
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequential removal of nitrate and sulfate in woodchip and hematite – coated biochar bioreactor†
Mohammed Masud
Parvage
and
Roger
Herbert
 *
*
Department of Earth Sciences, Uppsala University, Villavägen 16, SE-752 36 Uppsala, Sweden. E-mail: roger.herbert@geo.uu.se
First published on 9th December 2022
Abstract
Laboratory column experiments have been used to study the sequential removal of nitrate (NO3−) and sulfate (SO42−) from mine water, where NO3− was removed through denitrification and SO42− was removed through SO42− reduction and the subsequent precipitation of hydrogen sulfide (H2S) in a hematite-coated biochar (HCB) bioreactor. Denitrification and SO42− reduction were investigated in columns filled with pine woodchips and pine woodchips + biochar, both with and without the addition of lactate. Experimental results indicated that a >90% NO3− removal from 50 mg L−1 NO3−-N was achieved at a hydraulic residence time of 5 days without lactate addition, but that SO42− reduction was minimal after an initial startup period. Lactate was added to stimulate SO42− reduction, producing H2S with >90% SO42− removal from an initial concentration of 361 mg L−1 SO42−-S. Sulfate concentrations were reduced to a greater extent in the woodchip + biochar column, and NH4+ production was enhanced in both columns after lactate addition. After treatment in the HCB columns, H2S and NH4+ were removed to >95%. X-ray photoelectron spectroscopy (XPS) indicated that S2−, S22−, S0 and NH4+ were accumulating in the HCB columns and surface-bound iron was converted from Fe(III) to Fe(II). The XPS results suggested that the reductive dissolution of hematite preceded the precipitation of H2S as FeS, pyrite and elemental sulfur on the HCB surfaces.
Water impactThrough the release of NO3− and SO42−, mining operations can negatively impact aquatic ecosystems. We have investigated the sequential removal of NO3− and SO42− in woodchip bioreactors followed by H2S removal in hematite-coated biochar bioreactors. By adapting current denitrifying woodchip bioreactors for the removal of sulfate as well, mining companies can meet water quality criteria for NO3− and SO42−. |
Introduction
Heavy metal emissions and acidification are common environmental issues associated with mine drainage,1,2 but in mine wastes where the acid neutralization capacity of waste rock and tailings exceeds the acidity produced through sulfide oxidation and hydrolysis reactions, the resulting mine drainage will have a near-neutral pH. In the case of the mining of non-sulfidic magnetite-based ores in northern Sweden, heavy metal discharges to recipients are relatively low, and the abatement of nitrate (NO3−) and sulfate (SO42−) emissions to nutrient-poor recipients have become priorities for achieving environmental quality goals.Nitrogen compounds in mine drainage are commonly derived from the ammonium nitrate-based explosives used in the mining process. Upon detonation and exposure to the atmosphere, most nitrogen in the undetonated explosives either dissolves in groundwater or is weakly adsorbed to waste rock and is transported from the mine. In mine drainage, most of the nitrogen is in the form of NO3− where it is often detected at relatively high concentrations (>15 mg L−1 N).3,4 Excess release of NO3− to aquatic ecosystems can be hazardous as it may induce eutrophication leading to hypoxia, or may be transformed into ammonium (NH4+) or ammonia (NH3), the latter being toxic to aquatic organisms.5
Sulfate is a common constituent in mine drainage where it is typically derived from sulfide mineral oxidation. However, even in the absence of sulfide minerals, SO42−-bearing minerals such as gypsum and anhydrite are readily soluble and may be significant sources of SO42− to mine drainage. Sulfate may have a passive influence on surface water quality by increasing phosphate solubility,6,7 a key limiting factor for toxic algal blooms,8,9 and by promoting mercury methylation under sulfate-reducing conditions.10 Furthermore, chronic toxic effects have been exhibited on aquatic organisms when sulfate concentrations in water are sufficiently high; the British Columbia Ministry of Environment has based its long-term chronic sulfate water quality guideline on rainbow trout, one of the most sensitive species to sulfate exposure,11 where the sulfate guideline value is dependent on water hardness.
Because of the environmental impact of NO3− and SO42− emissions on aquatic ecosystems, environmental authorities in Sweden have proposed criteria for surface water quality that would, for mining companies, require a more than tenfold decrease in NO3− and SO42− concentrations discharged to surface water recipients.12 In response, the mining industry is investigating water treatment technologies that would enable them to meet these criteria. For the removal of NO3− from waste rock drainage, the mining company LKAB in Sweden has evaluated the performance of pilot-scale woodchip denitrifying bioreactors.13,14 The function of the woodchip bioreactor is to provide a carbon-rich anaerobic environment where denitrification can take place; mine drainage is allowed to flow through the permeable subsurface structure, where an abundance of denitrifying bacteria14 transform dissolved NO3− to nitrogen gas (N2). During the operation of the bioreactor, it has been noted that sulfate-reducing conditions are promoted when influent waters are depleted in NO3− and nitrite (NO2−).14 Dissimilatory sulfate reduction will occur in anaerobic environments where there is an abundance of organic matter:
| SO42− + 2CH3CH(OH)COO− = H2S + 2CH3COO− + 2HCO3− | (1) |
The potential therefore exists to adapt a denitrifying bioreactor for the removal of SO42− by promoting sulfate reduction and the subsequent precipitation of H2S. Iron sulfide (e.g., FeS or pyrite, FeS2) will precipitate following the reaction of dissolved H2S with ferrous iron:
| Fe2+ + H2S = FeS(s) + 2H+ | (2) |
| H2S + Fe2O3(s) + 4H+ → 2Fe2+ + S0 + 3H2O | (3) |
| FeS(s) + S0(s) → FeS2(s) | (4) |
| 2Fe2O3(s) + CH3CH(OH)COO− + 7H+ = 4Fe2+ + CH3COO− + 3H2O + HCO3− | (5) |
In this study, we have investigated the sequential removal of NO3− and SO42− during transport through a denitrifying woodchip bioreactor followed by a hematite-coated biochar bioreactor. Influent and effluent water quality were monitored to evaluate treatment efficiency, and the solid-phase surface chemistry of the hematite-coated biochar was analyzed using X-ray photoelectron spectroscopy (XPS). The treatment goal was to remove NO3− and SO42− without the excessive production of denitrification byproducts (e.g., nitrite) and with a high level of sulfur capture. While H2S removal by reaction with ferric hydroxides and oxides is not a unique solution for water treatment,18,19 it is a relatively novel method for the mining industry.20
Materials and methods
Column preparation
