
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEthyl cellulose-block-poly(benzyl glutamate) block copolymer compatibilizers for ethyl cellulose/poly(ethylene terephthalate) blends†
Abigail F.
Chinn‡
 ab,
Isabela
Trindade Coutinho‡
b,
Saipranavi Reddy
Kethireddy
a,
Noah R.
Williams
ab,
Kenneth M.
Knott
a,
Robert B.
Moore
*ab and
John B.
Matson
*ab
ab,
Isabela
Trindade Coutinho‡
b,
Saipranavi Reddy
Kethireddy
a,
Noah R.
Williams
ab,
Kenneth M.
Knott
a,
Robert B.
Moore
*ab and
John B.
Matson
*ab
aDepartment of Chemistry, Virginia Tech Center for Drug Discovery, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: jbmatson@vt.edu; rbmoore3@vt.edu
bMacromolecules Innovation Institute, Virginia Tech, Blacksburg, VA 24061, USA
First published on 8th August 2024
Abstract
Blends of petroleum-based polymers with bio-sourced polymers are an alternative to polymers derived from non-renewable resources. However, polymer blends are usually immiscible, and a compatibilizer, often a block copolymer, is required to improve mixing. In this work, we synthesized a block copolymer of ethyl cellulose (ECel) and poly(benzyl glutamate), termed ECel-block-poly(BG), and we applied it as a compatibilizer for ECel/poly(ethylene terephthalate) (ECel/PET) blends. To synthesize this block copolymer, two ECel-NH2 macroinitiators were evaluated for ring-opening polymerization of benzyl glutamate-N-thiocarboxyanhydride (BG-NTA), one with the amine directly attached to the ECel reducing chain end, and the other with a short PEG linker between ECel and the amine initiator. The PEG-containing macroinitiator led to the synthesis of a block copolymer that was unimodal by size-exclusion chromatography (SEC) while the other initiator led to uncontrolled homopolymerization of BG-NTA, presumably due to steric hindrance near the primary amine. A series of solvent studies revealed that polymerization of BG-NTA in CH2Cl2 was the best system for obtaining the ECel-block-poly(BG) block copolymer, achieving 95% conversion based on 1H NMR spectroscopy. The success of chain extension and molecular weight analysis were evaluated using SEC with multi-angle light scattering (SEC-MALS). Blends composed of 70% ECel and 30% PET with different weight percentages (wt%) of block copolymer compatibilizer were made via solvent casting from hexafluoroisopropanol. Phase contrast optical microscopy and small-angle laser light scattering were used to probe the effectiveness of the ECel-block-poly(BG) block copolymer as a compatibilizer (5–30 wt%) for the 70/30 ECel/PET blends. A decrease in average domain size from 15 ± 4 μm in the base blend (without compatibilizer) to 2 ± 1 μm in the blend containing 30 wt% ECel-block-poly(BG) indicated successful compatibilization of the blend.
1. Introduction
Greener alternatives to commonly used petroleum-based polymers, such as poly(ethylene terephthalate) (PET), are needed to increase biodegradability, renewability, and sustainability of commercial plastics.1,2 Among the greener alternatives are polysaccharides, including primarily cellulose-based materials.3–9 Many polysaccharide derivatives also hold potential property advantages over petroleum-based polymers such as transparency, high UV stability, and sustainability.10 While cellulose-based materials are more sustainable than those made from petroleum, they have limited applications due to poor solubility, processing difficulties, and moderate thermal stability, among other property deficits.11 Despite this, there exist several commercial polymers based on cellulose, and utilizing these cellulose derivatives in blends could reduce the amount of feedstock that comes from fossil fuels and lead to a more sustainable and renewable source of plastics. The need for new bio-derived polymers and new approaches to polymer sustainability is urgent—biopolymers made up around only 0.5% of all plastics produced in 2023,12 and growing this number would increase the sustainability of the plastic industry.Polysaccharides offer biodegradable and sustainable solutions for various applications, and blends of polysaccharides with existing commercial polymers may provide a route toward greater sustainability.7,13–15 While polysaccharides, including cellulose, are utilized in commercial applications, poor solubility and difficult purification and characterization have limited the expanded use of polysaccharides in blends. However, some recent reports highlight the benefits of polysaccharide-based polymer blends. For example, polysaccharide-containing polymer blends hold potential in food packaging,16 biomedical applications,17 and drug delivery.18 In particular, we imagined that ethyl cellulose (ECel) could be an ideal polymer for blends because it is a commercial polymer with several useful characteristics such as excellent film-forming properties and good UV stability. While new materials based on ECel have increased lately,19,20 it has not been heavily investigated in blends. Recent examples include the combination of ECel with hydroxyl-terminated polybutadiene to form partially miscible blends as well as its blending with butadiene-bridged polydimethylsiloxane for gas separation membranes.21,22
PET is a semicrystalline thermoplastic with thermal and mechanical properties suitable for food and beverage packaging.23 While the recycling of PET and biopolymer mixing with PET has improved in recent years,24 PET is still largely produced from petroleum, and mechanical recycling efforts have only a moderate impact.25 To enhance sustainability, polyester blends, including blends containing PET, have been studied in a variety of potential applications due to their transparency, toughness, and improved processability compared to PET alone.26–28 Creating blends of PET with biopolymers such as polysaccharides, including ECel, could further reduce reliance on petroleum for polyester-based plastics.
In this work, we sought to create a blend of ECel and PET. ECel, while slow to biodegrade compared with other biopolymers,29 is a cellulose-based polymer coming from a renewable and sustainable source. ECel has organic solubility, which increases the ease of synthetic modifications and blend preparation compared to native cellulose.3,4 Blending the two polymers, ECel and PET, would allow us to combine the desirable properties of both, increase the bio-derived content, and decrease the petroleum-based PET content in potential applications such as food packaging. However, because these two polymers are immiscible, a compatibilizer is needed to decrease the interfacial tension between the PET and ECel domains.
