that the 7Li signal of Li within the NMC811 structure is suppressed and the surface Li species will increasingly dominate. A shift in the 7Li resonance to −0.4 ppm was observed in the 7Li spectrum of the ALD NMC811 charged up to 4.4 VLi in the second cycle (Fig. S19(c), ESI†), which suggests that different species are present in the cycled ALD NMC811 sample compared to γ-LiAlO2 (0.5 ppm). Negligible signal attenuation of the −0.4 ppm 7Li resonance was observed in the 7Li{27Al} TRAPDOR experiment (Fig. S19(c), ESI†), using the same parameters as used for the reference material γ-LiAlO2, indicating that weaker 7Li–27Al dipolar interactions are present in the ALD sample. A 27Al{7Li} REDOR experiment was performed (in which a train of 7Li π pulses is applied, see Fig. S19(b) and (d), ESI†) to explore these couplings further. The much stronger (21%) signal attenuation of the 27Al resonance of γ-LiAlO2, compared to the cycled ALD NMC811 sample (where negligible attenuation was seen) supports the hypothesis of weaker 7Li–27Al interactions in the cycled ALD NMC811. Thus, these double resonance experiments are not consistent with the formation of a LiAlO2-like phase in the coating during charge.
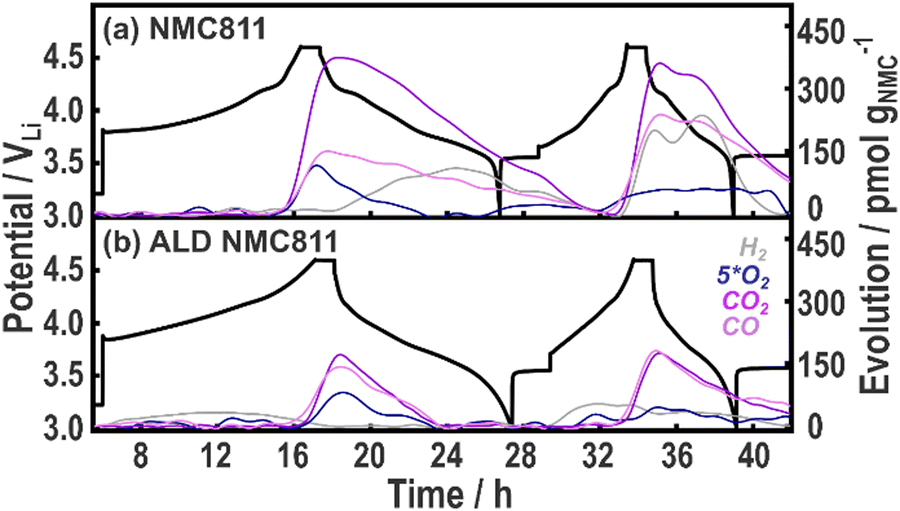 | ||
| Fig. 6 Online-electrochemical mass spectrometry (OEMS) and the corresponding voltage profiles of NMC811|Li (a) without and, (b) with an Al2O3 coating. Operando cells are cycled between 3–4.6 VLi at a C/10 and then C/5 rates (assuming 200 mA h g−1 practical capacity) with a 1 h hold at the top of charge and Ar being used as the carrier gas. | ||
For NMC811, the ratio of CO:CO2 at high potentials (∼4.31 VLi in Fig. 6(a)) is approximately 1:2.5. This CO:CO2 emission has previously been attributed to the chemical oxidation of carbonate solvents by either evolved singlet oxygen from the NMC811 surface reconstruction, or directly via reaction with surface oxygen.8,9 The increase in gas evolved on cycle 2 (Fig. S20, ESI†) is proposed to be a result of increased surface area in these polycrystalline NMC811 particles as a result of cracking.4 H2 evolution has an onset potential after the CO/CO2 evolution, suggesting that it originates from cross-talk and anodic reduction of soluble products generated during electrolyte oxidation at the NMC811 surface. On the other hand, the ALD NMC811 electrode shows a 1:1.2 CO:CO2 ratio in the first cycle (onset at ∼4.41 VLi in Fig. 6(b)). The release of H2 now precedes the onset of CO/CO2, suggesting that soluble protic species are released by the coating (chemisorbed water/hydroxyls) at potentials lower than 4.3 VLi (potentially reacting with LiPF6 to form other acidic species), which are all reduced on the Li metal counter electrode. The presence of the coating reduces the total gas evolution associated with the chemical oxidation of the carbonate solvents, but results in the generation of more protic species at lower potentials.
The impact of the coating on electrolyte degradation was also analysed after electrochemical cycling at high potentials (3.0–4.6 VLi with a 60 h hold at 4.6 VLi, Fig. S25, ESI†) with solution NMR. Delithiated LiFePO4 (LFP) was used as the counter electrode to avoid the reduction of any generated species due to its high operating potential of ∼3.5 VLi. Rinkel and co-workers have previously reported reactions between carbonate solvents in the electrolyte and reactive oxygen on the surface of layered transition metal oxides.8,10 Linear carbonates readily undergo hydrolysis resulting in alcohols that can be chemically oxidised by the reactive oxygen to form aldehydes. These aldehydes can undergo hydrolysis by either alcohols or the water to acetals and these acetals can also react with the reactive oxygen to form carboxylic acids. 1H NMR of the ALD NMC811 (Fig. S26(b), ESI†) after one cycle shows Li formate/formic acid (8.25/8.19 ppm) whereas the uncoated NMC811 (Fig. S26(a), ESI†) only shows acetals (5.69 and 5.80 ppm for methoxymethanol and methanediol respectively). After five cycles, the 1H solution NMR (Fig. S27, ESI†) shows that the range and relative concentrations of solution degradation products (including alcohols, aldehydes, acetals and formic acid) are similar between the NMC811 and ALD NMC811. A detailed summary of the 1H solution NMR assignments can be found in Table S16, ESI 5:† Supporting NMR experiments. The apparent lack of impact of the coating on the degradation of the electrolyte from the perspective of solution NMR after more than one cycle is explored further in the discussion.
The electronic state of the uncycled (ALD) NMC811 electrodes was first probed via Ni L-edge and O K-edge XAS in the total electron yield (TEY) mode. Two peaks are observed (528.3 and 529.4 eV) in the O K-edge that have previously been assigned to the excitation of electrons from O 1s to the hybridised TM 3d–O 2p orbital (Fig. S23, ESI†).91,92 In both samples, the presence of a small amount of Li2CO3 is evident (the peak at 533.5 eV).93 In the Ni L-edge (Fig. S24, ESI†), the main L3 peak has two components; a low energy peak at 853.7 eV is associated with low-spin Ni3+ with a 3d7 electronic configuration (and to a lesser degree Ni4+ if present) whilst the higher energy peak at 855.8 eV is present for all Ni2+, Ni3+ and Ni4+.93,94 The observed L3 splitting (1.87 eV) for the uncycled (ALD) NMC811 is consistent with the covalent nature of the hybridised Ni 3d–O 2p bond.95 As the 855.8 eV peak of the Ni L-edge is associated with oxidised Ni chemical states, the delithiated state of NMC811 (4.4 VLi) is selected to maximise changes in this feature.
On the first charge up to 4.4 VLi, the impurity Li2CO3 peak is no longer visible, confirming break down of carbonate surface impurity in the first charge. The Ni L3-edge (Fig. 7(c)) also evolves as the high energy feature (855.8 eV) becomes more prominent than the lower energy feature (853.7 eV), consistent with oxidation in the hybridised Ni 3d–O 2p bond.92,96 Analysing the L3 high energy:low energy (855.8 eV: 853.7 eV) area ratio ( in Fig. 7(c)) can therefore be a proxy for assessing the change of electron density in the hybridised Ni–O bond. The increased L3 ratio of the (ALD) NMC811 is consistent with the reported oxidation of the hybridised Ni 3d–O 2p bond at high SoC.92,97 Table S15 (ESI†) compares the L3 ratio for different SoCs to highlight how a large L3 ratio for samples in the charged state are an indicator of continued redox activity in the hybridised Ni 3d–O 2p bond. In addition, the growth in the L3–L2 splitting, from 1.87 eV to 2.34 eV, for the samples (with and without the Al2O3) with charging indicates an increase in the ionic nature of the hybridised bond.95 The O K-edge (∼531.6 eV) signatures of γ-Al2O3 and NiO overlap, making a reliable estimate of the relative contribution of the two components difficult. However, this peak clearly grows in intensity between the uncoated and ALD NMC811 sample, due to the Al2O3 coating (Fig. S23, ESI†). The intensity of this peak is stronger in the uncoated sample on charging to 4.4 V and on more extended cycling (Fig. 7(b)), this change being assigned to the more pronounced growth of the reduced surface (NiO-like) layers (during surface reconstruction) in the uncoated samples.93,98 In the Ni L-edge spectra (Fig. 7(d)), extensive cycling resulted in smaller area ratios relative to early cycling life. The values complement the O K-edge analysis of the aged samples showing a more reduced surface Ni environment compared to the early cycle stages. However, the ALD NMC811 shows the least reduced environment as it has a relatively high area ratio of 2.40 (initially 2.96). The electronic density in the surface Ni–O bonds of the Al2O3-coated NMC811 therefore indicates electrochemical activity is preserved after the electrochemical ageing and less surface degradation has occurred in the presence of the Al2O3. This is in agreement with the higher available capacity for the ALD NMC811 after 1042 cycles (Fig. 3(b)).
