
Effects of ozone and produced hydroxyl radicals on the transformation of graphene oxide in aqueous media†
Tingting
Du
a,
Adeyemi S.
Adeleye
 b,
Tong
Zhang
a,
Nan
Yang
a,
Rongjie
Hao
a,
Yao
Li
*a,
Weihua
Song
c and
Wei
Chen
*a
b,
Tong
Zhang
a,
Nan
Yang
a,
Rongjie
Hao
a,
Yao
Li
*a,
Weihua
Song
c and
Wei
Chen
*a
aCollege of Environmental Science and Engineering, Ministry of Education Key Laboratory of Pollution Processes and Environmental Criteria, Tianjin Key Laboratory of Environmental Remediation and Pollution Control, Nankai University, Tianjin 300350, China. E-mail: hkliyao@nankai.edu.cn; chenwei@nankai.edu.cn; Fax: +86 22 66229516; Tel: +86 22 23501117
bDepartment of Civil and Environmental Engineering, University of California, Irvine, CA 92697, California, USA
cDepartment of Environmental Science & Engineering, Fudan University, Shanghai 200433, China
First published on 27th June 2019
Abstract
The rapidly increasing applications of graphene oxide (GO) will lead to its release into the natural environment. To date, the fate and transformation of GO in water and wastewater treatment systems are not well understood. In this study, we investigate the mechanism of GO ozonation and probe the individual roles of oxidants such as ozone (O3) and hydroxyl radicals. We looked at the effects of the oxidants (ozone and hydroxyl radicals) on functionalized and unfunctionalized aromatic rings (using carbonaceous nanomaterials with different degrees of oxidation). Multiple characterization methods, including transmission electron microscopy (TEM), scanning electron microscopy (SEM), atomic force microscopy (AFM), X-ray photoelectron spectroscopy (XPS), Fourier transform infrared (FTIR) and Raman spectroscopy, were used to investigate the physicochemical changes of GO upon ozonation. The hydroxyl radical (˙OH)—formed from aqueous O3—hydroxylated GO. The combination of ˙OH/O3 further oxidized the functionalized aromatic rings of GO and their C–O groups (to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–CO), cleaving some of the aromatic rings of GO. When ˙OH was quenched, O3 had no observable effects on the unfunctionalized aromatic rings, which are more predominant on reduced GO and graphene. Microscopy analyses showed that ozonation led to crumpling of GO nanosheets, truncation of GO edges, formation of holes (diameter = 5–15 nm), and production of small-sized graphenic fragments. The changes in GO surface functional groups and hydrophobicity induced by ozonation affected the transport properties of GO in porous media, as well as its adsorption affinities for model organic contaminants.
O and O–CO), cleaving some of the aromatic rings of GO. When ˙OH was quenched, O3 had no observable effects on the unfunctionalized aromatic rings, which are more predominant on reduced GO and graphene. Microscopy analyses showed that ozonation led to crumpling of GO nanosheets, truncation of GO edges, formation of holes (diameter = 5–15 nm), and production of small-sized graphenic fragments. The changes in GO surface functional groups and hydrophobicity induced by ozonation affected the transport properties of GO in porous media, as well as its adsorption affinities for model organic contaminants.
Environmental significanceThe increasing application of nanoparticles leads to increasing environmental release of these novel materials, whose environmental implications are still not clearly understood. As shown in this study, the physicochemical properties of GO are remarkably changed by ozonation, a typical process in several wastewater treatment plants. The ozonation of GO can thus make it challenging to detect and quantify the nanomaterials in wastewater effluents for regulatory purposes. Similarly, regulatory limits will probably be established using fate and toxicity tests based on pristine GO. However, it is unclear whether these regulatory limits will be valid for the forms of GO in wastewater effluents if exposed to ozonation during treatment, which are crumpled, holey, more oxygenated, and have very small-sized fragments. |
1 Introduction
Graphene oxide (GO) is a structural analog of graphene and contains abundant O-containing functionalities such as epoxide (C–O–C), hydroxyl (C–OH), carbonyl (CO), and carboxyl groups (O–CO) covalently bound to either the basal planes (for epoxide and hydroxyl groups) or the edges (for carbonyl and carboxyl groups).1–3 GO is increasingly being used in commercial products, such as nano-paints and nanohybrid materials for water treatment, which can lead to its direct release into the environment.4–6 There are also many medical, energy-related, and engineering applications for GO.7–10 Thus, it is expected that wastes containing GO will be generated, released into natural surface waters, and finally flow into water treatment systems.11–13 Because of its small size (lateral size in the nanometer–micrometer range) and strong negative surface charge,14 GO may be challenging to remove via typical primary water treatment procedures such as coagulation and sedimentation.15 As a result, the nanomaterial may reach the secondary and tertiary water treatment steps, where it may be exposed to disinfectants. The exposure of GO to disinfectants (such as chlorine and chloramine) used in water/wastewater treatment changed the surface O-functionalities of GO and led to scrolling of the graphenic sheets.16–18
Ozone is one of the most commonly used disinfectants in water and wastewater treatments.19 Given the high oxidation efficiency of ozone (Ered0 = 2.08 V), it is reasonable to assume that GO with react with ozone (O3), similar to other oxidizing disinfectants.16,17 A limited number of studies have been conducted to investigate the effects of O3 on carbonaceous nanomaterials, and the results indicated that ozonation may increase and/or transform the O-functional groups on the surface of carbon nanomaterials. For instance, Fortner et al.20 showed that the reaction between C60 fullerene and dissolved O3 (3–6 mg L−1) resulted in the formation of water-soluble fullerene oxide species with hydroxyl and carboxyl functionalities. Peng et al.21 demonstrated that O3 enhanced the oxygenation of multiwall carbon nanotubes. Thus far, only a few studies are available on the ozonation of graphene-family nanomaterials. Wang et al.22 and Xu et al.23 reported that O3 oxidized the C–OH and CO groups on reduced graphene oxide (RGO) and thermally-exfoliated graphene nanosheets (T-GNS), respectively. Note that in previous studies, ozonation was done by continuously sparging O3 gas (0.1–2 L min−1) into the aqueous phase to achieve an aqueous O3 concentration of 20 mg L−1 or much higher,22,23 which may not necessarily reflect the realistic scenario in water and wastewater treatment processes. As such, the underlying mechanisms controlling the chemical transformation of GO by ozonation under environmentally relevant conditions (such as the disinfection concentration of O3) are still unclear. The transformation of GO may be different in real water treatment systems under relatively low O3 concentration. Moreover, although the ozonation of GO has been investigated in a few previous studies, the specific oxidants responsible for GO transformation and the detailed reactivity of the graphitic vs. edge GO domains remain unclear.
When O3 gas is injected into water, it forms different reactive oxygen species (ROS) and intermediate products. Most of the ROS and intermediates, including O2˙−, O3˙− and HO2˙−, have negligible effects on the oxidation of GO because of their low equilibrium concentration or poor oxidation potential.24–26 However, the hydroxyl radical (˙OH) has a high reactivity and can unselectively react with organic substances.26,27 As such ˙OH and O3 may both play an important role in the transformation of GO during ozonation.19,28–33 Even though the concentration of ˙OH is much lower than that of O3, ˙OH has a higher oxidation potential,27 which implies that it may be an important species during ozonation. The role of each important oxidant present during ozonation, such as O3 and ˙OH, on functionalized and unfunctionalized aromatic rings of GO remains unknown.
The objective of this study was to understand the underlying mechanisms controlling the physicochemical transformation of GO upon ozonation, under conditions similar to those used during water/wastewater treatment. We treated GO suspensions with an O3 level typically applied in water treatment (∼5 mg L−1).34 Fourier transform infrared (FTIR) and X-ray photoelectron spectroscopy (XPS) were used to discern the specific reactivities of O3 and generated ROS (˙OH) with different domains of GO. The mechanism of O3 and ˙OH-induced transformation of GO was explored and discussed. The implications of GO transformation from ozonation on its environmental fate and transport were illustrated using column transport and adsorption experiments.
