
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cavity-shaped cis-chelating P,N ligand for highly selective nickel-catalysed ethylene dimerisation†
Yang
Li
 a,
Katrin
Pelzer
a,
Damien
Sechet
b,
Geordie
Creste
a,
Dominique
Matt
b,
Pierre
Braunstein
*a and
Dominique
Armspach
*a
a,
Katrin
Pelzer
a,
Damien
Sechet
b,
Geordie
Creste
a,
Dominique
Matt
b,
Pierre
Braunstein
*a and
Dominique
Armspach
*a
aEquipe Confinement Moléculaire et Catalyse, Institut de Chimie de Strasbourg, UMR 7177 CNRS, Université de Strasbourg, 4, rue Blaise Pascal, CS 90032, 67081 Strasbourg Cedex, France. E-mail: d.armspach@unistra.fr
bLaboratoire de Chimie Inorganique Moléculaire et Catalyse, Institut de Chimie de Strasbourg, UMR 7177 CNRS, Université de Strasbourg, 4, rue Blaise Pascal, CS 90032, 67081 Strasbourg Cedex, France
First published on 21st July 2022
Abstract
The presence of a permethylated α-cyclodextrin (α-CD) cavity in a chelating P,N ligand promotes exclusive formation of 1 : 1 ligand/metal complexes. In MX2 complexes, one of the two halido ligands is forced to reside inside the CD hollow while the second one is pointing outside. Unlike its cavity-free analogue, a Ni(II) complex of the CD ligand is a highly selective precatalyst for ethylene dimerisation (96% C4 selectivity with up to 95% of 1-butene within the C4 fraction).
Inwardly directed donor atoms rigidly grafted on a macrocyclic host molecule are capable of facilitating a metal centred reaction to take place inside the cavity-shaped ligand.1–5 This feature is prone to produce high substrate recognition,6–14 catalytic chemo-15 and regioselectivity16–20 as well as enantioselectivity if the receptor is optically active.21 By connecting two C-6 carbon atoms of two glucose units of an α-, β- or γ-CD with a single donor atom such as nitrogen22 or phosphorus,23–25 (Fig. 1i) or a short NHC unit26–28 (Fig. 1ii), it was previously shown that the resulting bridge is sufficiently rigid to force the donor atom lone pair to point towards the CD interior. Doubly bridged ligands with a similar metal confining feature have also been prepared (Fig. 1iii).23,24 Almost all ligands of that type are monodentate or trans-chelating bidentate. However, most metal-catalysed reactions involve cis-chelate complexes. Increasing the structural diversity of