in Fig. 7(c)) can therefore be a proxy for assessing the change of electron density in the hybridised Ni–O bond. The increased L3 ratio of the (ALD) NMC811 is consistent with the reported oxidation of the hybridised Ni 3d–O 2p bond at high SoC.92,97 Table S15 (ESI†) compares the L3 ratio for different SoCs to highlight how a large L3 ratio for samples in the charged state are an indicator of continued redox activity in the hybridised Ni 3d–O 2p bond. In addition, the growth in the L3–L2 splitting, from 1.87 eV to 2.34 eV, for the samples (with and without the Al2O3) with charging indicates an increase in the ionic nature of the hybridised bond.95 The O K-edge (∼531.6 eV) signatures of γ-Al2O3 and NiO overlap, making a reliable estimate of the relative contribution of the two components difficult. However, this peak clearly grows in intensity between the uncoated and ALD NMC811 sample, due to the Al2O3 coating (Fig. S23, ESI†). The intensity of this peak is stronger in the uncoated sample on charging to 4.4 V and on more extended cycling (Fig. 7(b)), this change being assigned to the more pronounced growth of the reduced surface (NiO-like) layers (during surface reconstruction) in the uncoated samples.93,98 In the Ni L-edge spectra (Fig. 7(d)), extensive cycling resulted in smaller area ratios relative to early cycling life. The values complement the O K-edge analysis of the aged samples showing a more reduced surface Ni environment compared to the early cycle stages. However, the ALD NMC811 shows the least reduced environment as it has a relatively high area ratio of 2.40 (initially 2.96). The electronic density in the surface Ni–O bonds of the Al2O3-coated NMC811 therefore indicates electrochemical activity is preserved after the electrochemical ageing and less surface degradation has occurred in the presence of the Al2O3. This is in agreement with the higher available capacity for the ALD NMC811 after 1042 cycles (Fig. 3(b)).
 | ||
| Fig. 7 The O K-edge (a) and (c) Ni L-edge X-ray absorption spectroscopy of the NMC811 (black) and ALD NMC811 (red) electrodes charged to 4.4 VLi on the first cycle are presented with spectra from reference samples of γ-Al2O3 (magenta) and NiO (blue). O K-edge and Ni L-edge spectra of the materials in the electrochemically aged state (after cycle 1042) and charged up to 4.4 VLi are presented in (b) and (d) respectively. Ratios of the L3 high energy (I2, 855.8 eV): low energy (I1, 853.7 eV) integrated areas are expressed with R for the Ni-Ledge. | ||
Discussion
Peri,83 Knözinger and Ratnasamy102 have previously studied the 1H resonances of transition alumina phases and proposed five types of hydroxylated Al surface environments (Scheme 1). Case Ia is for tetrahedral Al with a terminal OH group; case Ib is for octahedral Al with a terminal OH group, case IIa consisting of a hydroxyl group that bridges one tetrahedral and one octahedral Al, case IIb with a hydroxyl group bridging two octahedral Al, and finally case III with an OH bridging three octahedral Al. The 1H chemical shift is extremely sensitive to the coordination number of the O in the O–H bond and the nature and number of Al ions coordinated it, along with any hydrogen bonding between clusters in aluminium hydroxide/aluminium oxide hydroxide phases.77 The three 1st cation coordination sphere Al3+ ions (relative to the proton) found within the Al3OH clusters are proposed to provide an induction effect that deshields these protons the most of all five cases (resulting in the most positive shift).77 In addition, hydrogen bonding to a nearby oxygen also results in positive shifts: the shorter the H-bond, the larger the degree of deshielding: Piedra and co-workers showed that the Al2OH clusters in Gibbsite Al(OH)3 have interlayer hydrogen bonds of 1.98 Å and an associated 1H resonance at 5.8 ppm, whilst environments with a longer intralayer hydrogen bonding (average of 2.3 Å) show resonances at 4.3 and 2.9 ppm.77 Therefore the highest frequency 1H resonances observed for the pristine ALD NMC811 (Fig. 2(d)) at 9.5, 7.5 and 4.5 ppm are tentatively assigned to case III (Al3OH clusters, 9.5 ppm vs. 10.8 ppm in Diaspore)77 and lower frequency resonances at 7.5 and 4.5 ppm to case IIa (or case IIb) type sites (Al2OH clusters), with varying degrees of H-bonding.
The Al sites in the ALD NMC811 show Lewis acidity103–105 in allowing chemisorption of Lewis-base trace water onto the NMC811 after exposure to the solvent (or electrolyte). Non-dissociative adsorption of water onto an Al3+ Lewis acid site followed either by a reaction with more water or by dissociative chemisorption of the OH− and H+ has previously been proposed as the mechanism for this reaction (Scheme 2).100,103 More generally, a model based on partial charges, entitled the multi-site complexation (MUSIC) model can be used to predict the affinity of a proton to bond to oxygen based on the expected electrostatic repulsion from nearby cation centres106,107 and the partial charges contributed by each bonded cation to the Al–O bond (see discussion in ESI 7:† Multi-site complexation (MUSIC) model). As such, the ability of the Al2O3 coating to sequester species like water from the electrolyte can be explained (using the MUSIC model as a framework) in terms of chemical characteristics of the coating i.e. the Lewis acid (undercoordinated Al) and Lewis basic (appropriate O) sites. Scheme 2 illustrates one possible series of reactions (of many possible sequences of reactions) involving dissociative chemisorption of water, cleavage of Al–O bonds in a OxAl–O–AlOx cluster to form a new hydroxylated site (red, case I type Alx–OH), and ultimately higher coordinate Al sites. Alternative mechanisms involving protonating nearby O (rather than Al–O–Al cleavage) are also possible, since protonation of bridging oxygens between two Al(V) sites in Al2OH clusters (Al––Al reported in dehydrated γ-Al2O3)104 are more favourable than protonation of terminal hydroxyl oxygens (Al–OH). These bridging oxygens are more basic than the terminal hydroxyl oxygens when considering a simple model that is based on oxygen partial charges (see ESI 7:† Multi-site complexation (MUSIC) model). However, the remaining undercoordinated Al site would be susceptible to further reactions with water as illustrated in Scheme 2, eventually forming two Al(VI) sites.
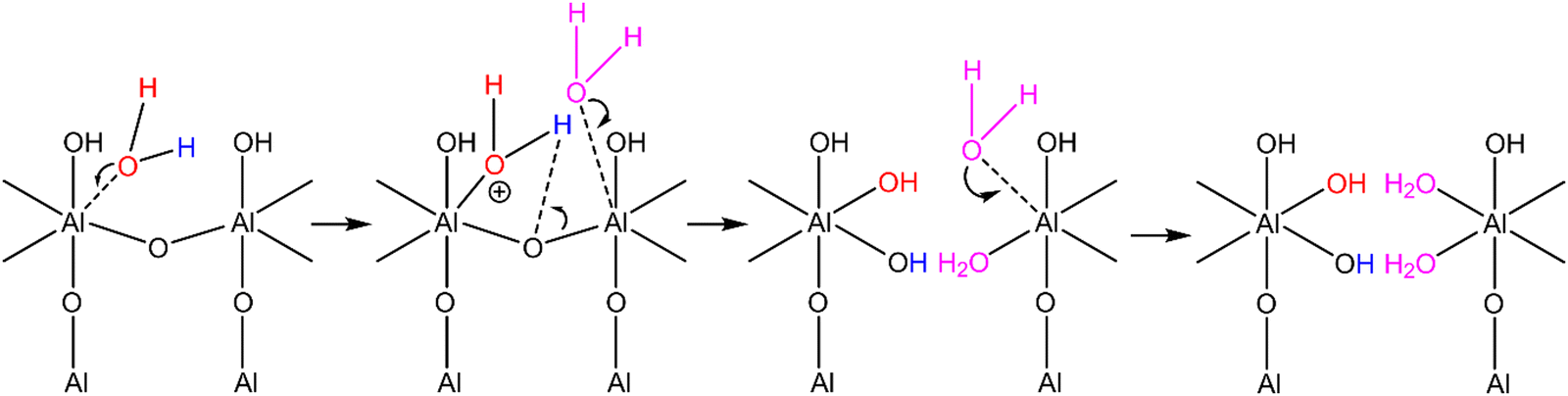 | ||
| Scheme 2 One example illustrating how water could adsorb onto a Lewis acidic Al(V) site. The dissociation and subsequent chemisorption of the molecule fragments (OH− in red and H+ in blue) results in the formation of a new Al(VI) site with new hydroxyl groups and an Al(V) site which can further react with other Lewis bases (as indicated by the two purple H2O water molecules shown in the next steps). Additional hydroxyl groups not involved in the reaction are omitted. | ||
These models are supported experimentally by the 27Al NMR with an increase in Al(VI) sites from the pristine ALD NMC811 to the EC/EMC-soaked material (Fig. 2) with chemisorption even of trace water. 27Al{1H} REDOR shows that the signal attenuation (from the 1H–27Al dipolar interactions) of the Al(IV), Al(V) and Al(VI) coordination sites changes depending on the sample state. In the pristine state, all Al3+ coordination sites are near 1H (Fig. S9(a), ESI†), but after the ALD NMC811 is soaked in the carbonate solvent and electrolyte, only the Al(VI) sites are close to 1H (Fig. S9(c) and S13(a), ESI†). A concomitant change is observed in the normalised 1H NMR (Fig. 2(d)–(f)) with the signal intensity growing from 600 a.u. (case II (Al2OH) at 4.5 ppm) to 1500 a.u. (case I (AlVIOH) at 1.5 ppm) which is consistent with Scheme 2.
