
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in transition metal-catalysed hydroacylation of alkenes and alkynes
Avipsa
Ghosh
,
Kirsten F.
Johnson
,
Kevin L.
Vickerman
,
James A.
Walker
Jr.
and
Levi M.
Stanley
*
Department of Chemistry, Iowa State University, Ames, IA 50011, USA. E-mail: lstanley@iastate.edu
First published on 11th February 2016
Abstract
This highlight discusses developments in transition metal-catalysed alkene and alkyne hydroacylation reactions over the past three years. The discussion summarizes the development of new catalyst systems for alkene and alkyne hydroacylation and applications to the synthesis of important ketone building blocks. The highlight captures transition metal-catalysed alkene and alkyne hydroacylation at a time of impressive growth when cobalt, nickel, ruthenium, and iridium catalysts are emerging as complements or replacements for traditional rhodium catalysts.
 Avipsa Ghosh, Kevin L. Vickerman, Kirsten F. Johnson, James A. Walker Jr., Levi M. Stanley (from left to right). | Levi Stanley received his B.A. in chemistry from Augustana College (SD) in 2001, and he received his Ph.D. from North Dakota State University in 2007 under the supervision of Professor Mukund P. Sibi. After postdoctoral studies at the University of Illinois at Urbana-Champaign with Professor John Hartwig, he joined the faculty at Iowa State University in 2012 as an assistant professor in the Department of Chemistry. Kirsten Johnson received her B.S. in chemistry and Spanish from the University of Wisconsin – Madison in 2011. Avipsa Ghosh received her B.Sc. in chemistry from the University of Calcutta in 2006, and she received her M.Sc. in organic chemistry from Bangalore University in 2008. Kevin Vickerman received his B.S. in chemistry and cellular and molecular biology from Winona State University in 2013. James Walker received his B.S. in chemistry from the University of Cincinnati in 2012. Johnson, Ghosh, Vickerman, and Walker are currently pursuing Ph.D. studies in chemistry at Iowa State University with Prof. Levi Stanley. Their research is focused on the development of new catalytic alkene and alkyne hydroacylation reactions. |
Introduction
Transition metal-catalysed alkene and alkyne hydroacylation, the formal addition of an aldehyde C–H bond across an unsaturated C–C bond, has emerged as a powerful, catalytic process to synthesize ketones over the past four decades.1–3 The identification and development of new catalysts has greatly expanded the scope and synthetic utility of these hydroacylation reactions in recent years. Intramolecular alkene hydroacylation reactions are now well-established methods to synthesize carbocyclic and heterocyclic ketones, often with high enantioselectivities. In addition, the development of intermolecular hydroacylations of new combinations of alkenes and alkynes with aldehydes continues to rapidly expand and provide direct routes to new ketones from simple starting materials.Despite recent advances in catalytic alkene and alkyne hydroacylation reactions, many challenges remain to fully harness the synthetic potential of these valuable processes. These challenges include: intramolecular hydroacylations to form medium and large rings in the absence of functional groups to stabilize catalytic intermediates; intermolecular hydroacylations that encompass a much broader array of substrates; and new catalyst types that improve activity, complement the selectivity, and reduce the cost of widely studied rhodium complexes of bisphosphine ligands.
Since the initial report by Sakai and co-workers in 1972,4 hydroacylation of alkenes and alkynes has evolved to encompass reactions catalysed by not only transition metal complexes, but also N-heterocyclic carbenes (NHCs)5,6 and photocatalysts.7–9 This highlight will focus on recent developments in transition metal-catalysed alkene and alkyne hydroacylation reactions over the past three years. The discussion is divided by catalyst type and includes highlights of rhodium-, cobalt-, ruthenium-, iridium-, and nickel-catalysed processes.
Rhodium-catalysed alkene and alkyne hydroacylation
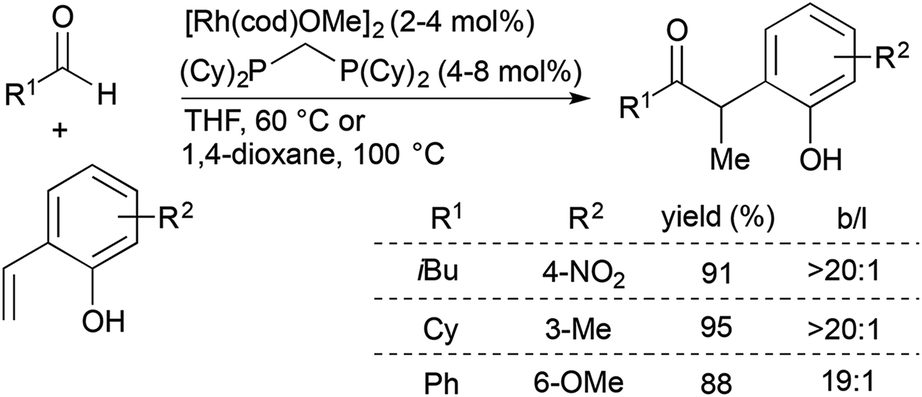
Since the initial report of transition metal-catalysed olefin hydroacylation,4 rhodium-catalysed alkene and alkyne hydroacylation reactions have remained at the forefront of studies in the area. While much progress has been made in the development of alkene and alkyne hydroacylation reactions with aldehydes containing functional groups capable of coordinating the rhodium center, intermolecular alkene hydroacylations with simple aromatic and aliphatic aldehydes remain challenging.Dong and co-workers recently reported highly branched-selective intermolecular hydroacylation reactions of a variety of non-chelating aliphatic, aromatic, and alkenyl aldehydes with ortho-vinylphenols (Scheme 1).10,11 [Rh(cod)(OMe)]2 and a small-bite-angle bisphosphine ligand, bis(dicyclohexylphosphino)methane (dcpm), combine with the ortho-vinylphenol substrate to generate an electron-rich and neutral rhodium complex that has a lower barrier for the turnover-limiting, non-directed oxidative addition of Rh to the aldehyde C–H bond. This catalyst promotes reactions of an array of non-chelating aldehydes with a wide range of substituted ortho-vinylphenols to form branched ketone products (typically >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 selectivity) in 50–99% yield. The α-aryl ketone products can readily undergo acid-catalyzed cyclocondensation to form benzofurans in high yields.