For the experiments, two columns were operated in series: denitrification and sulfate reduction were designed to occur in the first column, while ferric iron reduction and ferrous iron sulfide precipitation were designed to occur in the second column. All columns were constructed of Plexiglas with upward-directed flow; the dimensions of the first and second cylindrical columns were 40 cm × 10 cm (length × inner diameter) and 10 cm × 8.5 cm, respectively. The second column in the series was smaller as it was assumed that Fe2O3 dissolution and FeS precipitation would occur at a reaction front and not require a larger bioreactor volume. Two column systems were operated in parallel with each other, where the reactive material differed in the first columns. All experiments were conducted at room temperature (ca. 22 °C). See Fig. 1 for an illustration of the experimental system.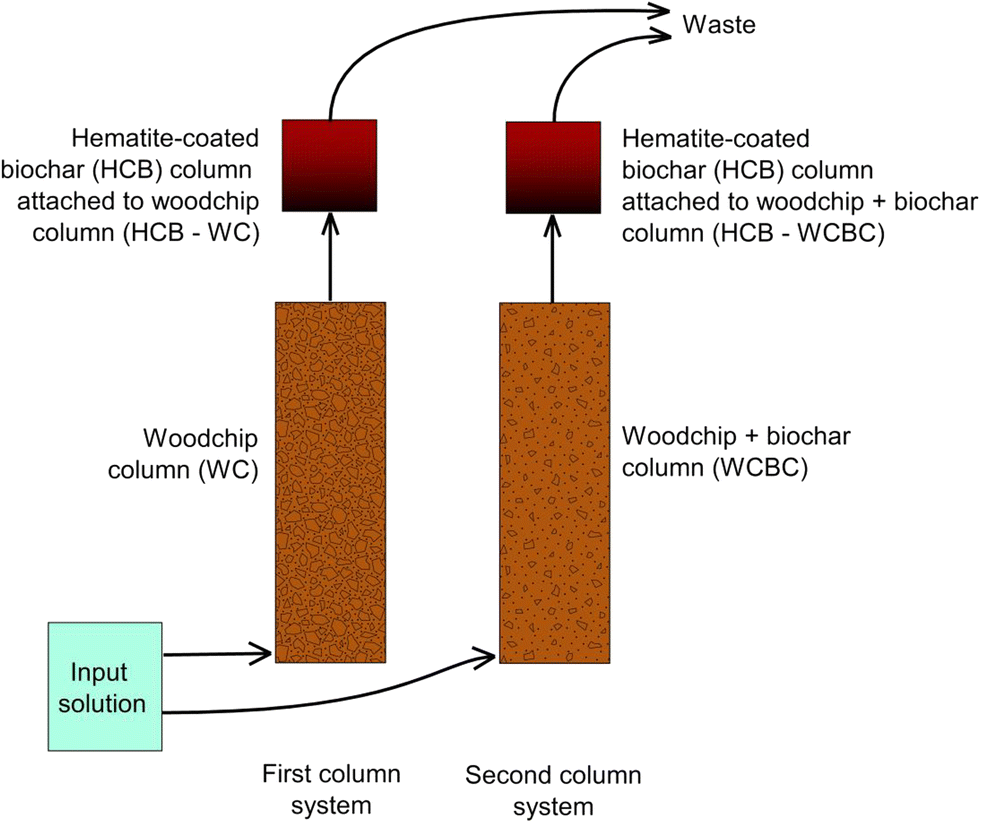 | ||
| Fig. 1 Experimental design used in column study. | ||
The first column in the first system was filled with one year-old pine woodchips (woodchip dimensions ca. 30 × 30 × 5 mm), while the first column in the second column system was filled with a mixture of one year-old pine woodchips and crop-residue biochar (ECOERA AB, Falkenberg, Sweden) mixed at a volume ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (see Table S1 in ESI† for physical characteristics). Biochar was selected as a column substrate since it is highly porous and may host a larger community of denitrifying and sulfate reducing bacteria21 relative to woodchips alone, and it provides a large surface area for adsorption and ion exchange.22
:1 (see Table S1 in ESI† for physical characteristics). Biochar was selected as a column substrate since it is highly porous and may host a larger community of denitrifying and sulfate reducing bacteria21 relative to woodchips alone, and it provides a large surface area for adsorption and ion exchange.22
The second column in each series (i.e., one following the woodchip-filled column, denoted HCB-WC, and one following the woodchip + biochar column, denoted HCB-WCBC; Fig. 1) was filled with hematite-coated biochar (HCB) that was manually prepared from a 1:1 mixture of hematite (99.1%, fine-grained powder, commercially produced as pigment) and the previously mentioned crop-residue biochar. To prepare this mixture, equal volumes of hematite and biochar were gently hand-mixed in a stainless-steel bowl for approximately one minute. The hematite composition was confirmed by X-ray diffractometry.
Prior to the start of the experiment, a consortium of bacteria was established in the woodchip columns by inoculating with diluted activated sludge collected from the Kungsängsverket wastewater treatment plant (Uppsala, central Sweden). It was assumed that the inoculant contained an elevated abundance of denitrifying bacteria but this was not verified. Wastewater-derived inoculants have been used in previous field-based studies13,14 where the rapid establishment of denitrification was obtained; such inoculants would most likely be used in a field application of the technology presented in the current study. The inoculant (2000 mL) consisted of 1250 mL of the activated sludge slurry mixed with a NO3− and SO42−-rich solution (final concentrations 13 mg NO3−-N per L and 90 mg SO42-S per L, respectively). To established a biofilm in the column bioreactors, the inoculant was pumped through the first columns and recirculated for 10 days at a rate of 14 mL per hour. While the selection of a 10 day period for inoculation was arbitrary, it was assumed that this period would be adequate for the initial establishment of a denitrifying community in the columns. After the inoculation period, the columns were flushed with three pore volumes of distilled water.
Experimental procedure
For the column experiments, the woodchip columns were first operated alone for 200 days, whereupon a hematite-coated biochar column was connected in series after each woodchip column for an additional 150 days. The column experiments were concluded on day 350. The woodchip columns consistently received an input solution consisting of 50 mg l−1 NO3−-N and 361 mg l−1 SO42-S (prepared from KNO3 and Na2SO4, respectively). The woodchip columns were first operated without the addition of an external carbon source to determine if the woodchips were sufficiently reactive to stimulate sulfate reduction. From day 173, sodium lactate was added as an external carbon source to the influent solution to the woodchip columns. Lactate was included in the input solution at a concentration of ca. 5000 mg L−1 lactate (prepared from 12.1 M lactic acid neutralized to pH 7 with the addition of 1 M NaOH), which was more than twice the amount required based on sulfate reduction stoichiometry (cf.reaction (1)) so that sufficient hematite reduction could be ensured as well (reaction (3)).The water flow to the bioreactor columns was delivered with a peristaltic pump calibrated to 14 mL h−1; for the woodchip column with an intergrain porosity of 0.537 (ESI,† Table S1), this corresponds to a ca. 5 day hydraulic residence time (HRT; where HRT = column volume × intergrain porosity/flow rate). This flow rate was used consistently throughout the experiment, with the exception of one period (days 141 to 172) when the HRT in the columns was shortened to 2 days.
Sampling and chemical analyses
Samples were collected weekly for the analysis of pH, NO3−-N, NO2−-N, NH4+-N, SO42−-S, S2−-S, Fe2+, and alkalinity. Before chemical analysis, samples were filtered with a 0.2 μm polyethersulfone membrane syringe filter. Samples for total organic carbon (TOC) and organic anions (lactate, acetate, formate) were collected every other week. All samples were stored at 4 °C until the analyses were performed.The pH was measured using a pH-electrode (VWR Collection pHenomenal 221 pH electrode) and the alkalinity by titration with 0.005 M H2SO4. Effluent concentrations of NO3−-N, NO2−-N, ammonium-nitrogen (NH4+-N), sulfide (S2-S), SO42−-S, and Fe2+ were determined colorimetrically using a HACH DR1900 spectrophotometer and HACH pre-dosed reagents having the following detection limits: 0.23 mg L−1 NO3−-N, 0.015 mg L−1 NO2−-N, 0.015 mg L−1 NH4+-N, 13 mg L−1 SO42−-S, 0.03 mg L−1 S−2−-S, and 0.02 mg L−1 Fe2+. Any dilution during analysis was performed with nitrogen-purged deionized water. It should be noted that the easily-oxidizable compounds S2− and Fe2+ were analyzed directly after sample collection, but that some oxidation may have occurred during sample collection and handling. The reported concentrations for S2− and Fe2+ should therefore be considered as minimum values. Nitrite and NH4+ are considered to be less sensitive to rapid oxidation and were analyzed within four hours of sample collection.
Organic acids were analyzed on a Metrohm IC system (883 Basic IC Plus and 919 Autosampler Plus). Separation was performed using a Metrosep A Supp 5 analytical column (250 × 4.0 mm) fitted with two guard columns (Dionex IonPac NG1 4 × 35 mm and an Metrosep A Supp 4/5) using a carbonate eluent (3.2 mM Na2CO3 + 1.0 mM NaHCO3) at a 0.7 mL min−1 flow rate. Unfiltered samples were used to measure TOC on a Shimadzu TOC-L TNM-L instrument.