Soaking the ALD NMC811 in the electrolyte (1 M LiPF6 in EC/EMC (3/7)) introduces acid–base reactions between the hydrated Al2O3 coating and species such as trace HF (generated upon PF6− hydrolysis). Fluorination of the coating is supported by the 27Al resonances shifting to lower values (Fig. 2(c), closer to AlF3 at −17 ppm), some of the Al(VI) being fluoridated as shown via the 27Al{19F} REDOR spectrum in (Fig. S13(c), ESI†), and a 19F resonance appearing at −180 ppm. Evidence of the involvement of protons from HF during this acid–base reaction is also reflected in the normalised 1H signal intensity increasing to ∼8000 a.u. (case II, Al2OH at 5.8 ppm in Fig. 2(f)). These 1H protons are also close to Al(VI) sites as demonstrated in 27Al{1H} REDOR and 1H{27Al} TRAPDOR (Fig. S13(a) and (b) respectively, ESI†).
Two reaction mechanisms describing the HF scavenging reaction are presented below – Scheme 3(I) involves an increase in the coordination number of Al sites that neighbour hydroxylated 6-coordinated Al which can undergo neutralisation with HF. During neutralisation, an Al–F bond forms in place of the Al–OH with cleavage of this bond releasing a water molecule: Al–F is more thermodynamically favourable with a bond dissociation energy of 675 kJ mol−1 compared to 547 kJ mol−1 for Al–OH.108 If there is a nearby undercoordinated site, the generated water can chemisorb to this site, thereby increasing the coordination number of that site and preventing further reactions triggered by “free” water. The second proton on the bound water has a greater affinity for bonding to bare bridging oxygens (in an Al––Al) than to the generated terminal Al–OH based on the simple MUSIC model analysis (ESI 7:† Multi-site complexation (MUSIC) model). This first scheme therefore enables both the formation of protons in an Al2OH environment (case II) and explains the fluorination of Al(VI).
 | ||
| Scheme 3 Two different scenarios in which HF scavenging could occur on the hydrated Al2O3 coating: (I) involves reaction with a terminal OH group that results in fluorination of an Al group with no change in coordination number. Water is released, which can react with a lower coordinate Al site nearby, resulting in an increase in the coordination number of this Al site and (II) a reaction involving fluorination of an undercoordinated site to increase its coordination number. The mechanisms presented here are based on basicity arguments, Al–F bonds strengths and the DFT predictions from Tebbe and co-workers.47 Hydroxyl groups on the Al groups not involved in the reaction are omitted. | ||
DFT calculations by Tebbe et al. of both bare (in their case LiCoO2) and alumina-coated surfaces47 showed that an Al–F(–OH) environment is formed in the acid–base reaction between HF and an undercoordinated Al–OH site on the alumina surface. They found that, unlike reactions with TM–OH groups on the bare Co oxide surface, reactions between HF and an undercoordinated Al–OH site nearby an undercoordinated Al–F site generate two Al–F(–OH) sites, rather than releasing water (water release triggering further reactions). We note that the energies they calculate are based on the highly reactive three-coordinate Al species, which we do not be believe to be present (on the basis of our NMR experiments) and thus the driving forces may be smaller than calculated. Our second proposed reaction type (Scheme 3(II)) is qualitatively similar to that of Tebbe et al.47 and results in no change to the coordination number. The proton generated binds to an oxygen atom in a nearby AlOx group and again no un-dissociated water is generated to cause further reactivity.
A chemical reaction (eqn (1)) is presented to describe the HF scavenging by the Al2O3 coating which is consistent with HF scavenging. It should be stressed that the illustrated mechanisms do not cover the full permutation space or possible reactivities, and further reactions between HF sites shown below likely also occur. However, the reactions do not proceed as far as to produce AlF3.
| Al2O2(OH)2 + xHF → Al2O2(OH)2−xFx(OH2)x | (1) |
A more well-defined 19F signal (−184 ppm) for the Al oxyfluoride appears in the sample charged to 4.4 VLi (Fig. 5(a)) compared with the electrolyte-soaked material (Fig. 5(a)). The reported chemical oxidation of EC by NMC811 at potentials >4.3 VLi that forms CO2, CO and water8–10,87 is verified in the gas evolution (Fig. 6(a)). This water participates in hydrolysis of LiPF6 to produce HF and this will react with the coating, forming more Al oxyfluoride than in the electrolyte-soaked state of the ALD NMC811. The chemisorbed protic species on the Al2O3 coating are not necessarily inactive with ion exchange between Li+ and H+ on the coating, liberating protons into the electrolyte. H+ (or H3O+) could diffuse to the Li counter electrode and electrochemically reduce during cycling of the ALD NMC811 as evidenced by H2 evolution as soon as charging begins (Fig. 6(b)). H2 evolution was only detected at high potentials (>4.3 VLi) for the NMC811 when water was generated (Fig. 6(a)).
Fitting the 27Al spectra of the ALD NMC811 at high SoC (II, Fig. 5(b) and V, Fig. S18(b), ESI†) showed additional minor Al(IV) and Al(VI) components (δiso,Al(IV)ii > 70 ppm and δiso,Al(VI)ii = 15 ppm). These were only required for the charged state (not for the sample discharged to 3.0 VLi, IV in Fig. S18(a), ESI†) and occur at higher chemical shifts than the major components (major components at δiso,Al(IV)i ≈ 61 ppm and δiso,Al(VI)i ≈ 0 ppm), indicating these environments are not fluorinated and there is some dependency of the Al local structure on SoC. Li+–H+ exchange on the coating is a chemical process, independent of electrochemical cycling, so the apparent SoC dependency suggests Li+–H+ exchange is not the underlying cause. Two hypotheses to account for these observations are discussed.
One hypothesis is the lithiation of alumina to form a new phase during electrochemical cycling.19,42,109,110 Since this cannot be a redox process, it would have to involve Li+–H+ exchange and possibly structural rearrangements. In this study, reference γ-LiAlO2 (82 ppm)111 was compared to a cycled ALD NMC811 (charged to 4.4 VLi on cycle 2) to test for lithiation; γ-LiAlO2 is appropriate as it crystallises first from an amorphous state of precursor materials.112 Unlike for the γ-LiAlO2 sample (Fig. S19(a) and (b), ESI†), the 7Li–27Al double resonance experiments of the ALD NMC811 charged up to 4.4 VLi in second cycle showed very little evidence for any 27Al–7Li dipolar couplings and thus no close Li–Al proximity (Fig. S19(c) and (d), ESI†). The magnitude of dipolar interactions (and thus signal attenuation when similar REDOR/TRAPDOR conditions are used) have an inverse cube relationship with the internuclear distance (∝1/r3). γ-LiAlO2 consists of LiO4 tetrahedra corner shared with AlO4 tetrahedra resulting in 27Al–7Li internuclear distances in the order of 3.09 or 3.17 Å (calculated from the structure).113 Therefore the lack of signal attenuation for the ALD NMC811 charged to 4.4 VLi in the second cycle in both REDOR and TRAPDOR indicates that any 27Al–7Li interactions present are likely occurring over distances much greater than 3.17 Å. It is thus unlikely that the 27Al resonances at 70 and 15 ppm correspond to phases structurally related to γ-LiAlO2 (83 ppm,114r = 3.09 or 3.17 Å),113 α-LiAlO2 (17 ppm, r = 2.87 Å)115 or LiAl5O8 with r = 2.76 Å.116 Haber and co-workers also used ssNMR to investigate their Al–F based coating on LiNi0.5Mn1.5O4.42 They similarly performed 7Li{19F} and 27Al{7Li} REDOR experiments to investigate the nature of the Li ions formed on the surface upon electrochemical cycling, which suggested a proximity of these interfacial Li ions to both F and Al sites. The authors propose Li intercalation into the coating/the formation of Li–Al–Ox phases to explain their experimental observations. We note that their coatings are more fluorinated and there will be a stronger driving force for the more acidic protons in the Al(OH)Fx groups to ion-exchange with Li (potentially also re-leaving protons), potentially explaining the differences between the two NMR studies.
Finally, the apparent SoC dependent resonances at higher chemical shifts (δiso,Al(IV)ii > 70 ppm and δiso,Al(VI)ii = 15 ppm) could be attributed to the effect of the paramagnetic ions on the Al ions at or on the NMC811–Al2O3 coating interface. These Al atoms, located within 5 Å of some Ni2/3+ or Mn3+/4+ cation centres, would experience through-space dipolar or through-bond interactions with unpaired electron density. However, reduced paramagnetism at high SoC (formally, Ni4+) may enable the observation of some of these resonances and the apparent SoC dependent behaviour of the 27Al spectra. Furthermore, some of these resonances may simply have been buried under the major, undercoordinated Al(IV) environments, and they become more visible as the sites nearer the surface of the coating react with water and fluoride ions and their coordination environments increase.
Al appears to be responsible for a lowering of energies away from the Fermi edge in the DOS of surface O (Fig. 4 left) compared to the uncoated case, whilst Bader charge analysis indicates an increase in the surface O charge with the Al (Fig. 4 right). Both observations suggest that the coating reduces the covalency between Ni 3d and O 2p, for O ions that are also coordinated to Al. The coating helps to hinder the phase transformation of the delithiated layered structure (e.g. NiO2) at high potentials to the more thermodynamically favourable rock salt (NiO) or rock salt like (Ni3O4) structure9,61,117 concomitant with oxygen loss. The products of organic carbonate oxidation are a sign of reactive oxygen at the surface of the NMC811 particle.
