
The quest for observation and isolation of oxyallyl derivatives
Vianney
Regnier
and
David
Martin
*
UMR CNRS 5250, “Département de Chimie Moléculaire”, Université Joseph Fourier, B. P. 53, 38041 Grenoble Cedex 9, France. E-mail: david.martin@ujf-grenoble.fr
First published on 18th August 2015
Abstract
This critical review narrates the story of 40 years of attempts to observe and isolate oxyallyl derivatives. These efforts have paved the way to recent achievements, among which are the establishment of the transition-state nature of the parent compound and the first observation of an alkyl-substituted oxyallyl. In recent years, this field of research has even reached the critical point, from which oxyallyl derivatives shouldn't be disregarded as chemical oddities anymore, but as attractive synthetic targets with potential applications in advanced materials science.
 Vianney Regnier | Vianney Regnier was born in 1992 in Grenoble, France. He obtained his master's degree in organic chemistry in 2015 at Joseph Fourier University. He is currently working as a Ph.D. student at the Molecular Chemistry Department of UJF. His research interests focus on the synthesis and characterization of new stable organic and inorganic molecules containing the oxyallyl pattern. |
 David Martin | David Martin was an undergraduate at Ecole Normale Supérieure of Lyon (France). After obtaining his PhD from the University of Toulouse in 2004, he moved to Switzerland as a postdoctoral assistant in the group of Prof. A. Alexakis in Geneva. In 2006 he became a CNRS researcher at the University of Marseille (France). He was a visiting researcher in the CNRS-UCSD joint chemistry lab of Prof. Guy Bertrand at San Diego from 2010 to 2014. His current research interests at the Department of Molecular Chemistry of Grenoble (France) span from the design of new families of paramagnetic species to the use of original redox active ligands in transition-metal catalysis. |
1. Introduction
R. Hoffmann first mentioned 2-(oxy)allyl 1 in 1968. He suggested that the recently synthesized1 cyclopropanone 2 could in fact adopt a more stable acyclic structure (Scheme 1).2 This hypothesis was essentially supported by extended Hückel calculations and was finally ruled out a year later, when microwave spectroscopy experiments ascertained the cyclic structure of 2.3 This short controversy had the virtue of drawing some attention to a new family of compounds, generally denoted as “oxyallyls”. In addition to the synthetic challenge they were offering to chemists, their study was also motivated by the need to investigate their intriguing electronic structure. Indeed oxyallyls belong to the class of non-Kekulé molecules. Their neutral π-system cannot be assigned to a classical Kekulé-type structure, but only to zwitterionic or diradical resonance forms, despite an even number of electrons.4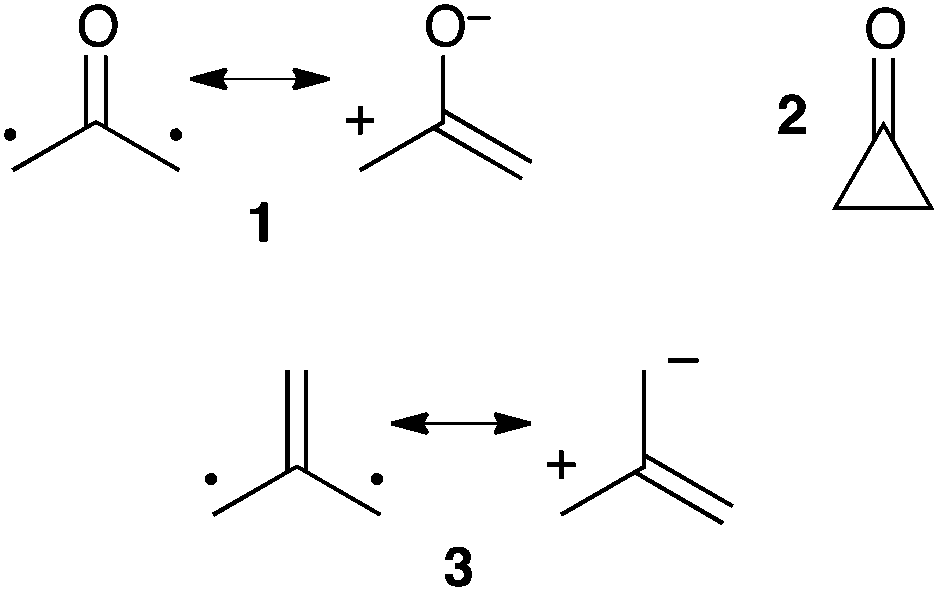 | ||
| Scheme 1 Oxyallyl 1, cyclopropanone 2 and tri(methylene)methane 3. | ||
However, it became rapidly obvious that oxyallyls were considerably more elusive than initially suggested by Hoffmann to the point that even the possibility of their observation had been questioned, not to mention their isolation. Indeed, over the years, oxyallyls have been postulated as intermediates in several rearrangement reactions,5 including the in vivo formation of prostaglandins.6 Importantly, they have been generated, trapped in situ by a large variety of dipolarophiles and found many synthetic applications.7 In spite of many clues of their existence, they had eluded isolation and even spectroscopic observation until very recently. In marked contrast, the related tri(methylene)methane 3 was observed as early as 1966 by EPR at 88 K.8
Over more than 40 years, successive publications have highlighted how especially challenging was the study of this class of compounds. Remarkable contributions allowed for a better comprehension of the issues at stake, and have paved the way to the design of observable versions. In this critical review, we tell the story of the quest for the observation and isolation of oxyallyl derivatives, from the early attempts in the 70–80s to the recent reports of stable forms.
2. Parent oxyallyl H2C(CO)CH2
2.1 Can the parent oxyallyl be observed?
The specific challenge of detecting the parent oxyallyl H2C(CO)CH21 has been interpreted in terms of singlet–triplet gap. Indeed, the disrotatory ring closure of the putative singlet 11 into cyclopropanone 2 had been predicted to occur with a negligible barrier, if any.9 Therefore only a triplet ground state is likely to be persistent enough to be easily observed.We already mentioned the early observation of the related tri(methylene)methane 3. This compound benefits from the triplet diradical ground state, which is well separated from the lowest singlet state by 16 kcal mol−1.10 This is a direct consequence of Hund's rule, as the two highest occupied molecular orbitals of 3 are degenerate, non-bonding and non-disjoint.11 The case of 1 is not so clear-cut. Indeed the lower symmetry of 1 results in a lifting of the degeneracy of the two highest occupied molecular orbitals, which favours the singlet state 11. In fact, several calculations predicted a singlet–triplet gap as small as 0.3 kcal mol−1, but still in favour of the triplet 31. Importantly, Schwarz et al. reported very small computed spin–orbit elements, which are expected to contribute the most to the triplet–singlet transition, and concluded that this may be sufficient to allow for the detection of 31. However, the attempts of these authors to identify 1 by neutralization–reionization mass spectroscopy were inconclusive.12,13
In 2009, Lineberger et al. reported a negative ion photoelectron spectroscopy study of the oxyallyl radical anion 1˙−, which could be generated by the gas phase reaction of acetone and the oxygen radical anion O˙− (Scheme 2).14 The adiabatic formation of both 11 and 31 could be unambiguously deduced from the photoelectron spectra. This remarkable set of experiments provided major fundamental answers about the nature of 1. First, the authors could determine that, contrary to the predictions of earlier theoretical studies, 11 is slightly more stable than 31 by 1.3 kcal mol−1. Moreover, significant broadening of the photoelectron spectra indicated that 11 corresponds in fact to a transition state! Therefore, 40 years after Hoffmann's initial proposal, it could be concluded that 1 is not a genuine intermediate, but an energetic maximum along the stereomutation of the methylenes of cyclopropanone 2.
