
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural principles of cation ordering and octahedral tilting in A-site ordered double perovskites: ferroelectric CaMnTi2O6 as a model system†
Elisabeth K.
Albrecht
and
Antti J.
Karttunen
 *
*
Department of Chemistry and Materials Science, Aalto University, P.O. Box 16100, FI-00076 Aalto, Finland. E-mail: antti.karttunen@aalto.fi
First published on 13th October 2022
Abstract
We have used quantum chemical methods to study the structural principles and energetics of A-site ordered AA′B2O6 double perovskites. 33 combinations of A-site ordering and Glazer tilting have been systematically studied for the ferroelectric CaMnTi2O6 model system. The used approach was able to predict the correct combination of A-site ordering and tilting of octahedra in comparison to the experimentally known CaMnTi2O6. The energy differences between the various combinations of A-site ordering and tilt systems show a large variation of tens of kJ mol−1 per formula unit, which suggests that the methodology used here can be used as a starting point for making reliable predictions on the structures of yet unknown A-site ordered double perovskites. The energy differences due to A-site ordering and octahedral tilting were larger compared to the energy difference arising from ferroelectric distortion in CaMnTi2O6. The energy differences between various hypothetical double perovskite structures could be explained by studying their structural characteristics in detail. The relative energies are closely correlated with the Mn–O distances and Mn coordination in the studied structures.
1 Introduction
Perovskites are a broad class of compounds showing a large variety of technologically relevant functionalities such as piezoelectricity, pyroelectricity, ferroelectricity, multiferroicity, and superconductivity.1 In the basic perovskite structure with the general structural formula ABX3 (space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), twelve-coordinated A cations are located at the corners of the primitive cubic unit cell, while the B cations are octahedrally coordinated by X anions located on the face centers of the unit cell. The anion octahedra are corner-linked. The most common anion is oxygen, leading to oxide perovskites ABO3, but other elements such as fluorine and chlorine can also be found at the X-site. By tuning the relative sizes and oxidation states of the cations and anions, the physical properties of perovskite materials can be modified and adapted to different requirements.1–3
m), twelve-coordinated A cations are located at the corners of the primitive cubic unit cell, while the B cations are octahedrally coordinated by X anions located on the face centers of the unit cell. The anion octahedra are corner-linked. The most common anion is oxygen, leading to oxide perovskites ABO3, but other elements such as fluorine and chlorine can also be found at the X-site. By tuning the relative sizes and oxidation states of the cations and anions, the physical properties of perovskite materials can be modified and adapted to different requirements.1–3
Most perovskite materials do not exhibit the perfect, ideal crystal structure in the space group Pmm. For example, a size mismatch of the cations can lead to the tilting of the BX6 octahedra or electronic effects can result in the distortion of the BX6 octahedra.4 Glazer proposed the most common notation for the tilting of octahedra in perovskites in his 1972 paper.5 In the Glazer notation, a, b, and c stand for the pseudocubic unit cell parameters in the three crystallographic directions. The pseudocubic unit cell, compared to the real unit cell, always refers to the cell around exactly one octahedron. If the octahedral tilts about two axes and therefore the lattice parameters in these directions are the same, they are described with the same letter. For example, in notation aac, the first two lattice parameters would be the same and the third would be different. Due to their linked corners, the rotation of one octahedron about a pseudocubic axis determines the rotations of all octahedra in the same plane. Along one axis, the octahedra can all be rotated in line, alternating, or not at all. These rotations are denoted by superscripts +, −, and 0, respectively. For example, notation a+a+c0 means that the pseudocubic lattice parameters are the same in the a and b directions, but different in the c direction, while the octahedra are tilted in line about the a and b axes but are not tilted about the c axis. Fig. 1 shows an ideal, untilted perovskite structure and two examples of perovskite structures with tilted octahedra. For a more detailed explanation of the Glazer notation and its symmetry relations, see the articles by Glazer5 and Woodward.6,7
 | ||
| Fig. 1 Perovskites with different Glazer tilt systems along the c direction: (a) ideal, untilted, (b) a0a0c+, and (c) a0a0c−. The A-site cations are in dark red, the B-site cations and their coordination polyhedra in blue, and the X-site anions in green. The unit cell in each system is drawn in black. All crystal structure illustrations in the manuscript have been prepared with the VESTA program.8 | ||
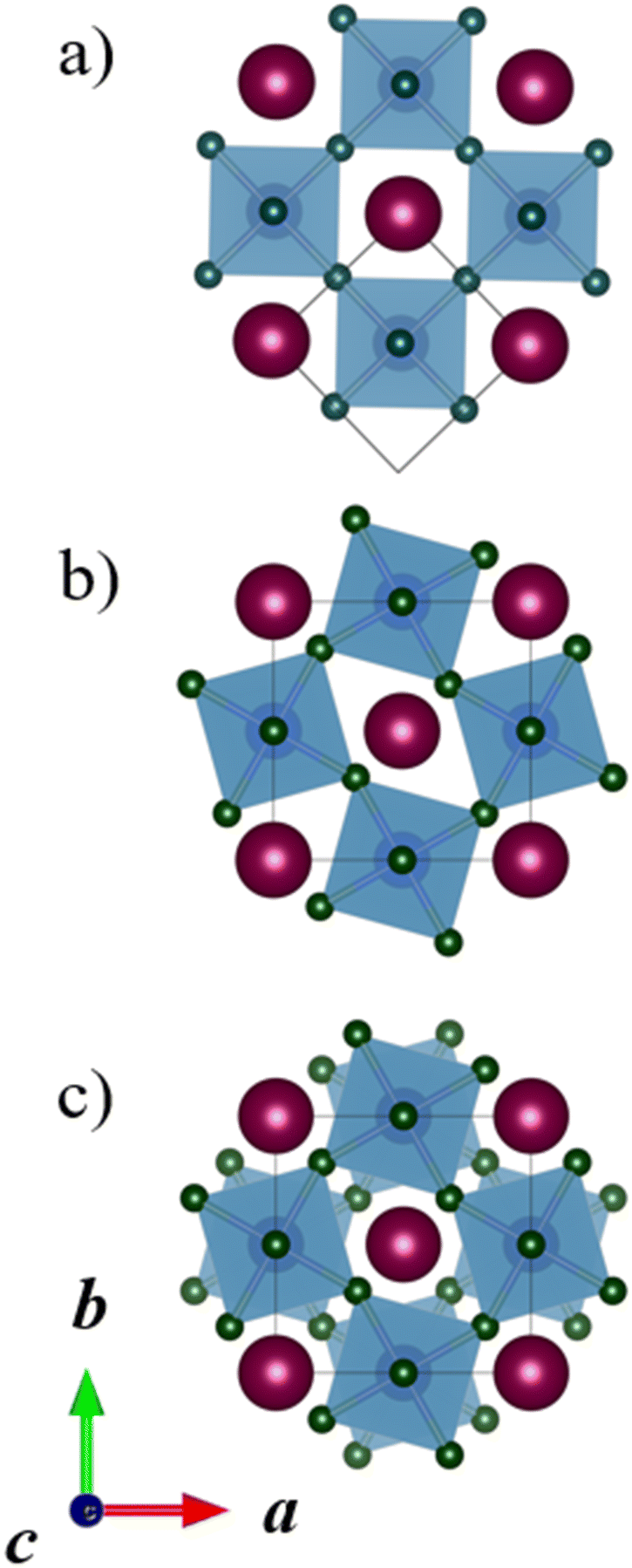