:1 selectivity) in 50–99% yield. The α-aryl ketone products can readily undergo acid-catalyzed cyclocondensation to form benzofurans in high yields.
 | ||
| Scheme 1 Hydroacylation of vinylphenols with non-chelating aldehydes. | ||
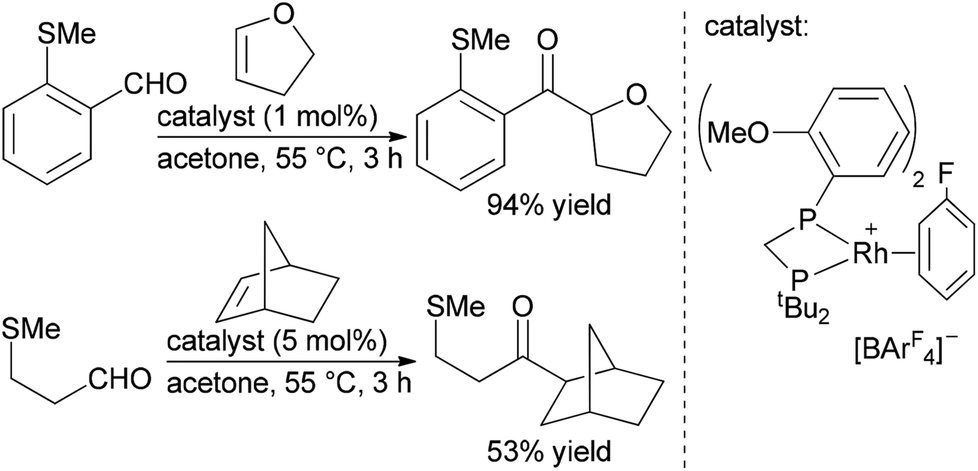
Development in the Willis and Weller labs of catalysts containing small-bite-angle bisphosphine ligands with the general structure [Rh(R2PCH2PR2)(η6-C6H5F)][BArF4] has substantially improved the activity and stability of rhodium catalyst in intermolecular hydroacylation of alkenes with β-substituted aldehydes.12–14 Recently, these groups reported a catalyst with this general structure based on the non-symmetrical ligand tBu2PCH2P(o-MeOC6H4)2 that enables intermolecular hydroacylation of a wide range of functionalized internal alkenes with β-substituted aldehydes (Scheme 2).14 Detailed mechanistic studies suggest that the o-MeOC6H4 group on phosphorous plays a key role in the stability and activity of the catalyst by increasing the rate of olefin insertion in to the rhodium hydride bond, which prevents decarbonylation by maintaining a low concentration of the acyl hydride intermediate. This catalyst system enables hydroacylations of a range of both 1,1- and 1,2-disubstituted alkenes, including cycloalkenes, cyclic and acyclic vinyl ethers, and α,β-unsaturated esters, with aryl and non-aryl aldehydes.
 | ||
| Scheme 2 Intermolecular hydroacylation of internal alkenes with β-substituted aldehydes. | ||
Inspired by Tanaka and Suemune's report on desymmetrization of β-bis(alkenyl)aldehydes to generate cyclopentanones bearing β-quaternary stereogenic centers,15,16 Dong and coworkers developed an enantioselective desymmetrization of α-trisubstituted aldehydes to form cyclopentanones with α-quaternary stereogenic centers (Scheme 3).17 A catalyst generated in situ from [Rh(coe)2Cl]2, (R)-DTBM-MeO-BIPHEP and AgBF4 facilitates the desymmetrization of the α-quaternary center through an isomerization–hydroacylation cascade. Key to this isomerization–hydroacylation cascade is a rare endocyclic β-hydride elimination for which this study provides experimental evidence that supports previous theoretical studies.18,19 The chiral Rh catalyst enables selective formation of a variety of α-vinylcyclopentanones (R = alkyl, aryl, heteroaryl) in high yields (73–91%) with excellent enantioselectivities (91–99% ee).