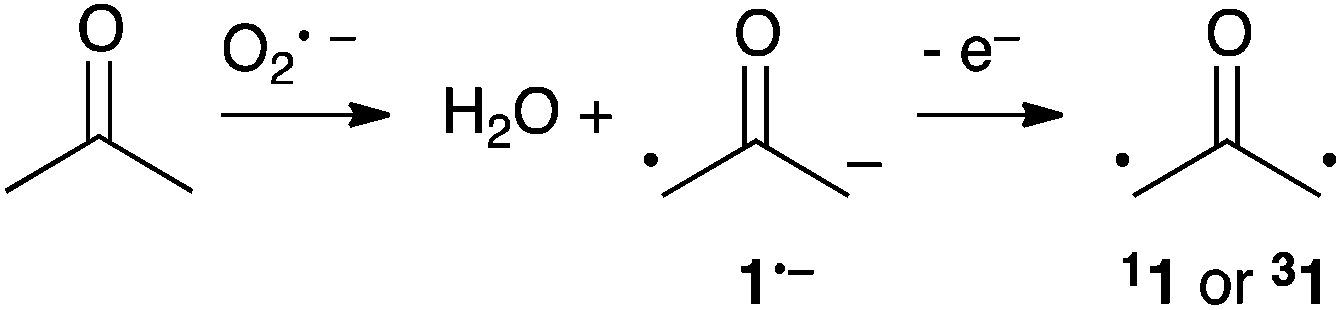 | ||
| Scheme 2 Observation of 11 and 31 by negative ion photoelectron spectroscopy. | ||
2.2 Transition metal complexes of 1
Complexation to transition metals is a classical strategy to isolate otherwise transient species. Several complexes featuring a 2-(oxy)allyl pattern have been reported in the literature. For instance, the dinickel complex 4, which features a CH2(CO)CH2 bridge, was synthesized by deprotonation of the corresponding cationic nickel(II)–acetone complex, isolated and fully characterized.15 Note that although 4 was presented as featuring a trapped oxyallyl ligand 1, it is certainly better described as two nickel(II) centres bridged by the bis-anion 12−. Similarly, osmium complex 5 is a di(metalla)cyclopentanone (Scheme 3).16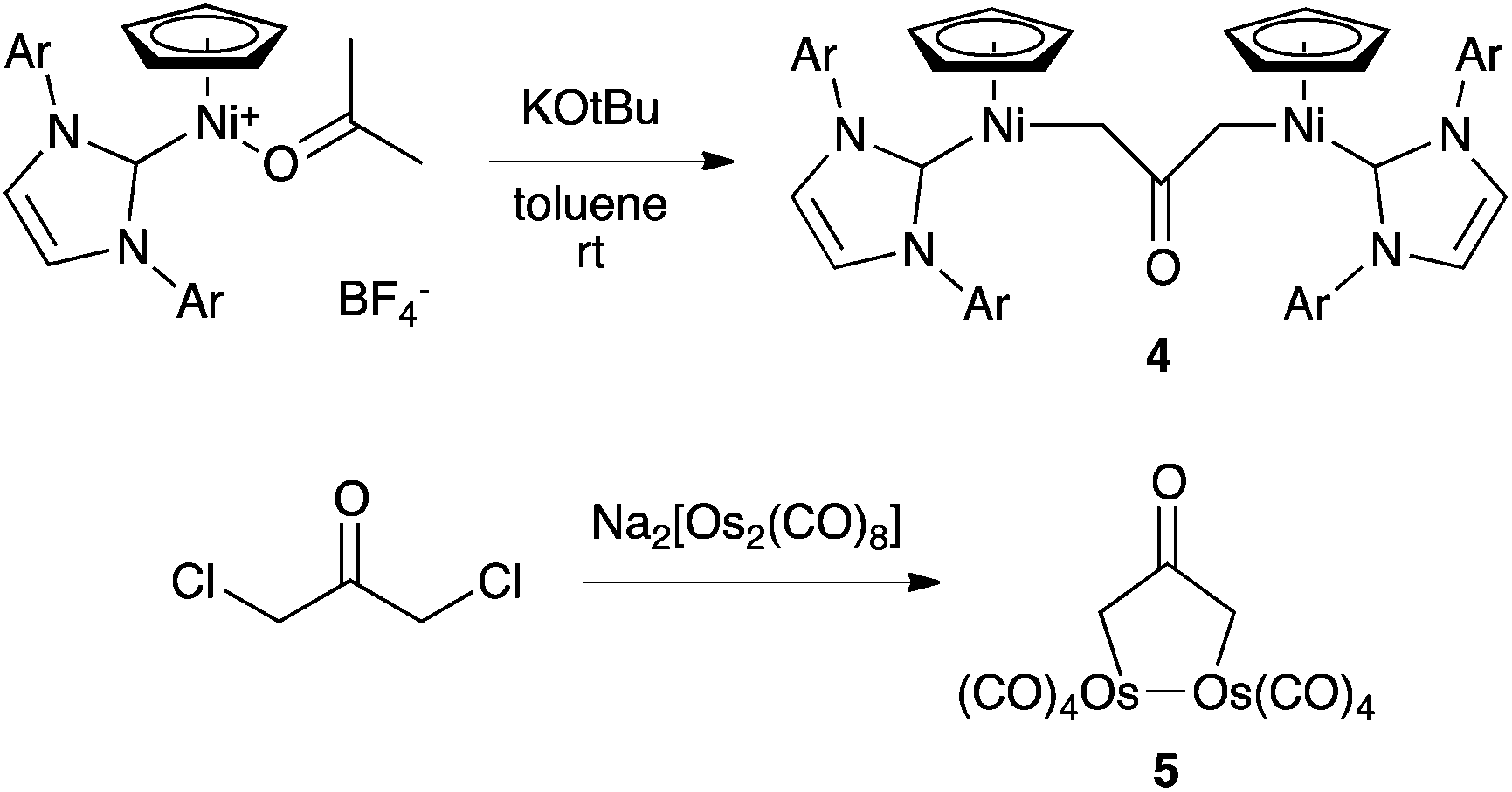 | ||
| Scheme 3 Synthesis of di(metalla)ketones 4 and 5. | ||
The 2-(hydroxy)allyl iron complex 6 was synthesized from silylenol ether and isolated. Its deprotonation (pKa = 5.2) in the presence of a bromine scavenger didn't afford complex 7, but its head-to-tail dimer 8 (Scheme 4). Interestingly, the tri(methylene)methane equivalent is monomeric.17
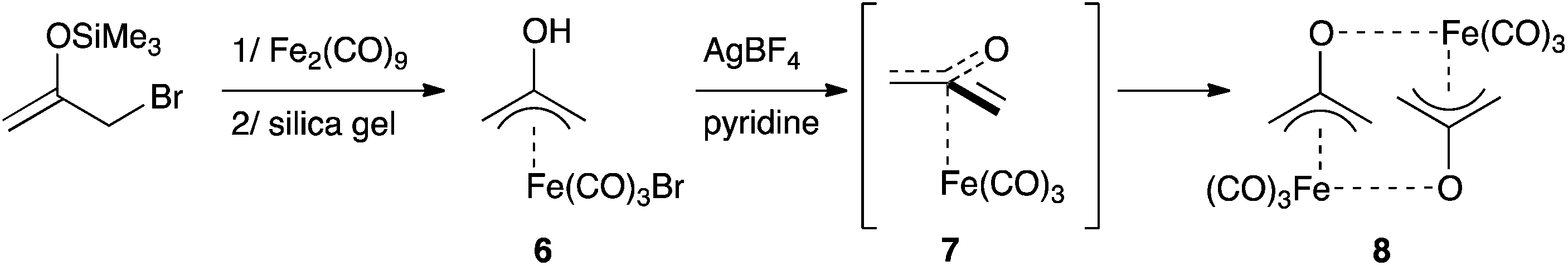 | ||
| Scheme 4 Synthesis of complexes 7 and 8. | ||
Related monomeric complexes of 12− with platinum(II) and palladium(II) have been reported (compounds 9 and 10, Scheme 5).18 Such π-allyl complexes may appear as more satisfying models for “stabilized” coordinated oxyallyls than 4 or 5 because they maintain a formal 4-centers π-system on the ligand. According to spectroscopic and structural studies, both π-allyl and metallacyclobutane mesomeric forms are significant in iron and platinum complexes (8 and 9). Palladium complexes 10 have a dominant π-allyl character.