Perovskites, where the A sites or B sites are occupied with two different cations ordering in a certain way are called double perovskites.1,9,10 A-site ordered double perovskites with the general formula AA′B2X6 have two different cations on the A-site, while B-site ordered double perovskites with the general formula A2BB′X6 have two different cations on the B-site. Three different orderings of the cations are typically considered: columnar, planar, and rock-salt. Fig. 2 illustrates these orderings for A-site ordered double perovskites. Columnar, one-dimensional ordering, means that the cations of one type (A) form columns in one direction and these columns are surrounded by columns of the other cation type (A′). In planar, two-dimensional ordering, planes of A and A′ cations are alternating. The rock-salt ordering is a three-dimensional ordering, where each A cation has only A′ cations as the nearest neighbours. While A-site ordered double perovskites are often in the planar ordering, in B-site ordered double perovskites the rock salt ordering is the most common one.9,11–13 A-site cation ordering is much less common than B-site cation ordering and in the majority of the known AA′B2X6 perovskites, the A and A′ cations do not order. The general principles of cation ordering in both A-site and B-site ordered double perovskites have been discussed in detail by King and Woodward.9
 | ||
| Fig. 2 A-site ordered double perovskites with (a) columnar, (b) planar, and (c) rock salt ordering. The two cation species on the A-site are shown in dark red and purple, the B-site cations and their coordination polyhedra in blue, and the X-site anions in green. The unit cell in each system is drawn in black. | ||
Various possibilities for cation ordering and octahedral tilting lead to a large number of structural varieties in the case of double perovskites, as illustrated by a systematic survey on B-site ordered A2BB′O6 double perovskites.13 This also complicates the efforts to predict the crystal structures of novel double perovskites just based on their elemental composition. Such predictive approaches would also be helpful when determining the exact crystal structure of newly synthesized double perovskite materials by powder X-ray diffraction, as the diffraction patterns of different structural alternatives may sometimes be too similar to confidently be distinguished. In fact, often after determining secondary properties, the cation ordering or octahedral tilting pattern can be deduced.
In the case of simple ABX3 perovskites, Goldschmidt proposed already in 1926 a simple expression for tolerance factor t that could be used to predict the crystal system of a perovskite based on the ionic radii of A, B, and X.14 The simple Goldschmidt tolerance does not take octahedral tilting into account in any way, nor does it offer any insight into the ordering of the cations in double perovskites. Despite this, it is still useful as a starting point in high-throughput screening studies for predicting potential perovskite compositions, where additional geometric criteria for octahedral tilting have been included.15 New tolerance factor τ, incorporating the oxidation states of the cations, has also been derived for A2BB′X6 double perovskites, and stability rankings of potential new double perovskites have been derived based on it.16 Similar to the original tolerance factor formula, the τ formula does not take octahedral tilting into account, nor does it offer any insights into the ordering of the cations.
Compared to structure prediction approaches based on geometric parameters such as ionic radii, quantum chemical calculations with Density Functional Theory (DFT) provide computationally more intensive, but at the same time, much more powerful structure prediction approaches. DFT methods can assess the relative stability of any existing or predicted perovskite structure, taking fully into account all structural variations such as cation ordering, tilting of octahedra, and displacement of A- or B-site cations. A recent example of the use of DFT methods is in the report of Shojaei and Yin, where they studied ABX3 perovskite halides (A = Cs, Rb, K; B = Pb, Sn; X = I, Br, Cl) to correlate their energetics and octahedral tilting energies with known stability descriptors of perovskite halides.17 Also, Ding et al. compared few different tilt systems for the B-site ordered double perovskite Sr2CoRuO6 to confirm its cation ordering.18 However, to our knowledge, systematic DFT studies have not been used to create stability rankings of A-site or B-site ordered double perovskites with different octahedral tilt patterns.
Here, we present a quantum chemical investigation of cation ordering and octahedra tilting in A-site ordered AA′B2O6 double perovskites. We systematically study all relevant combinations of cation ordering and tilting of octahedra to provide a solid foundation for structure prediction and structure elucidation of A-site ordered AA′B2O6 double perovskites with DFT methods. At the same time, we study in detail the energetic effects of structural variations for several known AA′B2O6 double perovskites. As our main benchmark system, we used CaMnTi2O6 which is a ferroelectric A-site ordered double perovskite oxide with potential piezoelectric and pyroelectric properties. It is also being investigated as a Pb- and Bi-free photovoltaic material.19–22 The ferroelectricity of CaMnTi2O6 also provides insight into the energetic effects of structural distortion beyond the tilting of the octahedra.