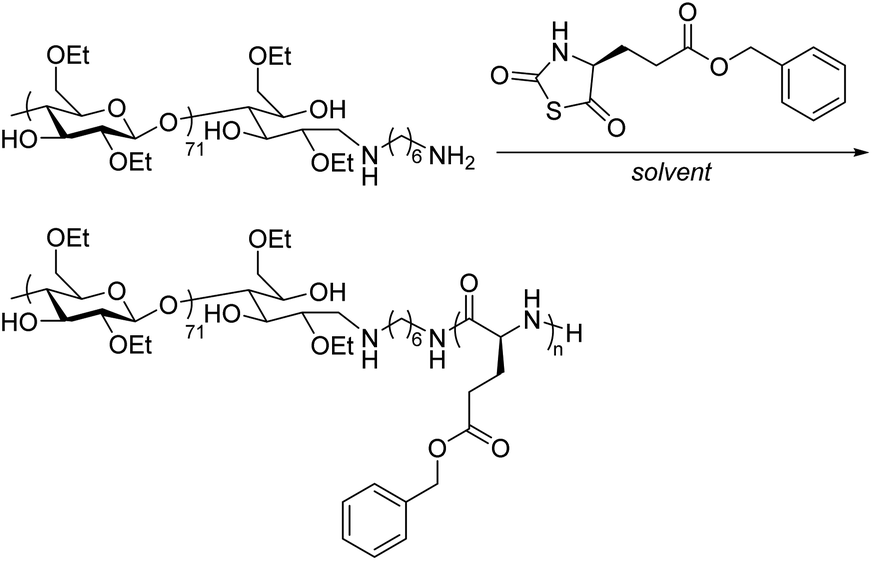
We envisioned that a block copolymer (BCP) compatibilizer, synthesized via a living chain polymerization such as ring-opening polymerization (ROP), could allow us to compatibilize ECel/PET blends while avoiding the poor solubility and difficult functionalization of PET itself. Poly(benzyl glutamate) (poly(BG)), derived from glutamic acid, can be made by ROP of benzyl glutamate-N-thiocarboxyanhydride (BG-NTA); poly(BG) possesses an aromatic group and an ester in the repeating unit, which is similar to PET (Fig. 1). Thus, we proposed that ECel could be functionalized with an amine on the reducing end through reductive amination and used to initiate polymerization of BG-NTA to synthesize an ECel-block-poly(BG) BCP. This BCP would then be used as a compatibilizer for ECel/PET blends. We hypothesized that this BCP compatibilizer would improve mixing in ECel/PET blends by decreasing the interfacial tension and average domain size for more a bio-derived, sustainable approach to traditional petroleum-based PET materials.
 | ||
| Fig. 1 Chemical structures of poly(BG) and PET. Both polymers possess an aromatic group and an ester in the repeating unit. | ||
2. Results and discussion
2.1 Block copolymer synthesis
We set out to synthesize ECel-block-poly(BG) BCP by first synthesizing an ECel-NH2 macroinitiator to initiate polymerization of BG-NTA monomer. Here, we chose ECel as the polysaccharide for polysaccharide/PET blends due to its thermal and mechanical stability as well as its solubility in organic solvents for ease of synthesis and blend preparation. We selected commercial ECel with a viscosity of 10 cps and ethoxyl content of 48% (degree of substitution, DS = 2.6). Size-exclusion chromatography with multi-angle light scattering (SEC-MALS) gave an Mn of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 g mol−1 in tetrahydrofuran (THF) and a Đ value of 1.9 (Fig. S1†) based on a measured dn/dc value of 0.060 mL g−1 (Fig. S2†).
200 g mol−1 in tetrahydrofuran (THF) and a Đ value of 1.9 (Fig. S1†) based on a measured dn/dc value of 0.060 mL g−1 (Fig. S2†).
Synthesis of polysaccharides with specific functional groups on one end can be accomplished by reductive amination, a slow but reliable method for functionalizing polysaccharides by utilizing the ring-chain equilibrium between a hemiacetal and an aldehyde on the reducing-end.30 In brief, we synthesized ECel with a single amine on the reducing end, termed ECel-NH2, in a reaction of ECel with a large excess of hexamethylene diamine in the presence of sodium cyanoborohydride. Successful reductive amination after 7 d was confirmed by 1H NMR spectroscopy in CDCl3 with the proton alpha to the coupled amine shifting up-field (Fig. S5†). The macroinitiator was purified via dialysis against acetone to remove unreacted diamine and residual salts from the reducing agent.
After successfully synthesizing the ECel macroinitiator, we aimed to identify polymerization conditions for the synthesis of an ECel-block-poly(BG) BCP (Scheme 1). NTAs can undergo polymerization to form polypeptides in a variety of solvents, including CH2Cl2, hexanes, and even water.31,32 NTAs are also more stable than N-carboxyanhydrides and less sensitive to moisture and heat,33,34 so we chose the make poly(BG) from BG-NTA rather than the N-carboxyanhydride derivative. However, initial attempts in CH2Cl2 using the ECel-NH2 macroinitiator yielded little to no conversion (Table 1, entry 1, Fig. S15†). These results led us to try a water suspension method. In this case, due to the insolubility of ECel-NH2, aqueous conditions also did not result in any conversion of the NTA monomer to polypeptide (Table 1, entry 2). To address this issue, we tried a biphasic water/CH2Cl2 mixture, which led to poor conversion (Table 1, entry 3). We attribute this result to the immiscibility between water and CH2Cl2. In response to this low conversion seen with an aprotic organic solvent with and without the addition of water, we moved to testing methanol (CH3OH) as an example of an organic protic solvent. Polymerization in CH3OH led to 100% hydrolysis/alcoholysis of the BG-NTA monomer after 24 h as indicated by 1H NMR spectroscopy but no measurable conversion to the target BCP (Table 1, entry 4). Similarly, trifluoroethanol (TFE), a fluorinated polar protic solvent, resulted in extensive hydrolysis/alcoholysis (90%) and very little polymerization (Table 1, entry 5). Mixtures of TFE with CH2Cl2 (50:50 and 75:25) led to minimal hydrolysis (1% and 0%, respectively) but also afforded minimal conversion (Table 1, entries 6 and 7, 6% and 7%, respectively).