Ni L-edge XAS provides experimental evidence of the stabilised surface with the Al2O3 coating when correlating features of the L3 peak (ratio of I2 peak at 855.8 eV and I1 peak at 853.7 eV,  ) to the health of the surface chemical state (capability to be redox active). In the first charge up to 4.4 VLi, the coating does not affect the ratio
) to the health of the surface chemical state (capability to be redox active). In the first charge up to 4.4 VLi, the coating does not affect the ratio  and becomes the fingerprint for a charge transfer insulator in a mixed d8L/d8L2 ligand hole state (Fig. 7(c), i.e. ∼Li0.2NiO2 in a non-degraded state). This notation is used rather than Ni3+/Ni4+ to convey the oxidation of the hybridised Ni 3d–O 2p bond at high SoC. However, after electrochemical ageing, the uncoated NMC811 charged to 4.4 VLi
and becomes the fingerprint for a charge transfer insulator in a mixed d8L/d8L2 ligand hole state (Fig. 7(c), i.e. ∼Li0.2NiO2 in a non-degraded state). This notation is used rather than Ni3+/Ni4+ to convey the oxidation of the hybridised Ni 3d–O 2p bond at high SoC. However, after electrochemical ageing, the uncoated NMC811 charged to 4.4 VLi shows a dramatic difference to the ALD NMC811
shows a dramatic difference to the ALD NMC811  in Fig. 7(d). This suggests that the surface environment of the uncoated NMC811 can no longer achieve the oxidised d8L/d8L2 state (∼80% delithiation), presumably due to surface reconstruction behaviour. In contrast, the ALD NMC811 shows preservation of the redox activity in the hybridised Ni–O bond. This implies that the additional charge on surface O, due to the presence of Al (Fig. 4 right), has stabilised the surface against the 1O2 evolution and surface reconstruction behaviour.
in Fig. 7(d). This suggests that the surface environment of the uncoated NMC811 can no longer achieve the oxidised d8L/d8L2 state (∼80% delithiation), presumably due to surface reconstruction behaviour. In contrast, the ALD NMC811 shows preservation of the redox activity in the hybridised Ni–O bond. This implies that the additional charge on surface O, due to the presence of Al (Fig. 4 right), has stabilised the surface against the 1O2 evolution and surface reconstruction behaviour.
Further evidence for reactive surface oxygen and surface reconstruction behaviour being affected by the Al2O3 coating can also be tracked via gas evolution at high potentials (>4.3 VLi).8,9,118 As detailed in the previous work by Jung and co-workers9 and Rinkel and co-workers,8 the reactive oxygen is a strong electrophile and can react with the organic carbonates (such as ethylene carbonate, EC). An overall reaction is presented in eqn (2), where the formate formed in eqn (3) continues to react further to generate a second CO2 molecule and water. The water generated in this reaction can contribute to hydrolysis of the LiPF6, adsorb to the NMC811 surface or hydrolyse the linear carbonate.8
OEMS measurements of the NMC811 (Fig. 6(a)) show a CO:CO2 ratio of 1:2.5 for the uncoated NMC811 in cycle 1 supporting the chemical oxidation reaction in eqn (2) – excess CO2 from EC chemical oxidation has also been observed in the literature.8,9 On the other hand, the lower volume of gases evolved for the ALD NMC811 is evidence that the coating has reduced the extent of EC chemical oxidation (Fig. 6(b)). Furthermore, the CO:CO2 ratio detected during electrochemical cycling of ALD NMC811 up to 4.6 VLi was 1:1.2, indicating a different oxidation pathway or extent of oxidation. Jung and co-workers9 previously suggested that the EC-intermediate formed from 1 mole of EC reacting with 1 mole of 1O2 (eqn (3)) could decompose into 1 mole of CO, 1 mole of CO2 and 1 mole of formaldehyde, but this reaction was not observed in their work. A lower extent of available oxidised oxygen sites in the presence of the Al2O3 coating may allow this reaction to out-compete the full reaction with both CH2 groups in EC viaeqn (2).
| EC + 21O2 → 2CO2 + CO + 2H2O | (2) |
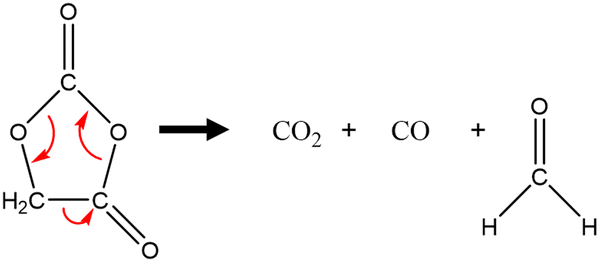 | (3) |
Secondary particle disintegration of uncoated samples under extreme charge–discharge conditions is widely documented.67,125 For example, an in-depth study by Parks et al., using ex situ nano computed tomography, showed that a 13–18% volume change in the NMC811 secondary particles occurred upon charging to 4.5 V.68 Microcracks developed in the bulk of the particles, leading to increased surface area and thus further increasing the volume change to 30–72%. Lee et al. similarly showed via cross sectional SEM that Ni-rich NMC cathode particles develop microcracks upon electrochemical cycling, which can expose uncoated cathode surfaces and allow parasitic reactions and isolate active cathode material from the conductive electrode matrix, resulting in impedance increase and capacity fade.125 As the coatings are limited to the secondary spherical particle surfaces and do not fully coat every primary particle, when the volume expansion occurs, new uncoated surfaces will become available for reactions with electrolyte and oxygen loss and surface reconstruction can occur. This growth in unprotected NMC811 surface will mask the benefit of a coating layer seen in initial cycles and helps explain the only marginal improvements in long term capacity retention, as the coating protection is a localised effect (Fig. 4, right), and ultimately continued electrolyte oxidation even in the coated material.
Finally, the compressive and tensile stresses felt by the particles as they expand and crack may lead to delamination or cracking of surface coatings, here the alumina layer. However, at the same time, the coating will also evolve on cycling, some of the fluorinated reaction products helping to passivate the surface.125 The alumina coating will also continue to “scrub” some of the reactive electrolyte degradation products (e.g., HF), which should help prevent these products from crossing over to the anode causing further degradation of the SEI, particularly in full cells.126
Conclusion
In this work, a mechanistic understanding of the evolution of Al2O3 coatings on cathode material NMC811 and the effect on the NMC811 surface stability was developed. Solid-state NMR has been instrumental in highlighting the changing nature of the coating as it responds to trace water and HF in the electrolyte system. The capability to scavenge such species is highly dependent on the presence of undercoordinated cation sites – namely tetrahedrally and penta-coordinated Al sites in Al2O3 (as seen by 27Al NMR) and the stronger Al–F vs. Al–OH bond. These sacrificial sites are important as water initiates a cycle of salt degradation that reduces the performance of Ni-rich layered oxides. Generation of water is especially concerning at high potentials for the Ni-rich layered oxides with oxidation of the electrolyte. Interestingly, the evidence indicates that in situ formation of lithium aluminate upon electrochemical cycling does not occur.The influence of the coating on the NMC811–electrolyte interactions was also demonstrated, with the coating lowering the amount and ratio of evolved CO and CO2 gases observed on charging. These gases are associated with the reaction between EC and reactive oxygen at the surface of NMC811 during surface reconstruction behaviour. Oxidation of the hybridised Ni 3d–O 2p bonds above 75% SoC generates reactive oxygen but different pathways to have been proposed to explain this degradation phenomenon of Ni-rich layered oxides.61,117 This prompted analysis of Bader charges and the surface O density of states in this work. Both metrics indicated a decrease in covalency of the surface Ni 3d–O 2p hybridised bonds for O due to charge transfer from the bonded Al. The reduced covalency at high SoC indicates a lower probability of formation of ligand holes on O and hence a reduction in O loss. Therefore, it appears that the Al2O3 coating fundamentally increases the chemical stability of nearby surface oxygens (<5 Å) by reducing hybridisation in the related Ni–O bonds. The localised protection offered by the coating could be expected to be compromised when degradation phenomena like cracking in polycrystalline particles occurs and dramatic increases in unprotected surface area arise.
These results highlight the dual chemical role of Al2O3 coatings on NMC811: scavenging species in the electrolyte and the local passivation of surface oxygen. Extended cycling to high potentials for more than 1000 cycles confirms the importance of these protective mechanisms. Despite the initial lower capacity, the Al2O3 NMC811 retained ∼71% capacity (and higher absolute capacity after electrochemical ageing) compared to ∼51% for no coating. Such insights mean that careful material design choices are needed to optimise the benefits to electrochemical performance. Desired chemical characteristics of coatings are that the cations can favourably form fluorinated bonds while also scavenging water. Additionally, the morphology of the electrode material also plays a role, coated single crystals potentially benefiting over polycrystalline agglomerates, due to a much-reduced tendency for crack formation, thereby resulting in much smaller increases in unprotected surface area as the cell is cycled. The results generated from our study of ALD coatings may have relevance in understanding the role that similar ALD coatings may have in preventing electrolyte degradation in, for example, cathodes for sodium–ion batteries or for helping to prevent dissolution in Mn and Fe-containing cathodes.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Faraday Institution degradation project (FIRG024, FIRG060). Samples were coated by Forge Nano with their proprietary atomic layer deposition process. R. L. B. C. gratefully acknowledges funding from the Cambridge Trust and the Cambridge Australia Scholarships. F. N. S. acknowledges funding from the Faraday Institution CATMAT project (FIRG016). H. B. acknowledges generous computing support provided by the CSD3 cluster of the University of Cambridge, Sulis HPC, Archer2 (access through Materials Chemistry Consortium), Michael cluster of UCL (Access through Faraday Institution Degradation project), and Brookhaven National Lab HPC Cluster (access through Centre for functional Nanomaterials). I. T. acknowledges support from a Beatriz Galindo senior fellowship (BG22/00148) from the Spanish Ministry of Science and Innovation. We thank Dr Pardeep K. Thakur and Dr Tien-Lin Lee for assistance with XAS experiments at the I-09 beamline of the Diamond Light Source, UK under proposal SI30759-1. F. N. S. is thankful to Prof. Robert S. Weatherup (University of Oxford) for discussion on XAS data. R. L. B. C. also acknowledges the help from Dr David Hall in initiating the collaboration with Forge Nano. The UK High-Field Solid-State NMR Facility is acknowledged for access to magnet time. Generous computing resources were provided by the Sulis HPC service (EP/T022108/1), and networking support by CCP-NC (EP/T026642/1), CCP9 (EP/T026375/1), and UKCP (EP/P022561/1).References
- W. Li, E. M. Erickson and A. Manthiram, High-nickel layered oxide cathodes for lithium-based automotive batteries, Nat. Energy, 2020, 5, 26–34 CrossRef CAS
.