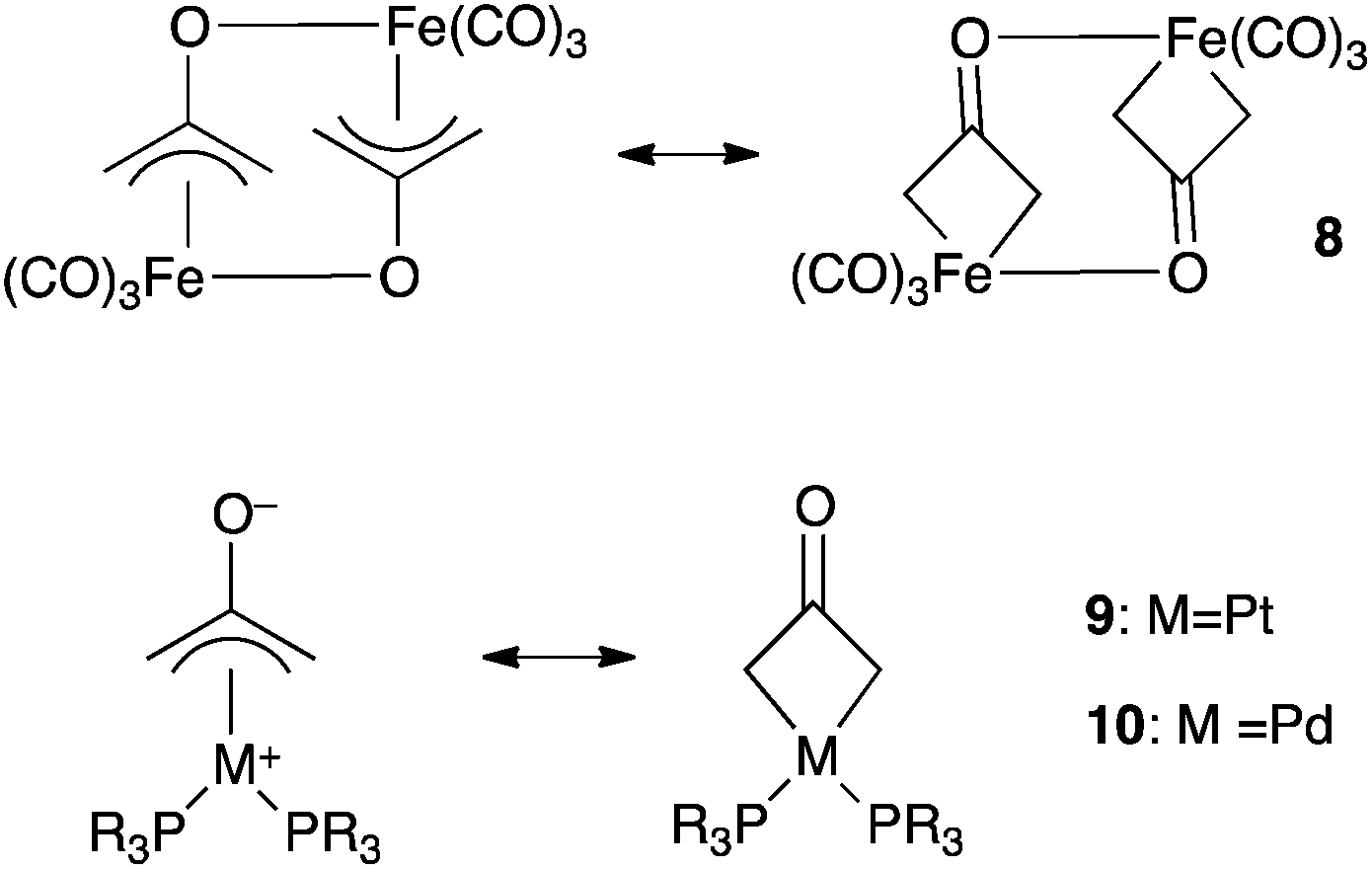 | ||
| Scheme 5 The metallacyclobutanone and π-allyl mesomeric forms of 8–10. | ||
3. Alkyl substituted oxyallyls
3.1 Decreasing the oxyallyl-cyclopropanone energy gap
Contrary to their parent compound 1, alkyl and aryl substituted oxyallyls are predicted to be genuine species, which unambiguously correspond to minima on energy hypersurfaces. However, because of their singlet ground state, their cyclisation into the corresponding cyclopropanone is predicted to occur with very low activation barriers.9d Therefore, any reasonable model for an isolable oxyallyl requires a situation in which the cyclopropanone form is made less stable than the oxyallyl.Estimates of the oxyallyl-cyclopropanone Gibbs energy difference (ΔG) have been deduced from studies of the stereomutation of cyclopropanones. Indeed, as the barrier for cyclisation of the oxyallyl ΔG≠1 is very small, ΔG can be reasonably approximated to the barrier for the ring opening into the corresponding oxyallyl ΔG≠2 (Scheme 6). In 1970, Greene et al. reported the partial resolution of trans-2,3-di(tert-butyl)cyclopropanone 11 by asymmetric destruction with D-amphetamine. Monitoring of the racemization of the enantioenriched samples of 11 afforded ΔG values of 27–29 kcal mol−1, depending on the solvent.19
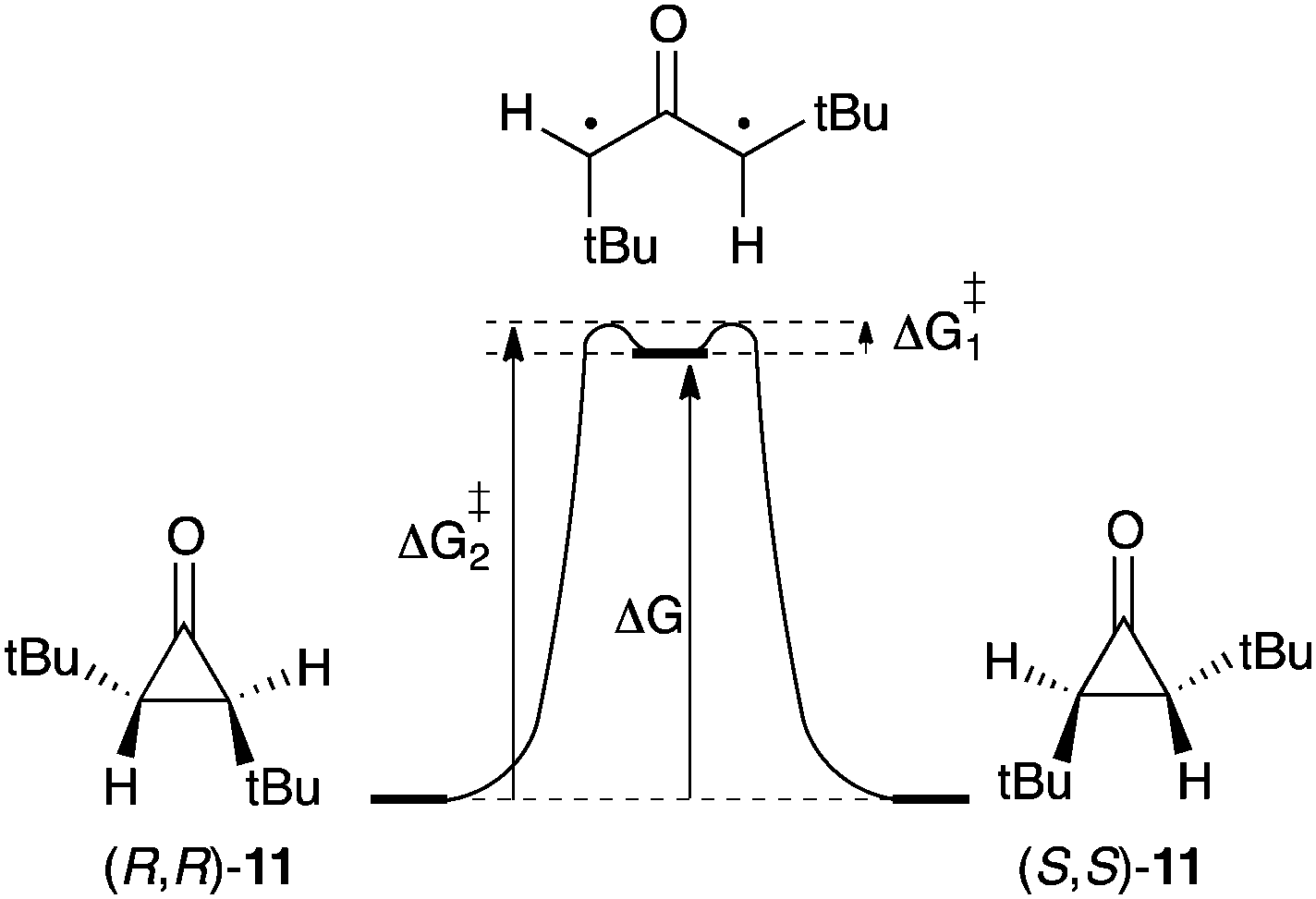 | ||
| Scheme 6 Thermal racemization of trans-2,3-di(tert-butyl)cyclopropanone 11. | ||
Twenty years later, Cordes and Berson reported a multi-step synthesis of two epimers of spiro(bicyclo[2.2.1]heptane-2,1′-cyclopropan)-2′-one from cyclopentadiene and acryloyl chloride (Scheme 7).20 The authors measured ΔG values of 16–19 kcal mol−1 for the interconversion of spiro-cyclopropanones 12 and 13 (through 14). Compared to 11, this corresponds to a decrease of about 45% for the oxyallyl-cyclopropanone energy gap.