 | ||
| Scheme 1 Polymerization of BG-NTA to synthesize ECel-block-poly(BG). | ||
| Entry | Solvent(s) | Concentration (mg mL−1) | % NTA remainingb | % NTA hydrolyzedb | Conversion to poly(BG)b (%) |
|---|---|---|---|---|---|
| a All polymerizations were conducted at rt for 24 h. b 1H NMR spectra of each polymerization reaction mixture appear in Fig. S15.† Intact NTA, hydrolyzed/alcoholized NTA, and conversion to poly(BG) were estimated using the method described in Fig. S16.† | |||||
| 1 | CH2Cl2 | 12 | 97 | 0 | 3 |
| 2 | H2O | 12 | 98 | 2 | 0 |
| 3 | 50:50 CH2Cl2:H2O |
12 | 96 | 1 | 3 |
| 4 | CH3OH | 12 | 0 | 100 | 0 |
| 5 | TFE | 12 | 11 | 89 | 0 |
| 6 | 50:50 TFE:CH2Cl2 |
12 | 93 | 1 | 6 |
| 7 | 75:25 TFE:CH2Cl2 |
12 | 93 | 0 | 7 |
| 8 | 60:40 acetone:H2O |
6 | 71 | 2 | 27 |
| 9 | 60:40 CH3CN:H2O |
6 | 70 | 2 | 28 |
| 10 | 50:50 CH3CN:H2O |
6 | 18 | 1 | 81 |
| 11 | 40:60 CH3CN:H2O |
6 | 29 | 2 | 69 |
Returning to our idea of utilizing a water/organic mixture to improve solubility, we evaluated two different water-miscible organic solvents, acetonitrile (CH3CN) and acetone. Initial trials included 60:40 mixtures of water with organic solvent, which afforded higher conversion than previously seen with organic solvents alone (Table 1, entries 8 and 9). The concentration for the polymerization was lowered to 6 mg mL−1 instead of the previous 12 mg mL−1 to minimize the potential aggregation of the ECel macroinitiator, which would hinder initiation. This higher conversion was promising but was still lower than desired. Because CH3CN led to slightly higher conversion compared to acetone (28% and 27%, respectively), we moved forward with CH3CN. Increasing the water content (50:50 and 40:60 CH3CN:H2O) resulted in even higher conversions (Table 1, entry 10–11, 81% and 69%, respectively), with the 50:50 mixture leading to the highest conversion of monomer to polymer in the set, as indicated by 1H NMR spectroscopy.
With a set of conditions leading to good monomer conversion to poly(BG), we set out to characterize the resulting polymer using SEC-MALS in THF. First, we characterized ECel-NH2, which showed a broad peak centered around 15 min elution time with Mn = 20.3 kg mol−1 and Đ = 1.7 (Fig. 2, grey trace), within a reasonable deviation from the values for as-received ECel before the addition of the chain end amine. For the polymerization of BG-NTA initiated by ECel-NH2 in CH3CN/H2O (50:50), we saw a broad, low intensity peak centered around 16.5 min elution time as well as sharper peaks centered around 18 and 19 min elution times (Fig. 2, green trace). The high elution time populations suggested homopolymerization of BG-NTA, likely due to hydrolysis of a small amount of BG-NTA in this solvent system, exposing a primary amine capable of initiating ROP of the remaining monomer. MALS analysis using a reported poly(BG) dn/dc value of 0.150 in THF35 showed that the molecular weights of the two sharp peaks were in the range of 1–3 kg mol−1. The peak near 16.5 min showed a higher elution time than the ECel-NH2, which is typically associated with a decrease rather than an increase in MW, but in this case, may be a result of a small amount of successful chain extension but with a higher elution time than ECel-NH2 due to interactions between poly(BG) and the column stationary phase. However, we were unable to accurately assess the molecular weight of this population because the ECel/poly(BG) molar ratio, and therefore the dn/dc value, was unknown.
 | ||
| Fig. 2 SEC traces of ECel-NH2 macroinitiator (grey) and polymerization of BG-NTA in 50:50 CH3CN:H2O initiated by either ECel-NH2 (green, Table 1 entry 10) or hexylamine (blue). 1H NMR spectra of macroinitiator ECel-NH2 and polymerizations can be found in Fig. S5, S11, and S15.† SEC eluent is THF. | ||
To confirm the suspected homopolymerization we observed in the SEC trace, we polymerized BG-NTA in a 50:50 CH3CN:H2O mixture, initiated by hexylamine (0.01 equiv.), to synthesize poly(BG) homopolymer (blue trace in Fig. 2). The 1H NMR spectrum of poly(BG) indicated high conversion (Fig. S11,† 93%), and we expected a peak with an Mn near 20 kg mol−1 based on this conversion. However, SEC-MALS revealed multimodal low molecular weight peaks again in the range of 1–3 kg mol−1, similar to the high elution time peaks observed in the trace for the attempted BCP synthesis (Fig. 2, green trace). This result again suggested hydrolysis of a small amount of BG-NTA leading to homopolymerization of the remaining BG-NTA rather than forming the desired BCP.
Most of the polymerization conditions tested using the ECel-NH2 macroinitiator indicated little to no conversion to polymer based on NMR studies (Table 1). The one set of polymerization conditions that led to significant conversion to polymer showed little to no BCP formation in the SEC trace. Instead, we observed homopolymerization of BG-NTA as indicated by SEC-MALS. Based on these disappointing results, we then theorized that initiation of NTA polymerization using ECel-NH2 was slow due to steric hindrances. Therefore, conversion of BG-NTA to polymer was minimal in organic solvents, but with the monomer remaining intact. However, in conditions that included water and a water-miscible organic solvent, the NTA monomer slowly hydrolyzed, revealing a free primary amine that was not sterically hindered, initiating homopolymerization of the remaining monomer. We hypothesized that by limiting the steric hindrance of the ECel macroinitiator and polymerizing BG-NTA in an organic solvent in which hydrolysis would be minimal, we would be able to produce the desired BCP. To test this hypothesis, we aimed to synthesize an ECel macroinitiator with a linker between the ECel backbone and the primary amine used for initiation. Ideally, this linker would then allow the primary amine to be more freely available for initiation.