2 Procedure and methods
2.1 Tilt systems and cation ordering
To study the different combinations of the A-site cation ordering and octahedral tilt system for CaMnTi2O6, suitable tilt systems were first selected. Glazer described 23 tilt systems in his original paper,5 but after a group theoretical analysis,23 15 simple distinct tilt systems remained. Out of these 15 tilt systems, 11 were taken into account for this work, as the remaining four tilt systems are low symmetry/transitional tilt systems that are observed as intermediates in a phase transition between higher symmetry structures.24 These 11 tilt systems are listed in Table 1 and they are in line with the current version of the SPuDS software that includes one additional tilt system in comparison to the original SPuDS paper.24| Tilt class | Tilts | Tilt number | Space group | |
|---|---|---|---|---|
| Zero-tilt | 000 | a 0 a 0 a 0 | 23 |
Pmm (221) |
| One-tilt | 00− | a 0 a 0 c − | 22 | I4/mcm (140) |
| 00+ | a 0 a 0 c + | 21 | P4/mbm (127) | |
| Two-tilt | 0−− | a 0 b − b − | 20 | Imma (74) |
| +0− | a + b 0 c − | 17 | Cmcm (63) | |
| 0++ | a 0 b + b + | 16 | I4/mmm (139) | |
| Three-tilt | −−− | a − a − a − | 14 |
Rc (167) |
| a − b − b − | 13 | C2/c (15) | ||
| −+− | a − b + a − | 10 | Pnma (62) | |
| ++− | a + a + c − | 5 | P42/nmc (137) | |
| +++ | a + a + a + | 3 |
Im (204) |
For each space group and tilt system, experimentally known perovskite crystal structures were searched from the structural databases and literature, and if an existing structure was available, then it was used to create the corresponding CaMnTi2O6 double perovskite structure template with columnar, planar, or rock salt ordering. If there was no existing perovskite crystal structure available for a certain tilt system, then the structure was built manually using the VESTA software.8 The tilt system a−a−a− (space group Rc) produced hexagonal unit cells. For the sake of comparability, calculations have been done with a pseudo-tetragonal (columnar, planar) or pseudo-cubic (rock salt) unit cell. These were obtained by deriving a unit cell with antiferromagnetic (AFM) ordering of the magnetic Mn(II) ions. For other tilt systems, the majority of the calculations were carried out for a ferromagnetic (FM) ground state, as there is no significant magnetic coupling between the Mn(II) ions. We found the FM and AFM ground states to show energy differences of less than 1 kJ mol−1. This finding is in line with the results of Gou et al., who studied the different spin configurations of CaMnTi2O6 with the HSE06 hybrid density functional method.19 For a few systems, the AFM ground state was used for purely technical reasons, to avoid convergence issues of the self-consistent field procedure during the DFT calculations (columnar a0b+b+ and a0a0c− tilt systems, planar a−b−b−, a0a0c+, and a0a0c− tilt systems, and rock salt a0b+b+ tilt system).
In addition to the hypothetical CaMnTi2O6 double perovskite structures described above, we included the experimentally determined crystal structure of CaMnTi2O6 in order to compare the theoretical results with the experimental results. The magnetic, electronic, and dielectric properties of the experimentally determined crystal structure were thoroughly studied by Gou et al.19
2.2 Computational details
The CRYSTAL1725 program package was used for all quantum chemical calculations. The hybrid PBE0 density functional method, (DFT-PBE0),26,27 was used in combination with all-electron, Gaussian-type basis sets based on Karlsruhe def2 sets.28 Triple-ζ-valence + polarization level basis sets were used for Mn, Ti, La, and O,29,30 while split-valence + polarization level basis sets were used for Ca and Ba (the Ca basis is included in the ESI†).31 The Monkhorst–Pack type k-meshes for sampling the reciprocal space are listed in the ESI.†32 Default DFT integration grids and optimization convergence thresholds of CRYSTAL17 were applied in all calculations. The calculations were carried out with the Coulomb and exchange integral tolerance factors (TOLINTEG) set to tight values of 8, 8, 8, 8, and 16. All calculations were carried out with the spin-polarized formalism due to the unpaired electrons of the Mn(II) ions. All reported relative energies are based on electronic energies obtained at 0 K, and Gibbs free energies have not been considered.CRYSTAL allows the use of the keyword FIXDEF to keep the ratio between two lattice parameters fixed throughout the geometry optimization or to fix a lattice parameter to a certain value. This option was used in some cases where the double perovskite space group arising from both tilting and A-site ordering allowed two lattice parameters to change independently, while the tilt system only would have restricted the two or more tilt angles and pseudocubic lattice parameters to be the same. For example, in the a−b−b− tilt systems, all A-site orderings lead to a monoclinic space group for the double perovskite, while the tilt system alone dictates the pseudocubic b and c lattice parameters to be the same. The need for the keyword FIXDEF is illustrated by the tilt system a−b−b− with planar A-site ordering (Fig. 3a), which requires the same monoclinic space group P2/c as the tilt system a−a−a− (Fig. 3b). These two systems have initially different lattice parameters, but as they have the same space group without the keyword FIXDEF, a full structural optimization would lead to the same optimized structure. So, FIXDEF was used in this case to constrain the structures to a certain tilt system; however, the energy difference between these two structures with and without the keyword FIXDEF would anyway be very small, clearly less than 1 kJ mol−1 per formula unit. Considering the accuracy of the used DFT method, these two tilt systems are thus anyway energetically and practically identical. In the case of rock salt A-site ordering, the tilt systems a−b−b− and a−a−a− could not be fully optimized with the default optimization convergence criteria in CRYSTAL and slightly looser criteria were applied (two to three times larger thresholds). This had no significant impact on the relative energies reported here.