- G. E. Blomgren, The Development and Future of Lithium Ion Batteries, J. Electrochem. Soc., 2017, 164, A5019–A5025 CrossRef CAS
- C. P. Grey and D. S. Hall, Prospects for lithium-ion batteries and beyond—a 2030 vision, Nat. Commun., 2020, 11, 6279, DOI:10.1038/s41467-020-19991-4
- S. Oswald, M. Bock and H. A. Gasteiger, Elucidating the Implications of Morphology on Fundamental Characteristics of Nickel-Rich NCMs: Cracking, Gassing, Rate Capability, and Thermal Stability of Poly- and Single-Crystalline NCM622, J. Electrochem. Soc., 2022, 169, 050501 CrossRef CAS
- T. M. M. Heenan, A. Wade, C. Tan, J. E. Parker, D. Matras, A. S. Leach, J. B. Robinson, A. Llewellyn, A. Dimitrijevic, R. Jervis, P. D. Quinn, D. J. L. Brett and P. R. Shearing, Identifying the Origins of Microstructural Defects Such as Cracking within Ni-Rich NMC811 Cathode Particles for Lithium-Ion Batteries, Adv. Energy Mater., 2020, 10, 2002655, DOI:10.1002/aenm.202002655
- H. Li, A. Liu, N. Zhang, Y. Wang, S. Yin, H. Wu and J. R. Dahn, An Unavoidable Challenge for Ni-Rich Positive Electrode Materials for Lithium-Ion Batteries, Chem. Mater., 2019, 31, 7574–7583 CrossRef CAS
- W. M. Dose, W. Li, I. Temprano, C. A. O’Keefe, B. L. Mehdi, M. F. L. De Volder and C. P. Grey, Onset Potential for Electrolyte Oxidation and Ni-Rich Cathode Degradation in Lithium-Ion Batteries, ACS Energy Lett., 2022, 3524–3530 CrossRef CAS PubMed
- B. L. D. Rinkel, J. P. Vivek, N. Garcia-Araez and C. P. Grey, Two electrolyte decomposition pathways at nickel-rich cathode surfaces in lithium-ion batteries, Energy Environ. Sci., 2022, 15, 3416–3438 RSC
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, Oxygen Release and Its Effect on the Cycling Stability of LiNixMnyCozO2 (NMC) Cathode Materials for Li-Ion Batteries, J. Electrochem. Soc., 2017, 164, A1361–A1377 CrossRef
- B. L. D. Rinkel, D. S. Hall, I. Temprano and C. P. Grey, Electrolyte oxidation pathways in lithium-ion batteries, J. Am. Chem. Soc., 2020, 142, 15058–15074 CrossRef
- J. Li, H. Liu, J. Xia, A. R. Cameron, M. Nie, G. A. Botton and J. R. Dahn, The Impact of Electrolyte Additives and Upper Cut-off Voltage on the Formation of a Rocksalt Surface Layer in LiNi0.8Mn0.1Co0.1O2 Electrodes, J. Electrochem. Soc., 2017, 164, A655–A665 CrossRef
- J. D. Steiner, L. Mu, J. Walsh, M. M. Rahman, B. Zydlewski, F. M. Michel, H. L. Xin, D. Nordlund and F. Lin, Accelerated Evolution of Surface Chemistry Determined by Temperature and Cycling History in Nickel-Rich Layered Cathode Materials, ACS Appl. Mater. Interfaces, 2018, 10, 23842–23850 CrossRef PubMed
- W. M. Dose, C. Xu, C. P. Grey and M. F. L. De Volder, Effect of Anode Slippage on Cathode Cutoff Potential and Degradation Mechanisms in Ni-Rich Li-Ion Batteries, Cell Rep. Phys. Sci., 2020, 1(11), 100253, DOI:10.1016/j.xcrp.2020.100253
- L. David, K. Dahlberg, D. Mohanty, R. E. Ruther, A. Huq, M. Chi, S. J. An, C. Mao, D. M. King, L. Stevenson and D. L. Wood, Unveiling the Role of Al2O3 in Preventing Surface Reconstruction during High-Voltage Cycling of Lithium-Ion Batteries, ACS Appl. Energy Mater., 2019, 2, 1308–1313 CrossRef CAS
- P. Karayaylali, R. Tatara, Y. Zhang, K.-L. Chan, Y. Yu, L. Giordano, F. Maglia, R. Jung, I. Lund and Y. Shao-Horn, Coating-Dependent Electrode-Electrolyte Interface for Ni-Rich Positive Electrodes in Li-Ion Batteries, J. Electrochem. Soc., 2019, 166, A1022–A1030 CrossRef CAS
- D. Mohanty, K. Dahlberg, D. M. King, L. A. David, A. S. Sefat, D. L. Wood, C. Daniel, S. Dhar, V. Mahajan, M. Lee and F. Albano, Modification of Ni-Rich FCG NMC and NCA Cathodes by Atomic Layer Deposition: Preventing Surface Phase Transitions for High-Voltage Lithium-Ion Batteries, Sci. Rep., 2016, 6, 26532, DOI:10.1038/srep26532
- R. S. Arumugam, L. Ma, J. Li, X. Xia, J. M. Paulsen and J. R. Dahn, Special Synergy between Electrolyte Additives and Positive Electrode Surface Coating to Enhance the Performance of Li[Ni0.6Mn0.2Co0.2]O2/Graphite Cells, J. Electrochem. Soc., 2016, 163, A2531–A2538 CrossRef
- R. S. Negi, E. Celik, R. Pan, R. Stäglich, J. Senker and M. T. Elm, Insights into the Positive Effect of Post-Annealing on the Electrochemical Performance of Al2O3-Coated Ni-Rich NCM Cathodes for Lithium-Ion Batteries, ACS Appl. Energy Mater., 2021, 4, 3369–3380 CrossRef
- M. J. Herzog, N. Gauquelin, D. Esken, J. Verbeeck and J. Janek, Facile Dry Coating Method of High-Nickel Cathode Material by Nanostructured Fumed Alumina (Al2O3) Improving the Performance of Lithium-Ion Batteries, Energy Technol., 2021, 9, 2100028, DOI:10.1002/ente.202100028
- Z. Chen and J. R. Dahn, Studies of LiCoO2 Coated with Metal Oxides, Electrochem. Solid-State Lett., 2003, 6, A221, DOI:10.1149/1.1611731
- Y. K. Sun, Y. S. Lee, M. Yoshio and K. Amine, Synthesis and electrochemical properties of ZnO-coated LiNi0.5Mn1.5O4 spinel as 5 V cathode material for lithium secondary batteries, Electrochem. Solid-State Lett., 2002, 5, A99, DOI:10.1149/1.1465375
- H.-J. Kweon and D. G. Park, Surface Modification of LiSr0.002Ni0.9Co0.1O2 by Overcoating with a Magnesium Oxide, Electrochem. Solid-State Lett., 2000, 3, 128–130 CrossRef
- J. Cho, Y. J. Kim and B. Park, Novel LiCoO2 cathode material with Al2O3 coating for a Li ion cell, Chem. Mater., 2000, 12, 3788–3791 CrossRef
- J. Cho, G. B. Kim, H. S. Lim, C.-S. Kim and S.-I. Yoo, Improvement of Structural Stability of LiMn2O4 Cathode Material on 55 °C Cycling by Sol–Gel Coating of LiCoO2, Electrochem. Solid-State Lett., 1999, 2, 607–609 CrossRef
- J. Cho, Y. J. Kim and B. Park, LiCoO2 Cathode Material That Does Not Show a Phase Transition from Hexagonal to Monoclinic Phase, J. Electrochem. Soc., 2001, 148, A1110 CrossRef
- Y. Cho and J. Cho, Significant Improvement of LiNi0.8Co0.15Al0.05O2 Cathodes at 60 °C by SiO2 Dry Coating for Li-Ion Batteries, J. Electrochem. Soc., 2010, 157, A625 CrossRef
- S. H. Lee, C. S. Yoon, K. Amine and Y. K. Sun, Improvement of long-term cycling performance of Li[Ni0.8Co0.15Al0.05]O2 by AlF3 coating, J. Power Sources, 2013, 234, 201–207 CrossRef
- M. J. Herzog, N. Gauquelin, D. Esken, J. Verbeeck and J. Janek, Increased Performance Improvement of Lithium-Ion Batteries by Dry Powder Coating of High-Nickel NMC with Nanostructured Fumed Ternary Lithium Metal Oxides, ACS Appl. Energy Mater., 2021, 4, 8832–8848 CrossRef CAS
- L. Zheng, T. D. Hatchard and M. N. Obrovac, A high-quality mechanofusion coating for enhancing lithium-ion battery cathode material performance, MRS Commun., 2019, 9, 245–250 CrossRef CAS
- Y. S. Jung, A. S. Cavanagh, L. A. Riley, S. H. Kang, A. C. Dillon, M. D. Groner, S. M. George and S. H. Lee, Ultrathin direct atomic layer deposition on composite electrodes for highly durable and safe Li-Ion batteries, Adv. Mater., 2010, 22, 2172–2176 CrossRef CAS
- Y. S. Jung, A. S. Cavanagh, A. C. Dillon, M. D. Groner, S. M. George and S.-H. Lee, Enhanced Stability of LiCoO2 Cathodes in Lithium-Ion Batteries Using Surface Modification by Atomic Layer Deposition, J. Electrochem. Soc., 2010, 157, A75 CrossRef CAS