Poly(ethylene glycol) (PEG) (Mn = 1 kg mol−1) was chosen as the linker due to its widescale availability, tunability, and organic solubility. A molecular weight of 1 kg mol−1 was chosen for the PEG linker to allow for the availability and flexibility of the primary amine on the macroinitiator. We envisioned that a larger PEG linker would potentially separate the ECel and poly(BG) blocks excessively, limiting the effectiveness of the BCP compatibilization in the eventual blend studies. To synthesize the ECel-PEG-NH2 macroinitiator, we first synthesized PEG diamine using previously published methods.36 In brief, PEG diol was mesylated, and then the chain-end mesylates were substituted in aqueous ammonium solution to generate chain-end amines (Fig. S6–S9†). The PEG diamine product was subsequently coupled to ECel using reductive amination to synthesize macroinitiator ECel-PEG-NH2 (Fig. S10†). The macroinitiator was purified using dialysis against acetone, allowing unreacted PEG diamine to escape. Synthesis of ECel-PEG-NH2 macroinitiator was confirmed using 1H NMR spectroscopy and SEC-MALS (Fig. S10 and S1†). The SEC-MALS trace in THF showed a small amount of unreacted PEG diamine (∼5%) that could not be removed by dialysis.
We then set out to use ECel-PEG-NH2 to initiate polymerization of BG-NTA to synthesize the target ECel-PEG-block-poly(BG) BCP compatibilizer (Scheme 2). To minimize hydrolysis, we aimed to choose an organic solvent in which BG-NTA monomer would be stable. Furthermore, to increase the rate of polymerization, we envisioned that acetic acid (AcOH) would help drive off COS gas. We chose CH2Cl2 as the solvent with the addition of AcOH due to previous success by Zhang with this approach in homopolymerization of NTA monomers initiated by hexylamine.31 While polymerization conditions in solely organic solvents led to limited conversion when using the ECel-NH2 macroinitiator, the polymerization reached 95% BG-NTA conversion to polymer when using the ECel-PEG-NH2 macroinitiator as indicated by 1H NMR spectroscopy (Fig. S13†). 1H NMR spectroscopy also did not suggest any hydrolysis as the previous polymerizations with ECel-NH2 exhibited. The BCP was then purified using dialysis against acetone (MWCO 6–8 kg mol−1) to remove any unreacted monomer (Fig. S14†).
 | ||
| Scheme 2 Polymerization of BG-NTA to synthesize ECel-PEG-block-poly(BG) using macroinitiator ECel-PEG-NH2. | ||
We first attempted to characterize the product using SEC-MALS in THF, but column interactions prevented accurate molecular weight analysis. Instead, SEC-MALS for this BCP was performed in DMAc/LiCl, which is a better solvent system for poly(BG) than THF. For ECel-PEG-block-poly(BG), SEC-MALS revealed a broad, unimodal peak centered around 15.5 min (Fig. 3, green trace). The SEC trace showed a shift to slightly higher retention time from the original ECel-PEG-NH2 peak (Fig. 3, grey trace), likely due to interactions between poly(BG) and the columns. The use of SEC-MALS was critical here to evaluate whether chain extension was successful despite the increase in retention time. By using 1H NMR spectroscopy to determine the polypeptide/polysaccharide ratio by moles and mass, we estimated the dn/dc for BCP ECel-PEG-block-poly(BG), to be 0.078 mL g−1. Applying this dn/dc value to the peak afforded an Mn value of 36.7 kg mol−1 and Đ value of 1.4. Therefore, despite the increase in retention time, which initially suggested a drop in molecular weight, SEC-MALS revealed the expected increase in molar mass and suggested successful chain extension. Furthermore, no homopolymerization of the BG-NTA monomer was detected in these traces, unlike what was observed when using the original ECel-NH2 macroinitiator that lacked the PEG linker.
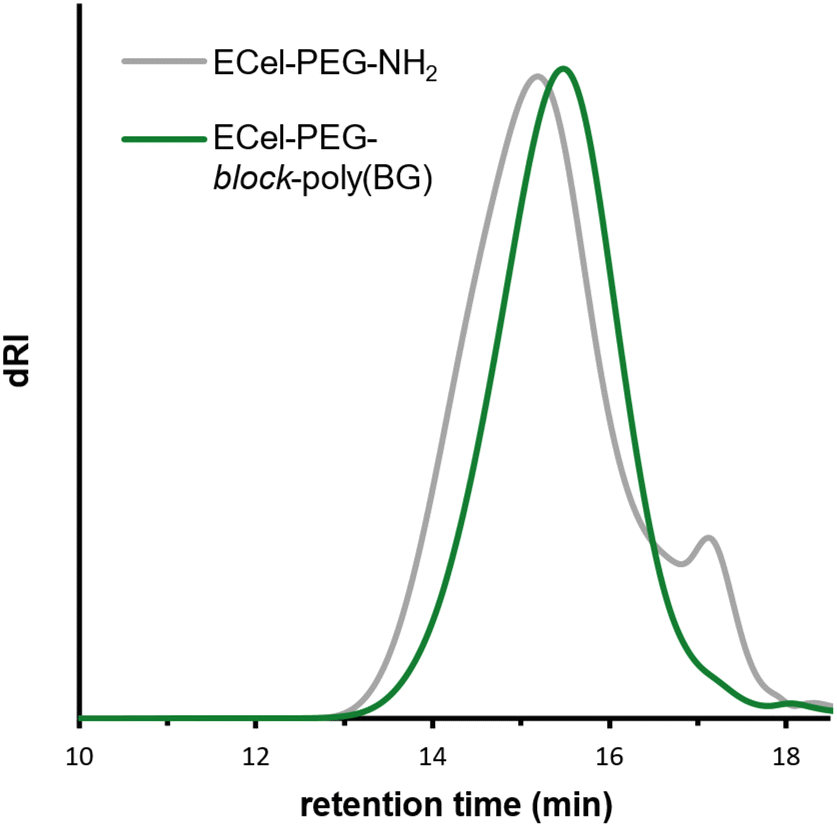 | ||
| Fig. 3 SEC-MALS traces (dRI trace shown) of polymerization of BG-NTA using macroinitiator ECel-PEG-NH2 (ECel-PEG-block-poly(BG), green) along with SEC trace of macroinitiator ECel-PEG-NH2 (grey). 1H NMR spectra can be found in Fig. S10 & S14.† SEC eluent was DMAc/LiCl. Mn (NMR) was estimated to be 40.4 kg mol−1 using poly(BG):ECel ratio. Mn (SEC-MALS) was estimated to be 36.7 kg mol−1 (Đ = 1.4) using a weighted ratio of dn/dc values for poly(BG) and ECel. | ||
Another complimentary characterization technique to confirm chain extension is diffusion ordered spectroscopy (DOSY). DOSY allows for differentiation of NMR signals in the spectrum by their diffusion coefficients, and differences in molecular weight can change the diffusion coefficients of small molecules and polymers. We envisioned that DOSY would corroborate the SEC-MALS data and confirm successful chain extension.