Cryogenic X-ray photoelectron spectroscopy
At the end of the experiment, the hematite-coated biochar columns were dismantled in a glovebag under continuous nitrogen flow; samples were collected as rapidly as possible and stored at −18 °C. The surface chemical composition of the hematite-coated biochar was investigated through cryogenic X-ray photoelectron spectroscopy (cryo-XPS). A detailed analysis protocol for cryo-XPS is described in Shchukarev & Ramstedt,23 and the general analytical procedure is described in Wan et al.24 In brief, samples were prepared in oxygen-free conditions and were placed on the sample holder and immediately fast-frozen to −170 °C under cryogenic conditions (using liquid nitrogen cooling) in the load-lock chamber of the spectrometer. The XPS spectra were recorded with an Axis Ultra DLD electron spectrometer (Kratos Analytical Ltd., UK). A monochromated Al Kα source operated at 150 W, a hybrid lens system with a magnetic lens, providing an analysis area of 0.3 mm by 0.7 mm, and a charge neutralizer were used for the measurements. Processing of the spectra was accomplished with Vision2 Kratos software. Survey spectra were collected from 1000 to 0 eV at a pass energy of 160 eV. High resolution spectra for Fe(2p), S(2p), O(1s), C(1s), and N(1s) were collected at a pass energy of 20 eV with a scan step of 0.1 eV. High-resolution XPS spectra were fitted using linear combinations of 70:30 G-Lorentz functions on Shirley background-subtracted spectra. The hydrocarbon C(1s) peak at 285.0 eV was used as the internal standard for binding energy (BE) scale calibration. The S(2p) spectra are fitted with a doublet representing the spin-orbit splitting of the S(2p1/2) and S(2p3/2) peaks, and are fitted with a S(2p1/2) component that is located 1.18 eV greater than the S(2p3/2) peak but with half the intensity.
Results and discussion
Woodchip column effluents before lactate addition
Effluent concentrations of both NO3−-N (Fig. 2a) and NO2−-N (ESI,† Fig. S1) were very low with a HRT of 5 days, while the lower 2 day HRT resulted in considerably greater NO3−-N and NO2−-N concentrations discharging from both columns. The greater HRT yielded treatment efficiencies of 90% and 95% for the woodchip (WC) and woodchips mixed with biochar (WCBC) columns, respectively (treatment efficiency calculated as the difference between influent NO3−-N and effluent NO3−-N + NO2−-N + NH4+-N concentrations, divided by influent NO3−-N concentration, multiplied by 100). Increasing the flow rate for a 2 day HRT drastically decreases the treatment efficiency of the WC column to 40%. In contrast, the efficiency of the WCBC column at higher flowrate was less affected (79%). Similar findings of the dependence of nitrate removal through denitrification on HRT have been reported in numerous studies,25–27 and Bock et al.25 have reported that the treatment efficiency of woodchip/biochar mixtures was less affected by HRT changes, compared with pure woodchip bioreactors.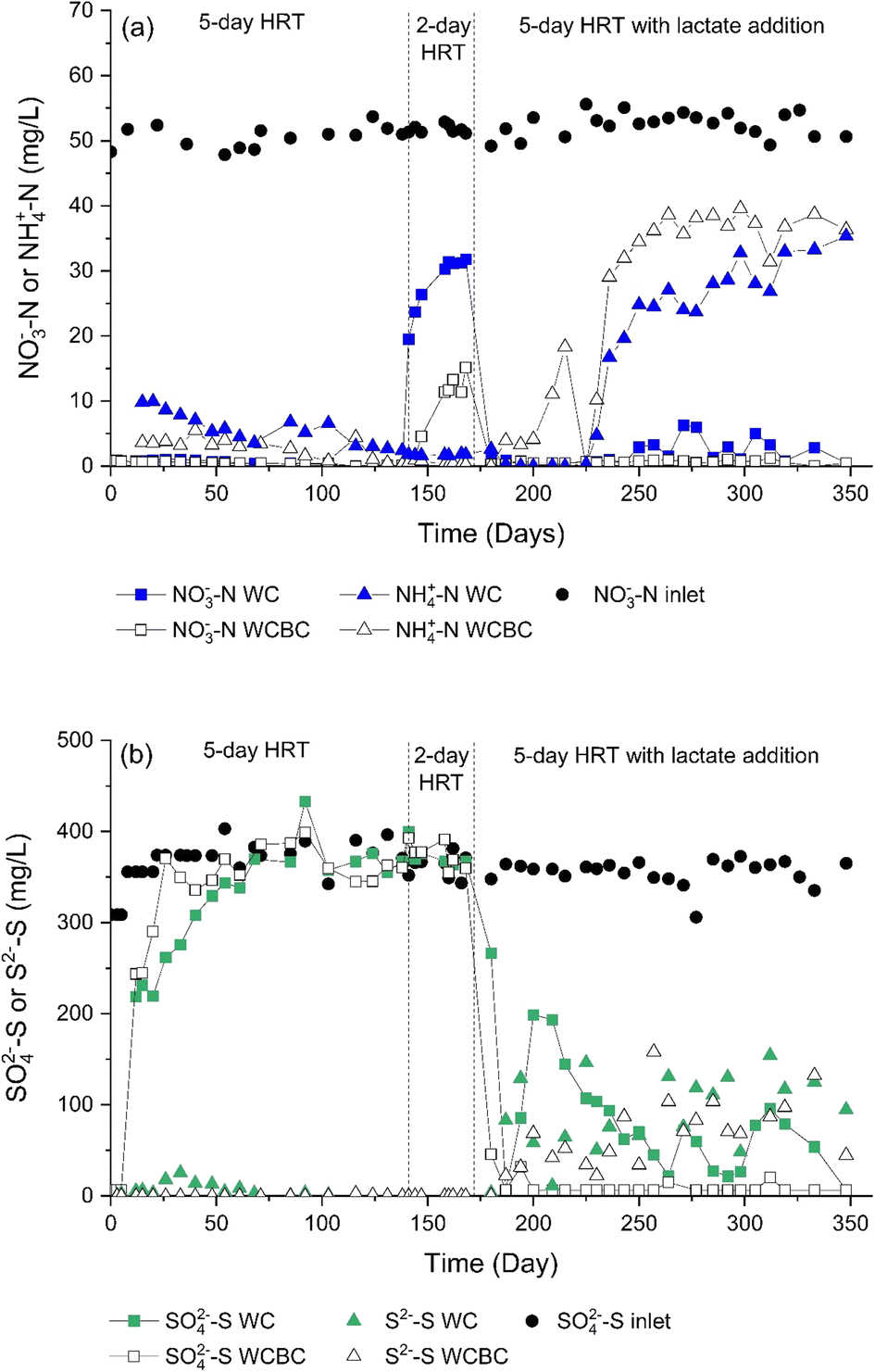 | ||
| Fig. 2 Concentrations of (a) NO3−-N and NH4+-N, and (b) SO42−-S and S2−-S measured in the influent and effluent from the woodchip + biochar (WCBC) and woodchip (WC) columns. | ||
During the first three weeks of operation, analyses of the WC and WCBC effluents (Fig. 2b) indicated that the SO42−-S concentrations were ca. 30% and 15% lower, respectively, in the outlets compared with the inlets. An odor of H2S was emitted from the WC column outlet and H2S (as S2−-S) was detected at low concentrations (Fig. 2b), such that the decrease in concentration was probably the result of dissimilatory sulfate reduction. Similar results for SO42−-S were obtained for the WCBC column, except that H2S was not detected in the effluent, suggesting that H2S was retained in the column. The initial but transient occurrence of sulfate-reducing conditions in both columns is likely due to the initially greater availability of organic carbon in the woodchip porewater; the TOC concentrations decreased from the start of the experiment (Fig. 3a) and coincided with a decrease in outlet water color. Prior to lactate addition, TOC consisted primarily of unidentified organic compounds, since the sum of lactate, acetate and formate concentrations did not exceed 0.15 mg L−1 C (Fig. 3b).