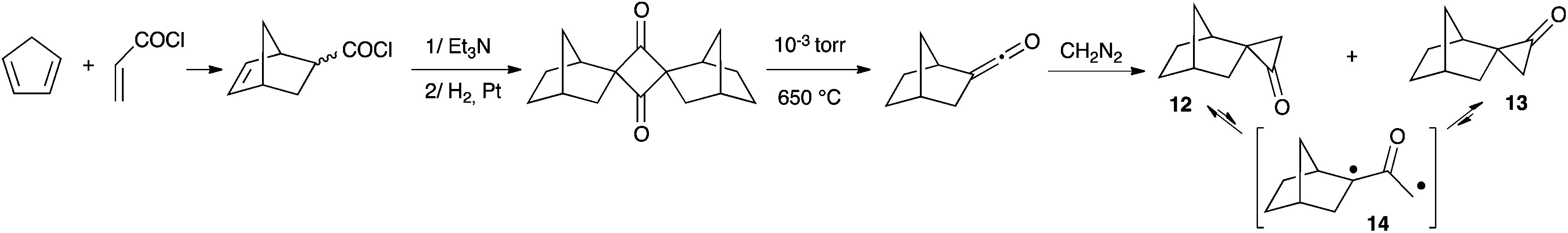 | ||
| Scheme 7 Synthesis and dynamic exchange of epimeric cyclopropanones 12 and 13. | ||
This latter result demonstrated that modulation of the steric effect could allow for significant destabilization of cyclopropanones with regard to their oxyallyl form. It prompted Sun and Sorensen to investigate a series of sterically hindered cis-cyclopropanones.21 In the case of cis-15 (with 2-(3,3,2-trimethyl)butyl substituents, see Scheme 9), they observed the coalescence of the 1H NMR signal of the two pairs of diastereotopic methyls, which are well resolved below 200 K. They attributed this phenomenon as being the result of the disrotatory opening of cis-15. Indeed, the resulting oxyallyl 16 can undergo disrotatory ring closure in two opposite directions, resulting in a formal scrambling of the pairs of methyls (Scheme 8).
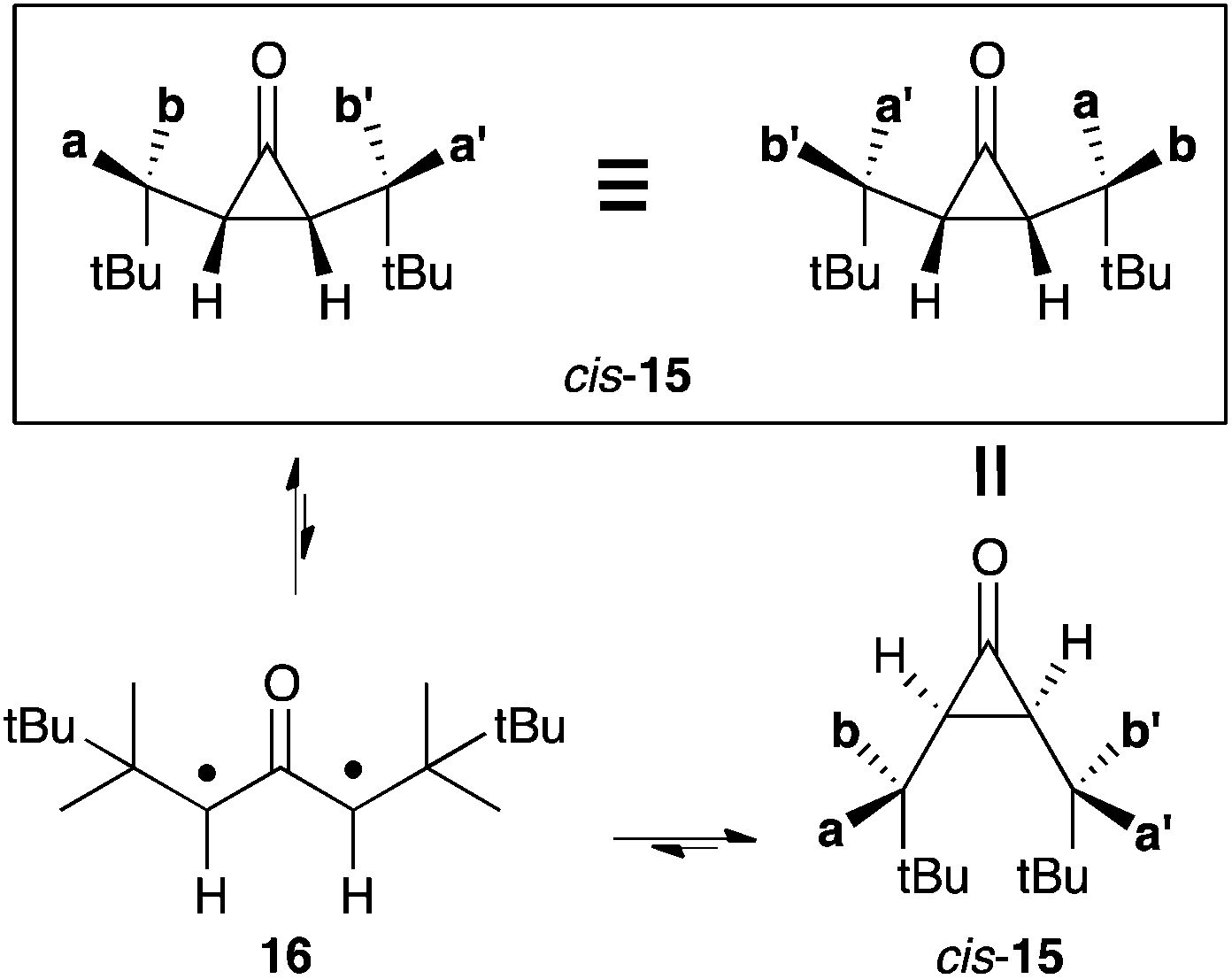 | ||
| Scheme 8 Exchange of diastereotopic methyls of cis-15 upon formation of oxyallyl 16. | ||
 | ||
| Scheme 9 Stability of fused cyclopropanones in respect of their oxyallyl forms. | ||
From the dynamic NMR study in chloroform, the authors deduced a Gibbs energy gap of 9.8 kcal mol−1 between cis-15 and 16. According to this remarkably low value, a 1 M solution of cis-15 should contain about 60 nM of 16 at 25 °C. In principle, oxyallyl 16 reaches detectable levels at such concentration. However, the experimental observation of this compound would require an ultra-pure solution of cis-15 to allow for an unambiguous identification.