2 Materials and methods
2.1 Materials and stock preparation
Graphene oxide (GO; purity >99%) was purchased from Plannano Materials Tech. Co. (Tianjin, China). Based on the information provided by the supplier, GO was produced using a modified Hummers method.35 Atomic force microscopy (AFM, Dimension Icon, Bruker, Karlsruhe, Germany) showed that the thickness of GO was about 0.8–1.2 nm (Fig. S1, in the ESI†), which is the typical thickness of single-layer GO on a SiO2 substrate.36 RGO was synthesized by reducing GO with hydrazine (N2H4). The detailed procedure is provided in ESI† section S1.0. Graphene was purchased from XF Nano Materials Tech. Co. (Nanjing, China). Graphite (99.99% carbon, size <45 μm) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Potassium indigo trisulfonate was purchased from Sigma-Aldrich (St. Louis, MO, USA). All the chemicals used in the study were of reagent grade or higher.The stock suspensions of GO, RGO, graphene, and graphite (all 200 mg L−1) were prepared by directly adding the dry powders to deionized (DI) water and stirring with a magnetic stirrer for 30 min, followed by ultrasonication at 100 W for 4 h in a water bath. The stock suspensions were kept in the dark at 4 °C until use. To use, the stocks were first ultrasonicated at 100 W for 30 min to ensure that the particles were well-dispersed. All aqueous samples were adjusted to pH 7 using NaOH (50 mM) and/or HCl (50 mM), similar to the pH during ozonation.
2.2 Ozonation experiments
O3 was generated from pure oxygen (99.995%) at a rate of 1.0 L min−1 using a 3S-A15 O3 generator (Tonglin Technology Co., Beijing, China) and was continuously introduced into 2 L of 4 °C DI water for 15 min through a ceramic sparger. The aqueous O3 concentration was determined using the 4500-O3 B: indigo colorimetric method.37 Then, each sample was quickly mixed with the O3-bearing water to achieve a final particle concentration of 10 mg L−1 and 5 mg L−1 aqueous O3 (which is within the range commonly used for water treatment34,38) in a stirred amber glass reactor. The reactor was kept at a constant temperature of 25 °C and the reaction was allowed to occur for 20 min (after which it was stopped by filtration using a suction filter). After ozonation, GO, RGO, graphene, and graphite were referred to as GO_O3, RGO_O3, Graphene_O3 and Graphite_O3, respectively.2.3 Material characterization
GO, RGO, graphene, and graphite were characterized via FTIR spectroscopy in transmission mode (110 Bruker TENSOR 27, Bruker Optics Inc., Karlsruhe, Germany), XPS (ESCALAB 250Xi, Thermo Fisher Scientific Inc., Waltham, USA), UV-vis spectrophotometry (UV-2401, Shimadzu Corp., Kyoto, Japan), and Raman spectroscopy (inVia, Renishaw, Wotton-under-Edge, UK) before and after ozonation. The morphology of GO, before and after ozonation, was obtained by scanning electron microscopy (SEM, ZEISS MERLIN Compact, Carl Zeiss AG, Aalen, Germany), transmission electron microscopy (TEM, JEM-2010FEF, JEOL Ltd., Tokyo, Japan), and atomic force microscopy (AFM, MMAFM/STM, D3100M, Digital Ltd., USA). The low molecular weight compounds formed due to the ozonation of GO were analyzed using UPLC-MS/MS (UPLC-Xevo TQ-S, Waters, USA) described in ESI† section S2.0. A ZetaSizer Nano ZS90 instrument (Malvern Instruments Ltd., Worcestershire, UK) was used to measure the hydrodynamic diameter (Dh) of GO in the aqueous phase. The relative hydrophobicity of GO/GO_O3 was estimated by measuring n-dodecane–water partition coefficients (KDW) as described in ESI† section S3.0.2.4 Transport experiments
Column transport experiments were conducted to investigate the influence of ozonation on the transport of GO in saturated quartz sand. Quartz sand was dry-packed into Omnifit borosilicate glass columns (10 cm × 0.66 cm, Bio-Chem Valve Inc., Boonton, NJ) with 10 μm stainless-steel screens (Valco Instruments Inc., Houston, TX) on both ends. The columns were operated in an upward direction using syringe pumps (KD Scientific, Holliston, MA). The sand-packed column was blown with carbon dioxide for 1 h to exhaust air, then the column was equilibrated by sequentially flushing it with 100 mL DI water at a flow rate of 3 mL h−1 and 100 mL DI water at a flow rate of 6.6 mL h−1 followed by 180 mL background electrolyte solution. To prepare the influents, aliquots of the GO or GO_O3 stock suspension were mixed with the electrolyte (final concentration of 35 mM NaCl or 0.3 mM CaCl2) to obtain a GO concentration of 10 mg L−1 and stirred for 2 h. The transport experiment was performed at pH 6, in order to keep Ca2+ in the ionic state. The procedure for a typical column experiment is as follows: the influent was loaded to the column from the bottom using a syringe pump, and the effluent was collected at every 4–5 pore volume. Then the column was flushed using a GO-free background electrolyte solution (until GO was no longer detectable in the effluent). The concentration of GO was determined by measuring the UV-vis absorbance at 230 nm.6,142.5 Adsorption experiments
Adsorption experiments were carried out by using a previously developed method.39 An aliquot of the GO stock suspension was diluted with DI water to achieve a final GO concentration of 50 mg L−1. Phenanthrene (a representative nonpolar, nonionic aromatic compound) and 1-naphthol (a representative polar aromatic compound) were selected as the model contaminants. The detailed procedure for adsorption isotherm experiments is described in ESI† section S4.0.The concentrations of phenanthrene and 1-naphthol in aqueous media were determined using a Waters high performance liquid chromatography (HPLC) system (Waters Corp., Milford, USA) equipped with a Waters symmetry reversed-phase C18 column (4.6 × 150 mm). Phenanthrene was detected with a Waters 2475 fluorescence detector at an excitation wavelength of 250 nm and an emission wavelength of 364 nm; the mobile phase was 80% acetonitrile and 20% DI water (v:v; 1.0 mL min−1). 1-Naphthol was detected with a Waters 2489 UV/visible detector at 328 nm; the mobile phase was 50% acetonitrile and 50% DI water (v:v; 1.0 mL min−1). No peaks for degraded/transformed products of the test compounds were detected in the spectra.
3 Results and discussion
3.1 Transformation of GO upon ozonation
Ozonation (5 mg L−1) resulted in remarkable changes in the physicochemical properties of GO. Fig. 1 shows the comparison of the TEM, SEM and AFM images of pristine GO and O3-treated GO. Relative to pristine GO, O3-treated GO was visibly crumpled, leading to a decrease in the lateral size (Fig. 1a and d). In addition, holes (with a diameter of 5–15 nm) were observed on O3-treated GO nanosheets (Fig. 1f), which suggested breaking of graphenic rings in the basal plane. The edges of GO also appeared truncated. The AFM micrographs clearly showed the disintegration of GO sheets to much smaller sizes similar to graphene quantum dots (Fig. 1c and f),40 while the sheets with lateral size similar to the pristine nanosheets had several holes within their basal plane. All these data showed that ozonation increased the defects on GO while also disintegrating the aromatic rings of the nanomaterial. The mass spectrometry spectra of GO before and after ozonation are shown in Fig. S2.† The data showed that more low molecular weight (m/z 100–400) compounds were formed after the ozonation of GO, and the intensity of high molecular weight chemical species (m/z 600–1000) decreased or disappeared. This further confirms the decomposition of GO by ozonation. | ||
| Fig. 1 Morphology of graphene oxide: (a) transmission electron microscopy (TEM), (b) scanning electron microscopy (SEM), and (c) atomic force microscopy (AFM) images of pristine GO. Morphology of O3-treated GO obtained via (d) TEM, (e) SEM, and (f) AFM. | ||
In addition to the changes in morphology, the chemical transformation of GO upon O3 treatment was also evident. The FTIR spectra (Fig. 2a) of pristine GO show four characteristic peaks located at 1055 cm−1, 1383 cm−1, 1628 cm−1, and 1735 cm−1, corresponding to the C–O–C stretching, C–O group, O–CO stretching, and CO stretching, respectively.41–43 After O3 treatment, the intensity of the C–O and C–O–C groups increased. The intensity of the CO and O–CO groups also increased, but to a lesser extent. The XPS spectra of pristine GO and O3-treated GO (GO_O3) are compared in Fig. 2b and the corresponding data are summarized in Table 1. The oxygen/carbon (O/C) ratio increased by almost 26% from 0.50 for GO to 0.63 for GO_O3. Since XPS is a surface-sensitive technique and is typically susceptible to contamination from adsorbed adventitious carbon, we also determined the O/C ratio of GO (before and after ozonation) using energy dispersive X-ray spectroscopy (EDS), which has a greater penetration depth and thus is less sensitive to surface contamination.44 According to EDS (Table S1†), the O/C ratio increased from 0.53 to 0.68 upon ozonation, in agreement with XPS analysis. In addition, the relative abundance of the different surface O-functional groups on GO changed upon ozonation, as shown by the deconvolution of the high resolution C1s XPS peak (Fig. 2b, Table 1).45 Upon ozonation, the relative abundance of the aromatic C–C/CC group (binding energy [BE] = 284.6 eV) decreased from 37% to 26%, while a slight increase in the abundance of the C–O–C/C–OH (BE = 286.6 eV), CO (BE = 287.8 eV) and O–CO groups (BE = 288.8 eV) was observed. The results of FTIR and XPS both indicated that oxidation occurred on the surface of GO after ozonation.