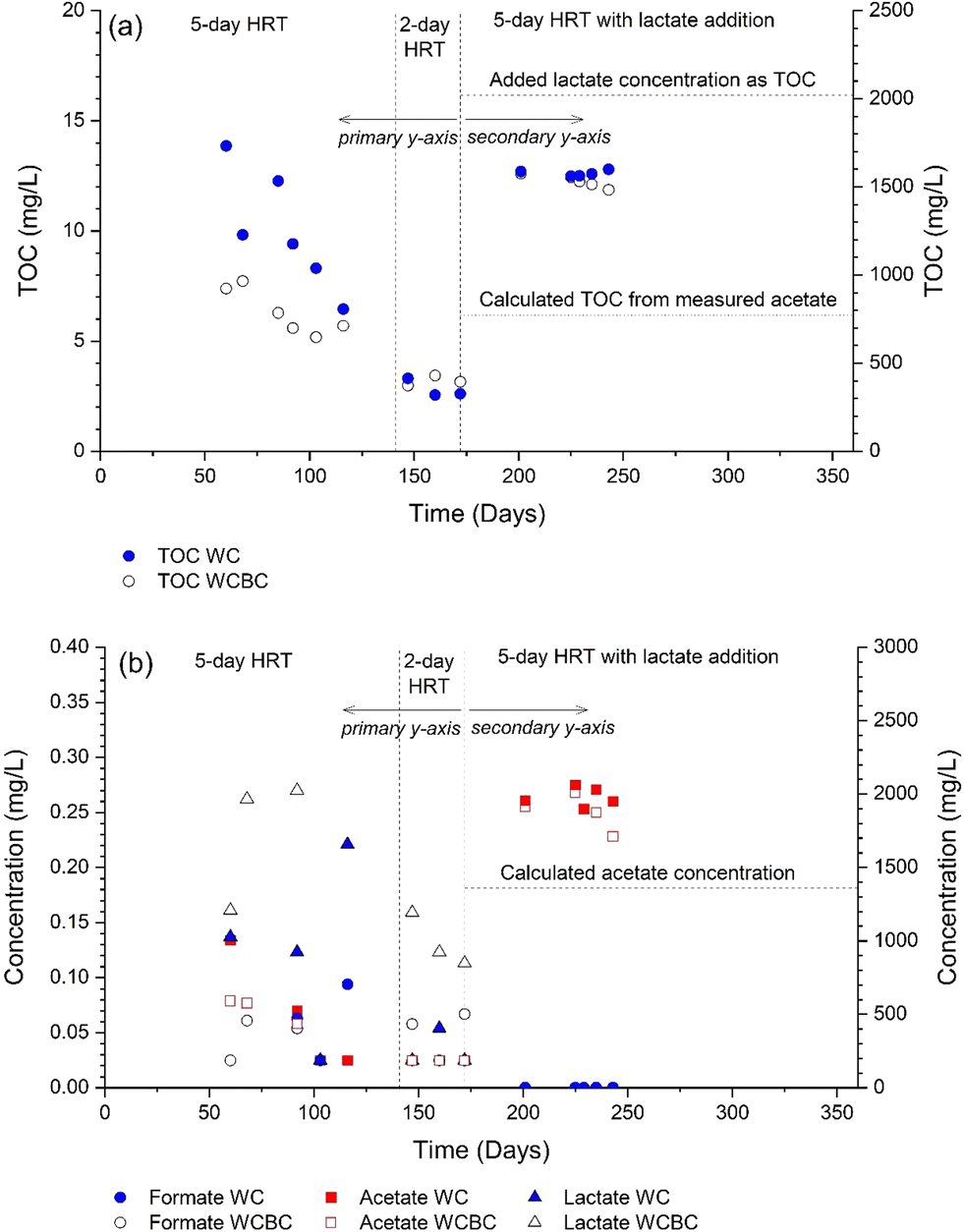 | ||
| Fig. 3 Concentration of (a) total organic carbon (TOC) and (b) carboxylates, measured from the effluent of the woodchip + biochar (WCBC) and woodchip (WC) columns. Primary y-axis applicable to day 172; secondary y-axis used after day 172. In (b), acetate concentration was calculated from SO42− concentration at the inlet and stoichiometry in reaction (1). | ||
During operations from day 61–85, flow through both columns was interrupted on several occasions due to gas generation and leakage from the columns; dissolved oxygen may have briefly entered the columns during this period but this does not seem to have had an impact on the long-term performance of the column system.
The effluent pH from the WC column was initially substantially lower (pH 6.0–6.3) than from the WCBC column (pH 7.9–8.0) (ESI,† Fig. S2). Initial pH conditions in the range 5–7 are common in column experiments27 and field studies13 of denitrifying bioreactors using pine woodchips, where the lower pH is attributed to fermentation reactions that control pH before denitrification becomes the dominating pH-determining process through alkalinity production (see ESI,† Fig. S2). In contrast, the initial pH of the WCBC column was ca. 8 and is explained by the basic pH of biochar. After about 50 days of operation, the pH of both column effluents was ca. 8.
Woodchip column effluents after lactate addition
With the addition of lactate to the column influents, NO3−-N (Fig. 2a) and NO2−-N (ESI,† Fig. S1) concentrations in the WC and WCBC column outlets were low and similar to the levels observed without lactate addition. However, after a lag time of ca. 60 days, NH4+-N concentrations increased greatly in the outlets, with concentrations of ca. 35 mg L−1 NH4+-N at the end of the experiment. Such an increase in ammonium production caused a substantial drop in the total N removing efficiency of the system, from 95% to 51% for the WCBC column and from 90% to 63% for the WC column. Ammonium production in both columns can be attributed to dissimilatory nitrate reduction to ammonia (DNRA), which is often stimulated by an increased supply of available carbon and hence C/N-ratio14,28,29 and elevated concentrations of H2S (see below).30The lactate addition stimulated the activity of the sulfate-reducing community in the columns, as evident by the decrease in SO42−-S concentrations to below detection limits in the outlet of the WCBC column, and to <200 mg L−1 SO42−-S in the WC column (Fig. 3b). Simultaneously, S2−-S concentrations increased to >150 mg L−1 at the outlets from both columns. These results are in line with many other studies31–33 that have observed a strong growth in the sulfate-reducing community and subsequent SO42− reduction to H2S following lactate addition.
After lactate addition, TOC concentrations were high in the column effluents (Fig. 3a) since lactate (initially ca. 5000 mg L−1, or 2022 mg L−1 C) is converted to acetate during sulfate reduction (cf.reaction (1)). Indeed, lactate and formate concentrations in the outlets were <2.5 mg L−1, while average acetate concentration was 1930 mg L−1 (Fig. 3b). Based on reaction stoichiometry (reaction (1)) and an initial SO42−-S concentration of 361 mg L−1, the resulting acetate concentration should be 1360 mg L−1; as shown in Fig. 3b, additional acetate was produced from other reactions such as denitrification. When converted to a TOC value, the average acetate concentration in the column effluent (772 mg L−1 C) was only 50% of the measured TOC (Fig. 3a); this suggests that there are other organic compounds contributing to TOC than lactate, acetate and formate.
The pH of the column effluents was in the range 7–8 (ESI,† Fig. S2), and the alkalinity was high (ESI,† Fig. S2) due to the presence of titratable carboxylates, primarily acetate (Fig. 3, cf.reaction (1)).