 | ||
| Fig. 3 Two tilt systems of CaMnTi2O6 with planar A-site ordering (space group P2/c): (a) a−b−b− and (b) a−a−a−. Ca ions colored in dark grey-blue, Mn ions in purple, Ti ions and their coordination polyhedra in blue, and oxygen anions in red. | ||
3 Results and discussion
Table 2 shows an overview of the tilt systems and A-site orderings studied for CaMnTi2O6, together with relative energies obtained with the DFT-PBE0 method. The ESI† also includes the absolute electronic energies of the studied systems and their optimized geometries in CIF format. The CaMnTi2O6 structure, in the experimentally observed P42mc space group, optimized by the same DFT-PBE0 method, was used as a reference (Erel = 0 kJ mol−1). In addition to the octahedral tilting and A-site ordering considered here, the experimental crystal structure in the polar space group P42mc shows further energy-lowering atomic displacements that are discussed in detail below. Concerning geometries, the DFT-PBE0 method performs very well when the experimental (300 K) lattice parameters of CaMnTi2O6 (P42mc) are compared with DFT-optimized (0 K) lattice parameters: a parameter is underestimated by −0.5% and c-parameter is overestimated by 1.0%.| Tilt system | Glazer space group | Double perovskite space group | Examples | E rel [kJ mol−1] |
|---|---|---|---|---|
| a + a + c − | 105 P42mc | CaMnTi2O6 | 0 | |
| Columnar A-site ordering | ||||
| a + a + a + | 204 Im |
71 Immm | 31 | |
| a + a + c − | 137 P42/nmc | 137 P42/nmc | CaFeTi2O6 | 9 |
| a − b + a − | 62 Pnma | 11 P21/m | 21 | |
| a − b − b − | 15 C2/c | 13 P2/c | 52 | |
| a − a − a − | 167 Rc |
13 P2/c | 52 | |
| a 0 b + b + | 139 I4/mmm | 139 I4/mmm/139 I4/mmm | 44 | |
| a + b 0 c − | 63 Cmcm | 25 Pmm2 | 20 | |
| a 0 b − b − | 74 Imma | 51 Pmma | 55 | |
| a 0 a 0 c + | 127 P4/mbm | 65 Cmmm | 91 | |
| a 0 a 0 c − | 140 I4/mcm | 132 P42/mcm/111 P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2m 2m |
87 | |
| a 0 a 0 a 0 | 221 Pmm |
123 P4/mmm | 184 | |
| Planar A-site ordering | ||||
| a + a + a + | 204 Im |
47 Pmmm | 65 | |
| a + a + c − | 137 P42/nmc | 115 Pm2 |
49 | |
| a − b + a − | 62 Pnma | 26 Pmc21 | NdBaMn2O6 | 26 |
| a − b − b − | 15 C2/c | 13 P2/c/3 P2 | 41 | |
| a − a − a − | 167 Rc |
13 P2/c | 41 | |
| a 0 b + b + | 139 I4/mmm | 123 P4/mmm | 70 | |
| a + b 0 c − | 63 Cmcm | 38 Amm2 | 38 | |
| a 0 b − b − | 74 Imma | 51 Pmma | 49 | |
| a 0 a 0 c + | 127 P4/mbm | 65 Cmmm/65 Cmmm | 90 | |
| a 0 a 0 c − | 140 I4/mcm | 125 P4/nbm/111 P2m |
82 | |
| a 0 a 0 a 0 | 221 Pmm |
123 P4/mmm | LaBaMn2O6 | 183 |
| Rock salt A-site ordering | ||||
| a + a + a + | 204 Im |
200 Pm |
63 | |
| a + a + c − | 137 P42/nmc | 115 Pm2 |
46 | |
| a − b + a − | 62 Pnma | 31 Pmn21 | 26 | |
| a − b − b − | 15 C2/c | 5 C2/3 P2 | 41 | |
| a − a − a − | 167 Rc |
5 C2/3 P2 | 46 | |
| a 0 b + b + | 139 I4/mmm | 123 P4/mmm/123 P4/mmm | 63 | |
| a + b 0 c − | 63 Cmcm | 38 Amm2 | 35 | |
| a 0 b − b − | 74 Imma | 44 Imm2 | 41 | |
| a 0 a 0 c + | 127 P4/mbm | 136 P42/mnm | 78 | |
| a 0 a 0 c − | 140 I4/mcm | 121 I2m |
71 | |
| a 0 a 0 a 0 | 221 Pmm |
225 Fmm |
NaBaLiNiF6 | 183 |
In the case of columnar A-site ordering, the correct final geometry a+b0c− tilt system could not be obtained. This combination of the tilt system and A-site ordering leads to a relatively low-symmetry space group, Pmm2, where the octahedra had the freedom to tilt in a different and more complex way compared to the ideal a+b0c− tilting. Here, we report the relative energy for the fully optimized structure which cannot be described by the Glazer notation, as this resulted in a relatively low-energy geometry with (Erel = 20 kJ mol−1) that is the second-lowest in energy after columnar A-site ordering with a+a+c− tilting (Erel = 9 kJ mol−1). This is also an example of a situation where a full DFT treatment may reveal structural alternatives that cannot be predicted simply by considering idealized tilt systems with empirical rules. However, the ideal tilt systems still provide the best starting point for a systematic study of the various structural alternatives.
The energy differences between the calculated A-site ordering/tilt system combinations and the experimental CaMnTi2O6 structure range from 9 kJ mol−1 to 184 kJ mol−1 per formula unit (Table 2). In all three orderings, the ideal perovskite structure, a0a0a0, has by far the highest energy difference of over 180 kJ mol−1 per formula unit. The non-tilted highest-energy structures are followed by the single tilt systems, a0a0c+ and a0a0c−. The energy differences between two- and three-tilt systems are already much smaller and both options can lead to low-energy structures. Two tilts such as a0b−b− already decreased the energy difference immensely compared to the untilted ideal perovskite structure, down to, for example, 41 kJ mol−1 in the rock salt ordering.
Table 2 shows that in the planar and rock salt ordering, the a−b+a−-tilts are the lowest energy ones and even in the columnar ordering, it is the second-lowest one if the a+b0c−-tilt is not taken into account (optimization in the low-symmetry space group led to a tilt system that cannot be described by the Glazer notation). Generally, the energy ordering of tilt systems clearly varies within all three A-site orderings. When we looked for correlations between the relative energy and the lattice parameters or the relative energy and the Ti–O distances, we did not find any. Since the tilt angles are related to the lattice parameters, they also did not show any reasonable correlation with the relative energies. Therefore, we looked into A-site cations to find correlations between the structural parameters and the relative energies of the studied systems.
The A-site cations play a major role in the tilting of octahedra in perovskites. Therefore, it is likely that the A-site cations are also a major driving force for the relative energy differences obtained for the different tilt systems. In particular, the coordination number and bond lengths of A atoms with the surrounding oxygen atoms play a key role. While in the specific case of CaMnTi2O6 the Ca–O interactions are strongly ionic (Pauling electronegativity difference 2.44), the Mn–O bonds may possess slightly more covalent nature (Pauling electronegativity difference 1.89). This leads to the coordination of Mn ions being the main driving force in CaMnTi2O6 for the relative energy differences. Even though a six-fold coordination is common for Mn2+, the preferred coordination of Mn in CaMnTi2O6 is four: in the experimental CaMnTi2O6 crystal structure, half of the Mn cations are square-planar coordinated and half of them tetrahedrally coordinated. Of the tilt systems studied here, the only tilt system that allows Mn to be six-fold coordinated is a+b0c−. The resulting coordination polyhedron can be considered as a distorted trigonal prism and this coordination for Mn is not energetically favorable.