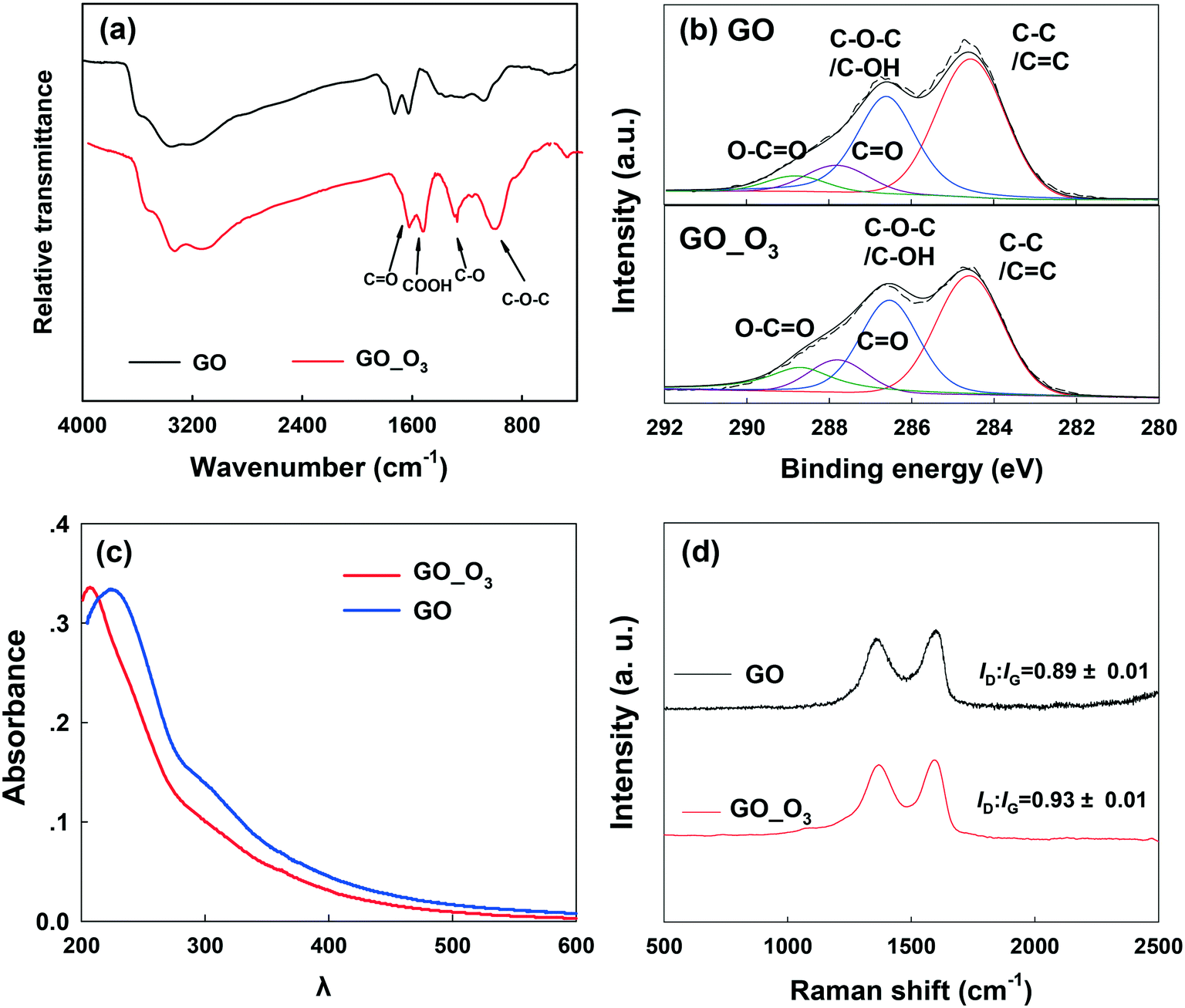 | ||
| Fig. 2 (a) Fourier transform infrared (FTIR) spectroscopy, (b) X-ray photoelectron spectroscopy (XPS), (c) UV-vis spectrophotometry, and (d) Raman spectroscopy of pristine GO and O3-treated GO (GO_O3). | ||
The UV-vis spectra of pristine and O3-treated GO are shown in Fig. 2c. The characteristic absorbance peak of GO at 230 nm was observed in pristine GO, which can be ascribed to π–π* transitions in small electronically conjugated domains present in the material.46 The absorbance peak blue-shifted to 210 nm in O3-treated GO, which indicates a decrease in the number of electronically conjugated domains, resulting from increased oxidation of GO. In addition, the specific ultraviolet absorbance obtained at 254 nm (SUVA254), which indicates the concentrations of aromatic rings,47 decreased upon the ozonation of GO (Fig. S3†).
Raman spectroscopy (Fig. 2d) showed the characteristic D band (around 1350 cm−1) and G band (around 1580 cm−1) in both GO and O3-treated GO. The D band is related to the vibration of carbon atoms with dangling bonds and ascribed to the edge of the carbon network while the G band is ascribed to the E2g mode of sp2 carbon atoms.48 The ratio of ID/IG (intensity of the D band to the G band) is usually employed to reflect the degree of defects on graphitic carbon materials.49 As shown in Fig. 2d, the ID/IG ratio of GO increased after O3 treatment, suggesting an increase in the defective sites on GO. This is consistent with the electron microscopy observations (Fig. 1).
The oxidation kinetics of GO during ozonation was also studied by measuring the absorbance of the GO suspension at 254 nm over time. As shown in Fig. S4a,† the transformation of the aromatic structure of GO during ozonation is a second-order reaction, with a rate constant of 0.247 g−1 L s−1 at pH 7. The rate constant increased as the pH increased, as shown in Fig. S4b,† from 0.216 g−1 L s−1 at pH 5 to 0.250 g−1 L s−1 at pH 8. According to Esplugas et al.,50 the reactivity of the ˙OH radical increases as the pH increases. The increased reactivity of the ˙OH radical can help increase the rate constant during the ozonation.
3.2 Effects of ozonation on functionalized versus graphitic domains of GO
The abovementioned characterization results indicate that oxidation occurred on the surface of GO, in agreement with other studies that investigated the reactions between O3 and graphene-based materials.22,23,32 However, the relative effects of ozonation on the functionalized domain (e.g., the edges of GO) versus the O-functionality-free domain are unclear. For example, even though holes were observed on GO nanosheets upon ozonation, the reason behind the increased proportion of CO and O–CO groups is not clear. To further understand the effects of ozonation on the graphitic domain of GO, ozonation of RGO and graphene, both of which contain very minimal functionalization (sp3 carbon), was performed.