Treatment in hematite-coated biochar columns
Chemical analysis of the effluents from the second (hematite-coated biochar, HCB) columns indicated that SO42−-S concentrations were generally low (<20 mg L−1 SO42−-S) in the column effluents (Fig. 4a). The decrease in SO42−-S concentration in the HCB column receiving effluents from the WCBC column (designated HCB-WCBC) also suggested that SO42− reduction occurred in the HCB column. The S2−-S concentrations were close to the detection limit at the HCB outlets, indicating the nearly complete removal of sulfur (Fig. 4b). This was supported by the absence of an H2S odor at the column outlets. The removal of S2− from solution in the HCB columns was most likely the result of the precipitation of sulfur compounds (e.g., FeS, FeS2), where the presence of Fe2+ at the outlets of the HCB columns was confirmed by chemical analyses. The Fe2+ concentrations differed between the columns; while Fe2+ concentrations at the HCB-WCBC outlet were generally <0.05 mg L−1, Fe2+ concentrations from HCB-WC were on average 2.7 mg L−1 until day 284, after which the average concentration declined to 0.12 mg L−1. Based on the initial stock SO42−-S concentration and final concentrations, a 96% and 91% SO42− removal was achieved in the HCB-WCBC and HCB-WC columns, respectively, after lactate addition (day 200–350). | ||
| Fig. 4 Concentrations of (a) SO42−-S and (b) S2−-S measured at the inlet and outlet of the hematite coated biochar columns (HCB), which are receiving influents from the woodchip + biochar column (WCBC) and woodchip column (WC) | ||
The capacity of HCB for sulfur accumulation can be estimated from the Fe content of the material, given the assumption that all iron is available for reaction with H2S and all sulfur is retained as FeS. If it is assumed that HCB is a 1:1 mixture of hematite and biochar with the total porosity of biochar (0.57, Table S1, ESI†), and that powdered hematite has a bulk density of 1.2 g cm−3, then 1 cm3 HCB contains 0.004 mol Fe that can precipitate 0.004 mol S. This implies a sulfur accumulation capacity of 0.004 moles S cm−3 HCB. For the flow rate, SO42−-S concentration and HCB column dimensions used in this study, iron in the HCB column would be consumed after 577 days.
The differences in sulfur accumulation in the HCB columns may potentially be linked to differences in the composition of the preceding columns and inlet water chemistry: SO42− reduction to H2S in the WC column was not as complete as in the WCBC column (Fig. 2b), such that SO42−-S concentrations at the outlet of WC varied between 20 and 150 mg L−1 SO42−-S and low SO42−-S concentrations were also detected at the outlet of HCB-WC (Fig. 4a). While solutions with similar S2−-S concentrations were pumped into both HCB columns (Fig. 4b), higher Fe2+ concentrations were detected in the outlet of HCB-WC, indicating that the S2−-S concentrations were not sufficient to precipitate all Fe2+. These combined observations suggest that the HCB-WCBC column combination produced a more reducing environment, possibly due to the presence of microenvironments in the biochar,21 promoting a higher rate of SO42− reduction and SO42− removal.
The NH4+ that was produced in the WC and WCBC columns (Fig. 2a) was subsequently removed in the HCB columns (ESI,† Fig. S3). While the removal mechanism has not been confirmed, NH4+ may have adsorbed to the abundant biochar surfaces in the HCB columns; this is supported by the XPS study (below). Biochar has been identified as an effective adsorbent of NH4+ in other studies.34,35
XPS
XPS spectra were obtained from unreacted biochar, unreacted hematite, the unreacted HCB mixture, and the reacted HCB material that treated effluents from the WC and WCBC columns (designated HCB-WC and HCB-WCBC, respectively). XPS spectra of the unreacted materials were used as reference spectra in order to ascertain changes in surface speciation following treatment. A comparison of surficial atomic concentrations from XPS survey scans (ESI,† Table S2) indicated a relative enrichment of C, N, Na and S on the reacted material surfaces, while Fe and O were depleted. Representative S(2p) spectra for the four reacted HCB materials are depicted in Fig. 5. XPS spectra for Fe(2p3/2), N(1s), C(1s) and O(1s) in most investigated materials are presented in Fig. S4–S7 in the ESI† and peak binding energies are listed in Table S3 (ESI†). The interpretation of the C(1s) and O(1s) spectra are included in the ESI.†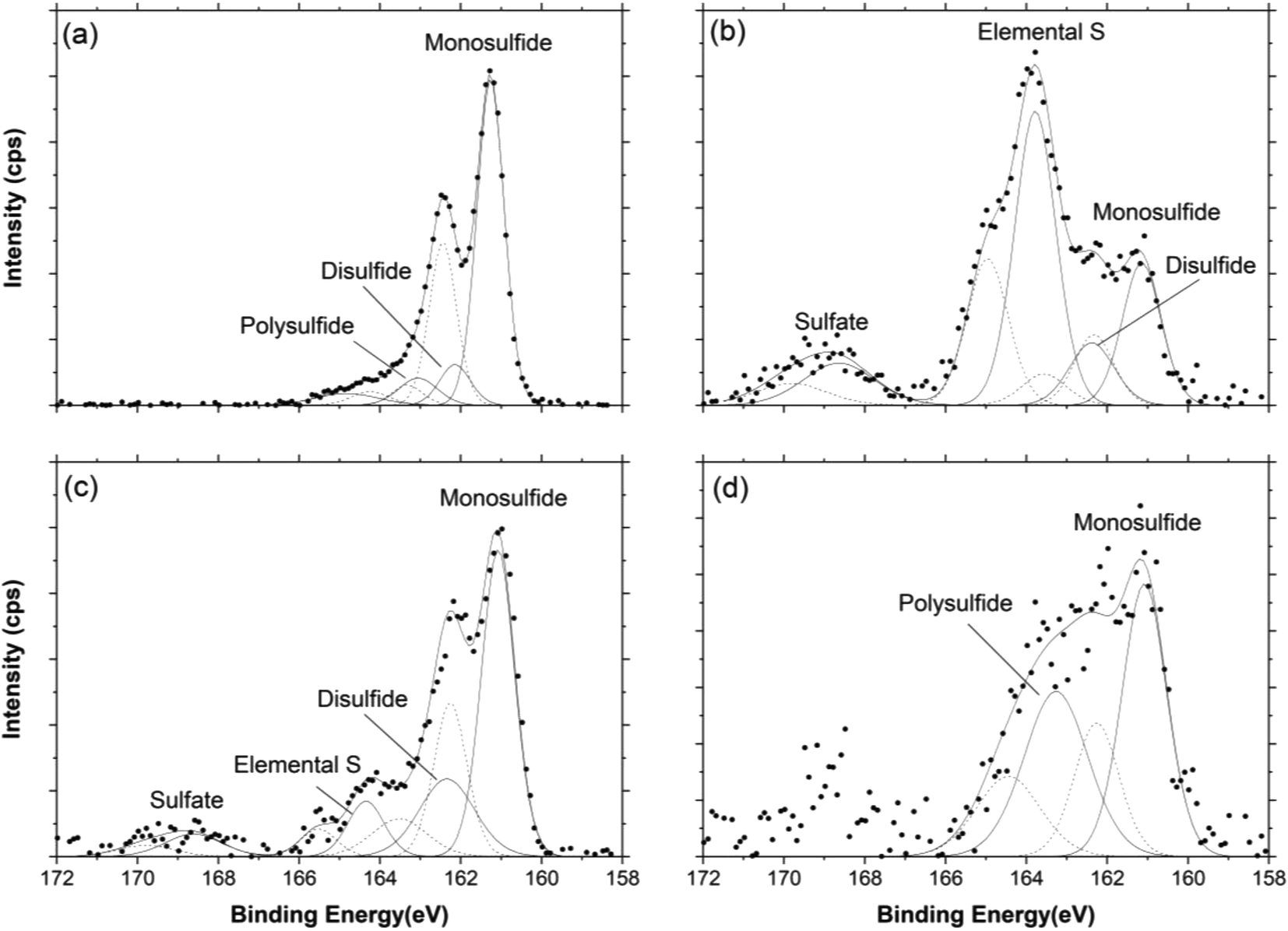 | ||
| Fig. 5 The X-ray photoelectron spectra of S(2p) line recorded from the sample surface of hematite-coated biochar (HCB). (a) near inlet of HCB-WCBC column, (b) near outlet of HCB-WCBC column, (c) near inlet of HCB-WC column, (d) near outlet of HCB-WC column. HCB columns fed with solutions from woodchip + biochar (WCBC) and woodchip (WC) columns. | ||
For the unreacted materials, sulfur peaks were not identified in any of the S(2p) spectra; hence, all S present in spectra for the reacted samples must be derived from S input to the HCB columns. For the unreacted HCB stock material, the Fe(2p3/2) spectrum is fitted with four multiplets with the primary peak centered at a binding energy (BE) of 711.2 eV (ESI,† Fig. S4), corresponding to Fe(III) in hematite.36,37 Surface-bound nitrogen in the HCB stock is inherited solely from the biochar (ESI,† Fig. S5), with a primary component in the N(1s) spectrum at 400.4 eV corresponding to carbon-bound nitrogen such as in amine groups. A lower intensity peak is fitted at 398.8 eV and is assigned to undefined organically-bound N.