cis-chelating ligands displaying a metal confining unit22,29–31 would considerably widen the scope of metal binding host molecules, in particular if catalytically relevant P(III) atoms could be incorporated into the macrocyclic structure. Self-assembled, cis-chelating P,N ligands consisting of a water-soluble phosphine ligand included in a mono-N,N-dialkylamino-β-CD were already prepared in water, but in these complexes, the metal centre is not located in the CD cavity.32,33
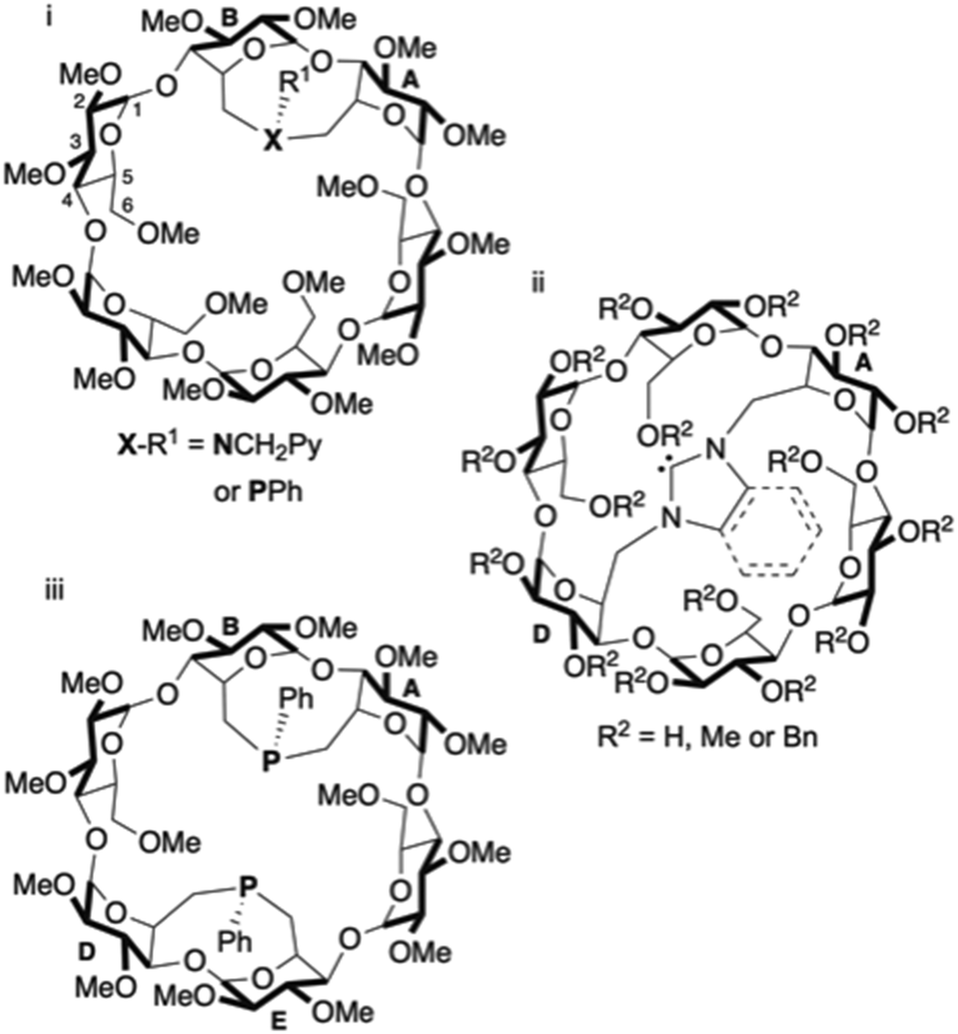 | ||
| Fig. 1 Examples of α-CD-based metal confining ligands with one (i and ii) or two (iii) bridging coordinating units. Bn stands for benzyl. | ||
Herein, we describe a rare example of P(III)-containing CD (1) capable of forcing the exclusive formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand/metal complex, showing cis-chelation inside the receptor hollow when associated with a N donor atom. A Ni(II) complex of this new P,N ligand was investigated for its potential as precatalyst in ethylene oligomerisation, a reaction of continuing considerable interest34,35 and its properties were compared to those of a cavity-free analogue.
:1 ligand/metal complex, showing cis-chelation inside the receptor hollow when associated with a N donor atom. A Ni(II) complex of this new P,N ligand was investigated for its potential as precatalyst in ethylene oligomerisation, a reaction of continuing considerable interest34,35 and its properties were compared to those of a cavity-free analogue.
An improved procedure (Scheme 1) was devised to synthesise diethyl [2-(N,N-dimethylamino)phenyl]phosphonate (3) from 2-bromoaniline (2).36 Its reduction afforded the key functional primary phosphine 4, which upon deprotonation reacted with dimesylate 5 to afford P,N ligand 1 in 63% yield. Although 1 is seemingly a very basic phosphine (δ31P = −28.4 ppm), this ligand is considerably more stable towards air than its PhP-bridged analogue (δ31P = −16.2 ppm)23 and can be purified by standard column chromatography without noticeable formation of phosphine oxide. For comparison, the related cavity-free ligand 737 was also synthesised in 40% yield from 2-bromo-N,N-dimethylaniline by ortho-lithiation followed by nucleophilic substitution of chlorodiethylphosphine.38 Resistance to oxidation was also observed for 7 (δ31P = −25.1 ppm) as in analogous P,N ligands of the Me-DalPhos family (Fig. 2).39