Fig. 3a and b show the FTIR analysis of RGO and graphene before and after ozonation. Similar to GO, all the O-containing functional groups on RGO and graphene increased notably. The XPS spectra of RGO and graphene before and after ozonation were also compared (Fig. S5a–d and Table S2†). Ozonation increased the O/C ratio of RGO by almost 94% (from 0.18 to 0.35) and that of graphene by 170% (from 0.10 to 0.27). In agreement with FTIR results, the relative abundance of the different O-functional groups on the surface of the materials increased upon ozonation. The results clearly show the functionalization of aromatic carbon rings by ozonation. However, these results alone cannot provide a definitive confirmation that the unfunctionalized (sp2) aromatic rings of GO can be readily oxygenated, in that it is possible that oxygenation from ozonation might only have occurred at the reactive sites that are adjacent to the existing O-functional groups on the aromatic rings.51
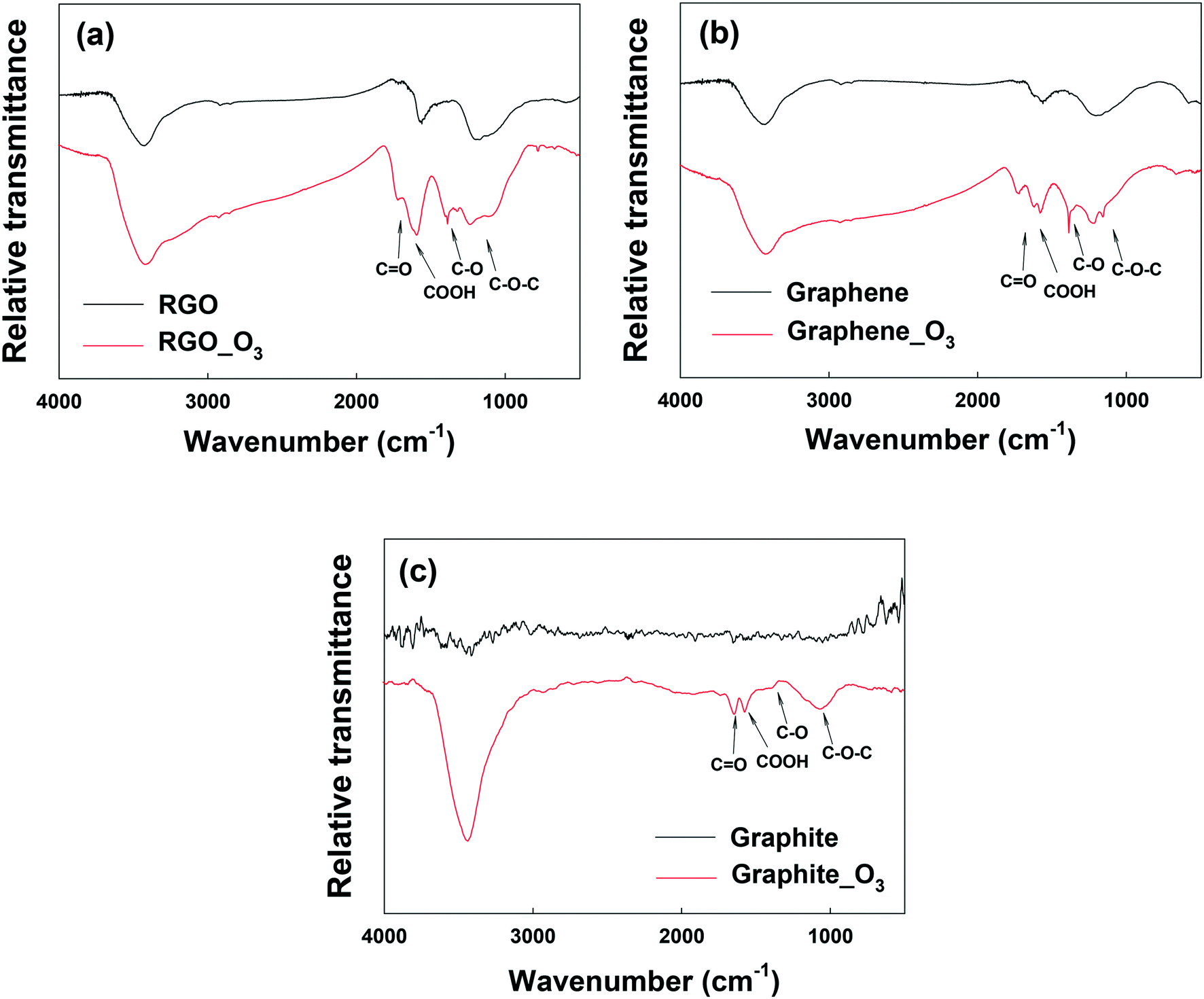 | ||
| Fig. 3 FTIR spectra of (a) reduced graphene oxide (RGO), (b) graphene, and (c) graphite before and after ozone treatment. | ||
To examine the effect of ozonation on the graphitic domains of GO, graphite was also ozonated and characterized. Although the size and layers of graphite are different from GO's, the exclusively unfunctionalized aromatic structure makes graphite ideal for determining whether ozonation can functionalize sp2 aromatic carbon. As shown in the FTIR spectrum of graphite in Fig. 3c, O-containing functional groups were clearly formed on the surface of graphite upon ozonation. In addition, XPS analysis showed that ozonation increased the atomic oxygen in graphite from approximately 1% (probably due to impurities) to nearly 13% (Fig. S5e and f, Table S2†), with the highest increase observed in the C–O group (that is, C–O–C/C–OH). Based on this result, we conclude that ozonation of GO can lead to the oxygenation of unfunctionalized aromatic rings regardless of their proximity to existing functionalized carbon groups. Moreover, Raman analyses showed increased defects on O3-treated RGO, graphene, and graphite (Fig. S6†).
3.3 Roles of O3 and ˙OH in GO transformation
To identify the roles of O3 and ˙OH radicals in the oxidation of GO, we added tert-butanol (t-BuOH) to GO suspensions before ozonation. t-BuOH is a scavenger that can selectively eliminate ˙OH radicals in suspension without reacting with O3,26 and can thus single out the specific role of O3 in the transformation of GO. Fig. 4a and b show the physicochemical characterization of O3-treated GO with and without t-BuOH. FTIR spectroscopy revealed that the O-functional groups changed substantially both without t-BuOH (i.e., when both O3 and ˙OH were active species) and when t-BuOH was present (i.e., in the absence of ˙OH) upon ozonation (Fig. 4a), indicating that both oxidants played important roles in GO transformation. An interesting observation, as shown in Fig. 4a, is that the O-functional groups of GO decreased when t-BuOH was present (i.e., in the absence of ˙OH). A possible explanation is that O3 can further oxidize the OH group to CO and O–CO groups, resulting in aromatic ring cleavage,51 and eventual decomposition of GO (i.e., loss of functional groups and the formation of low molecular weight compounds).29 This result was corroborated by XPS analysis (Fig. 4b, Table S3†), which confirmed the reduced oxygenation of GO in the absence of ˙OH. These results indicate that both ˙OH and O3 are important for the transformation of the O-functionality-containing domains of GO. We also observed crumpling of GO nanosheets and formation of holes on the basal plane of GO when ozonated in the absence of ˙OH, as shown in TEM (Fig. S7c†) and AFM (Fig. S7f†) micrographs. This supports our hypothesis that O3 alone (i.e. in the absence of hydroxyl radicals) can cause the decomposition and fragmentation of GO.