For the reacted HCB materials, the Fe(2p3/2) spectra indicate the presence of surface-bound Fe(II) and Fe(III). In addition to the Fe(III) multiplet identified in unreacted hematite, primary multiplet peaks at 707.1–707.7 eV were assigned to Fe(II)38 in the four reacted HCB materials (ESI,† Fig. S4, Table S2) indicating at least a partial reduction of hematite-derived Fe(III) at the surfaces. The reduction of surface-bound Fe(III) to Fe(II) could be potentially accomplished through either the abiotic or autotrophic reduction of Fe2O3 by H2S (reaction 3) or the heterotrophic microbial reduction of Fe2O3 in the presence of lactate (reaction 5). Indeed, dissimilatory iron reduction is favored in the presence of lactate, relative to other electron donors such as acetate,33 and Fe2O3 is reduced to a greater degree in the presence of fermentative and sulfate-reducing bacteria, suggesting that reaction 5 should be dominating.
The S(2p) spectra (Fig. 5) indicated that the HCB surfaces were enriched in sulfur, primarily as monosulfide, elemental sulfur and polysulfides. Since the Fe(2p3/2) spectra indicate the presence of ferrous iron at the HCB surface (see Fig. S4, ESI†), it is reasonable to assume that monosulfide is present as FeS surface precipitates. Disulfide was also identified in three of the samples and is likely present as FeS2 (pyrite). It should be noted that the actual content of elemental sulfur may be greater than obtained through peak fitting, since elemental sulfur may partially evaporate under high vacuum, despite the use of cryo-XPS.13 Differences in sulfur surface speciation between the inlet and outlet regions of the HCB columns were observed, as well as differences in the abundance of the various surface species (see Table 1, Fig. 5). Indeed, the S concentration at the HCB-WCBC inlet was more than nine times as large as at the outlet, and S concentrations in the HCB-WCB column exceeded those in the HCB-WC column (ESI,† Table S2). The surface speciation of sulfur is relevant in terms of the long-term stability of the sulfur compounds. Both FeS and S0 are considered as precursors to FeS2,16 which is the most stable of these three compounds. Hence, the formation of FeS2 surface species would suggest a more stable binding mechanism for sulfur that is more resistant to remobilization.
| Sample | BE (eV) | BE (eV) | ||||
|---|---|---|---|---|---|---|
| S(2p3/2) | Species | Fraction | N(1s) | Species | Fraction | |
| Stock | — | None detected | 398.8 | Organic N, C–NH2 | 0.18 | |
| 400.4 | Amino group (C–N) | 0.82 | ||||
| HCB-WCBC inlet | 161.3 | Monosulfide (e.g., FeS) | 0.76 | 400.4 | Amino group (C–N) | 0.86 |
| 162.1 | Disulfide (e.g., FeS2) | 0.11 | 402.1 | Ammonium (NH4+) | 0.14 | |
| 163.0 | Polysulfide | 0.13 | ||||
| HCB-WCBC outlet | 161.2 | Monosulfide (e.g., FeS) | 0.24 | 400.2 | Amino group (C–N) | 0.92 |
| 162.4 | Disulfide (e.g., FeS2) | 0.11 | 401.8 | Ammonium (NH4+) | 0.08 | |
| 163.8 | Elemental sulfur | 0.52 | ||||
| 168.6 | Sulfate | 0.13 | ||||
| HCB-WC inlet | 161.1 | Monosulfide (e.g., FeS) | 0.58 | 400.2 | Amino group (C–N) | 0.83 |
| 162.3 | Disulfide (e.g., FeS2) | 0.24 | 401.6 | Ammonium (NH4+) | 0.17 | |
| 164.4 | Elemental sulfur | 0.11 | ||||
| 168.6 | Sulfate | 0.07 | ||||
| HCB-WC outlet | 161.1 | Monosulfide (e.g., FeS) | 0.54 | 400.2 | Amino group (C–N) | 0.89 |
| 163.3 | Polysulfide | 0.46 | 401.8 | Ammonium (NH4+) | 0.11 | |
Adsorbed SO42− was fitted at a BE of ca. 169 eV in all spectra with the exception of near the inlet of HCB-WCBC (Fig. 5a); the occurrence of adsorbed SO42− in HCB-WC (Fig. 5c) was expected since SO42− was detected at both the inlet and outlet of HCB-WC. However, SO42−-S was generally below the detection limit (13 mg L−1 SO42−-S) at the inlet of HCB-WCBC (Fig. 4a), suggesting that reduced forms of sulfur must have been oxidized near the outlet of HCB-WCBC in order to produce adsorbed SO42− in this location. Oxidation near the outlet may have been induced to oxygen intrusion along the outlet tubing during periods of interrupted flow, which occurred on several occasions during days 200–350, or may indicate the occurrence of H2S-driven Fe(III) reduction resulting in SO42− instead of S0 (cf.reactions (3) and (6)). XPS studies by Neal et al.15 demonstrated that predominant SO42− contributions were resolved in S(2p) spectra of H2S-exposed Fe2O3 surfaces (i.e., reaction (6)), while monosulfide and disulfide components were predominant on Fe2O3 surfaces exposed to SO42−-reducing bacteria and lactate (reaction (5)).
| H2S + 2Fe2O3(s) + 2H+ → 4Fe2+ + SO42− + 2H2O | (6) |
Application of technology in mining environment
The results of this study provide a proof-of-concept that will enable the construction of a field-based treatment system in a neutral mine drainage environment. For the treatment of acid mine drainage, the acidity in the mine water would first have to be neutralized since full denitrification of nitrate to nitrogen gas is inhibited by low pH.42In a field-based system, the results demonstrate that the denitrification and sulfate reduction steps need to be physically separated, since NH4+ production may result through DNRA at high C/N – ratios43 if these processes are allowed to proceed concurrently. This could be achieved by passing the water first through a denitrifying woodchip bioreactor for nitrogen removal, without the addition of lactate, and then treating the water in a second bioreactor for sulfate reduction with the addition of lactate. Since the second bioreactor will be producing H2S, it will need to be a contained system such as a reactor tank where gas emissions can be carefully controlled. Also, by using a reactor tank, lactate can be precisely dosed and byproducts (e.g., acetate) can be monitored. While woodchips with biochar have been shown in this study to be a suitable supporting medium for the growth of sulfate-reducing bacteria, it is quite likely that other supporting media would be equally suited in a bioreactor. Following the sulfate reduction tank, a reactor with hematite-coated biochar would be installed to capture H2S in the water phase.