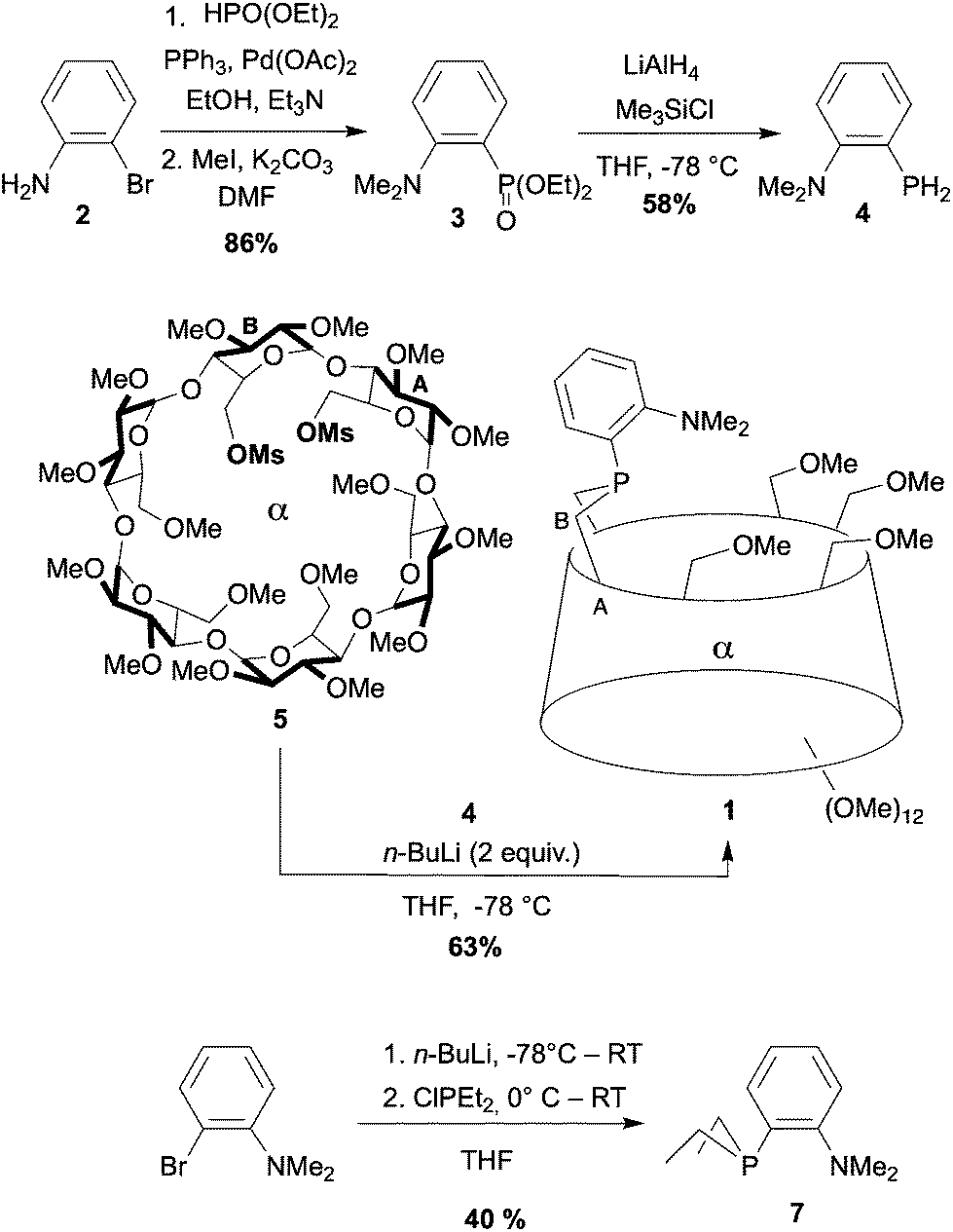 | ||
| Scheme 1 Synthesis of the confining ligand 1 and its cavity-free analogue 7. Ms stands for methylsulfonyl. | ||
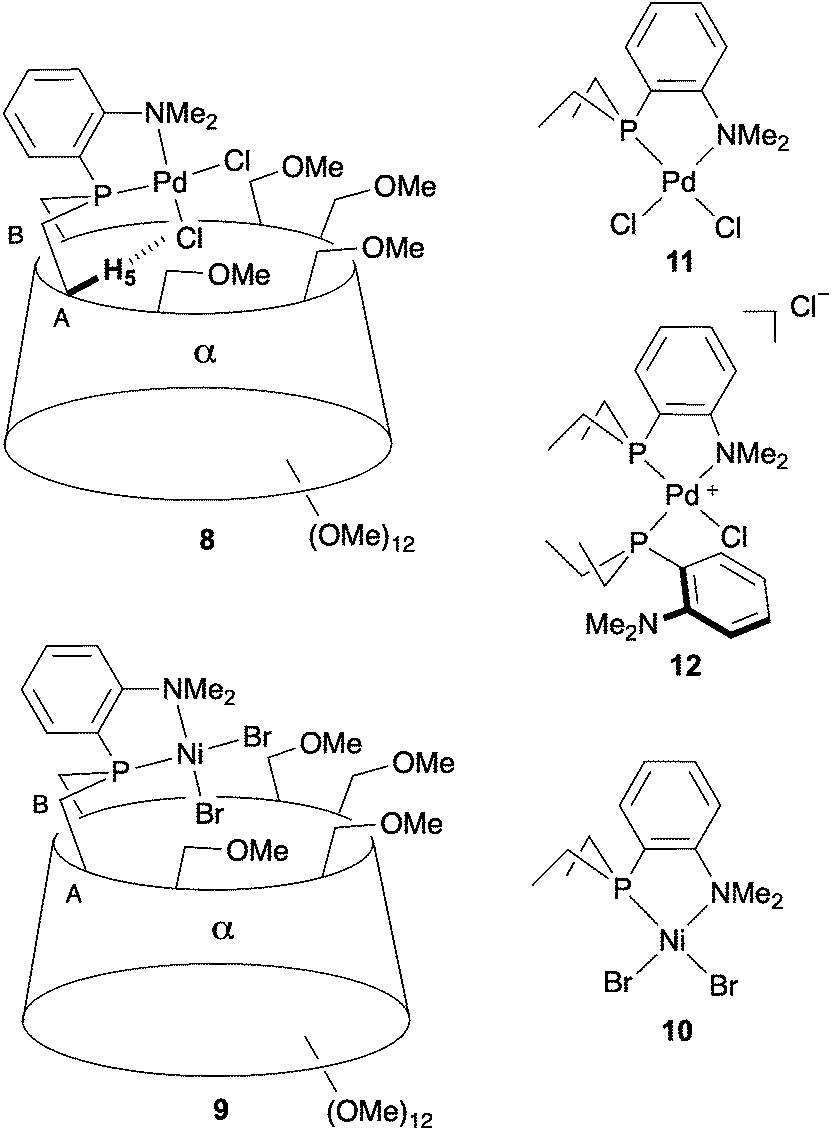 | ||
| Fig. 2 Cavity-shaped Pd and Ni chelate complexes 8 and 9, their cavity-free counterparts 11 and 10, and cationic Pd complex 12. | ||
When reacted with [PdCl2(cod)] (cod = 1,5-cyclooctadiene) in CH2Cl2, confining ligand 1 produced only chelate complex 8, even in the presence of excess ligand (Fig. S80†). Chelating behaviour was also observed when 1 or the cavity-free 7 were reacted with [NiBr2(dme)] (dme = 1,2-dimethoxymethane) to produce complexes 9 and 10, respectively.40 In stark contrast, the reaction of 7 with one equiv. of [PdCl2(cod)] in CH2Cl2 gave a 9:1 mixture of the chelate complex 11 and the cationic bis(phosphine) complex 12, respectively. As expected, raising the ligand/metal ratio to 2 : 1 caused the proportion of complex 12 to increase significantly (11/12 ratio of 3:7) (Fig. S80†). The structures of both 11 and 12 were established by X-ray diffraction analyses (Fig. 3). The single crystal X-ray structure of cationic 12 clearly shows that two phosphines but only one tertiary amine are coordinated to palladium (Fig. 3). Furthermore, line broadening upon heating 12 in CDCl3 (Fig. S66 and S67†) suggests fluxional behaviour possibly involving the slow coordination/decoordination of the NMe2 units on the NMR time scale (Fig. S1†). Confirmation of the metal confining character of ligand 1 in solution came from a detailed analysis of the 1H NMR spectrum of the square planar Pd(II) complex 8, the 31P NMR chemical shift of which is in the expected range (δ31P = 32.5 ppm). As previously reported for a CD-encapsulated M–X unit, the inner-cavity H-5 proton of bridging unit A in 8 is unusually downfield shifted (δ1H = 5.19 vs. 4.30 ppm for ligand 1) as a result of weak CH⋯Cl H-bonding within the CD hollow. The deshielding of this proton proves that 1 is indeed metal confining in solution. Such a feature was confirmed in the solid state by the single crystal X-ray structure of 8 (Fig. 4), which revealed a square planar PdCl2 unit seating just above the cavity and nearly orthogonal to the macrocyclic structure (86.05°).