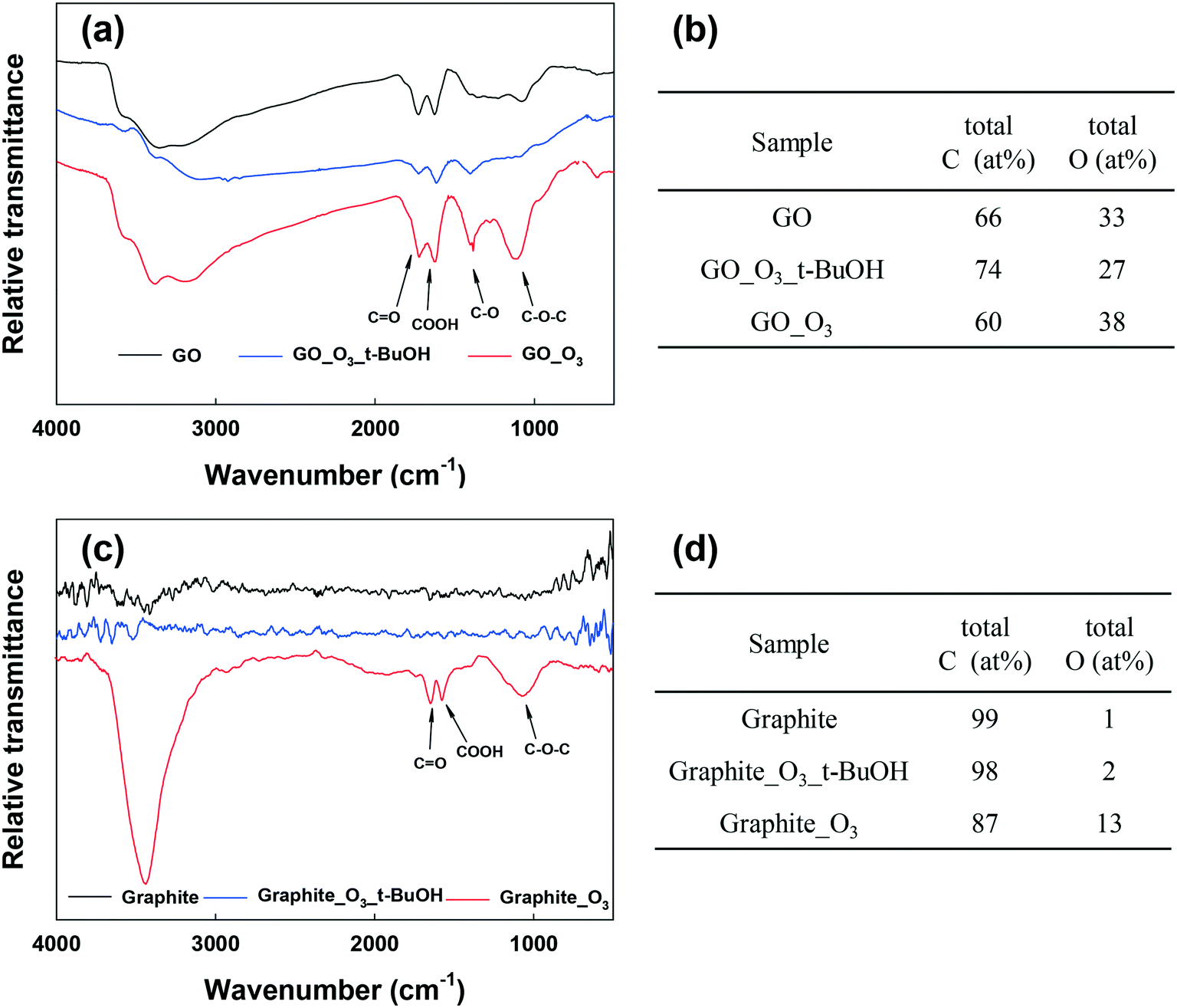 | ||
| Fig. 4 (a) FTIR spectra of GO, GO_O3 with t-BuOH and GO_O3. (b) C and O ratio of GO, GO_O3 and GO_O3 with t-BuOH obtained from XPS analysis. (c) FTIR spectra of graphite, Graphite_O3 with t-BuOH and Graphite_O3. (d) C and O ratio of graphite, Graphite_O3 and Graphite_O3 with t-BuOH obtained from XPS analysis. | ||
To understand the individual effects of O3 and ˙OH on the unfunctionalized aromatic domain of GO, an additional ˙OH-scavenging (by t-BuOH) experiment was conducted using graphite. Fig. 4c and d show the O3-induced transformation of graphite with and without t-BuOH. The FTIR spectrum of graphite after ozonation in the presence of t-BuOH was similar to the spectrum of untreated graphite. This shows that in the absence of ˙OH, O3 and other ROS species present cannot oxidize unfunctionalized aromatic carbon rings. This finding was also corroborated by XPS analysis (Fig. 4d and Table S4†). It is therefore clear that the addition of oxygen-functional groups onto the functionality-free domains of GO (e.g., certain regions on the basal plane) could only occur through ˙OH attack. This finding is in agreement with other studies, which indicated that ˙OH can attack the unfunctionalized aromatic ring of small molecular compounds.29–31
3.4 Mechanism of GO transformation by aqueous O3
Several conclusions can be made based on the abovementioned ˙OH-scavenging experiments. First, ˙OH can cause the addition of functional groups (especially the OH group) onto the surface of GO—probably both on functionalized and unfunctionalized aromatic carbon rings. Second, O3 (by itself) cannot oxidize unfunctionalized aromatic carbon rings. Third, the major effect of O3 is to oxidize the C–O–C/C–OH groups to CO and O–CO groups, which can cause aromatic ring cleavage.
Based on these results and the literature, we propose that the ozonation of GO occurred via two major pathways (Fig. 5). In pathway A, the ozonation of unfunctionalized aromatic rings, the aromatic double bonds on a graphitic basal plane are attacked by only ˙OH, resulting in the addition of C–OH (a1) by electrophilic addition. ˙OH and O3 can further oxidize aromatic rings by electrophilic addition (a2) or dipolar cycloaddition (b1). As such, C–OH groups on GO could be oxidized to CO (b2) and O–CO (b3) groups, cleaving the aromatic rings.28–31 Low molecular-weight compounds, CO2 and H2O, may form from the cleaved rings (b4).30 Pathway B occurred on the aromatic rings with functional groups. The ortho and para positions beside a C–OH group present the highest electronic densities, and so they are the priority sites for dipolar cycloaddition of O3 (b1) or electrophilic addition of ˙OH (c1). Then GO may undergo bond attack by O3 or ˙OH to form CO (b2) and/or O–CO (b3), with ring cleavage and decomposition to low molecular-weight compounds (b4) with further O3/˙OH oxidation. Reaction pathway A may be similar to pathway B after the addition of the OH group (a1).
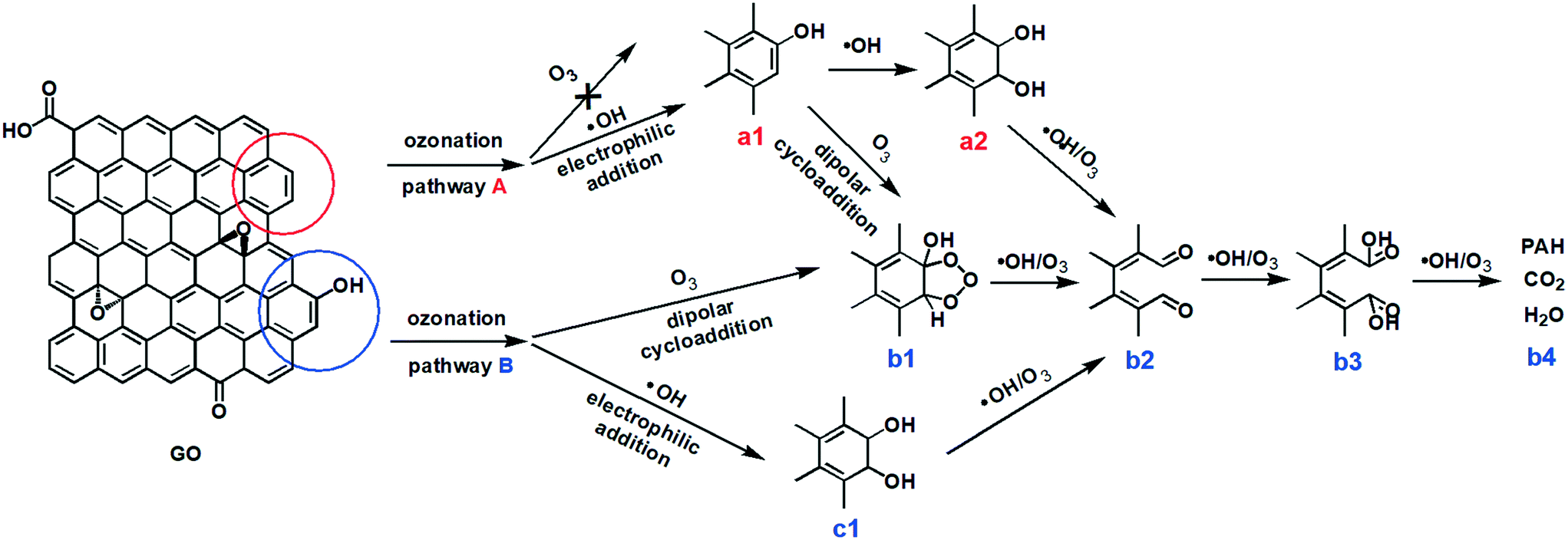 | ||
| Fig. 5 Proposed reaction pathways for the ozonation of GO in aqueous media. Pathway (A): unfunctionalized aromatic carbon–carbon double bonds, which are on the edge of the graphitic basal plane. Pathway (B): aromatic carbon functionalized with a hydroxyl group. | ||
3.5 Influence of ozonation on the environmental fate and effects of GO