Using the calculations previously discussed regarding the sulfur accumulation capacity of the HCB material (0.004 mol cm−3) and assuming a water flow of 0.5 L s−1 in a field-based system,13 iron in a 1 m3 HCB reactor would be consumed after 7 days. The relatively high consumption rate of the HCB material could be a major cost of the treatment system, since the consumption of the HCB material would require a method for either the disposal of the FeS sludge or for the recycling of the HCB material.
It is evident from the text above that a nitrate and sulfate-removing treatment system would no longer be a passive treatment technology such as a denitrifying woodchip bioreactor, but a system requiring energy input (e.g., pumps) and regular monitoring for FeS accumulation, H2S emissions, and Fe2+ and TOC release. For maintenance and monitoring purposes, it is not unlikely that the sulfate reduction and sulfide capture stages would be housed in an above-ground facility.
Ferrous iron release in a field system may be an issue. Since Fe2+ will precipitate in the presence of H2S, Fe2+ release probably occurred in the column experiments when H2S was depleted while high levels of organic carbon remained in solution (cf.reaction (5)). A decrease in lactate dosing would likely decrease the level of Fe2+ leaching, although additional studies are required to determine the conditions under which Fe2+ release is minimized.
This study indicates that there is a risk that the treatment system could release TOC (i.e., primarily acetate) to recipients. Lower TOC concentrations can be obtained by decreasing lactate dosing (see above), and also by allowing for variable lactate dosing to the reactor tank. Furthermore, the effluent stream from the bioreactor system could be partially recirculated to the inlet of the denitrifying bioreactor, providing denitrifiers with a readily-available carbon source.
Conclusions
Laboratory column experiments were used to study the sequential removal of NO3− and SO42−, where NO3− was removed through denitrification and SO42− was removed through SO42− reduction and the subsequent precipitation of solid-phase sulfur compounds such as FeS and pyrite. The experimental results indicated that a hydraulic residence time (HRT) of 5 days was adequate for the nearly complete removal of NO3− in a woodchip substrate, while a shorter HRT of 2 days led to incomplete denitrification. Ammonium production, most likely through DNRA, occurred during all phases of the experiment, but NH4+ concentrations reached relatively high levels (25–40 mg L−1 NH4+-N) after the addition of lactate as an external carbon source. Ammonium was an undesirable byproduct and a treatment scheme utilizing both denitrification and SO42− reduction should be designed for the physical separation of these processes.Sulfate reduction in the WC and WCBC columns resulted in only the production of low concentrations of H2S without the addition of an external carbon source. Once lactate was added to the input solution, the biochar-containing column reduced SO42− concentrations to a greater degree than the column with only woodchips; one explanation for this effect may be that the biochar has a high specific surface area with microenvironments where SO42−- reduction is promoted. X-ray photoelectron spectroscopy indicated that sulfur was accumulating in the HCB columns as multiple solid phases, including FeS, pyrite and elemental sulfur, where ferrous iron was likely produced from the conversion of Fe(III) in hematite through dissimilatory reductive Fe2O3 dissolution. Hematite was hence consumed in this reaction, but the iron and sulfur will be retained in the column as long as reducing conditions are maintained. As treatment proceeds, the HCB material will develop a relatively thick layer of sulfur compounds so that the material can no longer effectively remove H2S from solution. The actual capacity of the HCB material for sulfur retention has not been accurately determined, but the material will eventually need to be replaced or regenerated, and disposal options must be investigated.
Author contributions
M. Parvage contributed to project conceptualization, validation, data acquisition, laboratory work, and writing the original draft. R. Herbert contributed with methodology, supervision, resources, validation, review and writing final version of manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Many thanks are extended to Andrey Shchukarev, Umeå University, who performed the XPS analyses and fitted the spectra. The research project presented in this manuscript was conducted as part of the EIT RawMaterials-funded NITREM project (project number 17013). This activity has received funding from the European Institute of Innovation and Technology (EIT). This body of the European Union received support from the European Union's Horizon 2020 research and innovation programme.References
- D. Banks, P. Younger, R. T. Arnesen, R. E. Iversen and S. B. Banks, Mine-water chemistry: the good, the bad and the ugly, Environ. Geol., 1997, 32, 157–174 CrossRef.
- A. S. Ek and I. Renberg, Heavy metal pollution and lake acidity changes caused by one thousand years of copper mining at Falun, central Sweden, J. Paleolimnol., 2001, 26, 89–107 CrossRef.
- B. L. Bailey, L. J. D. Smith, D. W. Blowes, C. J. Ptacek, L. Smith and D. C. Sego, The Diavik Waste Rock Project: Persistence of contaminants from blasting agents in waste rock effluent, Appl. Geochem., 2013, 36, 256–270 CrossRef CAS.
- S. Chlot, A. Widerlund, E. Husson, B. Öhlander and F. Ecke, Effects on nutrient regime in two recipients of nitrogen-rich mine effluents in northern Sweden, Appl. Geochem., 2013, 31, 12–24 CrossRef CAS.
- Environmental Protection Agency, Aquatic Life Ambient Water Quality Criteria for Ammonia –freshwater, Report EPA 822-R-13-001, United States Environmental Protection Agency, Washington, DC, 2013 Search PubMed.
- S. Katsev, I. Tsandev, I. L'Heureux and D. G. Rancourt, Factors controlling long-term phosphorus efflux from lake sediments: Exploratory reactive-transport modeling, Chem. Geol., 2006, 234, 124–147 CrossRef.
- D. Zak, A. Kleeberg and M. Hupfer, Sulphate-mediated phosphorus mobilization in riverine sediments at increasing sulphate concentration, River Spree, NE Germany, Biogeochemistry, 2006, 80, 109–119 CrossRef CAS.
- B. Boström, G. Persson and B. Broberg, Bioavailability of different phosphorus forms in freshwater systems, Hydrobiologia, 1988, 170, 133–155 CrossRef.
- H. L. Golterman and N. T. de Oude, Eutrophication of Lakes, Rivers and Coastal Seas. in The Handbook of Environmental Chemistry, Volume 5 Part A Water Pollution, ed. O. Hutzinger, Springer-Verlag Berlin Heidelberg GmbH, 1991, pp. 80–124 Search PubMed.
- J. K. King, J. E. Kostka, M. E. Frischer and F. M. Saunders, Sulfate-Reducing Bacteria Methylate Mercury at Variable Rates in Pure Culture and in Marine Sediments, Appl. Environ. Microbiol., 2000, 66, 2430–2437 CrossRef CAS PubMed.
- British Columbia Ministry of Environment, Ambient Water Quality Guidelines for Sulphate, Technical Appendix, Ministry of Environment, Province of British Columbia, 2013, https://www2.gov.bc.ca/assets/gov/environment/air-land-water/water/waterquality/water-quality-guidelines/approved-wqgs/sulphate/bc_moe_wqg_sulphate.pdf Search PubMed.
- Land and Environmental Court of Appeals (Swedish, Mark- och miljööverdomstolen), Decision of the Land and Environmental Court of Appeals, Case no. M 4160-15, Land and Environmental Court of Appeals, Stockholm, Sweden, 2016, Available from https://www.domstol.se/globalassets/filer/domstol/markochmiljooverdomstolen/avgoranden/2016/m-4160-15.pdf (in Swedish) (accessed 2022-06-29) Search PubMed.
- A. Nordström and R. Herbert, Determination of major biogeochemical processes in a denitrifying woodchip bioreactor for treating mine drainage, Ecol. Eng., 2018, 110, 54–66 CrossRef.