Fig. 4 shows the distance between Mn and O atoms for all 11 tilt systems and also the sum of the ionic radii of Mn(II) and O(-II) as a reference point for an “ideal” Mn–O distance.33 For each A-site ordering, the x-axis in the plot lists the tilt systems from the lowest (left) to the highest (right) relative energy with respect to the optimised CaMnTi2O6 structure, in the experimentally observed P42mc space group. Different coordination numbers are also denoted in addition to the Mn–O distances.
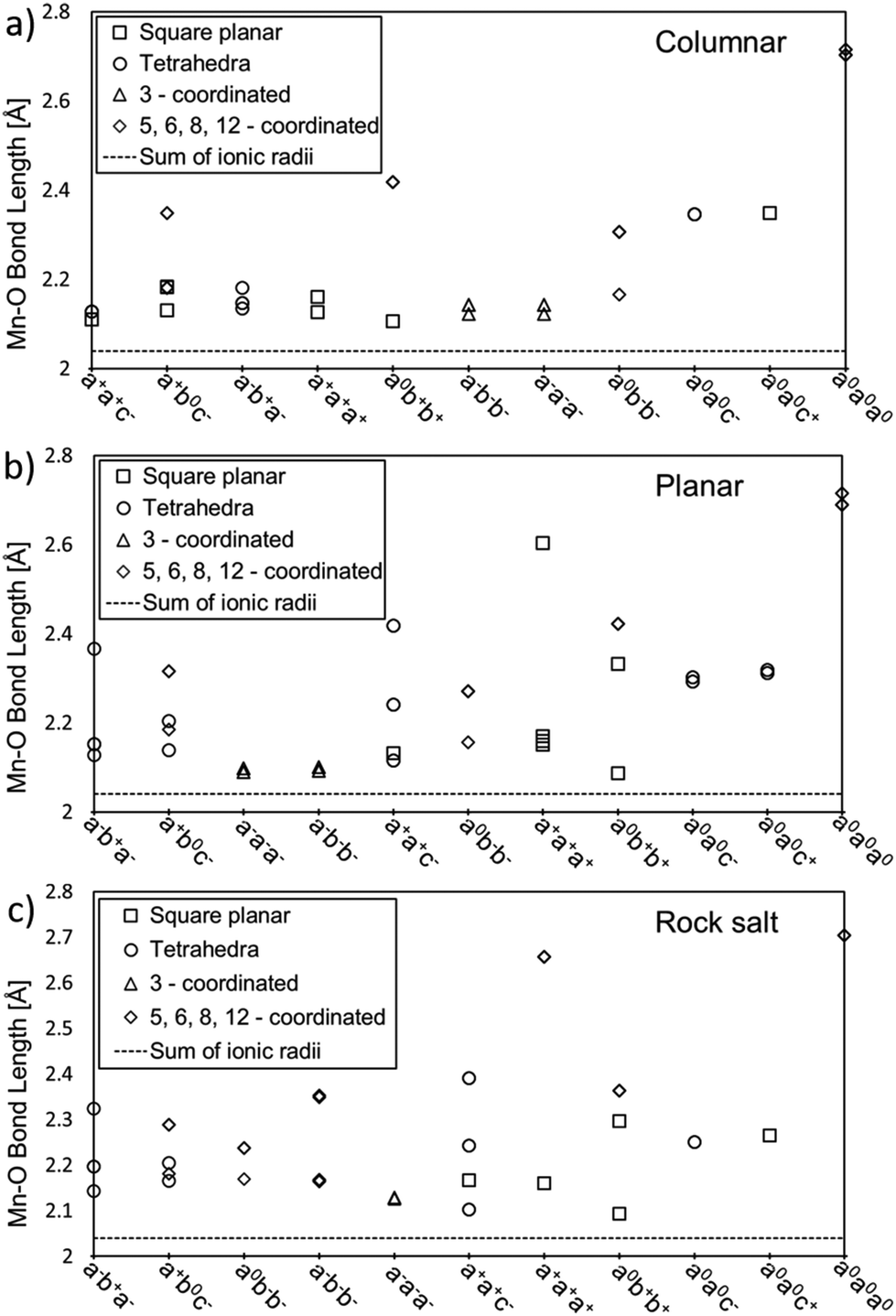 | ||
| Fig. 4 Mn–O distances for all studied tilt systems and A-site ordering: (a) columnar, (b) planar, and (c) rock salt. The x-axis lists the tilt systems from the lowest (left) to the highest (right) relative energy with respect to the experimental CaMnTi2O6 structure. Different coordinations are depicted by squares for square planar coordination, circles for tetrahedral coordination, triangles for three-fold coordination and diamonds for more than four-fold coordination. The sum of ionic radii of Mn(II) (0.66 Å) and O(-II) (1.38 Å) is shown by a dotted line at the bottom of each plot.33 | ||
The distances between Mn and O atoms range from 2.09 Å in the planar a0b+b+ system to 2.72 Å in the columnar and planar a0a0a0 systems. For all three high-energy a0a0a0 systems, the Mn–O distances are way too long for any covalent interaction. Overall, there is a tendency for higher-energy tilt systems to have longer Mn–O distances. However, there are also exceptions. For example, in columnar A-site ordering, the tilt system a0b+b+ has square-planar coordinated Mn cations with Mn–O distances close to the sum of the ionic radii and hence rather optimal bond lengths, but still the tilt system is not among the lowest-energy ones. The reason for this is that there are also Mn ions which are 8-coordinated with a long Mn–O distance of about 2.4 Å.
In the case of planar A-site ordering, the Mn–O distances in a−a−a− and a−b−b− are rather short and even closer to the sum of ionic radii than in the lowest-energy tilt system a−b+a−, but the coordination of the Mn atoms is 3-fold and not the optimal 4-fold. Another good example of the planar A-site ordering is the high-energy a+a+a+ tilt system where all Mn atoms have square-planar coordination and most Mn–O distances are rather close to the ideal sum of ionic radii. However, there is one Mn–O distance of about 2.6 Å, which drives the energy of this tilt system up.