3.2 Destabilization of cyclopropanones with fused polycyclic structures
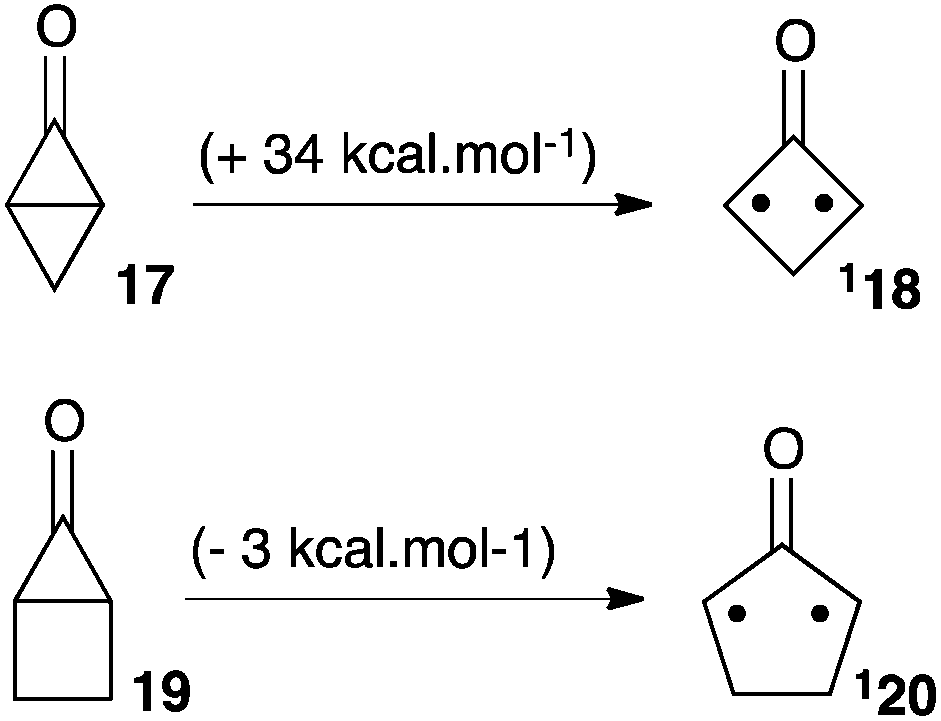
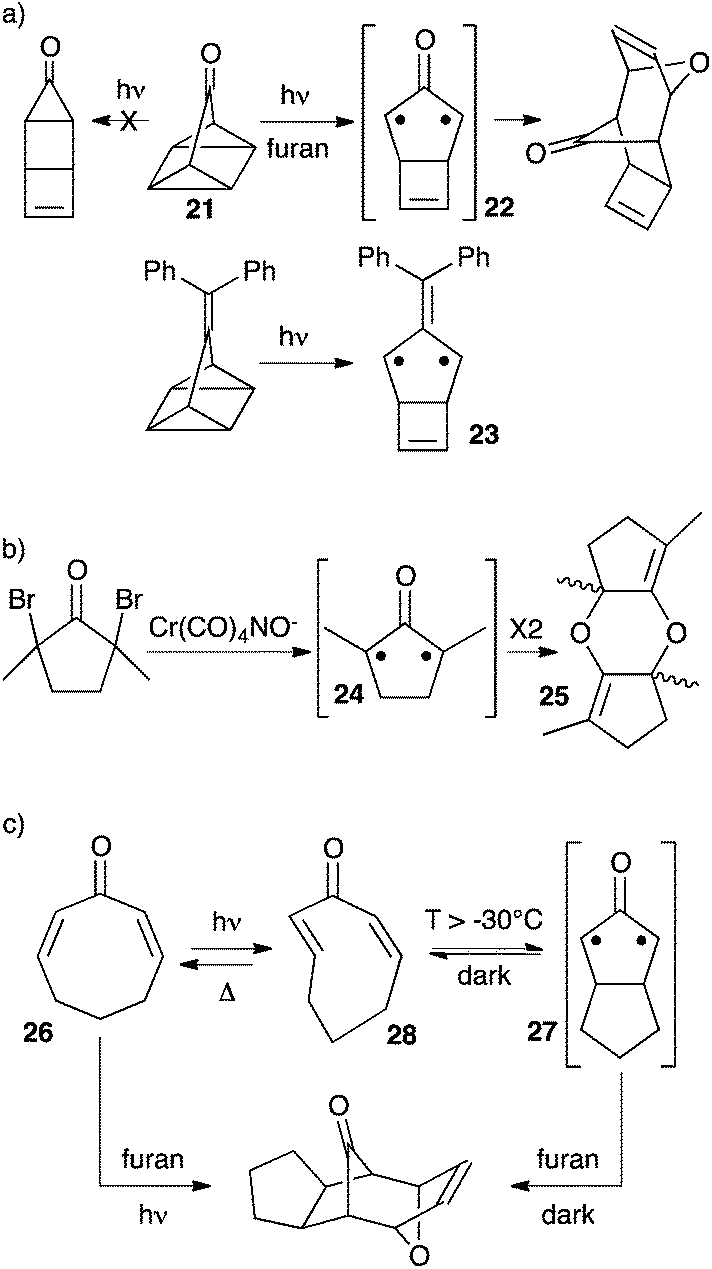
In 1990, Lahti et al. reported a computational study of fused bicyclic cyclopropanones.22 Bicyclo[1.1.0]butanone 17 was found significantly more stable than its corresponding oxyallyl 18 by 34 kcal mol−1. On the other hand, bicyclo[2.1.0]pentan-5-one 19 laid +3 kcal mol−1 above the oxyallyl form 20 (Scheme 9). This can be interpreted as the result of the difference in the ring-strain situation in the two systems. Indeed, the case of 20 is optimal: it features a five-membered ring with no major ring strain, whereas the fused 3-membered and 4-membered rings thermodynamically disfavour the corresponding cyclopropanone 19. On the contrary, 18 remains involved in a strained 4-membered ring. Of note, both oxyallyls were predicted to have a singlet diradicaloid ground state and a modest singlet–triplet gap of 4–6 kcal mol−1.There are several reports of attempts to observe oxyallyls that are related to 20. Ikegami et al. showed that photolysis of the polycyclic compound 21 generated oxyallyl 22, which could be trapped by various dipolarophiles, including furan (Scheme 10a). However, all attempts to observe 22 failed. In marked contrast, the related tri(methylene)methane derivative 23 could be generated under the exact same conditions and observed by EPR in a methylcyclohexane matrix at 77 K.23
 | ||
| Scheme 10 Reported attempts to observe oxyallyls related to 20. | ||
Sorensen et al. demonstrated the formation of oxyallyl 24 by several trapping experiments upon in situ reduction of the corresponding dibromoketone (Scheme 10b).24 Needless to say, 24 lacks the destabilizing fused cyclobutene ring of 22, and therefore is a far better candidate for a persistent oxyallyl. Unfortunately, in the absence of a trapping agent the authors couldn't observe 24, but the “instantaneous” formation of its head-to-tail dimer 25. They could only conclude that the t1/2 for the dimerization of 24 must be lower than 10 minutes at −120 °C.