 | ||
| Scheme 3 Desymmetrization of α-quaternary centers by isomerization–hydroacylation. | ||
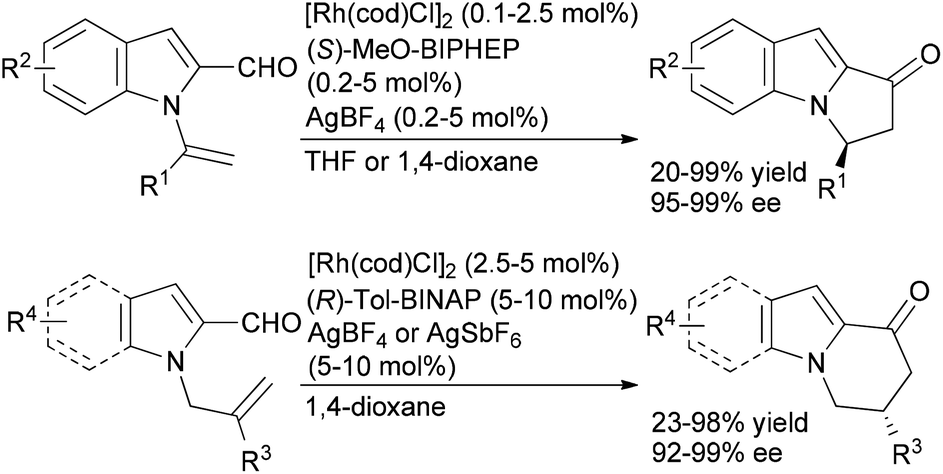
Our group and others have investigated alkene and alkyne hydroacylation reactions as a strategy to synthesize polycyclic nitrogen heterocycles. We initially reported highly enantioselective, intramolecular hydroacylation of N-vinylindole-2-carboxaldehydes to generate dihydropyrroloindolones in high yield with excellent enantioselectivities (Scheme 4).20 More recently, we reported catalytic, enantioselective hydroacylations of N-allylindole-2-carboxaldehydes and N-allylpyrrole-2-carboxaldehydes to form dihydropyridoindolones and dihydroindolizinones in moderate-to-high yields with high enantioselectivities.21 These reactions represent the first examples of highly enantioselective, transition metal-catalyzed hydroacylation to form six-membered rings in the absence of chelation-assistance and encompass an array of N-allylindole- and N-allylpyrrole-2-carboxaldehydes substrates. The ability to form six-membered rings in the absence of chelation-assistance led to the development of a related catalyst system for endo- and enantioselective hydroacylation of o-allylbenzaldehydes.22
 | ||
| Scheme 4 Enantioselective hydroacylation to access polycyclic nitrogen heterocycles. | ||
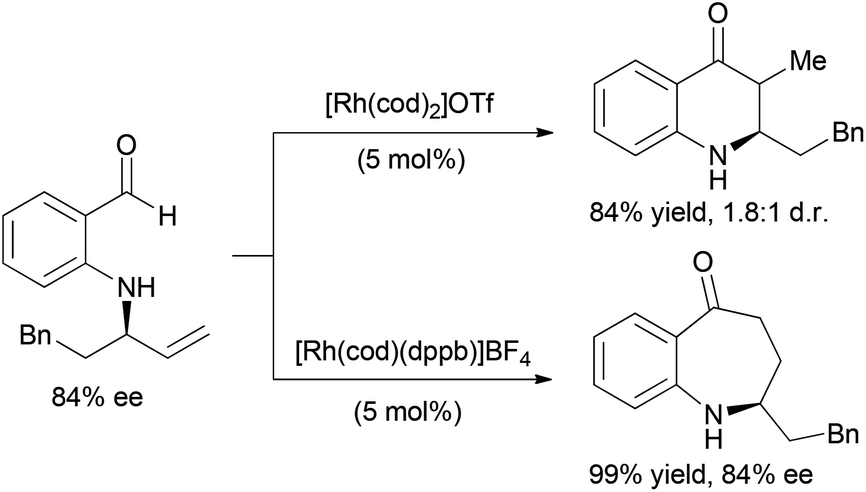
The coupling of alkene and alkyne hydroacylation reactions in sequence or in tandem with additional bond-forming processes has expanded the types of heterocyclic ketones that are readily accessible from simple starting materials. Nguyen and coworkers reported a sequential approach to the synthesis of dihydroquinolinones and tetrahydrobenzo[b]azepinones involving Rh-catalyzed asymmetric allylic amination followed by intramolecular alkene hydroacylation reactions.23 The enantioenriched allylic amine, formed by Rh-catalyzed dynamic kinetic asymmetric transformation (DYKAT) of racemic allylic trichloroacetimidates with 2-aminobenzaldehyde, is a suitable substrate for alkene hydroacylation to form six- or seven-membered ring aza-ketones based on the choice of the rhodium catalyst (Scheme 5).
 | ||
| Scheme 5 Sequential allylic amination and hydroacylation reactions to access six- and seven-membered nitrogen heterocycles. | ||
Aldehydes containing β-heteroatoms are privileged substrates in transition metal-catalyzed alkene and alkyne hydroacylation reactions due their ability to coordinate acylmetal hydride intermediates and suppress catalyst deactivation pathways. Often these β-chelating units are viewed as a limitation to the scope of alkene and alkyne hydroacylation processes. Recent reports from Willis and our laboratory demonstrated sequential and tandem reaction strategies that utilize these β-heteroatom functional groups as synthetic handles for the construction of dihydroquinolones and chromanones (Scheme 6). Willis and coworkers reported a sequential protocol for the hydroacylation of alkynes with 2-aminobenzaldehydes followed by Lewis acid-catalysed, intramolecular aza-Michael addition that gave rapid access to dihydroquinolones.24 Later, our group developed a tandem process for the synthesis of trans-2,3-disubstituted chroman-4-ones involving hydroacylation of symmetrical alkynes with salicylaldehydes followed by oxo-Michael addition reactions.25 The trans-2,3-disubstituted chroman-4-ones could be readily fluorinated to form trans-3-fluoro-2,3-disubstituted chroman-4-ones in high yields (67–93%) and excellent diastereoselectivities (up to >20:1).