- A. Nordström, M. Hellman, S. Hallin and R. Herbert, Microbial controls on net production of nitrous oxide in a denitrifying woodchip bioreactor, J. Environ. Qual., 2021, 50, 228–240, DOI:10.1002/jeq2.20181.
- A. L. Neal, S. Techkarnjanaruk, A. Dohnalkova, D. McCready, B. M. Peyton and G. G. Geesey, Iron sulfides and sulfur species produced at hematite surfaces in the presence of sulfate-reducing bacteria, Geochim. Cosmochim. Acta, 2001, 65, 223–235 CrossRef CAS.
- D. Rickard and G. W. Luther III, Kinetics of pyrite formation by the H2S oxidation of iron(II) monosulfide in aqueous solutions between 25 and 125°C: The mechanism, Geochim. Cosmochim. Acta, 1997, 61, 135–147 CrossRef CAS.
- C. M. Hansel, S. G. Benner, P. Nico and S. Fendorf, Structural constraints of ferric (hydr)oxides on dissimilatory iron reduction and the fate of Fe(II), Geochim. Cosmochim. Acta, 2004, 68, 3217–3229 CrossRef CAS.
- O. Lahav, G. Ritvo, I. Slijper, G. Hearne and M. Cochva, The potential of using iron-oxide-rich soils for minimizing the detrimental effects of H2S in freshwater aquaculture systems, Aquaculture, 2004, 238, 263–281 CrossRef CAS.
- Q. Zhou, X. Jiang, X. Li and W. Jiang, The control of H2S in biogas using iron ores as in situ desulfurizers during anaerobic digestion process, Appl. Microbiol. Biotechnol., 2016, 100, 8179–8189 CrossRef CAS PubMed.
- H. Runtti, E.-T. Tolonen, S. Tuomikoski, T. Luukkonen and U. Lassi, How to tackle the stringent sulfate removal requirements in mine water treatment—A review of potential methods, Environ. Res., 2018, 167, 207–222 CrossRef CAS PubMed.
- J. Lehmann, M. C. Rilig, J. Thies, C. A. Masiello, W. C. Hockaday and D. Crowley, Biochar effects on soil biota: A review, Soil Biol. Biochem., 2011, 43, 1812–1823 CrossRef CAS.
- M. Lawrineko and D. A. Laird, Anion exchange capacity of biochar, Green Chem., 2015, 17, 4628–4636 RSC.
- A. Shchukarev and M. Ramstedt, Cryo-XPS: probing intact interfaces in nature and life, Surf. Interface Anal., 2017, 49, 349–356 CrossRef CAS.
- M. Wan, A. Shchukarev, R. Lohmayer, B. Planer Fridrich and S. Peiffer, Occurrence of surface polysulfides during the interaction between ferric (hydr)oxides and aqueous sulfide, Environ. Sci. Technol., 2014, 28, 5076–5084 CrossRef PubMed.
- E. M. Bock, B. Coleman and Z. M. Easton, Effect of biochar on nitrate removal in a pilot-scale denitrifying bioreactor, J. Environ. Qual., 2016, 45, 762–771 CrossRef CAS PubMed.
- J. Audet, A. J'Eglot, L. Elsgaard, A. Ledet Maagaard, S. Reinhold Sørensen, D. Zak and C. C. Hoffmann, Nitrogen removal and nitrous oxide emissions from woodchip bioreactors treating agricultural drainage waters, Ecol. Eng., 2021, 169, 106328 CrossRef.
- A. Nordström and R. Herbert, Denitrification in a low temperature bioreactor system at two different hydraulic residence times: laboratory column studies, Environ. Technol., 2017, 38, 1362–1375 CrossRef PubMed.
- A. J. Burgin and S. K. Hamilton, Have we over emphasized the role of denitrification in aquatic ecosystem? A Review of Nitrate Removal Pathways, Front. Ecol. Environ., 2007, 5, 89–96 CrossRef.
- B. Kraft, H. E. Tegetmeyer, R. Sharma, M. G. Klotz, T. G. Ferdelman, R. L. Hettich, J. S. Geelhoed and M. Strous, Nitrogen cycling: The environmental controls that govern the end product of bacterial nitrate respiration, Science, 2014, 345(6197), 676–679, DOI:10.1126/science.1254070.
- R. C. Barnet and L. J. Garcia-Gill, Sulfide-induced dissimilatory nitrate reduction to ammonia in anaerobic freshwater sediments, FEMS Microbiol. Ecol., 1996, 21, 131–138 CrossRef.
- P. N. L. Lens, A. Visser, A. J. H. Janssen, L. W. Hulshoff Pol and G. Lettinga, Biotechnological treatment of sulfate-rich wastewaters, Crit. Rev. Environ. Sci. Technol., 1998, 28(1), 41–88 CrossRef CAS.
- Y. Zhao, N. Ren and A. Wang, Contributions of fermentative acidogenic bacteria and sulfate-reducing bacteria to lactate degradation and sulfate reduction, Chemosphere, 2008, 72, 233–242 CrossRef CAS PubMed.
- C. J. Lentini, S. D. Wankel and C. M. Hansel, Enriched iron(III)-reducing bacterial communities are shaped by carbon substrate and iron oxide mineralogy, Front. Microbiol., 2012, 3, 404, DOI:10.3389/fmicb.2012.00404.
- D. V. Sarkot, T. A. Ghezzehei and A. A. Berhe, Effectiveness of Biochar for Sorption of Ammonium and Phosphate from Dairy Effluent, J. Environ. Qual., 2014, 42, 1545–1554 CrossRef PubMed.
- J. Tian, V. Miller, P. C. Chiu, J. A. Maresca, M. Guo and P. T. Imhoff, Nutrient release and ammonium sorption by poultry litter and wood biochars in stormwater treatment, Sci. Total Environ., 2016, 553, 596–606 CrossRef CAS PubMed.
- P. Mills and J. L. Sullivan, A study of the core level electrons in iron and its three oxides by means of photoelectron spectroscopy, J. Phys. D: Appl. Phys., 1983, 16, 723–732 CrossRef CAS.
- A. Shchukarev and J.-F. Boily, XPS study of the hematite- aqueous solution interface, Surf. Interface Anal., 2008, 40, 349–353 CrossRef CAS.
- A. R. Pratt, I. J. Muir and H. W. Nesbitt, X-ray photoelectron and Auger electron spectroscopic studies of pyrrhotite and mechanism of air oxidation, Geochim. Cosmochim. Acta, 1994, 58, 827–841 CrossRef CAS.
- K. Shimizu, A. Shchukurev and J.-F. Boily, X-ray photoelectron spectroscopy of fast-frozen hematite colloids in aqueous solutions. 3. Stabilization of ammonium species by surface (hydr)oxo groups, J. Phys. Chem. C, 2011, 115, 6796–6801 CrossRef CAS.
- J. A. Davis and D. B. Kent, Surface complexation modeling in aqueous geochemistry, Rev. Mineral., 1990, 23, 177–260 Search PubMed.
- A. Mukherjee, A. R. Zimmerman and W. Harris, Surface chemistry variations among a series of laboratory-produced biochars, Geoderma, 2011, 163, 247–255 CrossRef CAS.
- R. M. Maier, Chapter 14 - Biogeochemical Cycling, in Environmental Microbiology, ed. R. M. Maier, I. L. Pepper and C. P. Gerba, 2nd edn, Academic Press, 2009, pp. 287–318 Search PubMed.
- E. M. Van Den Berg, U. Van Dongen, B. Abbas and M. C. Van Loosdrecht, Enrichment of DNRA bacteria in a continuous culture, ISME J., 2015, 9, 2153–2161, DOI:10.1038/ismej.2015.26.
Footnote |
| † Electronic supplementary information (ESI) available: Additional material is available through ESI. See DOI: https://doi.org/10.1039/d2ew00499b |
| This journal is © The Royal Society of Chemistry 2023 |