As early as 1968, Crandall et al. noticed that irradiation of 2,7-cyclooctadienone 26 at −78 °C followed by addition of furan in the absence of light afforded the same cycloadduct that was obtained upon irradiation of 26 in the presence of furan (Scheme 10c).25,26 This indicated the formation of a persistent reactive intermediate. Of course, in light of the above-mentioned computational studies, it is tempting to hypothesize the formation of oxyallyl 27. In 1999, Raulerson et al. reinvestigated this reaction and showed that the intermediate was in fact the strained trans,cis-cyclooctadienone 28, which can undergo thermal cyclisation above −30 °C to afford oxyallyl 27.27 Note that 26–28 are in equilibrium under irradiation and about 20% of 26 is converted into 28 at the photostationary state. Therefore it is likely that 27 is unobservable because its fast thermal ring-opening in the absence of light recreates the most stable cyclooctadienone 26.
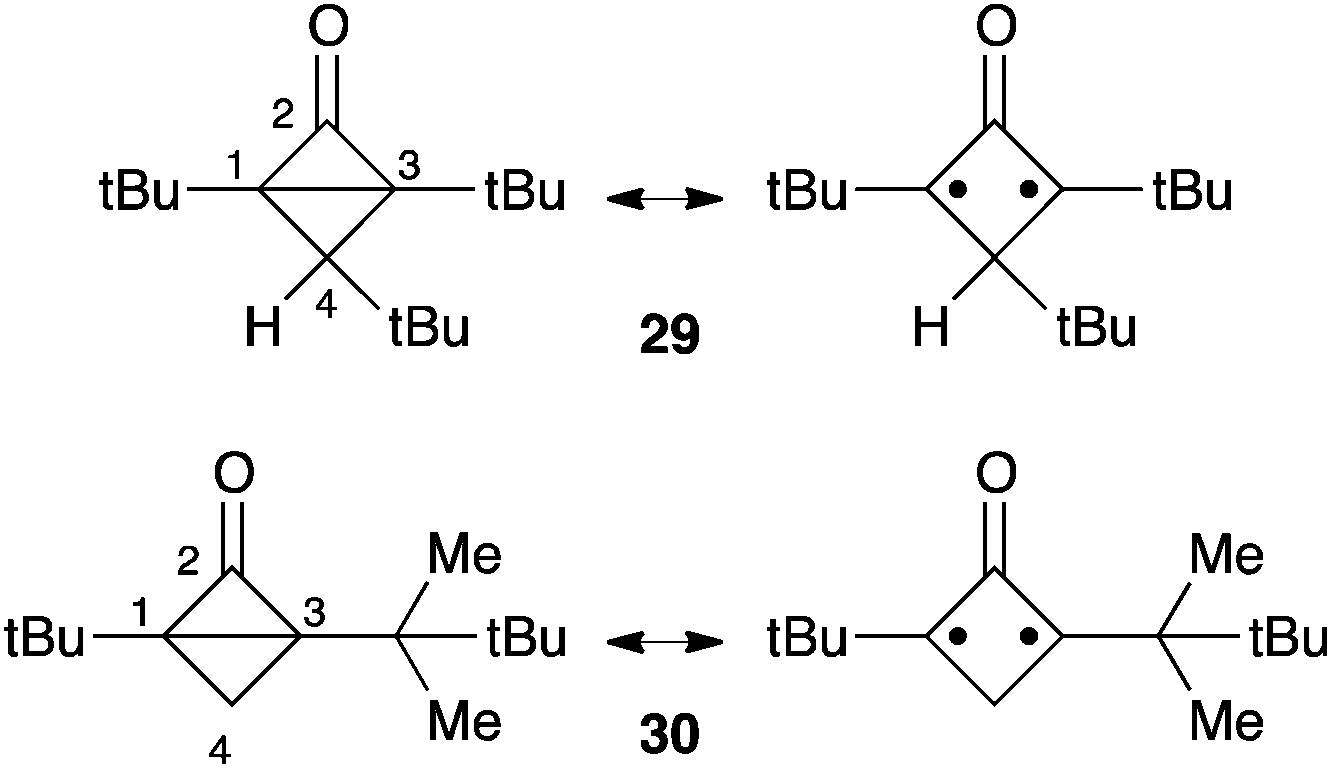
Bicyclo[1.1.0]butanones had been disregarded for long as potential entries to oxyallyls, because the ring-opening of 17 was predicted to be strongly thermodynamically disfavoured. Another obstacle to the study of these bicyclic compounds, and not the least, is their high reactivity and the ensuing difficulty in their synthesis. The first X-ray structure of isolable representatives, the tert-butyl substituted bicyclo[1.1.0]butanone 29 and 30, was reported in 2005 by Sorensen et al.28,29 Ironically, spectroscopic and structural data clearly show that bicyclo[1.1.0]butanones have a hybrid structure with a non-negligible oxyallyl character (Scheme 11). For instance, the NMR chemical shifts of the fused carbons C1 and C3 are significantly deshielded (67–81 ppm) compared to monocyclic cyclopropanones (29–36 ppm). Furthermore, compounds 29 and 30 feature especially long C1–C3 bonds (about 170 pm, compared to 157 pm in 1)3 and a distinctly pyramidal ketone functionality (“out of plane” angle: 12–21°). Unsurprisingly, the thermal evolution of bicyclo[1.1.0]butanones is reminiscent of oxyallyls.
 | ||
| Scheme 11 The oxyallyl–bicyclo[1.1.0]butanone hybrid structures of 29 and 30. | ||
3.3 Observation of alkyl-substituted oxyallyls in the solid state
In 2011 Garcia-Garibay et al. reported a new strategy to tackle the issue of the ultra-fast formation of cyclopropanones. They envisioned that oxyallyls generated in the solid state should undergo slower ring-closures, and therefore may be easier to detect. They studied the photo-induced decarbonylation of the crystalline samples of spiro-dione 31 and observed a deep blue product, which could be identified as the di(cyclohexyl)oxyallyl 32. The decay of this compound in the solid state was remarkably slow, with a half-life of 42 minutes at 298 K! Note that the ensuing cyclopropanone was observed, but couldn't be isolated due to its decomposition under irradiation to afford dicyclohexylidene by the loss of CO (Scheme 12).30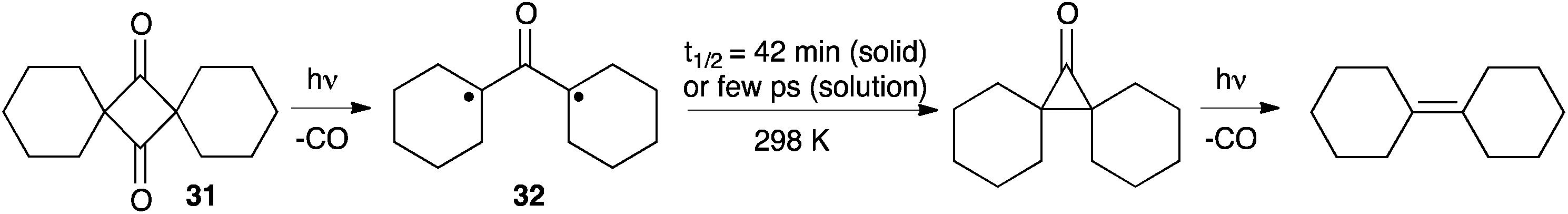 | ||
| Scheme 12 Generation and evolution of oxyallyl 32. | ||
Interestingly, the authors also measured the transient absorption spectra of 31 in solution upon excitation by 150 fs laser pulses. Once again they could observe the formation of the oxyallyl and measured half-lives of 1–8 ps, depending on the solvent. These results mark a pivotal milestone in the study of alkyl-substituted oxyallyl intermediates. They also illustrate the specific challenges of detecting these species. Indeed, in contrast with tri(methylene)methanes, EPR cannot allow for easy and unambiguous observations. As a matter of fact, without the guidance of the univocal spectroscopic observation of 32 in the solid state, it is likely that the interpretation of transient absorption spectra in solution would have been inconclusive.