In a similar fashion to the rock salt A-site ordering, the a0b+b+ tilt system has the shortest Mn–O distance of about 2.1 Å in square planar coordination but is still far from the lowest energy tilt system in energy. The reason is that this tilt system also contains 8-coordinated manganese cations with a long Mn–O distance of about 2.4 Å. A particularly interesting case in the rock salt A-site ordering is the tilt system a0a0c+. In principle, the Mn atoms have optimal square-planar coordination and the Mn–O distances are not excessively long, but still this tilt system has the second-highest relative energy. In this case, other structural factors may also play a role: there are Ca–O distances of 2.27 Å, which are significantly shorter compared to typical Ca–O distances.
Comparing the lowest-energy tilt systems of planar and rock salt A-site ordering (a−b+a− in both cases) with the lowest energy tilt system in columnar ordering (a+a+c−), it is clear that the difference in energy here seems to originate from the different Mn–O distances. While in all three A-site orderings the lowest-energy tilt systems contain only 4-coordinated Mn cations, in planar and rock salt ordering some of the bond lengths range between 2.3 Å and 2.4 Å, whereas in columnar ordering all Mn–O distances are about 2.1 Å long and close to the sum of the ionic radii.
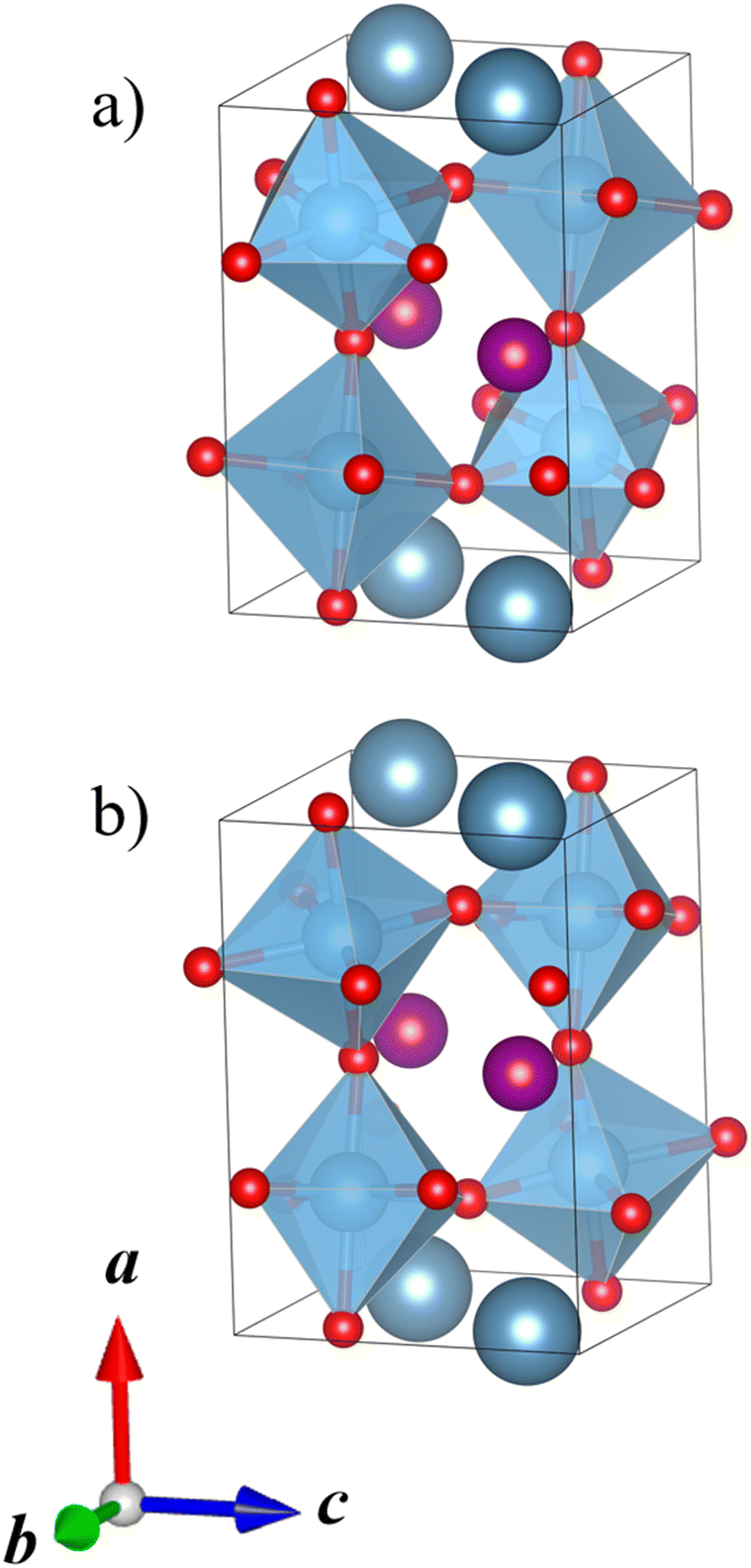
Our DFT calculations correctly predict that the lowest-energy combination of A-site ordering and tilt systems is the columnar-ordered a+a+c− tilt system (Fig. 5a) that has been found in the experimental crystal structure of CaMnTi2O6 (Fig. 5b).21 However, even though the experimental and computational A-site ordering and the tilt system are in agreement, the optimised CaMnTi2O6 structure in the experimentally observed P42mc space group is 9 kJ mol−1 lower in energy compared to the lowest-energy calculated structure. This non-negligible difference arises from the atomic displacement of the square-planar Mn ions, together with the slight displacement of Ti ions, discussed in detail by Gou et al.19 In the experimental structure, the Mn cations are displaced slightly out of the square plane and the symmetry is decreased from the non-polar space group P42/nmc to the polar space group P42mc. In our screening of the A-site ordering and tilt systems, we used the non-polar CaFeTi2O6 crystal structure (P42/nmc) as a starting point for the columnar a+a+c− tilt system. In this structure, the square-planar Mn ions stay in the square plane and Ti ions stay in the center of the octahedra. If the symmetry of the structure is lowered to P42mc and the Mn atoms are allowed to be displaced from the square plane, the energy is further lowered.