‡ As expected from the solution studies, one of the two chlorido ligands points toward the interior of the cavity and displays short contact with the H-5 proton of bridging unit A (2.840 Å). The other is clearly pointing outside and is at a much longer distance to palladium (2.368 Å) than the encapsulated one (2.295 Å). This is to be expected because of the larger trans influence of the P(III) donor atom. Both complexes 9 and 10 are diamagnetic in keeping with their square planar structures (Fig. 4 and 3 respectively) and low temperature NMR studies in CD2Cl2 revealed at −60 °C a well-defined 31P singlet at 10.4 and 41.4 ppm, respectively.§ It is noteworthy that the single crystal X-ray structures of 8 and 9, which are very similar, clearly establish that each halido ligand experiences a very different steric environment, an unprecedented feature in metal confining ligands that could have a great impact on catalytic properties. In the Pd complex 8, the PNMX2 unit is slightly more included in the cavity than in its Ni counterpart 9 (angle between CD O-4 atoms and the C6–P1–C16 planes = 50.27° vs. 54.82° respectively). Moreover, both cavity-free and cavity-shaped complexes of d8 metals have a very similar first coordination sphere, the only significant difference between the two types of complexes being an elongation of the external Ni–Br bond by ca. 0.02 Å on going from 10 to 9 and a shortening of the corresponding M–X bond by ca. 0.04 Å on going from Pd complexes 11 to 8 (Fig. 3 and 4). The gold(I) complex 13 was also synthesised quantitatively by reacting 1 with [AuCl(tht)] (tht = tetrahydrothiophene). Unlike its d8 counterparts, the d10 metal cation is not chelated by the ligand but only bound to the P(III) atom, leaving the NMe2 unit uncoordinated above the encapsulated metal centre, as revealed by its single crystal X-ray structure (Fig. 4). As in monophosphine analogues,23,24 the phosphorus donor atom imposes the overall orientation of the metal with respect to the cavity. The steric protection provided by the CD cavity is responsible for a remarkable control of the metal coordination sphere and prevents the formation of bis(phosphine) complexes analogous to 12. Such a feature is also expected to be retained under catalytic reaction conditions.