We performed DOSY at 21 °C on as-received ECel, the ECel-PEG-NH2 macroinitiator, and the ECel-PEG-block-poly(BG) BCP (Fig. S17–S19†). Each revealed a diffusion coefficient consistent with a high molecular weight species (i.e., a polymer) in the range of 10−11 to 10−10 m2 s−1. For ECel, we observed a single high molecular weight species with a diffusion coefficient of 6.7 ± 0.5 × 10−11 m2 s−1. DOSY results from the ECel-PEG-NH2 macroinitiator showed a slightly faster diffusion coefficient of 8 ± 3 × 10−11 m2 s−1. The large error is consistent with a mixture of the desired product along with a small amount of residual PEG diamine that was not removed by dialysis, as suggested by the SEC data. The diffusion plots for these signals also revealed two diffusing species. Finally, the ECel-PEG-block-poly(BG) BCP showed a diffusion coefficient of 8 ± 1 × 10−11 m2 s−1. The diffusion plots suggested only one diffusing species, consistent with the SEC data. The similar diffusion coefficients among all three samples likely reflect molecular weight dispersity in each and the moderate 2-fold molecular weight increase, which translates to only an expected ∼10% change in diffusion coefficient, which is within the error of these measurements. We then set out to apply the ECel-PEG-block-poly(BG) BCP in compatiblization studies on ECel and PET blends.
2.2 Compatibilization of ECel/PET blends
An effective compatibilizer operates at the ECel/PET interface, causing a reduction in the interfacial tension between the polymers and resulting in a decrease in domain size. In order to have the compatibilization effect, each block of the compatibilizing BCP must have specific interactions or miscibility with one of the polymers of the blend.37,38 To assess the miscibility of each block of our BCP with ECel and PET, preliminary tests were performed by mixing ECel/ECel-PEG-NH2 and PET/Poly(BG) at a 70/30 ratio. We chose this ratio because preliminary tests on the base blend (ECel and PET only) without compatibilizer revealed that this ratio had large enough domains in order to see an impact of compatibilization but smaller domains in blends with less PET; ECel was chosen as the major component in order to create blends with mostly bio-derived content. Phase contrast optical microscopy (PCOM) and small angle laser light scattering (SALLS) (Fig. S20†) showed that ECel-PEG-NH2 and poly(BG) were miscible with ECel and PET, respectively. Therefore, these preliminary results demonstrated the potential of the ECel-block-poly(BG) BCP to be used as a compatibilizer for ECel/PET blends.To test our hypothesis that the ECel-block-poly(BG) BCP would compatibilize ECel/PET blends, we prepared several films at this 70/30 ratio with a range of amounts of additional BCP compatibilizer. The polymer mixture was prepared and then dissolved in HFIP at 1 wt/v%. Films with thicknesses of below 10 μm were prepared by solvent casting and then drying at rt for 15 min.
PCOM images of both the base blend (without compatibilizer) and the compatibilized blends as well as their respective SALLS scattering patterns are shown in Fig. 4. The base blend 70/30-0 (70% ECel, 30% PET, 0 wt% BCP) exhibited large domains (Fig. 4a), characteristic of the later stages of spinodal decomposition, after coalescence of the initial bi-continuous morphology.39 This blend also revealed a significant population of smaller domains (droplets) dispersed homogeneously among the larger domains. With increasing amounts of BCP compatibilizer (Fig. 4b–e), the size and relative population of the large domains decreased significantly. At 30 wt% compatibilizer (Fig. 4e), the large domains observed in the previous blends (with lower compatibilizer content) were generally absent, and the very small domains retained a more characteristic, bi-continuous spinodal texture (Fig. 4f). Evidently, 30 wt% compatibilizer inhibited domain coalescence (i.e., by steric stabilization) during solvent evaporation.
 | ||
| Fig. 4 Phase contrast optical microscopy (PCOM) images and small angle laser light (SALLS) scattering patterns of ECel/PET-wt% compatibilizer blends (a) 70/30-0, (b) 70/30-5, (c) 70/30-10, (d) 70/30-20, (e) 70/30-30 and (f) higher magnification of 70/30-30. Domain sizes decreased and the scattering patterns (halos) became more defined and larger by the addition of increasing amounts of ECel-block-poly(BG). | ||
The reduction of the domain size with increasing compatibilizer content is plotted in Fig. 5 and listed in Table 2. These results highlight the significant decrease in the size of the large domains with increasing compatibilizer content. In contrast, the size of the smaller domains only decreased modestly across the series. Given that the phase-separated morphology of the 70/30-30 blend did not contain discrete domains (Fig. 4f), a spherical droplet approximation to quantify the domain size was no longer reasonably valid. Alternatively, a measure of interdomain distances using SALLS provided a more characteristic comparison in evaluating the compatibilization effectiveness.