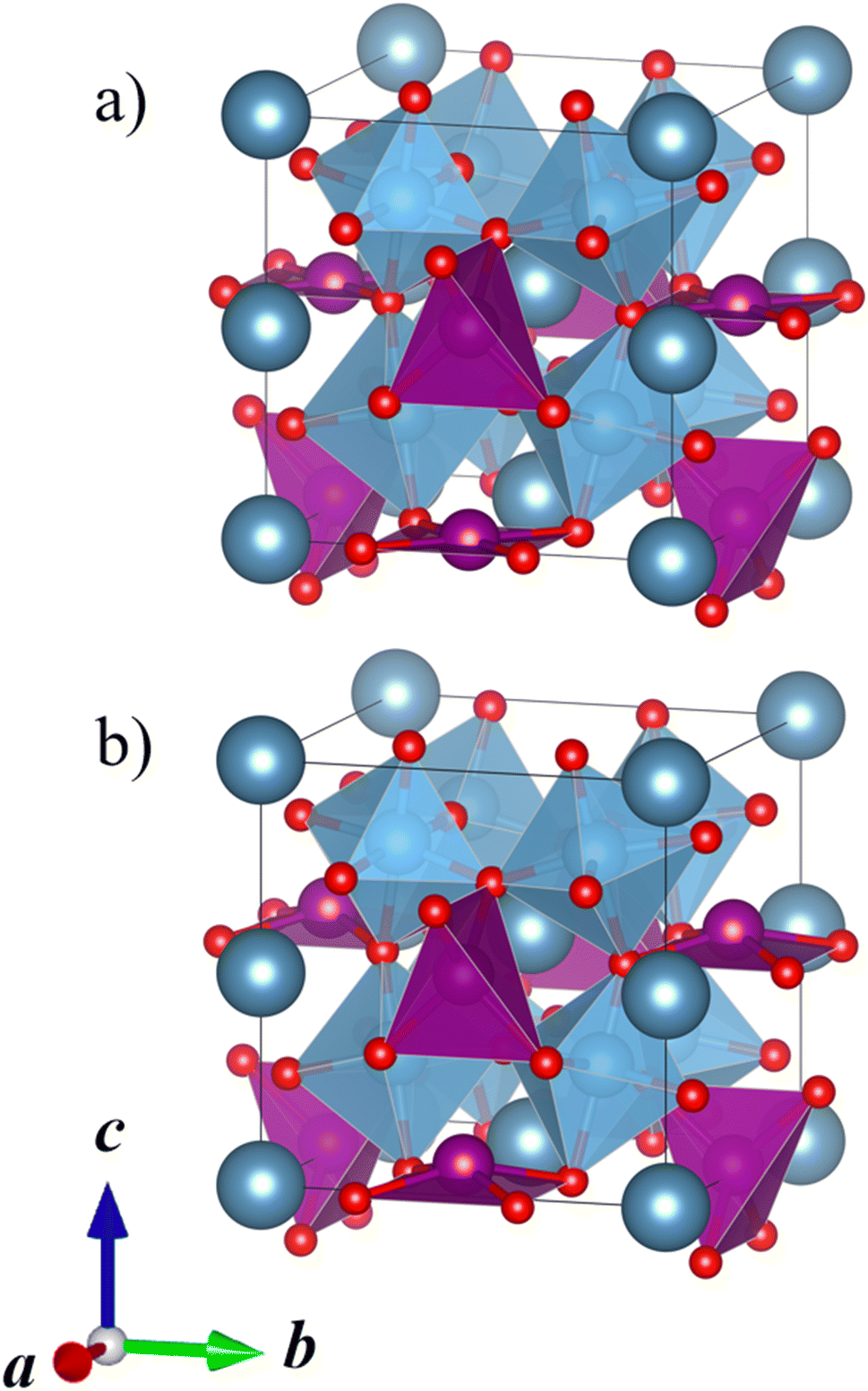 | ||
| Fig. 5 (a) Calculated lowest-energy structure of CaMnTi2O6 (P42/nmc, Erel = 9 kJ mol−1 per formula unit). (b) Experimental ferroelectric CaMnTi2O6 structure with the square-planar Mn cations displaced (P42mc, Erel = 0 kJ mol−1 per formula unit).21 Ca ions colored in dark grey-blue, Mn ions in purple, Ti ions and their coordination polyhedra in blue, and oxygen anions in red. | ||
In this work, we have focused on the A-site ordering and tilting of the octahedra and have not systematically studied the effect of atomic displacements such as ferroelectric distortion. Group-theoretical analyses of octahedral tilting in ferroelectric perovskites have been carried out, showing that the number of structural alternatives increases significantly even for simple perovskites.34,35 For double perovskites, the A- or B-site ordering further has to be taken into account. A systematic DFT study of such ferroelectric distortion in A-site ordered perovskites is expected to provide similar insights into their structural principles and energetics as the present investigation. In the case of CaMnTi2O6, the energetic effect of the ferroelectric displacements is 9 kJ mol−1 per formula unit, which is still less than the energy difference between the lowest energy and second lowest energy tilt systems studied in this work (11 kJ mol−1 per formula unit). Therefore, already the study of A-site ordering and tilting of octahedra seems to provide reasonable and useful structural predictions even for ferroelectric double perovskites.
We also benchmarked our computational approach for LaBaMn2O6, which is an A-site ordered double perovskite crystallizing in the space group P4/mmm (a0a0a0 tilt system, planar A-site ordering).36 Four structural alternatives were calculated, mainly to study the energetic effect of the A-site ordering (Table 3). In our calculations, the experimentally found combination of the tilt system and A-site ordering comes out as the lowest energy one. The columnar A-site ordering is only 3 kJ mol−1 per formula unit higher in energy, while the rock salt ordering already has a clear difference from the planar ordering. In this double perovskite, La and Ba cations do not provoke tilting of the octahedra and the non-tilted ideal structure is less prone to the influence of the A-site ordering (planar vs. columnar). When LaBaMn2O6 is optimized in the experimental CaMnTi2O6 crystal structure with tilted octahedra (P42mc), the structure is higher in energy compared to the non-tilted structure. However, the energy difference is only 6 kJ mol−1 per formula unit.