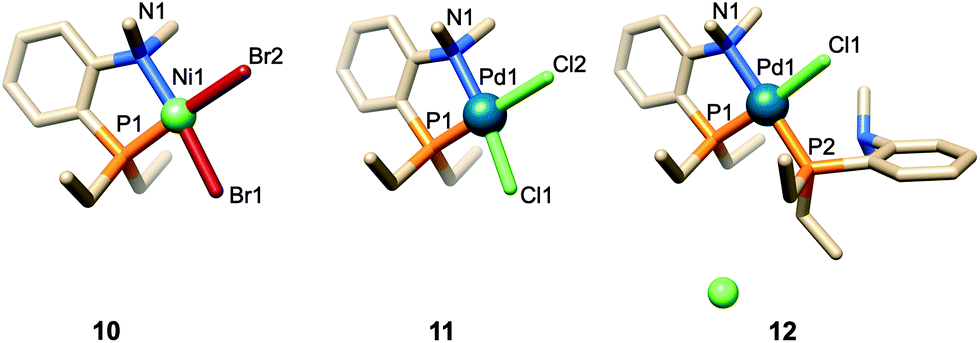 | ||
| Fig. 3 Single crystal X-ray structures of cavity-free Pd complexes 11 and 12 as well as Ni complex 10. Solvent molecules have been omitted for clarity. Selected bond lengths (Å) and angles (°): 10, Ni1–P1 2.1248(10), Ni1–N1 2.010(3), NI1–Br1 2.3122(6), Ni1–Br2 2.3626(5); P1–Ni1–N1 88.63(9), P1–Ni1–Br1 85.25(3), N1–Ni1–Br2 94.48(8), Br1–Ni1–Br2 91.64(2); 11, Pd1–P1 2.1857(9), Pd1–N1 2.111(3), Pd1–Cl1 2.2982(9), Pd1–Cl2 2.4087(9); P1–Pd1–N1 86.68(8), P1–Pd1–Cl1 87.20(3), N1–Pd1–Cl2 94.33(8), Cl1–Pd1–Cl2 91.81(3); 12, Pd1–P1 2.2326(15), Pd1–P2 2.2724(14), Pd1–N1 2.185(5), Pd1–Cl1 2.3728(14); P1–Pd1–N1 84.71(17), P1–Pd1–P2 95.45(5), N1–Pd1–Cl1 91.36(17), P2–Pd1–Cl1 88.28(5). | ||
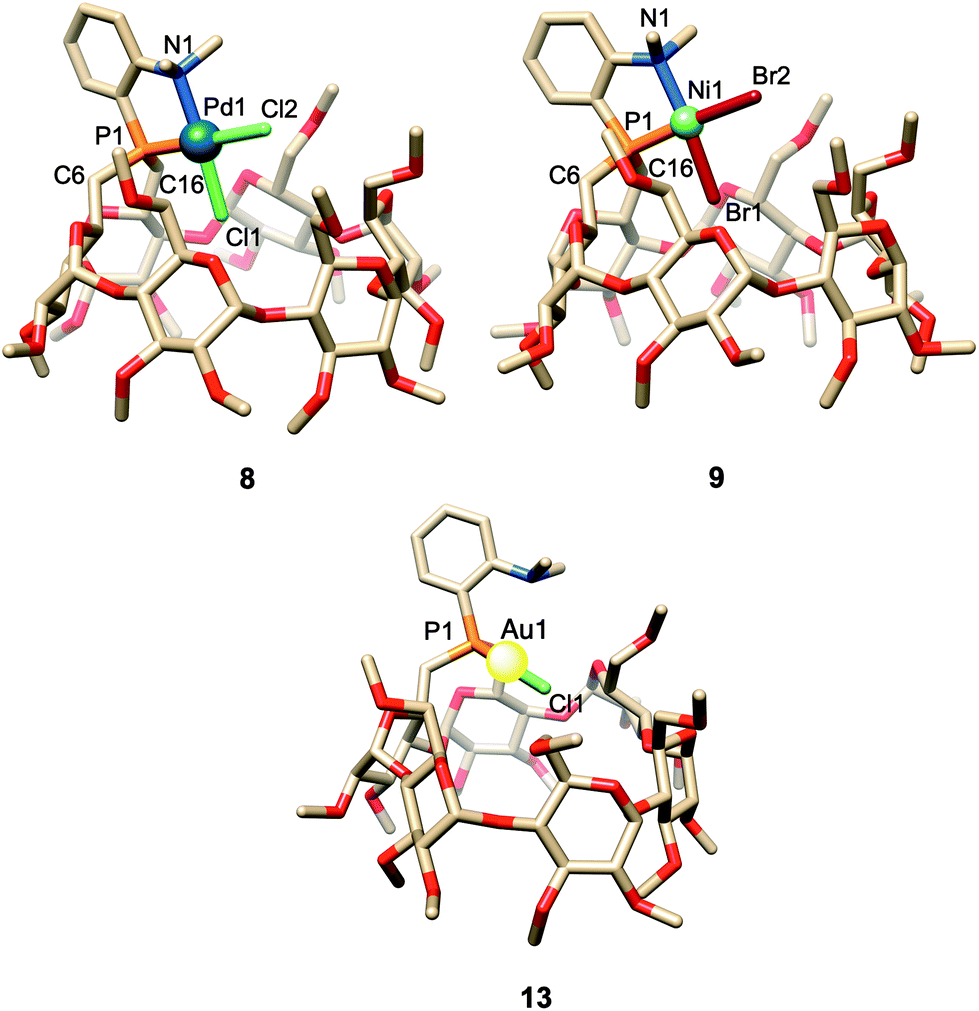 | ||
| Fig. 4 Single crystal X–ray structures of chelate Pd complex 8 and Ni complex 9, and monophosphine Au complex 13 displaying metal encapsulation. Solvent molecules have been omitted for clarity. Selected bond lengths (Å) and angles (°): 8, Pd1–P1 2.1947(14), Pd1–N1 2.125(4), Pd1–Cl1 2.2949(14), Pd1–Cl2 2.3676(15); P1–Pd1–N1 86.83(12), P1–Pd1–Cl1 88.49(5), N1–Pd1–Cl2 92.05(12), Cl1–Pd1–Cl2 92.63(6); 9, Ni1–P1 2.1263(15), Ni1–N1 2.007(6), Ni1–Br1 2.3063(10), Ni1–Br2 2.3807(9); P1–Ni1–N1 88.53(15), P1–Ni1–Br1 84.06(5), N1–Ni1–Br2 95.01(15), Br1–Ni1–Br2 92.40(3); 13, Au1–P1 2.2329(18), Au1–Cl1 2.2755(19); P1–Au1–Cl1 178.16(7). | ||
Numerous Ni complexes derived from P,N ligands, most of them of the pyridine–phosphine type,41–43 have been evaluated in the catalytic oligomerisation of ethylene.44,45 Many studies have focused on varying the electronic properties of both P- and N-donor atoms, but steric factors have also been found to play a crucial role in the metal-catalysed oligomerisation of ethylene to α-olefins.46,54 In particular, the presence of steric bulk at the metal axial sites tends to inhibit termination reactions, thus favouring chain growth.47 The cavity-shaped complex 9 exhibits a unique steric environment, in which the phosphorus atom is heavily congested and forces the chelated metal to be encapsulated in the CD with both axial sites protected by CD 6-methoxy groups. Moreover, unlike their cavity-free analogues, the two active coordination sites involved in the oligomerisation process, generated by bromide abstraction and methylation of 9, will experience very different steric environments. While one of the active sites is deeply buried in the CD cavity, the other is much less protected. The unusual steric environment around the metal in Ni complex 9 prompted us to evaluate it as a precatalyst in ethylene oligomerisation. For comparison, the cavity-free analogue 10 was also tested under similar reaction conditions.