As we have shown, ozonation led to the oxidation and aromatic ring cleavage of GO. In addition, there was an overall increase in the oxygen content of GO after ozonation, which may further increase the hydrophilicity of the nanomaterial. All these O3-treatment induced changes may affect the behavior of GO in the environment. To understand the influence of ozonation on the environmental fate of GO, we examined how the changes of GO's physicochemical properties from ozonation would affect its transport in saturated porous media and its adsorption affinities for common organic contaminants.The transport of GO and GO_O3 suspensions in porous media was studied in the presence of 35 mM NaCl or 0.3 mM CaCl2 (in duplicate), similar to a previous study.17 The breakthrough curves of the GO and GO_O3 transport studies are shown in Fig. 6a and b. In the presence of 35 mM NaCl, GO_O3 exhibited greater mobility in saturated quartz sand than the untreated GO. The greater mobility of O3-treated GO is consistent with the increased hydrophilicity (indicated by the decrease in KDW from 0.47 for GO to 0.22 for GO_O3), which decreased the tendency of attachment to porous media.52 Moreover, the hydrodynamic diameter (Dh) of GO decreased considerably upon ozonation, from 186 nm for the pristine GO to 120 nm for GO_O3 (in 35 mM NaCl, Table S5†), which would weaken the van der Waals attraction between GO nanosheets and sand grains. Ozonation inhibited the transport of GO in the presence of Ca2+. Even though the eluted concentration of GO_O3 reached the maximum at the same time as the pristine GO, the breakthrough concentration of GO_O3 declined more quickly afterwards (Fig. 6b). The seemingly counterintuitive observation is consistent with the ozonation-induced changes in GO surface O-functionality. Specifically, ozonation resulted in more than 100% increase of the O–CO group on the GO surface (Table 1), which facilitated the deposition of GO nanosheets to sand grains through cation-bridging,53–56 wherein Ca2+ served as the bridging agent by forming complexes between the O–CO group on GO and the OH group on sand grains.
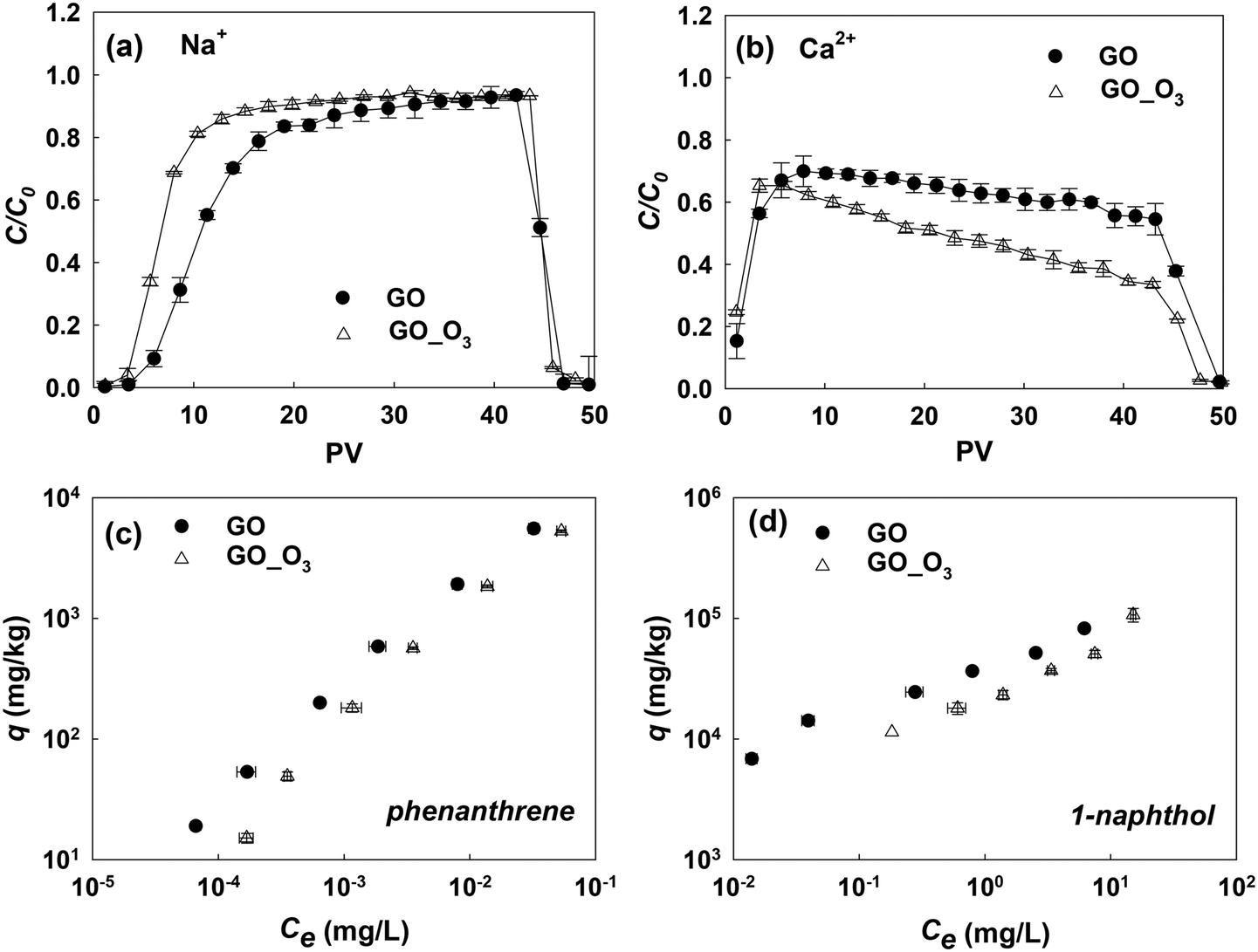 | ||
| Fig. 6 Transport of GO and GO_O3 in saturated quartz sand at (a) 35 mM NaCl (pH 6.0) and (b) 0.3 mM CaCl2 (pH 6.0). C = concentration of GO in the effluent; C0 = concentration of GO in the influent; C/C0, the ratio of the effluent concentration to the influent concentration. The data of breakthrough curves between 5 and 25 PV in the presence of Na+ and after 10 PV in the presence of Ca2+ were statistically significant by statistical analysis (t-test, P < 0.05) performed with SPSS 22.0; adsorption isotherms of (c) phenanthrene and (d) 1-naphthol adsorbed onto GO and GO_O3. q (mg kg−1) and Ce (mg L−1) are equilibrium concentrations of contaminants on GO and in aqueous solution, respectively. | ||
Fig. 6c and d show the comparison of the adsorption isotherms of phenanthrene and 1-naphthol on pristine GO and O3-treated GO. The isotherm experiments were conducted in duplicate, and the data were fitted with the Freundlich adsorption model (Table S6†). Clearly, ozonation impaired the adsorption affinities of GO for both model compounds. Adsorption of PAHs to GO is mainly driven by hydrophobic effects and π–π interactions.39,57–59 The impact of hydrogen bonding on the polar aromatic compound (1-naphthol) was not significant in this study. Thus, the decrease in the adsorption of phenanthrene and 1-naphthol was a direct consequence of the increased hydrophilicity and damaged graphitic structure (which decreased the π-electron density) of O3-treated GO.