- L. A. Riley, S. Van Atta, A. S. Cavanagh, Y. Yan, S. M. George, P. Liu, A. C. Dillon and S. H. Lee, Electrochemical effects of ALD surface modification on combustion synthesized LiNi1/3Mn1/3Co1/3O2 as a layered-cathode material, J. Power Sources, 2011, 196, 3317–3324 CrossRef
- V. Riesgo-González, D. S. Hall, K. Märker, J. Slaughter, D. S. Wright and C. P. Grey, Effect of Annealing on the Structure, Composition, and Electrochemistry of NMC811 Coated with Al2O3 Using an Alkoxide Precursor, Chem. Mater., 2022, 34(21), 9722, DOI:10.1021/acs.chemmater.2c02580
- G. L. Xu, Q. Liu, K. K. S. Lau, Y. Liu, X. Liu, H. Gao, X. Zhou, M. Zhuang, Y. Ren, J. Li, M. Shao, M. Ouyang, F. Pan, Z. Chen, K. Amine and G. Chen, Building ultraconformal protective layers on both secondary and primary particles of layered lithium transition metal oxide cathodes, Nat. Energy, 2019, 4, 484–494 CrossRef
- J. Ahn, E. K. Jang, S. Yoon, S. J. Lee, S. J. Sung, D. H. Kim and K. Y. Cho, Ultrathin ZrO2 on LiNi0.5Mn0.3Co0.2O2 electrode surface via atomic layer deposition for high-voltage operation in lithium-ion batteries, Appl. Surf. Sci., 2019, 484, 701–709 CrossRef
- J. Xie, A. D. Sendek, E. D. Cubuk, X. Zhang, Z. Lu, Y. Gong, T. Wu, F. Shi, W. Liu, E. J. Reed and Y. Cui, Atomic Layer Deposition of Stable LiAlF4 Lithium Ion Conductive Interfacial Layer for Stable Cathode Cycling, ACS Nano, 2017, 11, 7019–7027 CrossRef
- J. R. Croy, D. C. O’Hanlon, S. Sharifi-Asl, M. Murphy, A. Mane, C. W. Lee, S. E. Trask, R. Shahbazian-Yassar and M. Balasubramanian, Insights on the Stabilization of Nickel-Rich Cathode Surfaces: Evidence of Inherent Instabilities in the Presence of Conformal Coatings, Chem. Mater., 2019, 31, 3891–3899 CrossRef
- A. F. Harper, S. P. Emge, P. C. M. M. Magusin, C. P. Grey and A. J. Morris, Modelling amorphous materials via a joint solid-state NMR and X-ray absorption spectroscopy and DFT approach: application to alumina, Chem. Sci., 2023, 14, 1155–1167 RSC
- B. Han, B. Key, A. S. Lipton, J. T. Vaughey, B. Hughes, J. Trevey and F. Dogan, Influence of Coating Protocols on Alumina-Coated Cathode Material: Atomic Layer Deposition versus Wet-Chemical Coating, J. Electrochem. Soc., 2019, 166, A3679–A3684 CrossRef
- B. Han, A. R. Dunlop, S. E. Trask, B. Key, J. T. Vaughey and F. Dogan, Tailoring Alumina Based Interphases on Lithium Ion Cathodes, J. Electrochem. Soc., 2018, 165, A3275–A3283 CrossRef
- B. Han, B. Key, S. H. Lapidus, J. C. Garcia, H. Iddir, J. T. Vaughey and F. Dogan, From Coating to Dopant: How the Transition Metal Composition Affects Alumina Coatings on Ni-Rich Cathodes, ACS Appl. Mater. Interfaces, 2017, 9, 41291–41302 CrossRef PubMed
- S. Haber, N. Solomatin, A. Shapira, T. Bendikov, O. Brontvein, Y. Ein-Eli and M. Leskes, Lithium exchange across a lithium-less coating for high energy cathodes, J. Power Sources, 2023, 560, 232693, DOI:10.1016/j.jpowsour.2023.232693
- B. Han, T. Paulauskas, B. Key, C. Peebles, J. S. Park, R. F. Klie, J. T. Vaughey and F. Dogan, Understanding the Role of Temperature and Cathode Composition on Interface and Bulk: Optimizing Aluminum Oxide Coatings for Li-Ion Cathodes, ACS Appl. Mater. Interfaces, 2017, 9, 14769–14778 CrossRef
- S. T. Myung, K. Izumi, S. Komaba, Y. K. Sun, H. Yashiro and N. Kumagai, Role of alumina coating on Li–Ni–Co–Mn–O particles as positive electrode material for lithium-ion batteries, Chem. Mater., 2005, 17, 3695–3704 CrossRef
- Z. Chen, Y. Qin, K. Amine and Y. K. Sun, Role of surface coating on cathode materials for lithium-ion batteries, J. Mater. Chem., 2010, 20, 7606–7612 RSC
- M. Aykol, S. Kirklin and C. Wolverton, Thermodynamic aspects of cathode coatings for lithium-ion batteries, Adv. Energy Mater., 2014, 4(17), 1400690, DOI:10.1002/aenm.201400690
- J. L. Tebbe, A. M. Holder and C. B. Musgrave, Mechanisms of LiCoO2 Cathode Degradation by Reaction with HF and Protection by Thin Oxide Coatings, ACS Appl. Mater. Interfaces, 2015, 7, 24265–24278 CrossRef
- Y. Tesfamhret, R. Younesi and E. J. Berg, Influence of Al2O3 Coatings on HF Induced Transition Metal Dissolution from Lithium-Ion Cathodes, J. Electrochem. Soc., 2022, 169, 010530 CrossRef
-
R. E. Dinnebier, A. Leineweber and J. S. O. Evans, Practical Powder Diffraction Pattern Analysis using TOPAS, De Gruyter, 2019, pp. 16–88 Search PubMed
-
A. Coelho, Academic TOPAS (v.6.1) software, 2015 Search PubMed
- C. V. Chandran, C. E. A. Kirschhock, S. Radhakrishnan, F. Taulelle, J. A. Martens and E. Breynaert, Alumina: discriminative analysis using 3D correlation of solid-state NMR parameters, Chem. Soc. Rev., 2019, 48, 134–156 RSC
- C. P. Grey, W. S. Veeman and A. J. Vega, Rotational echo 14N/13C/1H triple resonance solid-state nuclear magnetic resonance: a probe of 13C–14N internuclear distances, J. Chem. Phys., 1993, 98, 7711–7724 CrossRef
- T. Gullion and A. J. Vega, Measuring heteronuclear dipolar couplings for I = 1/2, S > 1/2 spin pairs by REDOR and REAPDOR NMR, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 47, 123–136 CrossRef
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. Ferrero Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef
- Z. Lebens-Higgins, H. Chung, I. Temprano, M. Zuba, J. Wu, J. Rana, C. Mejia, M. A. Jones, L. Wang, C. P. Grey, Y. Du, W. Yang, Y. S. Meng and L. F. J. Piper, Electrochemical Utilization of Iron IV in the Li1.3Fe0.4Nb0.3O2 Disordered Rocksalt Cathode, Batteries Supercaps, 2021, 4, 771–777 CrossRef
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed
- A. R. Genreith-Schriever, H. Banerjee, A. S. Menon, E. N. Bassey, L. F. Piper, C. P. Grey and A. J. Morris, Oxygen hole formation controls stability in LiNiO2 cathodes, Joule, 2023, 7, 1623–1640 CrossRef CAS
- A. I. Liechtenstein, V. I. Anisimov and J. Zaanen, Density-functional theory and strong interactions: orbital ordering in Mott–Hubbard insulators, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, R5467–R5470 CrossRef CAS PubMed
- R. Dronskowski and P. E. Bloechl, Crystal orbital Hamilton populations (COHP): energy-resolved visualization of chemical bonding in solids based on density-functional calculations, J. Phys. Chem., 1993, 97, 8617–8624 CrossRef CAS
- R. Nelson, C. Ertural, J. George, V. L. Deringer, G. Hautier and R. Dronskowski, LOBSTER: local orbital projections, atomic charges, and chemical-bonding analysis from projector-augmented-wave-based density-functional theory, J. Comput. Chem., 2020, 41, 1931–1940 CrossRef CAS PubMed
- R. F. W. Bader, Atoms in molecules, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS
- R. Sim, S. Lee, W. Li and A. Manthiram, Influence of Calendering on the Electrochemical Performance of LiNi0.9Mn0.05Al0.05O2 Cathodes in Lithium-Ion Cells, ACS Appl. Mater. Interfaces, 2021, 13, 42898–42908 CrossRef CAS PubMed