After activation with MMAO, the Ni complex 9 catalysed the dimerisation of ethylene with high C4 (96%) and 1-butene (95%) selectivities (Table 1, entry 2), even after 24 h reaction time (Table 1, entry 3), indicating that chain-walking and post-isomerisation reactions do not readily take place. Moreover, the proportion of 1-hexene in the small C6 fraction is also much higher with the cavity-shaped ligand (70% on average for 9). Even if such level of selectivity is not uncommon for early transition metals,48 it is very high for a nickel-catalysed reaction.44,45 For comparison, the related nickel complex 10 (Table 1, entry 4), which has similar electronic properties, but lacks the CD cavity is about 15 times more active for short reaction times, but its activity rapidly drops with time as it is only 3 fold higher than that of 9 after 24 h (Table 1, entry 6). It is much less selective for C4 (77%) and 1-butene (71%) as well as 1-hexene (8%), whether for short (Table 1, entry 4) or longer reaction times (Table 1, entries 5 and 6). These poor selectivities are also observed for many other P,N Ni complexes except for ligands displaying sterically hindered nitrogen or phosphorus atoms49,50 Although moderately active (TOF = 5780 mol(C2H4) mol(Ni)−1 h−1) (Table 1, entry 1), the catalyst derived from 9 proved to be very robust, since loss of activity is marginal over a prolonged period of time in stark contrast with cavity-free 10. Despite the steric protection of axial positions, which is known to retard the rate of chain transfer relative to chain propagation and improve thermal stability,47 including in macrocyclic complexes,51 hardly any C6-olefins and higher oligomers were formed. The high selectivity for 1-butene strongly suggests that metal encapsulation in the rather small α-CD cavity does not allow further chain growth and largely prevents isomerisation although the ligand electronic properties as revealed by slight changes in the UV spectra of complexes 8–11 (Fig. S87 and S88†) on going from cavity-free to cavity-shaped systems may also have an impact on the catalytic outcome.52,53 Further cis-chelating and confining ligands with different donor atoms and larger cavities will be tested next in our group to see whether access to longer oligomers with a narrow mass distribution can be achieved. The present study also revealed that encapsulation exclusively leads to complexes with a 1:1 ligand/metal ratio, allowing the possibility to introduce in the potentially chelating unit a much weaker donor group susceptible to display catalytically relevant hemilability in solution. This unusual behaviour constitutes a promising development in the design of hybrid functional ligands.
| Entry | Precatalyst | Time | Activity [g(C2H4) g(Ni)−1 h−1] | TOF [mol(C2H4) mol(Ni)−1 h−1] | Products weight distributionb [%] | ||
|---|---|---|---|---|---|---|---|
| C4 (α)c | C6 (α)d | C8 | |||||
| a Conditions: amount of catalyst: 1 × 10−5 mol, amount of cocatalyst (MMAO-12): 4 × 10−3 mol (400 equiv.), T = 30–35 °C, solvent: toluene, total volume: 20 mL, 10 bar C2H4, every test was repeated at least twice. b % calculated by GC analysis. c 1-Butene vs. total butenes formed. d 1-Hexene vs. total hexenes formed. | |||||||
| 1 | 9 | 35 min | 2760 | 5780 | 92 (95) | 6 (70) | 2 |
| 2 | 9 | 3 h | 2190 | 4580 | 96 (95) | 3 (75) | <1 |
| 3 | 9 | 24 h | 1910 | 4000 | 85 (92) | 12 (65) | 3 |
| 4 | 10 | 35 min | 41570 |
86970 |
77 (71) | 22 (8) | 1 |
| 5 | 10 | 3 h | 17920 |
37490 |
79 (81) | 19 (8) | 2 |
| 6 | 10 | 24 h | 5740 | 12000 |
75 (67) | 24 (6) | 1 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the China Scholarship Council for a PhD studentship awarded to Y. L. (CSC201808420261).Notes and references
- R. Gramage-Doria, D. Armspach and D. Matt, Coord. Chem. Rev., 2013, 257, 776–816 CrossRef.