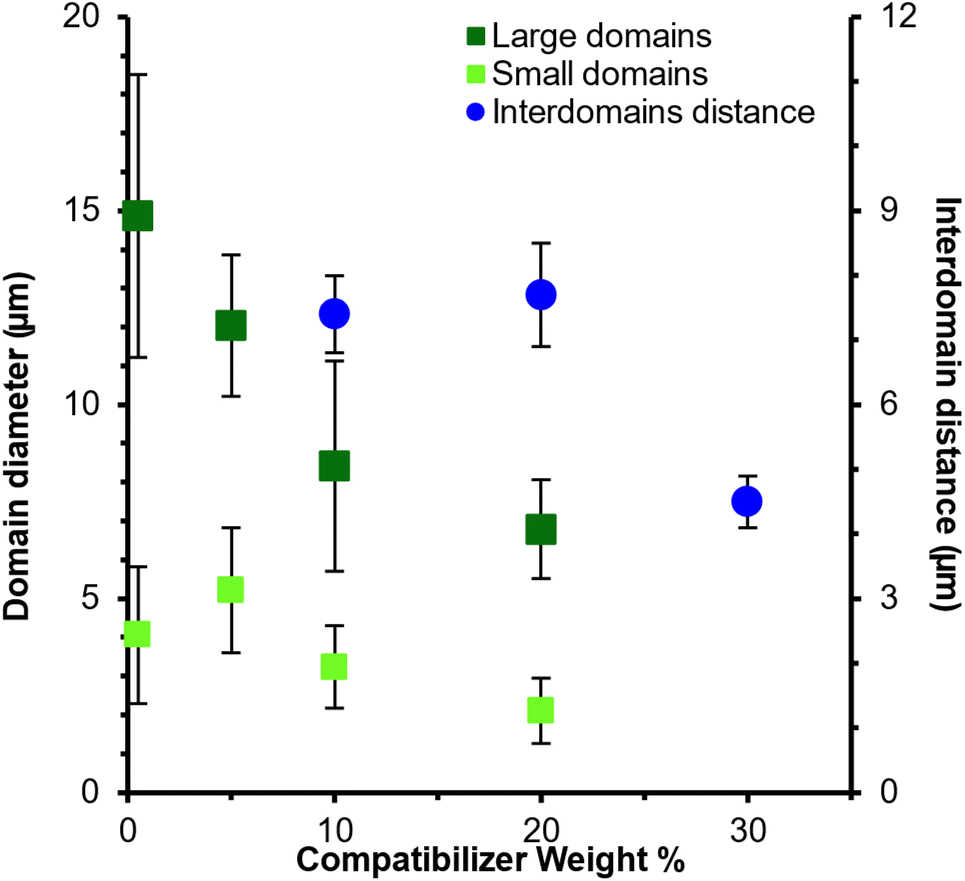 | ||
| Fig. 5 Average domain diameter (PCOM) and interdomain distance (SALLS) of ECel/PET 70/30 blends as a function of ECel-block-poly(BG) concentration. Diameters and interdomain distance decreased as BCP compatibilizer content increased. | ||
| Compatibilizer wt% | Domain diameter (μm) (PCOM) | Interdomain distance (μm) (SALLS) | |
|---|---|---|---|
| Large domains | Small domains | ||
| a,b,c,dDifferent letters across the same columns indicate significant differences using Tukey's test with an alpha value of 0.05. | |||
| 0 | 14.9 ± 3.6a | 4.1 ± 1.8a | — |
| 5 | 12.0 ± 1.8b | 5.2 ± 1.6b | — |
| 10 | 8.4 ± 2.7c | 3.2 ± 1.1c | 7.4 ± 0.6a |
| 20 | 6.8 ± 1.3d | 2.1 ± 0.8d | 7.7 ± 0.7a |
| 30 | — | — | 4.5 ± 0.4b |
SALLS is a complementary technique to PCOM, allowing a global average measurement of the center-to-center interdomain distances in a phase-separated blend. The presence of a distinct scattering halo in the 2D Vv SALLS pattern (e.g., inset of Fig. 4c) is attributed to interparticle interferences within the phase-separated morphology, whereby the radius of the halo at maximum intensity is inversely proportional to the center-to-center interdomain distance.40–42 As shown in Fig. 4a and b, the SALLS patterns for the 70/30-0 base blend and the 70/30-5 compatibilized blend did not present well defined halos. We attribute this scattering behavior to the comparable populations of both large and small domains yielding overlapping distributions of two interparticle interferences, obscuring any evidence of a discrete scattering maximum. In contrast, blend samples with a compatibilizer content of 10 wt% or more contained a relatively minor population of large domains, and thus presented distinct scattering halos (Fig. 4c–e). For these samples, the radius of the halos increased with compatibilizer content, consistent with a corresponding decrease in the respective interdomain distances as plotted in Fig. 5. It is also important to note the significant decrease in scattering intensity for the 70/30-30 blend. We attribute this behavior to an elevated degree of compositional mixing that lowered the scattering contrast in this highly compatibilized blend. Therefore, the observed decrease in both domain size (PCOM) and interparticle distance (SALLS) suggested that the compatibilized blends contained more, smaller domains as the compatibilizer content increased. Collectively, these complementary techniques indicate that the ECel-block-poly(BG) BCP is a remarkably effective compatibilizer capable of establishing a well-dispersed morphology in these highly incompatible blends of ECel and PET. Future work focusing on optimizing compatibilizer structure (e.g., polymer topology, molecular weight of each block), homopolymer ratios, and processing conditions may enable effective compatibilization with lower amounts of copolymer.