4 Conclusions
Release of GO nanosheets into water and wastewater treatment plants will cause some physicochemical transformations of the nanomaterials. As shown in this study, GO is transformed in the presence of a relatively low O3 concentration (5 mg L−1), similar to O3 levels used during disinfection or tertiary treatment in drinking water/wastewater plants. Specifically, we observed cleaving of GO nanosheets and increased oxidation (including the oxidation of hydroxyl groups to carboxyl and carbonyl groups). This study showed that both the O3 molecule and generated ˙OH radicals were involved in the oxidation and transformation reactions. O3 could only oxidize functionalized aromatic carbon rings in the absence of ˙OH, while ˙OH was involved in the oxidation of both functionalized and unfunctionalized aromatic carbon rings. After O3 treatment, the increased hydrophilicity and decreased lateral size of GO led to an increase in the mobility of GO in Na+ solution, but the mobility decreased in Ca2+ solution because of bridging effects that were intensified by increased O–CO groups. The adsorption of phenanthrene and 1-naphthol by GO both decreased due to increased hydrophilicity. One limitation of this study is that we only used pristine nanomaterials. Future studies should consider using nanomaterials incorporated into ‘real-world’ matrices (like polymers) and perform experiments under more environmentally relevant conditions (e.g. involving soils or sediments).
Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
This project was supported by the Ministry of Science and Technology of China (Grant 2015CB459000), the National Natural Science Foundation of China (Grants 21677078 and 21425729), 111 program, Ministry of Education, China (Grant T2017002) and Basic Scientific Research Program (Grant 7954).References
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, The chemistry of graphene oxide, Chem. Soc. Rev., 2010, 39, 228–240 RSC
.
- W. Gao, L. B. Alemany, L. Ci and P. M. Ajayan, New insights into the structure and reduction of graphite oxide, Nat. Chem., 2009, 1, 403–408 CrossRef CAS PubMed
- X. Ren, J. Li, C. Chen, Y. Gao, D. Chen, M. Su, A. Alsaedi and T. Hayat, Graphene analogues in aquatic environments and porous media: Dispersion, aggregation, deposition and transformation, Environ. Sci.: Nano, 2018, 5, 1298–1340 RSC
- A. S. Adeleye, J. R. Conway, K. Garner, Y. Huang, Y. Su and A. A. Keller, Engineered nanomaterials for water treatment and remediation: Costs, benefits, and applicability, Chem. Eng. J., 2016, 286, 640–662 CrossRef CAS
- K. Krishnamoorthy, K. Jeyasubramanian, M. Premanathan, G. Subbiah, H. S. Shin and S. J. Kim, Graphene oxide nanopaint, Carbon, 2014, 72, 328–337 CrossRef CAS
- A. S. Adeleye, K. T. Ho, M. Zhang, Y. Li and R. M. Burgess, Fate and transformation of graphene oxide in estuarine and marine waters, Environ. Sci. Technol., 2019, 53, 5858–5867 CrossRef CAS PubMed
- K. Yang, J. Wang, X. Chen, Q. Zhao, A. Ghaffar and B. Chen, Application of graphene-based materials in water purification: From the nanoscale to specific devices, Environ. Sci.: Nano, 2018, 5, 1264–1297 RSC
- X. Zhu, K. Yang and B. Chen, Membranes prepared from graphene-based nanomaterials for sustainable applications: A review, Environ. Sci.: Nano, 2017, 4, 2267–2285 RSC
- X. Zeng, G. Wang, Y. Liu and X. Zhang, Graphene-based antimicrobial nanomaterials: Rational design and applications for water disinfection and microbial control, Environ. Sci.: Nano, 2017, 4, 2248–2266 RSC
- V. Georgakilas, J. N. Tiwari, K. C. Kemp, J. A. Perman, A. B. Bourlinos, K. S. Kim and R. Zboril, Noncovalent functionalization of graphene and graphene oxide for energy materials, biosensing, catalytic, and biomedical applications, Chem. Rev., 2016, 116, 5464–5519 CrossRef CAS PubMed
- S. Perez, M. L. Farre and D. Barcelo, Analysis, behavior and ecotoxicity of carbon-based nanomaterials in the aquatic environment, TrAC, Trends Anal. Chem., 2009, 28, 820–832 CrossRef CAS
- N. C. Mueller and B. Nowack, Exposure modeling of engineered nanoparticles in the environment, Environ. Sci. Technol., 2008, 42, 4447–4453 CrossRef CAS PubMed
- H. H. Liu and Y. Cohen, Multimedia environmental distribution of engineered nanomaterials, Environ. Sci. Technol., 2014, 48, 3281–3292 CrossRef CAS PubMed
- D. G. Goodwin, Jr., A. S. Adeleye, L. Sung, K. T. Ho, R. M. Burgess and E. J. Petersen, Detection and quantification of graphene-family nanomaterials in the environment, Environ. Sci. Technol., 2018, 52, 4491–4513 CrossRef PubMed
- L. Duan, R. Hao, Z. Xu, X. He, A. S. Adeleye and Y. Li, Removal of graphene oxide nanomaterials from aqueous media via coagulation: Effects of water chemistry and natural organic matter, Chemosphere, 2017, 168, 1051–1057 CrossRef CAS PubMed
- T. Du, A. S. Adeleye, A. A. Keller, Z. Wu, W. Han, Y. Wang, C. Zhang and Y. Li, Photochlorination-induced transformation of graphene oxide: Mechanism and environmental fate, Water Res., 2017, 124, 372–380 CrossRef CAS PubMed
- Y. Li, N. Yang, T. Du, X. Wang and W. Chen, Transformation of graphene oxide by chlorination and chloramination: Implications for environmental transport and fate, Water Res., 2016, 103, 416–423 CrossRef CAS PubMed
- T. Du, Y. Wang, X. Yang, W. Wang, H. Guo, X. Xiong, R. Gao, X. Wuli, A. S. Adeleye and Y. Li, Mechanisms and kinetics study on the trihalomethanes formation with carbon nanoparticle precursors, Chemosphere, 2016, 154, 391–397 CrossRef CAS PubMed
- U. von Gunten, Ozonation of drinking water: Part I. Oxidation kinetics and product formation, Water Res., 2003, 37, 1443–1467 CrossRef CAS PubMed
- J. D. Fortner, D.-I. Kim, A. M. Boyd, J. C. Falkner, S. Moran, V. L. Colvin, J. B. Hughes and J.-H. Kim, Reaction of water-stable C60 aggregates with ozone, Environ. Sci. Technol., 2007, 41, 7497–7502 CrossRef CAS PubMed
- K. Peng, L. Liu, H. Li, H. Meyer and Z. Zhang, Room temperature functionalization of carbon nanotubes using an ozone/water vapor mixture, Carbon, 2011, 49, 70–76 CrossRef CAS
- Y. Wang, Y. Xie, H. Sun, J. Xiao, H. Cao and S. Wang, Efficient catalytic ozonation over reduced graphene oxide for p-hydroxylbenzoic acid (PHBA) destruction: Active site and mechanism, ACS Appl. Mater. Interfaces, 2016, 8, 9710–9720 CrossRef CAS PubMed
- Z. Xu, M. Yue, L. Chen, B. Zhou, M. Shan, J. Niu, B. Li and X. Qian, A facile preparation of edge etching, porous and highly reactive graphene nanosheets via ozone treatment at a moderate temperature, Chem. Eng. J., 2014, 240, 187–194 CrossRef CAS
- M. S. Elovitz and U. von Gunten, Hydroxyl radical/ozone ratios during ozonation processes. I. The Rct concept, Ozone: Sci. Eng., 1999, 21, 239–260 CrossRef CAS
- M.-O. Buffle, J. Schumacher, E. Salhi, M. Jekel and U. von Gunten, Measurement of the initial phase of ozone decomposition in water and wastewater by means of a continuous quench-flow system: Application to disinfection and pharmaceutical oxidation, Water Res., 2006, 40, 1884–1894 CrossRef CAS PubMed
- J. Staehelin and J. Hoigne, Decomposition of ozone in water in the presence of organic solutes acting as promoters and inhibitors of radical chain reactions, Environ. Sci. Technol., 1985, 19, 1206–1213 CrossRef CAS PubMed