- S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC.
- V. Mouarrawis, R. Plessius, J. I. van der Vlugt and J. N. H. Reek, Front. Chem., 2018, 6, 623 CrossRef PubMed.
- D. Matt and J. Harrowfield, ChemCatChem, 2021, 13, 153–168 CrossRef.
- G. R. F. Orton, B. S. Pilgrim and N. R. Champness, Chem. Soc. Rev., 2021, 50, 4411–4431 RSC.
- B.-Y. Wang, T. Žujović, D. A. Turner, C. M. Hadad and J. D. Badjić, J. Org. Chem., 2012, 77, 2675–2688 CrossRef PubMed.
- A. Cavarzan, J. N. H. Reek, F. Trentin, A. Scarso and G. Strukul, Catal. Sci. Technol., 2013, 3, 2898–2901 RSC.
- M. Otte, P. F. Kuijpers, O. Troeppner, I. Ivanović-Burmazović, J. N. H. Reek and B. de Bruin, Chem. – Eur. J., 2014, 20, 4880–4884 CrossRef.
- P. F. Kuijpers, M. Otte, M. Dürr, I. Ivanović-Burmazović, J. N. H. Reek and B. de Bruin, ACS Catal., 2016, 6, 3106–3112 CrossRef.
- M. Otte, ACS Catal., 2016, 6, 6491–6510 CrossRef.
- D. Zhang, J.-P. Dutasta, V. Dufaud, L. Guy and A. Martinez, ACS Catal., 2017, 7, 7340–7345 CrossRef.
- T. Chavagnan, C. Bauder, D. Sémeril, D. Matt and L. Toupet, Eur. J. Org. Chem., 2017, 70–76 CrossRef.
- C. Tugny, N. del Rio, M. Koohgard, N. Vanthuyne, D. Lesage, K. Bijouard, P. Zhang, J. Meijide Suárez, S. Roland and E. Derat, ACS Catal., 2020, 10, 5964–5972 CrossRef.
- J. Meijide Suárez, O. Bistri-Aslanoff, S. Roland and M. Sollogoub, ChemCatChem, 2022, 14, e202101411 CrossRef.
- G. Xu, S. Leloux, P. Zhang, J. Meijide Suárez, Y. Zhang, E. Derat, M. Ménand, O. Bistri-Aslanoff, S. Roland, T. Leyssens, O. Riant and M. Sollogoub, Angew. Chem., Int. Ed., 2020, 59, 7591–7597 CrossRef.
- T. Gadzikwa, R. Bellini, H. L. Dekker and J. N. H. Reek, J. Am. Chem. Soc., 2012, 134, 2860–2863 CrossRef.
- M. Jouffroy, R. Gramage-Doria, D. Armspach, D. Sémeril, W. Oberhauser, D. Matt and L. Toupet, Angew. Chem., 2014, 126, 4018–4021 CrossRef.
- C. García-Simón, R. Gramage-Doria, S. Raoufmoghaddam, T. Parella, M. Costas, X. Ribas and J. N. Reek, J. Am. Chem. Soc., 2015, 137, 2680–2687 CrossRef PubMed.
- P. Zhang, J. Meijide Suárez, T. Driant, E. Derat, Y. Zhang, M. Ménand, S. Roland and M. Sollogoub, Angew. Chem., Int. Ed., 2017, 56, 10821–10825 CrossRef.
- N. Endo, M. Inoue and T. Iwasawa, Eur. J. Org. Chem., 2018, 1136–1140 CrossRef.
- Z. Kaya, E. Bentouhami, K. Pelzer and D. Armspach, Coord. Chem. Rev., 2021, 445, 214066 CrossRef.
- D. Sechet, Z. Kaya, T. A. Phan, M. Jouffroy, E. Bentouhami, D. Armspach, D. Matt and L. Toupet, Chem. Commun., 2017, 53, 11717–11720 RSC.
- E. Engeldinger, L. Poorters, D. Armspach, D. Matt and L. Toupet, Chem. Commun., 2004, 634–635 RSC.
- R. Gramage-Doria, D. Rodriguez-Lucena, D. Armspach, C. Egloff, M. Jouffroy, D. Matt and L. Toupet, Chem. – Eur. J., 2011, 17, 3911–3921 CrossRef PubMed.
- M. Jouffroy, D. Armspach and D. Matt, Dalton Trans., 2015, 44, 12942–12969 RSC.
- M. Guitet, P. Zhang, F. Marcelo, C. Tugny, J. Jiménez-Barbero, O. Buriez, C. Amatore, V. Mouriès-Mansuy, J. P. Goddard and L. Fensterbank, Angew. Chem., Int. Ed., 2013, 52, 7213–7218 CrossRef.