3. Conclusions
In summary, ECel-block-poly(BG) BCP was successfully synthesized through ROP of BG-NTA using ECel-PEG-NH2 as a macroinitiator. An ECel-NH2 macroinitiator lacking a PEG linker afforded very low conversion due to steric hindrances near the amine except in the presence of water, which resulted in slow monomer hydrolysis leading to homopolymerization of the remaining BG-NTA. The addition of a PEG linker, i.e. ECel-PEG-NH2, allowed the primary amine to smoothly initiate ROP. The resulting BCP, ECel-block-poly(BG), was then exploited for compatibilization of ECel/PET blends. PCOM and SALLS analysis demonstrated the efficacy of the ECel-block-poly(BG) BCP as a compatibilizer for ECel/PET 70/30 blends. As the compatibilizer content increased, both domain size and interdomain distance decreased, reaching the smallest dimensions at a composition with 30 wt% of the ECel-block-poly(BG) compatibilizer. The morphological information from these complementary techniques demonstrated that ECel-block-poly(BG) effectively compatibilizes ECel/PET blends. Ultimately, we envision that this work can be translated to other polysaccharides to afford more bio-derived alternatives to traditional petroleum-based polymers.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by GlycoMIP, a National Science Foundation (NSF) Materials Innovation Platform funded through Cooperative Agreement DMR-1933525. This work was supported by the Glycomaterial Education Experience (GlycoTREE) Research Experiences for Undergraduates (REU) program funded by the NSF and US Department of Defense (DMR-2244483). This work used shared facilities at the Nanoscale Characterization and Fabrication Laboratory, which is funded and managed by Virginia Tech's Institute for Critical Technology and Applied Science. Additional support is provided by the Virginia Tech National Center for Earth and Environmental Nanotechnology Infrastructure (NanoEarth), a member of the National Nanotechnology Coordinated Infrastructure (NNCI), supported by NSF (ECCS 1542100 and ECCS 2025151). We thank Anna Steele and Jon Mase for SEC-MALS analysis as well as measurements of off-line dn/dc values; we also thank Prof. Kevin Edgar for helpful suggestions.References
- R. Koshti, L. Mehta and N. Samarth, Biological Recycling of Polyethylene Terephthalate: A Mini-Review, J. Polym. Environ., 2018, 26(8), 3520–3529 CrossRef CAS.
- R. Muthuraj, M. Misra and A. K. Mohanty, Biodegradable compatibilized polymer blends for packaging applications: A literature review, J. Appl. Polym. Sci., 2018, 135(24), 45726 CrossRef.
- P. Ahmadi, A. Jahanban-Esfahlan, A. Ahmadi, M. Tabibiazar and M. Mohammadifar, Development of Ethyl Cellulose-based Formulations: A Perspective on the Novel Technical Methods, Food Rev. Int., 2022, 38(4), 685–732 CrossRef CAS.
- N. Osaka, Preparation and Properties of Immiscible Poly (lactic acid)/Ethyl Cellulose Bioplastic Blends with Good Transparency and Optical Control by Drawing, J. Macromol. Sci., Part B: Phys., 2023, 1–19 Search PubMed.
- S. Z. Rogovina and G. A. Vikhoreva, Polysaccharide-based polymer blends: Methods of their production, Glycoconjugate J., 2006, 23(7), 611–618 CrossRef CAS PubMed.
- V. D. Hiremani, T. Gasti, S. P. Masti, R. B. Malabadi and R. B. Chougale, Polysaccharide-based blend films as a promising material for food packaging applications: physicochemical properties, Iran. Polym. J., 2022, 31(4), 503–518 CrossRef CAS.
- K. J. Arrington, J. V. Haag IV, E. V. French, M. Murayama, K. J. Edgar and J. B. Matson, Toughening Cellulose: Compatibilizing Polybutadiene and Cellulose Triacetate Blends, ACS Macro Lett., 2019, 8(4), 447–453 CrossRef CAS PubMed.
- B. Yavuzturk Gul, E. Pekgenc, V. Vatanpour and I. Koyuncu, A review of cellulose-based derivatives polymers in fabrication of gas separation membranes: Recent developments and challenges, Carbohydr. Polym., 2023, 321, 121296 CrossRef CAS PubMed.
- P. Sakellariou, R. C. Rowe and E. F. T. White, Polymer/polymer interaction in blends of ethyl cellulose with both cellulose derivatives and polyethylene glycol 6000, Int. J. Pharm., 1986, 34(1), 93–103 CrossRef CAS.
- H. M. P. C. Kumarihami, N. Kumar, P. Pratibha and A. T. Neeraj, Effect of Natural Materials on Thermal Properties of Food Packaging Film: An Overview, in Natural Materials for Food Packaging Application, 2023, pp. 255–274 Search PubMed.
- M. Sarkar, A. Upadhyay, D. Pandey, C. Sarkar and S. Saha, Cellulose-Based Biodegradable Polymers: Synthesis, Properties, and Their Applications, in Biodegradable Polymers and Their Emerging Applications, Springer, 2023, pp. 89–114 Search PubMed.
- Bioplastics market development update 2023, europeanbioplastics, 2023, (accessed April 4, 2024).
- A. S. Volokhova, J. B. Waugh, K. J. Arrington and J. B. Matson, Effects of graft polymer compatibilizers in blends of cellulose triacetate and poly(lactic acid), Polym. Int., 2019, 68(7), 1263–1270 CrossRef CAS.
- F. Seidi, M. Khodadadi Yazdi, M. Jouyandeh, M. Dominic, H. Naeim, M. N. Nezhad, B. Bagheri, S. Habibzadeh, P. Zarrintaj and M. R. Saeb, et al. Chitosan-based blends for biomedical applications, Int. J. Biol. Macromol., 2021, 183, 1818–1850 CrossRef CAS PubMed.
- J. P. Pinto, O. J. D’Souza, V. D. Hiremani, N. P. Dalbanjan, P. K. SK, S. S. Narasagoudr, S. P. Masti and R. B. Chougale, Functional properties of taro starch reinforced polysaccharide based films for active packaging, Food Biosci., 2023, 56, 103340 CrossRef CAS.
- C. Liu, J. Huang, X. Zheng, S. Liu, K. Lu, K. Tang and J. Liu, Heat sealable soluble soybean polysaccharide/gelatin blend edible films for food packaging applications, Food Packag. Shelf Life, 2020, 24, 100485 CrossRef.
- F. Seidi, M. K. Yazdi, M. Jouyandeh, M. Dominic, H. Naeim, M. N. Nezhad, B. Bagheri, S. Habibzadeh, P. Zarrintaj and M. R. Saeb, Chitosan-based blends for biomedical applications, Int. J. Biol. Macromol., 2021, 183, 1818–1850 CrossRef CAS PubMed.
- K. Łupina, D. Kowalczyk and W. Kazimierczak, Gum arabic/gelatin and water-soluble soy polysaccharides/gelatin blend films as carriers of astaxanthin—A comparative study of the kinetics of release and antioxidant properties, Polymers, 2021, 13(7), 1062 CrossRef PubMed.
- Y. Yang, Y. Shi, X. Cao, Q. Liu, H. Wang and B. Kong, Preparation and functional properties of poly (vinyl alcohol)/ethyl cellulose/tea polyphenol electrospun nanofibrous films for active packaging material, Food Control, 2021, 130, 108331 CrossRef CAS.