- D. Zhang, S. Yan and W. Song, Photochemically induced formation of reactive oxygen species (ROS) from effluent organic matter, Environ. Sci. Technol., 2014, 48, 12645–12653 CrossRef CAS PubMed
- E. Perraudin, H. Budzinski and E. Villenave, Identification and quantification of ozonation products of anthracene and phenanthrene adsorbed on silica particles, Atmos. Environ., 2007, 41, 6005–6017 CrossRef CAS
- P. N. Chen, G. A. Junk and H. J. Svec, Reactions of organic pollutants. 1. Ozonation of acenaphthylene and acenaphthene, Environ. Sci. Technol., 1979, 13, 451–454 CrossRef CAS
- J.-J. Yao, Z.-H. Huang and S. J. Masten, The ozonation of pyrene: Pathway and product identification, Water Res., 1998, 32, 3001–3012 CrossRef CAS
- J.-J. Yao, Z.-H. Huang and S. J. Masten, The ozonation of benz[a]anthracene: pathway and product identification, Water Res., 1998, 32, 3235–3244 CrossRef CAS
- N. Leconte, J. Moser, P. Ordejon, H. Tao, A. Lherbier, A. Bachtold, F. Alsina, C. M. Sotomayor Torres, J.-C. Charlier and S. Roche, Damaging graphene with ozone treatment: A chemically tunable metal-insulator transition, ACS Nano, 2010, 4, 4033–4038 CrossRef CAS PubMed
- L. V. Liu, W. Q. Tian and Y. A. Wang, Ozonization at the vacancy defect site of the single-walled carbon nanotube, J. Phys. Chem. B, 2006, 110, 13037–13044 CrossRef CAS PubMed
- S. G. Zimmermann, M. Wittenwiler, J. Hollender, M. Krauss, C. Ort, H. Siegrist and U. von Gunten, Kinetic assessment
and modeling of an ozonation step for full-scale municipal wastewater treatment: Micropollutant oxidation, by-product formation and disinfection, Water Res., 2011, 45, 605–617 CrossRef CAS PubMed
- V. C. Tung, M. J. Allen, Y. Yang and R. B. Kaner, High-throughput solution processing of large-scale graphene, Nat. Nanotechnol., 2009, 4, 25–29 CrossRef CAS PubMed
- C. Gomez-Navarro, R. T. Weitz, A. M. Bittner, M. Scolari, A. Mews, M. Burghard and K. Kern, Electronic transport properties of individual chemically reduced graphene oxide sheets, Nano Lett., 2007, 7, 3499–3503 CrossRef CAS PubMed
-
APHA, AWWA and WEF, Standard Methods for the Examination of Water and Wastewater, American Public Health Association, American Water Works Association and Water Environment Federation, Washington, DC, 1998 Search PubMed
- R. S. Kingsbury and P. C. Singer, Effect of magnetic ion exchange and ozonation on disinfection by-product formation, Water Res., 2013, 47, 1060–1072 CrossRef CAS PubMed
- F. Wang, F. Wang, D. Zhu and W. Chen, Effects of sulfide reduction on adsorption affinities of colloidal graphene oxide nanoparticles for phenanthrene and 1-naphthol, Environ. Pollut., 2015, 196, 371–378 CrossRef CAS PubMed
- S.-H. Choi, Unique properties of graphene quantum dots and their applications in photonic/electronic devices, J. Phys. D: Appl. Phys., 2017, 50, 103002 CrossRef
- C. Nethravathi, T. Nisha, N. Ravishankar, C. Shivakumara and M. Rajamathi, Graphene-nanocrystalline metal sulphide composites produced by a one-pot reaction starting from graphite oxide, Carbon, 2009, 47, 2054–2059 CrossRef CAS
- S. Park, K.-S. Lee, G. Bozoklu, W. Cai, S. T. Nguyen and R. S. Ruoff, Graphene oxide papers modified by divalent ions - Enhancing mechanical properties via chemical cross-linking, ACS Nano, 2008, 2, 572–578 CrossRef CAS PubMed
- Y. Jiang, R. Raliya, P. Liao, P. Biswas and J. D. Fortner, Graphene oxides in water: Assessing stability as a function of material and natural organic matter properties, Environ. Sci.: Nano, 2017, 4, 1484–1493 RSC
- H. R. Thomas, S. P. Day, W. E. Woodruff, C. Valles, R. J. Young, I. A. Kinloch, G. W. Morley, J. V. Hanna, N. R. Wilson and J. P. Rourke, Deoxygenation of graphene oxide: Reduction or cleaning?, Chem. Mater., 2013, 25, 3580–3588 CrossRef CAS
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I.-C. Hwang and K. S. Kim, Water-dispersible magnetite-reduced graphene oxide composites for arsenic removal, ACS Nano, 2010, 4, 3979–3986 CrossRef CAS PubMed
- L. Guardia, S. Villar-Rodil, J. I. Paredes, R. Rozada, A. Martinez-Alonso and J. M. D. Tascon, UV light exposure of aqueous graphene oxide suspensions to promote their direct reduction, formation of graphene-metal nanoparticle hybrids and dye degradation, Carbon, 2012, 50, 1014–1024 CrossRef CAS
- J. Wenk, M. Aeschbacher, E. Salhi, S. Canonica, U. von Gunten and M. Sander, Chemical oxidation of dissolved organic matter by chlorine dioxide, chlorine, and ozone: Effects on its optical and antioxidant properties, Environ. Sci. Technol., 2013, 47, 11147–11156 CrossRef CAS PubMed
- A. C. Ferrari and J. Robertson, Interpretation of Raman spectra of disordered and amorphous carbon, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS
- S. Huh, J. Park, Y. S. Kim, K. S. Kim, B. H. Hong and J.-M. Nam, UV/ozone-oxidized large-scale graphene platform with large chemical enhancement in surface-enhanced Raman scattering, ACS Nano, 2011, 5, 9799–9806 CrossRef CAS PubMed
- S. Esplugas, J. Gimenez, S. Contreras, E. Pascual and M. Rodriguez, Comparison of different advanced oxidation processes for phenol degradation, Water Res., 2002, 36, 1034–1042 CrossRef CAS PubMed
- E. Mvula and C. von Sonntag, Ozonolysis of phenols in aqueous solution, Org. Biomol. Chem., 2003, 1, 1749–1756 RSC
- T. Xia, P. Ma, Y. Qi, L. Zhu, Z. Qi and W. Chen, Transport and retention of reduced graphene oxide materials in saturated porous media: Synergistic effects of enhanced attachment and particle aggregation, Environ. Pollut., 2019, 247, 383–391 CrossRef CAS PubMed
- T. Xia, J. D. Fortner, D. Zhu, Z. Qi and W. Chen, Transport of sulfide-reduced graphene oxide in saturated quartz sand: Cation-dependent retention mechanisms, Environ. Sci. Technol., 2015, 49, 11468–11475 CrossRef CAS PubMed
- Y. Li, N. Yang, T. Du, T. Xia, C. Zhang and W. Chen, Chloramination of graphene oxide significantly affects its transport properties in saturated porous media, NanoImpact, 2016, 3–4, 90–95 CrossRef
- K. L. Chen and M. Elimelech, Influence of humic acid on the aggregation kinetics of fullerene (C60) nanoparticles in monovalent and divalent electrolyte solutions, J. Colloid Interface Sci., 2007, 309, 126–134 CrossRef CAS PubMed
- P. Yi and K. L. Chen, Influence of surface oxidation on the aggregation and deposition kinetics of multiwalled carbon nanotubes in monovalent and divalent electrolytes, Langmuir, 2011, 27, 3588–3599 CrossRef CAS PubMed
- Z. Pei, L. Li, L. Sun, S. Zhang, X. Shan, S. Yang and B. Wen, Adsorption characteristics of 1,2,4-trichlorobenzene, 2,4,6-trichlorophenol, 2-naphthol and naphthalene on graphene and graphene oxide, Carbon, 2013, 51, 156–163 CrossRef CAS
- J. Wang, Z. Chen and B. Chen, Adsorption of polycyclic aromatic hydrocarbons by graphene and graphene oxide nanosheets, Environ. Sci. Technol., 2014, 48, 4817–4825 CrossRef CAS PubMed
- J. Xu, L. Wang and Y. Zhu, Decontamination of bisphenol A from aqueous solution by graphene adsorption, Langmuir, 2012, 28, 8418–8425 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9en00361d |
| This journal is © The Royal Society of Chemistry 2019 |