3.4 Perspective and potential synthetic targets
The quest for the observation of alkyl-substituted oxyallyls hasn't reached its conclusion yet. The above-mentioned results clearly paved the way for the design of persistent versions. For example, it is likely that derivatives of 24 can be sheltered from dimerization by more sterically demanding substituents. Interestingly, theoretical studies have suggested several potential alternative synthetic targets, such as compounds 33, in which the stretch effect of a macrocyclic ring is predicted to push the hybrid structure of bicyclo[1.1.0]butanones towards a genuine oxyallyl form (Scheme 13).31 Another possible strategy is to stabilize a zwitterionic form by introducing groups that stabilize positive charges. Hess reported a computational study of derivatives with cyclopropenyl, cyclopentadienyl and cycloheptatrienyl moieties. He found oxyallyl 34 more stable than its cyclopropanone and allene oxide forms, by 5.4 and 10.7 kcal mol−1, respectively, and suggested that it could be observable in an appropriate environment.32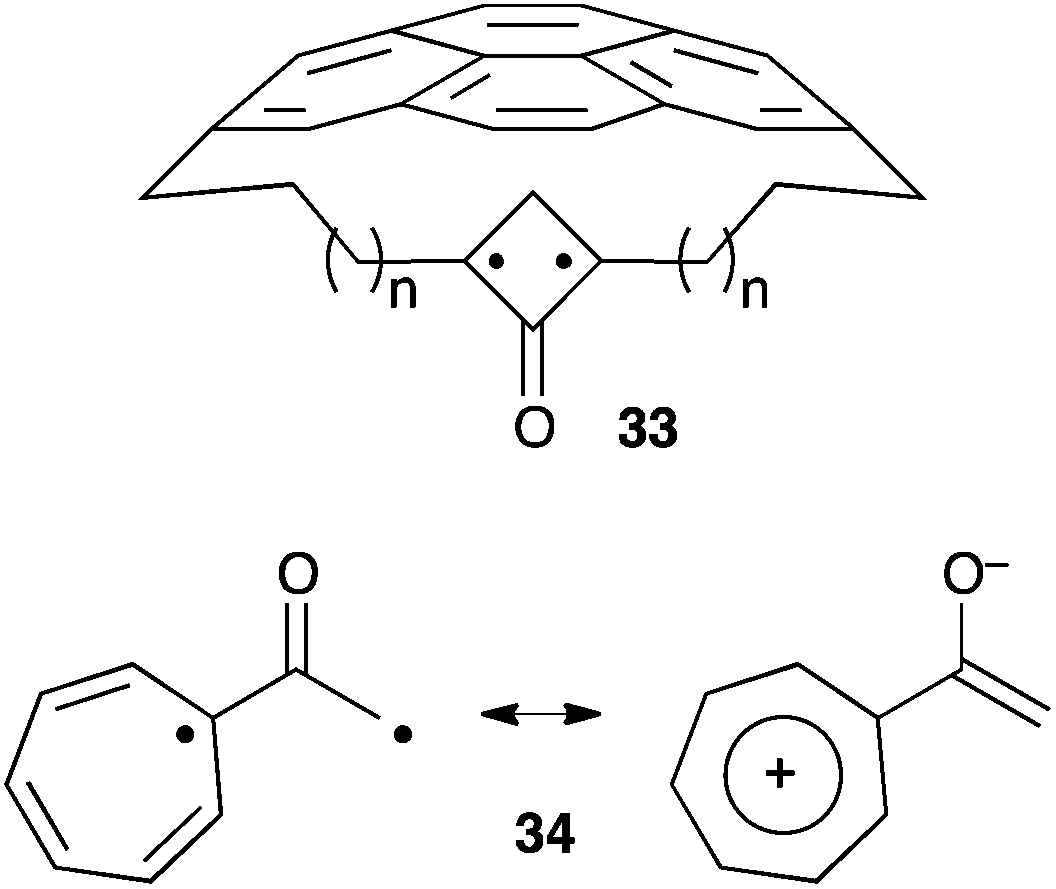 | ||
| Scheme 13 Possible synthetic targets suggested by computational studies. | ||
4. Stable hetero-substituted derivatives
4.1 Donor-stabilized oxyallyls
Extension of delocalized π-systems is a traditional method to achieve significant stability. In the case of oxyallyls, the introduction of +M donating groups seems the most appropriate, since it strongly stabilizes the cationic allylic moieties in the zwitterionic resonance form (Scheme 14). In addition, push–push patterns are likely to preclude the formation of the cyclopropanone forms.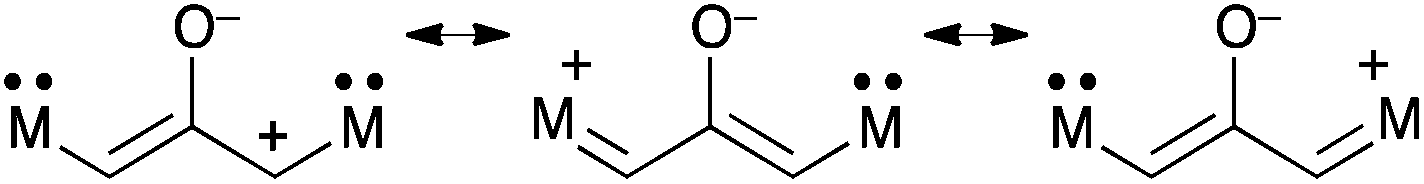 | ||
| Scheme 14 Stabilization of oxyallyls by +M donating substituents. | ||
Scarce examples of such species can be found in the literature. In 2003, Tian et al. reported the synthesis of a series of compounds 35 from the thermal rearrangement of some croconaines featuring 4-amino-2,6-(dihydroxy)phenyl substituents (Scheme 15).33 These compounds are near IR-dyes that are attractive for the design of non-linear optic materials, due to their propensity to form J-aggregates on spin-coated films.34
 | ||
| Scheme 15 Synthesis of 35 from low-yield rearrangement of croconaines. | ||
Molecules 35 could be seen as examples of oxyallyl centres with two +M donating 4-aminophenyl groups. However, the sharing of two protons by three hydroxy groups blurs the genuine structure of 35. Interestingly, examination of X-ray diffraction data reveals that the C–O bonds in the phenoxy and the formal oxyallyl moieties have similar lengths (132–133 pm). Therefore, although they have been depicted as a H-bond stabilized oxyallyl (structure 35Ox), they can be described just as well as protonated oxyallyl cations interacting with phenoxy groups (structure 35OxH).35
The rearrangement of croconaines is of limited practical interest because of low yields and the narrow range of accessible structures. However, the stability of 35 suggests that the introduction of stabilizing H-bonds must be a good strategy for the design of new stable hetero-substituted oxyallyl derivatives.