- J. K. Morzy, W. M. Dose, P. E. Vullum, M. C. Lai, A. Mahadevegowda, M. F. L. De Volder and C. Ducati, Origins and Importance of Intragranular Cracking in Layered Lithium Transition Metal Oxide Cathodes, ACS Appl. Energy Mater., 2024, 7, 3945–3956 CrossRef CAS PubMed
- H. C. W. Parks, A. M. Boyce, A. Wade, T. M. M. Heenan, C. Tan, E. Martínez-Pañeda, P. R. Shearing, D. J. L. Brett and R. Jervis, Direct observations of electrochemically induced intergranular cracking in polycrystalline NMC811 particles, J. Mater. Chem. A, 2023, 11, 21322–21332 RSC
- S. K. Lee, S. Y. Park, Y. S. Yi and J. Moon, Structure and disorder in amorphous alumina thin films: insights from high-resolution solid-state NMR, J. Phys. Chem. C, 2010, 114, 13890–13894 CrossRef CAS
- D. R. Neuville, L. Cormier and D. Massiot, Al environment in tectosilicate and peraluminous glasses: a 27Al MQ-MAS NMR, Raman, and XANES investigation, Geochim. Cosmochim. Acta, 2004, 68, 5071–5079 CrossRef CAS
- J. B. d’Espinose de Lacaillerie, C. Fretigny and D. Massiot, MAS NMR spectra of quadrupolar nuclei in disordered solids: the Czjzek model, J. Magn. Reson., 2008, 192, 244–251 CrossRef PubMed
- N. M. Trease, I. D. Seymour, M. D. Radin, H. Liu, H. Liu, S. Hy, N. Chernova, P. Parikh, A. Devaraj, K. M. Wiaderek, P. J. Chupas, K. W. Chapman, M. S. Whittingham, Y. S. Meng, A. Van Der Van and C. P. Grey, Identifying the distribution of Al3+ in LiNi0.8Co0.15Al0.05O2, Chem. Mater., 2016, 28, 8170–8180 CrossRef CAS
- F. Dogan, J. T. Vaughey, H. Iddir and B. Key, Direct Observation of Lattice Aluminum Environments in Li Ion Cathodes LiNi1−y−zCoyAlzO2 and Al-Doped LiNiiMnyCozO2 via 27Al MAS NMR Spectroscopy, ACS Appl. Mater. Interfaces, 2016, 8, 16708–16717 CrossRef CAS PubMed
- J. Borau-Garcia, D. V. Gutsulyak, R. J. Burford and W. E. Piers, Selective hydration of nitriles to amides catalysed by PCP pincer supported nickel(II) complexes, Dalton Trans., 2015, 44, 12082–12085 RSC
- G. Yu, F. Hu, H. Huo, W. Ding and L. Peng, Probing local structure of paramagnetic Ni–Al layered double hydroxides with solid-state 2H NMR spectroscopy, Chem. Phys. Lett., 2018, 706, 47–52 CrossRef CAS
- Z. Zhang, H. Huo, Z. Yu, L. Xiang, B. Xie, C. Du, J. Wang and G. Yin, Unraveling the reaction mechanism of low dose Mn dopant in Ni(OH)2 supercapacitor electrode, J. Energy Chem., 2021, 61, 497–506 CrossRef CAS
- G. Piedra, J. J. Fitzgerald, N. Dando, S. F. Dec and G. E. Maciel, Solid-State 1H NMR Studies of Aluminum Oxide Hydroxides and Hydroxides, Inorg. Chem., 1996, 35, 3474–3478 CrossRef CAS
- J. J. Fitzgerald, G. Piedra, S. F. Dec, M. Seger and G. E. Maciel, Dehydration Studies of a High-Surface-Area Alumina (Pseudo-boehmite) Using Solid-State 1H and 27Al NMR, J. Am. Chem. Soc., 1997, 119, 7832–7842 CrossRef CAS
- T. Isobe, T. Watanabe, J. B. D’Espinose De La Caillerie, A. P. Legrand and D. Massiot, Solid-state 1H and 27Al NMR studies of amorphous aluminum hydroxides, J. Colloid Interface Sci., 2003, 261, 320–324 CrossRef CAS PubMed
- M. Forsyth and D. R. Macfarlane, A Study of Hydrogen Bonding in Concentrated Diol/Water Solutions by Proton NMR. Correlations with Glass Formation, J. Phys. Chem., 1990, 94(17), 6889 CrossRef CAS
- S. Tsushima, K. Teranishi and S. Hirai, Water diffusion measurement in fuel cell SPE membrane by NMR, Energy, 2005, 30(2), 235–245 CrossRef CAS
- S. Ishihara, K. Deguchi, H. Sato, M. Takegawa, E. Nii, S. Ohki, K. Hashi, M. Tansho, T. Shimizu, K. Ariga, J. Labuta, P. Sahoo, Y. Yamauchi, J. P. Hill, N. Iyi and R. Sasai, Multinuclear solid-state NMR spectroscopy of a paramagnetic layered double hydroxide, RSC Adv., 2013, 3, 19857–19860 RSC
- J. B. Peri, A Model for the Surface of γ-Alumina, J. Phys. Chem., 1965, 69, 220–230 CrossRef CAS
- A. V. Plakhotnyk, L. Ernst and R. Schmutzler, Hydrolysis in the system LiPF6 – Propylene carbonate – Dimethyl carbonate – H2O, J. Fluorine Chem., 2005, 126, 27–31 CrossRef CAS
- C. L. Campion, W. Li and B. L. Lucht, Thermal Decomposition of LiPF6-Based Electrolytes for Lithium-Ion Batteries, J. Electrochem. Soc., 2005, 152, A2327 CrossRef CAS
- Z. W. Lebens-Higgins, D. M. Halat, N. V. Faenza, M. J. Wahila, M. Mascheck, T. Wiell, S. K. Eriksson, P. Palmgren, J. Rodriguez, F. Badway, N. Pereira, G. G. Amatucci, T. L. Lee, C. P. Grey and L. F. J. Piper, Surface Chemistry Dependence on Aluminum Doping in Ni-rich LiNi0.8Co0.2−yAlyO2 Cathodes, Sci. Rep., 2019, 9(1), 17720, DOI:10.1038/s41598-019-53932-6
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, Chemical versus electrochemical electrolyte oxidation on NMC111, NMC622, NMC811, LNMO, and conductive carbon, J. Phys. Chem. Lett., 2017, 8, 4820–4825 CrossRef CAS PubMed
- H. Huang, Y. C. Chang, Y. C. Huang, L. Li, A. C. Komarek, L. H. Tjeng, Y. Orikasa, C. W. Pao, T. S. Chan, J. M. Chen, S. C. Haw, J. Zhou, Y. Wang, H. J. Lin, C. Te Chen, C. L. Dong, C. Y. Kuo, J. Q. Wang, Z. Hu and L. Zhang, Unusual double ligand holes as catalytic active sites in LiNiO2, Nat. Commun., 2023, 14, 2112, DOI:10.1038/s41467-023-37775-4
- H. Banerjee, M. Aichhorn, C. P. Grey and A. J. Morris, Insulating behaviour in room temperature rhombohedral LiNiO2 cathodes is driven by dynamic correlation, J. Phys. Energy, 2024, 6, 045003 CrossRef CAS
- H. Banerjee, C. P. Grey and A. J. Morris, Stability and Redox Mechanisms of Ni-Rich NMC Cathodes: Insights from First-Principles Many-Body Calculations, Chem. Mater., 2024, 36(13), 6575 CrossRef CAS
- C. Tian, D. Nordlund, H. L. Xin, Y. Xu, Y. Liu, D. Sokaras, F. Lin and M. M. Doeff, Depth-Dependent Redox Behavior of LiNi0.6Mn0.2Co0.2O2, J. Electrochem. Soc., 2018, 165, A696–A704 CrossRef CAS
- M. Mirolo, C. A. F. Vaz, P. Novák and M. El Kazzi, Multi-length-scale X-ray spectroscopies for determination of surface reactivity at high voltages of LiNi0.8Co0.15Al0.05O2vs. Li4Ti5O12, J. Chem. Phys., 2020, 152(18), 184705, DOI:10.1063/5.0006269
- E. Björklund, C. Xu, W. M. Dose, C. G. Sole, P. K. Thakur, T.-L. Lee, M. F. L. De Volder, C. P. Grey and R. S. Weatherup, Cycle-Induced Interfacial Degradation and Transition-Metal Cross-Over in LiNi0.8Mn0.1Co0.1O2 – Graphite Cells, Chem. Mater., 2022, 18, 10 Search PubMed
- I. M. DiMucci, C. J. Titus, D. Nordlund, J. R. Bour, E. Chong, D. P. Grigas, C.-H. Hu, M. D. Kosobokov, C. D. Martin, L. M. Mirica, N. Nebra, D. A. Vicic, L. L. Yorks, S. Yruegas, S. N. MacMillan, J. Shearer and K. M. Lancaster, Scrutinizing formally Ni(IV) centers through the lenses of core spectroscopy, molecular orbital theory, and valence bond theory, Chem. Sci., 2023, 14, 6915, 10.1039/D3SC02001K
- H. Wang, C. Y. Ralston, D. S. Patil, R. M. Jones, W. Gu, M. Verhagen, M. Adams, P. Ge, C. Riordan, C. A. Marganian, P. Mascharak, J. Kovacs, C. G. Miller, T. J. Collins, S. Brooker, P. D. Croucher, K. Wang, E. I. Stiefel and S. P. Cramer, Nickel L-edge soft X-ray spectroscopy of nickel-iron hydrogenases and model compounds – Evidence for high-spin nickel(II) in the active enzyme, J. Am. Chem. Soc., 2000, 122, 10544–10552 CrossRef CAS