- P. Zhang, C. Tugny, J. Meijide Suárez, M. Guitet, E. Derat, N. Vanthuyne, Y. Zhang, O. Bistri, V. Mouriès-Mansuy, M. Ménand, S. Roland, L. Fensterbank and M. Sollogoub, Chem, 2017, 3, 174–191 Search PubMed.
- Z. Kaya, L. Andna, D. Matt, E. Bentouhami, J. P. Djukic and D. Armspach, Chem. – Eur. J., 2018, 24, 17921–17926 CrossRef.
- I. Tabushi and Y. Kuroda, J. Am. Chem. Soc., 1984, 106, 4580–4584 CrossRef.
- O. Sénèque, M. Campion, B. Douziech, M. Giorgi, Y. Le Mast and O. Reinaud, Dalton Trans., 2003, 4216–4218 RSC.
- M. Jouffroy, D. Sémeril, D. Armspach and D. Matt, Eur. J. Org. Chem., 2013, 6069–6077 CrossRef.
- C. Machut, J. Patrigeon, S. Tilloy, H. Bricout, F. Hapiot and E. Monflier, Angew. Chem., Int. Ed., 2007, 46, 3040–3042 CrossRef PubMed.
- J. Patrigeon, F. Hapiot, M. Canipelle, S. Menuel and E. Monflier, Organometallics, 2010, 29, 6668–6674 CrossRef.
- O. L. Sydora, Organometallics, 2019, 38, 997–1010 CrossRef.
- Z. Wang, Q. Liu, G. A. Solan and W.-H. Sun, Coord. Chem. Rev., 2017, 350, 68–83 CrossRef.
- F. Y. Xu, O. M. Duke, D. Rojas, H. M. Eichelberger, R. S. Kim, T. B. Clark and D. A. Watson, J. Am. Chem. Soc., 2020, 142, 11988–11992 CrossRef PubMed.
- F. G. Mann and H. R. Watson, J. Chem. Soc., 1957, 3945–3949 RSC.
- F. Benvenuti, C. Carlini, M. Marchionna, R. Patrini, A. M. Raspolli Galletti and G. Sbrana, J. Mol. Catal. A: Chem., 1999, 140, 139–155 CrossRef.
- R. J. Lundgren, B. D. Peters, P. G. Alsabeh and M. Stradiotto, Angew. Chem., Int. Ed., 2010, 49, 4071–4074 CrossRef PubMed.
- W. Levason and K. G. Smith, Inorg. Chim. Acta, 1980, 41, 133–141 CrossRef CAS.
- P. Espinet and K. Soulantica, Coord. Chem. Rev., 1999, 193–195, 499–556 CrossRef CAS.
- F. Speiser, P. Braunstein and L. Saussine, Acc. Chem. Res., 2005, 38, 784–793 CrossRef CAS PubMed.
- A. Kermagoret and P. Braunstein, Organometallics, 2008, 27, 88–99 CrossRef CAS.
- H. Olivier-Bourbigou, P. A. R. Breuil, L. Magna, T. Michel, M. F. Espada Pastor and D. Delcroix, Chem. Rev., 2020, 120, 7919–7983 CrossRef CAS PubMed.
- G. E. Bekmukhamedov, A. V. Sukhov, A. M. Kuchkaev and D. G. Yakhvarov, Catalysts, 2020, 10, 498 CrossRef CAS.
- F. Wang and C. Chen, Polym. Chem., 2019, 10, 2354–2369 RSC.
- D. Zhang, E. T. Nadres, M. Brookhart and O. Daugulis, Organometallics, 2013, 32, 5136–5143 CrossRef CAS.
- F. L. Grasset, R. Welter, P. Braunstein, H. Olivier-Bourbigou and L. Magna, ChemCatChem, 2021, 13, 2167–2178 CrossRef CAS.
- J. Flapper, H. Kooijman, M. Lutz, A. L. Spek, P. W. N. M. van Leeuwen, C. J. Elsevier and P. C. J. Kamer, Organometallics, 2009, 28, 3272–3281 CrossRef CAS.
- J. Flapper, P. W. N. M. van Leeuwen, C. J. Elsevier and P. C. J. Kamer, Organometallics, 2009, 28, 3264–3271 CrossRef CAS.
- D. H. Camacho, E. V. Salo, J. W. Ziller and Z. Guan, Angew. Chem., Int. Ed., 2004, 43, 1821–1825 CrossRef CAS PubMed.
- P. W. N. M. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Chem. Rev., 2000, 100, 2741–2769 CrossRef CAS PubMed.
- K. Yamamoto, K. Higuchi, M. Ogawa, H. Sogawa, S. Kuwata, Y. Hayashi, S. Kawauchi and T. Takata, Chem. – Asian J., 2020, 15, 356–359 CrossRef CAS.
- L. Cao, Z. Cai and M. Li, Macromolecules, 2022, 55, 3513–3521 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data. CCDC 2163655 (8), 2163663 (9), 2163657 (10), 2163751 (11), 2163658 (12) and 2163666 (13). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt01553f |
| ‡ Angle between the CD O-4 atoms plane and the 5-membered chelate ring. |
| § Line broadening at room temperature is common with this type of complexes. |
| This journal is © The Royal Society of Chemistry 2022 |