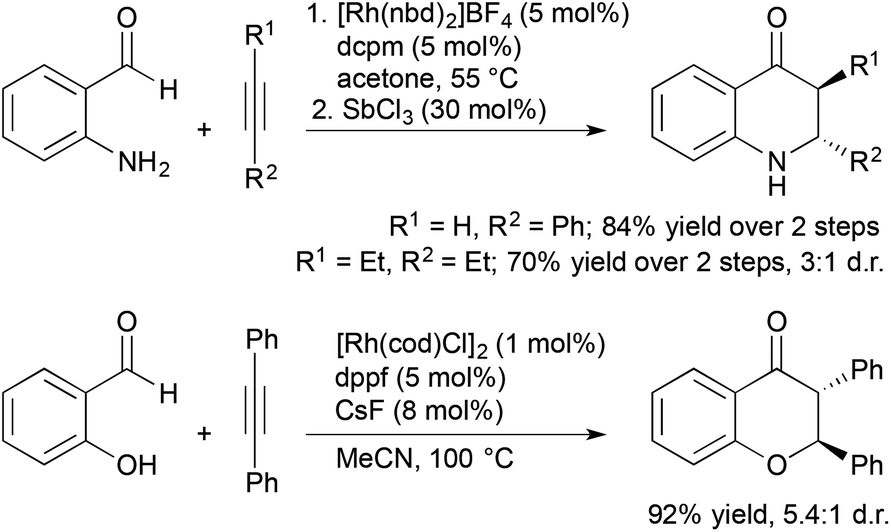 | ||
| Scheme 6 Sequential and tandem alkyne hydroacylation aza- and oxo-Michael addition reactions to form dihydroquinolones and chromanones. | ||
Willis, Weller and co-workers also reported a traceless hydroacylation cascade involving Rh-catalysed hydroacylation of alkenes, allenes, and alkynes coupled to a Rh-catalysed, silane-mediated sulfide reduction (Scheme 7).26 These cascades utilise the methyl sulfide functionality in the aldehyde substrate to stabilize the acylrhodium hydride intermediate formed during the hydroacylation cycle, and remove the chelating sulfide group in the Rh-catalyzed reduction. The same group successfully sequenced alkyne hydroacylation to Suzuki coupling with the methyl sulfide group functioning as a pseudohalide for cross-coupling with aryl and alkenyl boronic acids.27 The utility of this approach was further illustrated through a three-component hydroacylation/Suzuki coupling cascade using a single rhodium catalyst.
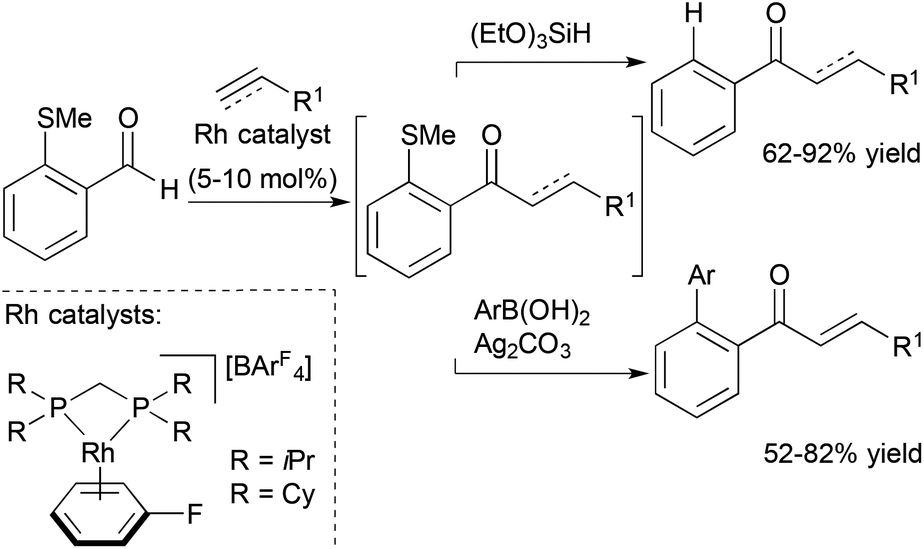 | ||
| Scheme 7 Traceless chelation-assisted hydroacylation and alkyne hydroacylation/Suzuki coupling cascade. | ||
Cobalt-catalysed alkene and alkyne hydroacylation
The use of cobalt complexes as catalysts for alkene and alkyne hydroacylation is attractive due to the abundance and cost of cobalt relative to the 2nd- and 3rd-row group 9 metals rhodium and iridium. Brookhart and coworkers demonstrated the first examples of cobalt-catalysed alkene hydroacylations nearly twenty years ago.28,29 However, the utility of cobalt catalysts in alkene and alkyne hydroacylation reactions remained limited until a recent flurry of reports.In 2014, Dong and coworkers developed Co-catalysed hydroacylations of 1,3-dienes that occur with regioselectivity that is distinct from previously reported ruthenium catalysts.30 Based on the identity of the bisphosphine ligand, cobalt complexes catalyse the C1-selective hydroacylations of 1,3-dienes with aromatic and aliphatic aldehydes to form products of 1,4-addition (1 and 3) and 1,2-addition (2 and 4), respectively (Scheme 8). These cobalt-catalysed hydroacylations proceed in the absence of chelation assistance without significant decarbonylation of the aldehyde starting materials. Mechanistic studies support a pathway involving an oxidative cyclization step, which contrasts the traditional mechanism of transition metal-catalysed hydroacylation and prevents decarbonylation of the aldehyde substrate by avoiding the formation of an acylmetal hydride intermediate.