- H. Wang, S. Friedrich, L. Li, Z. Mao, P. Ge, M. Balasubramanian and D. S. Patil, L-edge sum rule analysis on 3d transition metal sites: from d10 to d0 and towards application to extremely dilute metallo-enzymes, Phys. Chem. Chem. Phys., 2018, 20, 8166–8176 RSC
- H. Wang, C. Y. Ralston, D. S. Patil, R. M. Jones, W. Gu, M. Verhagen, M. Adams, P. Ge, C. Riordan, C. A. Marganian, P. Mascharak, J. Kovacs, C. G. Miller, T. J. Collins, S. Brooker, P. D. Croucher, K. Wang, E. I. Stiefel and S. P. Cramer, Nickel L-edge soft X-ray spectroscopy of nickel–iron hydrogenases and model compounds – Evidence for high-spin nickel(II) in the active enzyme, J. Am. Chem. Soc., 2000, 122, 10544–10552 CrossRef CAS
- W. S. Yoon, O. Haas, S. Muhammad, H. Kim, W. Lee, D. Kim, D. A. Fischer, C. Jaye, X. Q. Yang, M. Balasubramanian and K. W. Nam, In situ soft XAS study on nickel-based layered cathode material at elevated temperatures: a novel approach to study thermal stability, Sci. Rep., 2014, 4, 6827, DOI:10.1038/srep06827
- A. Ionescu, A. Allouche, J. P. Aycard, M. Rajzmann and F. Hutschka, Study of γ-alumina surface reactivity: adsorption of water and hydrogen sulfide on octahedral aluminum sites, J. Phys. Chem. B, 2002, 106, 9359–9366 CrossRef CAS
- D. J. Coster, J. J. Fripiat, M. Muscas and A. Auroux, Effect of Bulk Properties on the Rehydration Behavior of Aluminas, Langmuir, 1995, 11, 2615–2620 CrossRef CAS
- C. S. John, N. C. M. Alma and G. R. Hays, Characterization of transitional alumina by solid-state magic angle spinning aluminium NMR, Appl. Catal., 1983, 6, 341–346 CrossRef CAS
- H. Knozinger and P. Ratnasamy, Catalytic Aluminas: Surface Models and Characterization of Surface Sites, Catal. Rev., 1978, 17, 31–70 CrossRef
- A. Ionescu, A. Allouche, J.-P. Aycard, M. Rajzmann and F. Hutschka, Study of γ-Alumina Surface Reactivity: Adsorption of Water and Hydrogen Sulfide on Octahedral Aluminum Sites, J. Phys. Chem. B, 2002, 106, 9359–9366 CrossRef CAS
- A. A. Tsyganenko and P. P. Mardilovichb, Structure of alumina surfaces, J. Chem. Soc., Faraday Trans., 1996, 4843–4852 RSC
- D. Coster, A. L. Blumenfeld and J. J. Fripiat, Lewis Acid Sites and Surface Aluminum in Aluminas and Zeolites: A High-Resolution NMR Study, J. Phys. Chem., 1994, 98, 6201–6211 CrossRef CAS
- T. Hiemstra, P. Venema and W. H. Van Riemsdijk, Intrinsic Proton Affinity of Reactive Surface Groups of Metal (Hydr)oxides: The Bond Valence Principle, J. Colloid Interface Sci., 1996, 184, 680–692 CrossRef CAS PubMed
- P. Venema, T. Hiemstra, P. G. Weidler and W. H. Van Riemsdijk, Intrinsic Proton Affinity of Reactive Surface Groups of Metal (Hydr)oxides: Application to Iron (Hydr)oxides, J. Colloid Interface Sci., 1998, 198, 282–295 CrossRef CAS
-
Y.-R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRC Press, 1st edn, 2002 Search PubMed
- S. C. Jung and Y. K. Han, How do Li atoms pass through the Al2O3 coating layer during lithiation in Li-ion batteries?, J. Phys. Chem. Lett., 2013, 4, 2681–2685 CrossRef CAS
- Y. Liu, N. S. Hudak, D. L. Huber, S. J. Limmer, J. P. Sullivan and J. Y. Huang, In situ transmission electron microscopy observation of pulverization of aluminum nanowires and evolution of the thin surface Al2O3 layers during lithiation-delithiation cycles, Nano Lett., 2011, 11, 4188–4194 CrossRef CAS PubMed
-
D. Müller, W. Gessner and G. Scheler, Chemical shift and quadrupole coupling of the 27Al NMR spectra of LiAlO2 polymorphs, 1983, vol. 2 Search PubMed
- V. P. Isupov, O. A. Kharlamova, L. E. Chupakhina, M. R. Sharafutdinov, D. F. Khabibulin and O. B. Lapina, Mechanochemical synthesis of γ-LiAlO2 studied by 6Li and 27Al NMR and synchrotron X-ray diffraction, Inorg. Mater., 2011, 47, 763–767 CrossRef CAS
- D. Wiedemann, S. Indris, M. Meven, B. Pedersen, H. Boysen, R. Uecker, P. Heitjans and M. Lerch, Single-crystal neutron diffraction on γ-LiAlO2: structure determination and estimation of lithium diffusion pathway, Z. Kristallogr. - Cryst. Mater., 2016, 231, 189–193 CrossRef CAS
- D. Müller, W. Gessner and G. Scheler, Chemical shift and quadrupole coupling of the 27Al NMR spectra of LiAIO2 polymorphs, Polyhedron, 1983, 2, 1195–1198 CrossRef
- M. Marezio and J. P. Remeika, J. Chem. Phys., 1966, 44, 3143–3144 CrossRef CAS
- R. Famery and F. Queyroux, Etude structurale de la forme ordonnbe de LiAlsOs, J. Solid State Chem., 1979, 30, 257–263 CrossRef CAS
- G. J. Páez Fajardo, E. Fiamegkou, J. A. Gott, H. Wang, I. Temprano, I. D. Seymour, M. J. W. Ogley, A. S. Menon, I. E. L. Stephens, M. Ans, T. L. Lee, P. K. Thakur, W. M. Dose, M. F. L. De Volder, C. P. Grey and L. F. J. Piper, Synergistic Degradation Mechanism in Single Crystal Ni-Rich NMC//Graphite Cells, ACS Energy Lett., 2023, 8, 5025–5031 CrossRef
- Y. Zhang, Y. Katayama, R. Tatara, L. Giordano, Y. Yu, D. Fraggedakis, J. G. Sun, F. Maglia, R. Jung, M. Z. Bazant and Y. Shao-Horn, Revealing electrolyte oxidation: via carbonate dehydrogenation on Ni-based oxides in Li-ion batteries by in situ Fourier transform infrared spectroscopy, Energy Environ. Sci., 2020, 13, 183–199 RSC
- B. Jørgensen, S. Egholm Christiansen, M. L. Dahl Thomsen and C. H. Christensen, Aerobic oxidation of aqueous ethanol using heterogeneous gold catalysts: efficient routes to acetic acid and ethyl acetate, J. Catal., 2007, 251, 332–337 CrossRef
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Reactivity of the Gold/Water Interface During Selective Oxidation Catalysis, Science, 2010, 330, 74–78 CrossRef CAS PubMed
- G. Y. Popova, T. V. Andrushkevich, Y. A. Chesalov and E. S. Stoyanov, In situ FTIR Study of the Adsorption of Formaldehyde, Formic Acid, and Methyl Formiate at the Surface of TiO2 (Anatase), Kinet. Catal., 2000, 41, 805–811 CrossRef CAS
- K. Märker, P. J. Reeves, C. Xu, K. J. Griffith and C. P. Grey, Evolution of Structure and Lithium Dynamics in LiNi0.8Mn0.1Co0.1O2 (NMC811) Cathodes during Electrochemical Cycling, Chem. Mater., 2019, 31, 2545–2554 CrossRef
- C. Xu, K. Märker, J. Lee, A. Mahadevegowda, P. J. Reeves, S. J. Day, M. F. Groh, S. P. Emge, C. Ducati, B. Layla Mehdi, C. C. Tang and C. P. Grey, Bulk fatigue induced by surface reconstruction in layered Ni-rich cathodes for Li-ion batteries, Nat. Mater., 2021, 20, 84–92 CrossRef CAS
- H. Huang, L. Qiao, H. Zhou, Y. Tang, M. J. Wahila, H. Liu, P. Liu, G. Zhou, M. Smeu and H. Liu, Efficacy of atomic layer deposition of Al2O3 on composite LiNi0.8Mn0.1Co0.1O2 electrode for Li-ion batteries, Sci. Rep., 2024, 14, 18180 CrossRef CAS PubMed
- S. Lee, L. Su, A. Mesnier, Z. Cui and A. Manthiram, Cracking vs. surface reactivity in high-nickel cathodes for lithium-ion batteries, Joule, 2023, 7, 2430–2444 CrossRef CAS
- B. L. D. Rinkel, D. S. Hall, I. Temprano and C. P. Grey, Electrolyte oxidation pathways in lithium-ion batteries, J. Am. Chem. Soc., 2020, 142, 15058–15074 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee03444a |
| This journal is © The Royal Society of Chemistry 2025 |