- Y. Zhou, Y. Hu, Z. Tan and T. Zhou, Cellulose extraction from rice straw waste for biodegradable ethyl cellulose films preparation using green chemical technology, J. Cleaner Prod., 2024, 439, 140839 CrossRef CAS.
- Z. Kordjazi and A. Ajji, Partially miscible polymer blends of ethyl cellulose and hydroxyl terminated polybutadiene, Polymer, 2020, 211, 123067 CrossRef CAS.
- S. Xu, W. Ma, H. Zhou, Y. Zhang, H. Jia, J. Xu, P. Jiang, X. Wang and W. Zhao, Preparation of butadiene-bridged polymethylsiloxane (BBPMS)/ethyl cellulose (EC) hybrid membranes for gas separation, Eur. Polym. J., 2021, 157, 110679 CrossRef CAS.
- M.-P. Belioka, G. Markozanne, K. Chrissopoulou and D. S. Achilias, Advanced Plastic Waste Recycling—The Effect of Clay on the Morphological and Thermal Behavior of Recycled PET/PLA Sustainable Blends, Polymers, 2023, 15(14), 3145 CrossRef CAS PubMed.
- A. M. Torres-Huerta, D. Palma-Ramírez, M. A. Domínguez-Crespo, D. Del Angel-López and D. de la Fuente, Comparative assessment of miscibility and degradability on PET/PLA and PET/chitosan blends, Eur. Polym. J., 2014, 61, 285–299 CrossRef CAS.
- F. Welle, Twenty years of PET bottle to bottle recycling—An overview, Resour., Conserv. Recycl., 2011, 55(11), 865–875 CrossRef.
- L. Ju, J. M. Dennis, K. V. Heifferon, T. E. Long and R. B. Moore, Compatibilization of Polyester/Polyamide Blends with a Phosphonated Poly(ethylene terephthalate) Ionomer: Comparison of Monovalent and Divalent Pendant Ions, ACS Appl. Polym. Mater., 2019, 1(5), 1071–1080 CrossRef CAS.
- G. Aravinthan and D. Kale, Blends of poly (ethylene terephthalate) and poly (butylene terephthalate), J. Appl. Polym. Sci., 2005, 98(1), 75–82 CrossRef CAS.
- P. Visakh, Polyethylene terephthalate: blends, composites, and nanocomposites–state of art, new challenges, and opportunities, Polyethylene-Based Blends, Compos. Nanocompos., 2015, 1–14 CAS.
- N. B. Erdal and M. Hakkarainen, Degradation of Cellulose Derivatives in Laboratory, Man-Made, and Natural Environments, Biomacromolecules, 2022, 23(7), 2713–2729 CrossRef CAS PubMed.
- A. S. Volokhova, K. J. Edgar and J. B. Matson, Polysaccharide-containing block copolymers: synthesis and applications, Mater. Chem. Front., 2020, 4(1), 99–112 RSC.
- D. Siefker, A. Z. Williams, G. G. Stanley and D. Zhang, Organic acid promoted controlled ring-opening polymerization of α-amino acid-derived N-thiocarboxyanhydrides (NTAs) toward well-defined polypeptides, ACS Macro Lett., 2018, 7(10), 1272–1277 CrossRef CAS.
- J. Cao, D. Siefker, B. A. Chan, T. Yu, L. Lu, M. A. Saputra, F. R. Fronczek, W. Xie and D. Zhang, Interfacial ring-opening polymerization of amino-acid-derived N-thiocarboxyanhydrides toward well-defined polypeptides, ACS Macro Lett., 2017, 6(8), 836–840 CrossRef CAS.
- N. Gangloff, J. Ulbricht, T. Lorson, H. Schlaad and R. Luxenhofer, Peptoids and Polypeptoids at the Frontier of Supra- and Macromolecular Engineering, Chem. Rev., 2016, 116(4), 1753–1802 CrossRef CAS PubMed.
- X. Tao, B. Zheng, H. R. Kricheldorf and J. Ling, Are N-substituted glycine N-thiocarboxyanhydride monomers really hard to polymerize?, J. Polym. Sci., Part A: Polym. Chem., 2017, 55(3), 404–410 CrossRef CAS.
- J. P. Kratohvil, On solution properties of poly(γ-benzyl-L-glutamate), Kolloid Z. Z. Polym., 1970, 238(1), 455–459 CrossRef CAS.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, RAFT aqueous dispersion polymerization yields poly (ethylene glycol)-based diblock copolymer nano-objects with predictable single phase morphologies, J. Am. Chem. Soc., 2014, 136(3), 1023–1033 CrossRef CAS PubMed.
- A. Ajitha and S. Thomas, Introduction: Polymer blends, thermodynamics, miscibility, phase separation, and compatibilization, in Compatibilization of polymer blends, Elsevier, 2020, pp. 1–29 Search PubMed.
- A. Ajitha and S. Thomas, Compatibilization of polymer blends: micro and nano scale phase morphologies, interphase characterization, and properties, Elsevier, 2019 Search PubMed.
- M. F. Butler and M. Heppenstall-Butler, Phase separation in gelatin/dextran and gelatin/maltodextrin mixtures, Food Hydrocolloids, 2003, 17(6), 815–830 CrossRef CAS.
- K. Nishida, H. Ogawa, G. Matsuba, T. Konishi and T. Kanaya, A high-resolution small-angle light scattering instrument for soft matter studies, J. Appl. Crystallogr., 2008, 41(4), 723–728 CrossRef CAS.
- J. Laeuger, R. Lay, S. Maas and W. Gronski, Structure development of a polybutadiene/polyisoprene blend during spinodal decomposition. Comparison between light scattering and optical microscopy, Macromolecules, 1995, 28(20), 7010–7015 CrossRef CAS.
- K. Shah, J. L. White and K. Min, A study of the miscibility of blends of polybutadiene and styrene–butadiene copolymers, Polym. Eng. Sci., 1989, 29(9), 586–592 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00688g |
| ‡ Indicates co-first author. |
| This journal is © The Royal Society of Chemistry 2024 |