 | ||
| Scheme 8 Intermolecular hydroacylation of 1,3-dienes. | ||
Yoshikai and Yang reported the first enantioselective, Co-catalysed intramolecular hydroacylations of alkenes and ketones.31 A catalyst generated by in situ reduction of a Co(II)–BDPP complex with zinc metal promotes highly enantioselective hydroacylations of 2-alkenylbenzaldehydes to form indanone derivatives (Scheme 9). Although the scope of enantioselective, Co-catalysed alkene hydroacylation is limited relative to related enantioselective, rhodium-catalysed hydroacylations of 2-alkenylbenzaldehydes, this work is likely to serve as a foundation for the development of new Co-catalyzed, enantioselective alkene hydroacylation reactions.
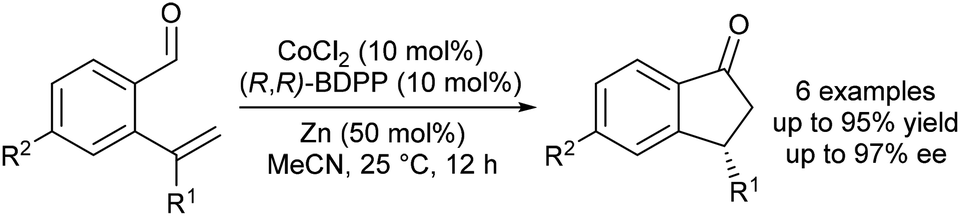 | ||
| Scheme 9 Enantioselective hydroacylation of 2-alkenylbenzaldehydes. | ||
Yoshikai and coworkers subsequently developed chelation-assisted, Co-catalysed intermolecular hydroacylation of alkenes with aldimines generated from 2-amino-3-methylpyridine (Scheme 10).32 The formal hydroacylation process encompasses a wide range of alkenes, including styrenes, vinylsilanes, allylsilanes, and simple alkenes, and forms, after hydrolysis, the corresponding ketone products in moderate-to-good yields with high linear-to-branched ratios.
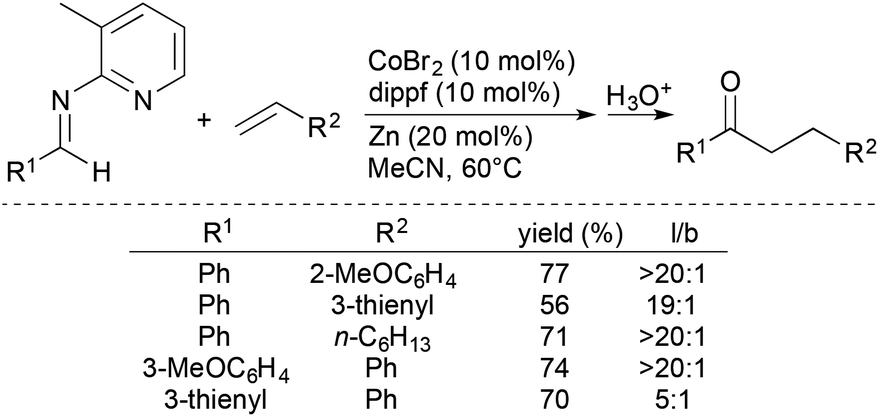 | ||
| Scheme 10 Cobalt-catalyzed hydroacylation of alkenes with aldimines. | ||
Cheng and coworkers recently reported a cobalt-catalysed cyclization of 1,6-enynes with aldehydes (Scheme 11).33 The scope of these reactions included a variety of N-, O-, and malonate-tethered enynes and an array of aromatic, heterocyclic, and aliphatic aldehydes. The formal hydroacylation is proposed to proceed via a novel cobaltacycle intermediate that is generated from the reaction of enyne substrate with cobalt catalyst. Interestingly, the cobalt catalyst system demonstrated switchable C–H functionalization activity depending on the electronic nature of the ancillary ligand enabling the authors to achieve either hydroacylation or hydroarylation processes.