| Structure | Tilt system | Space group | E rel [kJ mol−1] |
|---|---|---|---|
| Planar LaBaMn2O6 (exp.) | a 0 a 0 a 0 | P4/mmm | 0 |
| Columnar LaBaMn2O6 | a 0 a 0 a 0 | P4/mmm | 3 |
| Rock salt LaBaMn2O6 | a 0 a 0 a 0 | P4/mmm | 30 |
| CaMnTi2O6 (exp.) | a + a + c − | P42mc | 6 |
4 Conclusions
We have used quantum chemical methods to study systematically all relevant combinations of cation ordering and tilting in A-site ordered AA′B2O6 double perovskites. The used DFT-PBE0 method is able to predict the correct combination of A-site ordering and tilting of octahedra in comparison to the experimentally known CaMnTi2O6 double perovskite. The energy differences between the various combinations of A-site ordering and tilt systems show a large variation of tens of kJ mol−1 per formula unit, which suggests that the methodology used here can be used as a starting point for making reliable predictions on the structures of yet unknown A-site ordered double perovskites. The energy differences due to A-site ordering and octahedral tilting were larger compared to the energy difference arising from ferroelectric distortion in CaMnTi2O6. We investigated the relative energies of various CaMnTi2O6 structural alternatives in detail and the main driving forces for the energy differences are Mn–O distances and Mn coordination. Similar structural analysis can be readily extended to A-site ordered double perovskites with other compositions. The methodology used here can be further improved and extended to several directions. Consideration of phonon properties and thermodynamics (Gibbs free energies) would further improve the accuracy of the predictions. To enable full investigation of thermodynamics, the computational efficiency probably needs to be increased by using for example semi-empirical tight-binding density functional methods,37 but benchmarking with respect to standard DFT methods is needed first. The methodology can be extended towards ferroelectric distortion with the help of previous group-theoretical groundwork.34,35 Finally, a similar approach can also be used to make predictions for B-site ordered double perovskites, where group-theoretical groundwork has also been laid,34,35 but magnetism will play a more significant role compared to the work presented here.Author contributions
Elisabeth K. Albrecht: Conceptualization, investigation, visualization, writing – original draft preparation, and writing – review and editing. Antti. J. Karttunen: Conceptualization, writing – review and editing, supervision, and funding acquisition. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Academy of Finland for funding (Grant No. 317273) and CSC – The Finnish IT Center for Science for computational resources.References
- R. J. D. Tilley, Perovskites: structure-property relationships, Wiley, Chichester, West Sussex, United Kingdom, 1st edn, 2016 Search PubMed.
- N. Xie, J. Zhang, S. Raza, N. Zhang, X. Chen and D. Wang, J. Phys.: Condens. Matter, 2020, 32, 315901 CrossRef CAS PubMed.
- H. W. Eng, P. W. Barnes, B. M. Auer and P. M. Woodward, J. Solid State Chem., 2003, 175, 94–109 CrossRef CAS.
- M. W. Lufaso and P. M. Woodward, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 10–20 CrossRef PubMed.
- A. M. Glazer, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 3384–3392 CrossRef CAS.
- P. M. Woodward, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 44–66 CrossRef.
- P. M. Woodward, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 32–43 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- G. King and P. M. Woodward, J. Mater. Chem., 2010, 20, 5785 RSC.
- M. C. Knapp and P. M. Woodward, J. Solid State Chem., 2006, 179, 1076–1085 CrossRef CAS.
- P. Davies, H. Wu, A. Borisevich, I. Molodetsky and L. Farber, Annu. Rev. Mater. Res., 2008, 38, 369–401 CrossRef CAS.
- A. A. Belik, Dalton Trans., 2018, 47, 3209–3217 RSC.
- S. Vasala and M. Karppinen, Prog. Solid State Chem., 2015, 43, 1–36 CrossRef CAS.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef CAS.
- M. R. Filip and F. Giustino, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5397–5402 CrossRef CAS PubMed.
- C. J. Bartel, C. Sutton, B. R. Goldsmith, R. Ouyang, C. B. Musgrave, L. M. Ghiringhelli and M. Scheffler, Sci. Adv., 2019, 5, eaav0693 CrossRef CAS PubMed.
- F. Shojaei and W.-J. Yin, J. Phys. Chem. C, 2018, 122, 15214–15219 CrossRef CAS.
- J. Ding, Y. Liu and Y. Zhang, J. Magn. Magn. Mater., 2021, 535, 168035 CrossRef CAS.
- G. Gou, N. Charles, J. Shi and J. M. Rondinelli, Inorg. Chem., 2017, 56, 11854–11861 CrossRef CAS.
- J. Herrero-Martín, J. Ruiz-Fuertes, T. Bernert, M. Koch-Müller, E. Haussühl and J. L. García-Muñoz, Phys. Rev. B, 2018, 97, 235129 CrossRef.
- A. Aimi, D. Mori, K.-i. Hiraki, T. Takahashi, Y. J. Shan, Y. Shirako, J. Zhou and Y. Inaguma, Chem. Mater., 2014, 26, 2601–2608 CrossRef CAS.
- Z. Li, Y. Cho, X. Li, X. Li, A. Aimi, Y. Inaguma, J. A. Alonso, M. T. Fernandez-Diaz, J. Yan, M. C. Downer, G. Henkelman, J. B. Goodenough and J. Zhou, J. Am. Chem. Soc., 2018, 140, 2214–2220 CrossRef CAS PubMed.
- C. J. Howard and H. T. Stokes, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 782–789 CrossRef.
- M. W. Lufaso and P. M. Woodward, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 725–738 CrossRef CAS PubMed.
- R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich-Wilson, B. Civalleri, L. Maschio, M. Rérat, S. Casassa, J. Baima, S. Salustro and B. Kirtman, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, 1–36 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- M. S. Kuklin, K. Eklund, J. Linnera, A. Ropponen, N. Tolvanen and A. J. Karttunen, Molecules, 2022, 27, 1–26 CrossRef PubMed.
- A. J. Karttunen, T. Tynell and M. Karppinen, Nano Energy, 2016, 22, 338–348 CrossRef CAS.
- S. I. Ivlev, A. J. Karttunen, M. R. Buchner, M. Conrad, R. V. Ostvald and F. Kraus, Crystals, 2018, 8, 1–14 Search PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- H. T. Stokes, E. H. Kisi, D. M. Hatch and C. J. Howard, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 934–938 CrossRef PubMed.
- K. S. Aleksandrov and J. Bartolomé, Phase Transitions, 2001, 74, 255–335 CrossRef CAS.
- T. Nakajima, H. Kageyama, H. Yoshizawa, K. Ohoyama and Y. Ueda, J. Phys. Soc. Jpn., 2003, 72, 3237–3242 CrossRef CAS.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1493 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal structures of the studied double perovskite systems in CIF format, the absolute electronic energies of the studied systems, and additional computational details. See DOI: https://doi.org/10.1039/d2dt02283d |
| This journal is © The Royal Society of Chemistry 2022 |