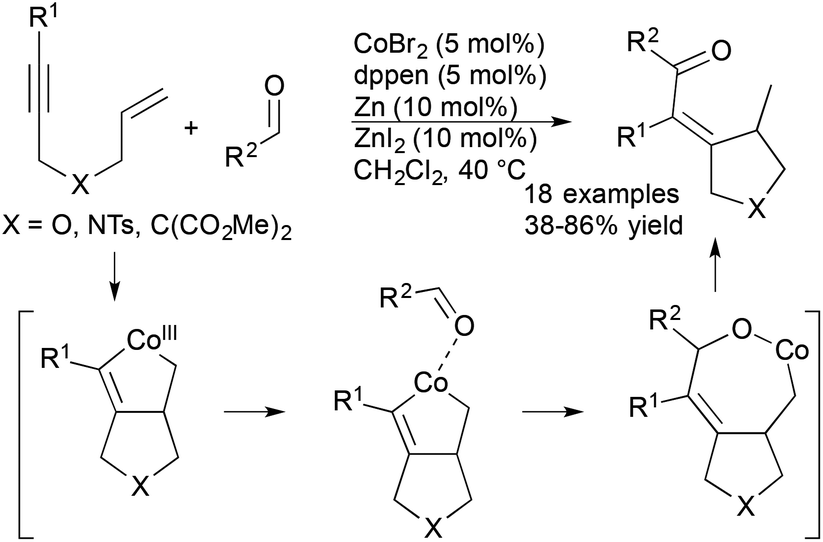 | ||
| Scheme 11 Cobalt-catalyzed formal hydroacylation of enynes. | ||
Ruthenium-, iridium- and nickel-catalyzed alkene and alkyne hydroacylation
Although rhodium complexes are the most common transition metal catalysts employed for alkene and alkyne hydroacylation reactions, the competition between hydroacylation and decarbonylation pathways observed with rhodium catalysts has led to a growing interest in the use of alternative transition metals for these processes. In particular, recent developments have been made in ruthenium-, iridium- and nickel-catalysed alkene and alkyne hydroacylations.In 2013, Inoue, Wada and coworkers reported the intermolecular hydroacylation of internal alkynes catalysed by a ceria-supported ruthenium complex.34 The authors propose that a Ru(IV)-oxo species is generated at the CeO2 surface. The Ru(IV)-oxo species is reduced to a Ru(II)H2 by sodium formate, and hydrogenation of excess alkyne leads to the catalytically active Ru(0) species. These alkyne hydroacylation reactions occur in moderate to high yields (19–93% yield) and represent a rare example of alkyne hydroacylation using a recyclable catalyst.
Previous work on Ru-catalysed hydroacylation of dienes by Krische and coworkers showed that these processes do not proceed through an acylmetal hydride intermediate.35 The absence of this intermediate precludes the decarbonylation pathway that has historically plagued olefin hydroacylation reactions. Building on this precedent, Dong and coworkers developed ruthenium-catalysed hydroacylation of alkynes via a tandem catalysis strategy for the formation of β,γ-unsaturated ketones (Scheme 12).36 A ruthenium-hydride species generated in situ from RuHCl(CO)(PPh3) and Josiphos promotes both the formation of an allene intermediate and catalyses the subsequent hydroacylation reaction to form β,γ-unsaturated ketone products. Aryl-, heteroaryl- and selected alkyl-substituted propynes are well-tolerated, and the β,γ-unsaturated hydroacylation products are isolated in moderate-to-high yields (61–95%) with excellent selectivities (typically >20:1 rr). However, alkynes substituted with longer alkyl chains (e.g. 1-phenyl-1-butyne) did not lead to the desired hydroacylation product, likely due to increased allylic strain that disfavors formation of the key allene intermediate.
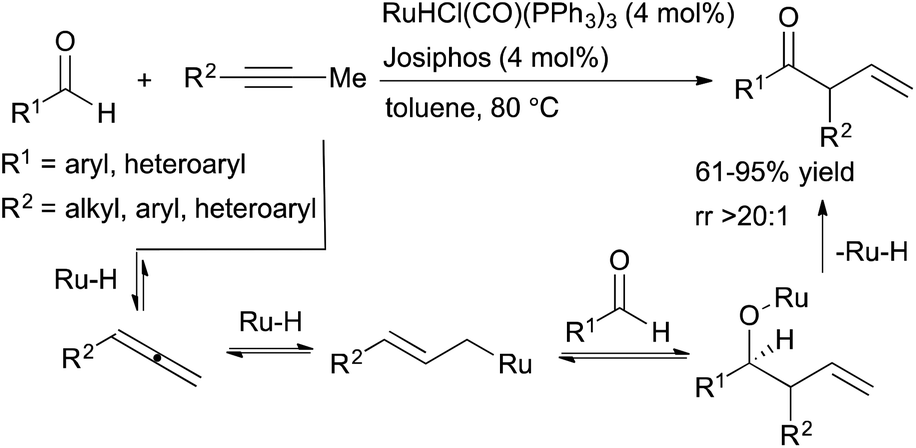 | ||
| Scheme 12 Ruthenium-catalysed hydroacylation of alkynes to form β,γ-unsaturated ketones. | ||
More recently, Nagamoto and Nishimura reported the iridium-catalysed hydroacylation of bicyclic alkenes with salicylaldehydes (Scheme 13).37 A readily available iridium complex, [Ir(OH)(cod)]2, catalyses the hydroacylation of a variety of bicyclic alkenes with salicylaldehydes, and the ketone products are formed in high yields (91–99%) with nearly perfect exo-selectivity. The authors note that in contrast to the previously reported rhodium-catalysed hydroacylation of norbornadiene with salicylaldehyde,38 the iridium-catalysed reaction is highly exo-selective and generates the mono-acylation product in 85% yield. The authors also demonstrate an enantioselective variant of these reactions using an iridium catalyst containing a chiral diene ligand to form the corresponding ketone product in 97% yield with 86% ee.
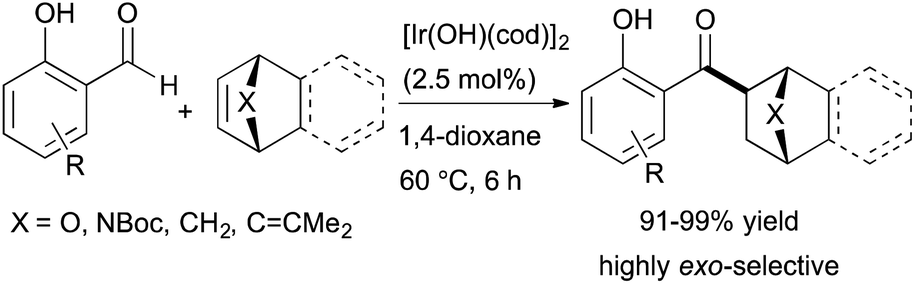 | ||
| Scheme 13 Iridium-catalysed hydroacylation of bicyclic alkenes. | ||
Reports on nickel-catalysed alkene and alkyne hydroacylation are becoming more frequent.39–41 In 2012, Yamamoto and co-workers reported the nickel-catalyzed hydroacylation of 2-(prop-2-ynyl)benzaldehydes to form 2,3-dihydro-1H-inden-1-ones using a catalyst generated from Ni(cod)2 and P(iPr)3.39 Also that year, Ogoshi and coworkers reported exo-selective hydroacylations of o-allyl- and o-homoallylbenzaldehydes in the presence of a Ni(0)/NHC complex (Scheme 14).41 α-Substituted indanones and tetralones are readily prepared from these reactions, including products containing a quaternary carbon center. However, an enantioselective variant has not yet been reported.
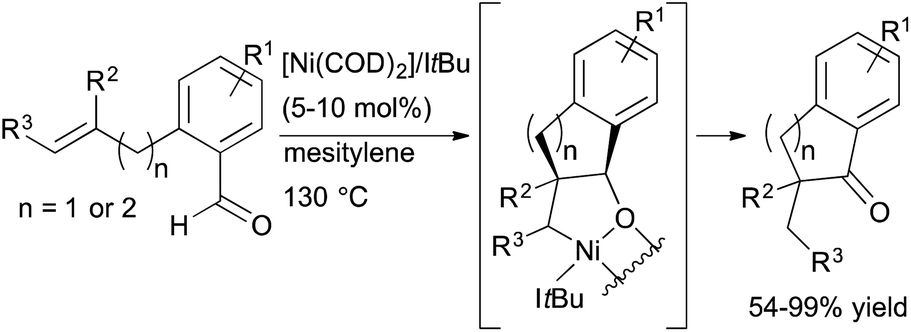 | ||
| Scheme 14 Nickel-catalysed hydroacylation of o-allyl- and o-homoallylbenzaldehydes. | ||
Conclusions
The development of new transition metal-catalysed alkene and alkyne hydroacylation reactions has progressed rapidly in the past three years. Rhodium-catalysed hydroacylation remains an active area of study with new intermolecular alkene and alkyne hydroacylation reactions and applications to the synthesis of new heterocyclic ketones emerging in recent years. However, the field of alkene and alkyne hydroacylation is rapidly expanding beyond rhodium catalysts. Cobalt catalysts hold great promise for the discovery of new hydroacylation reactions and as a replacement for traditional rhodium catalysts. In addition, ruthenium, iridium, and nickel catalysts of alkene and alkyne hydroacylation have emerged to improve upon or complement previous catalyst systems. We look forward to exciting new developments that are sure to emerge in the coming years.Acknowledgements
We thank the National Science Foundation (CHE-CAREER 1353819) for supporting our studies on olefin hydroacylation reactions.Notes and references
- M. C. Willis, Chem. Rev., 2010, 110, 725–748 CrossRef CAS PubMed.
- S. K. Murphy, A. Bruch and V. M. Dong, Angew. Chem., Int. Ed., 2014, 53, 2455–2459 CrossRef CAS PubMed.
- J. C. Leung and M. J. Krische, Chem. Sci., 2012, 3, 2202–2209 RSC.
- K. Sakai, O. Oda, N. Nakamura and J. Ide, Tetrahedron Lett., 1972, 13, 1287–1290 CrossRef.
- A. Biju, S. Yetra and A. Patra, Synthesis, 2015, 1357–1378 CrossRef.
- D. Janssen-Müller, M. Schedler, M. Fleige, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 12492–12496 CrossRef PubMed.
- V. Chudasama, RSC Adv., 2015, 5, 44423–44426 RSC.
- V. Chudasama, A. R. Akhbar, K. A. Bahou, R. J. Fitzmaurice and S. Caddick, Org. Biomol. Chem., 2013, 11, 7301–7317 CAS.
- V. Chudasama, R. J. Fitzmaurice and S. Caddick, Nat. Chem., 2010, 2, 592–596 CrossRef CAS PubMed.
- S. K. Murphy and V. M. Dong, Chem. Commun., 2014, 50, 13645–13649 RSC.
- S. K. Murphy, A. Bruch and V. M. Dong, Chem. Sci., 2015, 6, 174–180 RSC.
- A. B. Chaplin, J. F. Hooper, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 4885–4897 CrossRef CAS PubMed.
- I. Pernik, J. F. Hooper, A. B. Chaplin, A. S. Weller and M. C. Willis, ACS Catal., 2012, 2, 2779–2786 CrossRef CAS.
- A. Prades, M. Fernandez, S. D. Pike, M. C. Willis and A. S. Weller, Angew. Chem., Int. Ed., 2015, 54, 8520–8524 CrossRef CAS PubMed.
- M. Tanaka, M. Imai, M. Fujio, E. Sakamoto, M. Takahashi, Y. Eto-Kato, X. M. Wu, K. Funakoshi, K. Sakai and H. Suemune, J. Org. Chem., 2000, 65, 5806–5816 CrossRef CAS PubMed.
- M. Tanaka, M. Takahashi, E. Sakamoto, M. Imai, A. Matsui, M. Fujio, K. Funakoshi, K. Sakai and H. Suemune, Tetrahedron, 2001, 57, 1197–1204 CrossRef CAS.
- J. W. Park, K. G. M. Kou, D. K. Kim and V. M. Dong, Chem. Sci., 2015, 6, 4479–4483 RSC.
- I. F. D. Hyatt, H. K. Anderson, A. T. Morehead and A. L. Sargent, Organometallics, 2008, 27, 135–147 CrossRef CAS.
- S. Y. Y. Yip and C. Aïssa, Angew. Chem., Int. Ed., 2015, 54, 6870–6873 CrossRef CAS PubMed.
- A. Ghosh and L. M. Stanley, Chem. Commun., 2014, 50, 2765–2768 RSC.
- X. W. Du, A. Ghosh and L. M. Stanley, Org. Lett., 2014, 16, 4036–4039 CrossRef CAS PubMed.
- K. F. Johnson, A. C. Schmidt and L. M. Stanley, Org. Lett., 2015, 17, 4654–4657 CrossRef CAS PubMed.
- J. S. Arnold, E. T. Mwenda and H. M. Nguyen, Angew. Chem., Int. Ed., 2014, 53, 3688–3692 CrossRef CAS PubMed.
- M. Castaing, S. L. Wason, B. Estepa, J. F. Hooper and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 13280–13283 CrossRef CAS PubMed.
- X. W. Du and L. M. Stanley, Org. Lett., 2015, 17, 3276–3279 CrossRef CAS PubMed.
- J. F. Hooper, R. D. Young, A. S. Weller and M. C. Willis, Chem. – Eur. J., 2013, 19, 3125–3130 CrossRef CAS PubMed.
- J. F. Hooper, R. D. Young, I. Pernik, A. S. Weller and M. C. Willis, Chem. Sci., 2013, 4, 1568–1572 RSC.
- C. P. Lenges and M. Brookhart, J. Am. Chem. Soc., 1997, 119, 3165–3166 CrossRef CAS.
- C. P. Lenges, P. S. White and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 6965–6979 CrossRef CAS.
- Q. A. Chen, D. K. Kim and V. M. Dong, J. Am. Chem. Soc., 2014, 136, 3772–3775 CrossRef CAS PubMed.
- J. Yang and N. Yoshikai, J. Am. Chem. Soc., 2014, 136, 16748–16751 CrossRef CAS PubMed.
- J. Yang, Y. W. Seto and N. Yoshikai, ACS Catal., 2015, 5, 3054–3057 CrossRef CAS.
- R. Santhoshkumar, S. Mannathan and C.-H. Cheng, J. Am. Chem. Soc., 2015, 137, 16116–16120 CrossRef CAS PubMed.
- H. Miura, K. Wada, S. Hosokawa and M. Inoue, Chem. – Eur. J., 2013, 19, 861–864 CrossRef CAS PubMed.
- F. Shibahara, J. F. Bower and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 14120–14122 CrossRef CAS PubMed.
- Q. A. Chen, F. A. Cruz and V. M. Dong, J. Am. Chem. Soc., 2015, 137, 3157–3160 CrossRef CAS PubMed.
- M. Nagamoto and T. Nishimura, Chem. Commun., 2015, 51, 13791–13794 RSC.
- R. T. Stemmler and C. Bolm, Adv. Synth. Catal., 2007, 349, 1185–1198 CrossRef CAS.
- F. Yang, T. A. Jin and Y. Yamamoto, Tetrahedron, 2012, 68, 5223–5228 CrossRef CAS.
- H. Taniguchi, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2009, 131, 11298–11299 CrossRef CAS PubMed.
- Y. Hoshimoto, Y. Hayashi, H. Suzuki, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2012, 51, 10812–10815 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2016 |