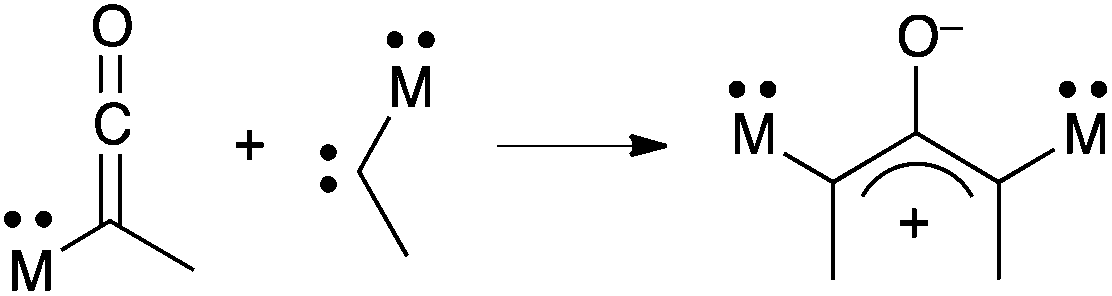
Another synthetic strategy could rely on the reaction of carbenes with ketenes, which is a known method for generating transient oxyallyls.36 In principle, this reaction could be extended to the formation of stabilized 1,3-di(hetero-substituted)oxyallyls, especially 1,3-di(amino)oxyallyls (Scheme 16). However, aminoketenes are only available from the addition of stable (amino)carbenes on carbon monoxide.37–41 The scope of this reaction is limited to few stable electrophilic carbenes and doesn't include the classical cyclic di(amino)carbenes, which don't react with CO.42 Furthermore, the steric bulk of stable carbenes usually precludes the addition of a second equivalent, which would afford the corresponding oxyallyl.
 | ||
| Scheme 16 Hetero-substituted oxyallyl from the addition of carbenes to ketenes. | ||
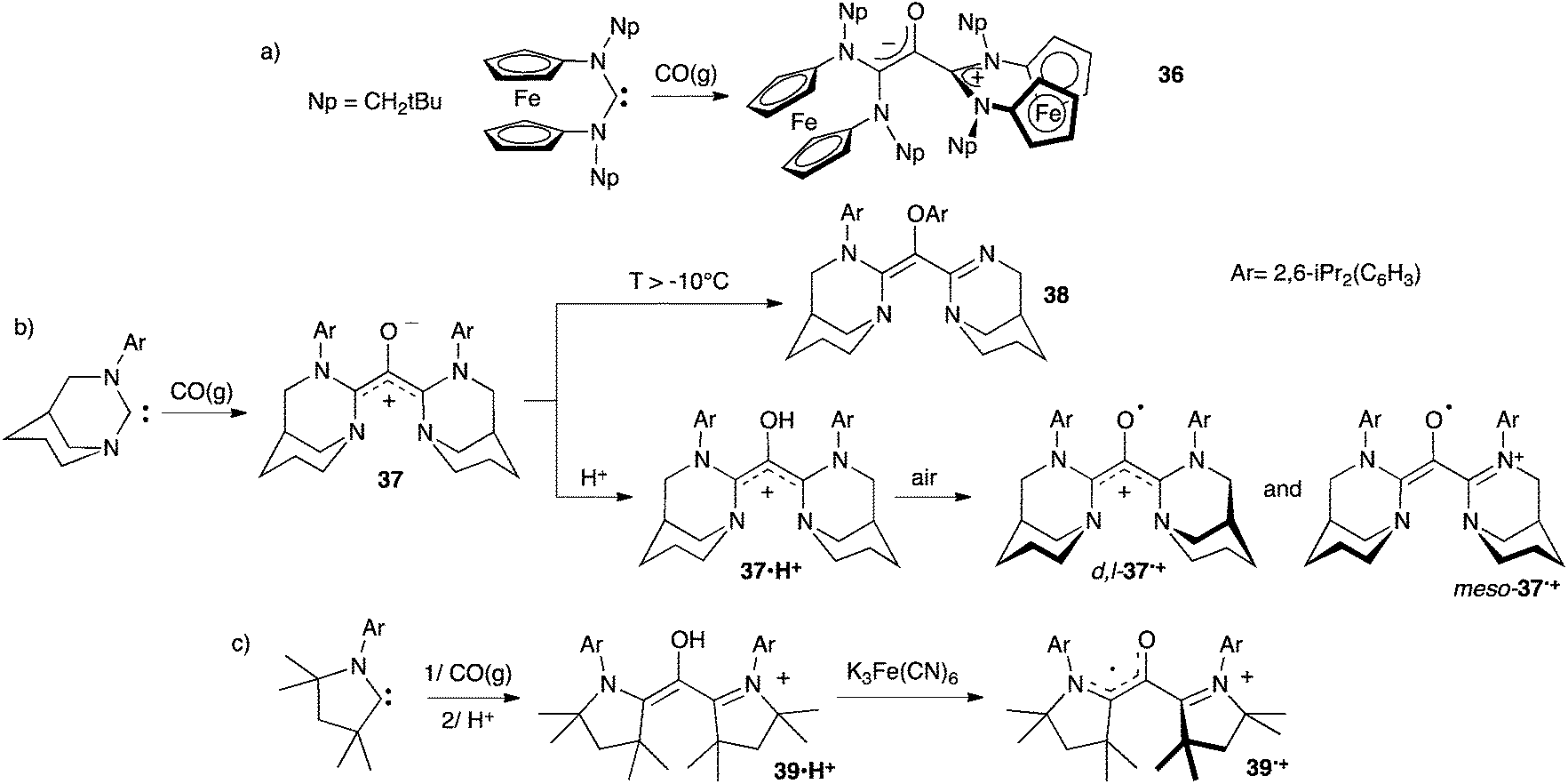
Siemeling et al. first reported the addition of carbon monoxide to two equivalents of a ferrocene-based carbene in 2010.38a However, the resulting product 36 features two orthogonal carbene units, only one being conjugated with the carbonyl. Therefore 36 has the structure of a zwitterionic enolate, not an oxyallyl (Scheme 17a).
 | ||
| Scheme 17 Reaction of stable unhindered electrophilic cyclic carbenes with carbon monoxide. | ||
In 2013 we showed that bubbling CO in a solution of an anti-Bredt diaminocarbene at −78 °C affords the deep-blue amino-substituted oxyallyl 37.39 Compound 37 was characterized by NMR, but couldn't be isolated, as it rearranges into 38 above −10 °C. Protonation of 37 yields the oxyallyl cation 37˙H+, which was fully characterized, including a single crystal diffraction study (Scheme 17b). Similarly the oxyallyl cation 39˙H+ could be obtained from a cyclic (alkyl)(amino)carbene featuring small substituents (Scheme 17c).40
Oxyallyl cations 37˙H+ and 39˙H+ were oxidized into their corresponding radical cations 37˙+ and 39˙+, respectively.39,40,43 Surprisingly, these compounds are very stable toward air and moisture. Radical 39˙+ has been stored for years as a solid and has a half-life of about a week in aerated technical solutions. Even more remarkably, both diastereomers of 37˙+ could be crystallized under aerobic conditions from refluxing technical toluene solutions. Note that only one carbene unit of meso-37˙+ and 39˙+ is involved in the radical stabilization, the π-system of the second unit being orthogonal to the SOMO. Thus, they are better depicted as (amino)(carboxy)radicals with a cationic spectator substituent. In contrast, D,L-37˙+ is a genuine one-electron oxidized oxyallyl, with a fully delocalized π-system on the carbonyl and both carbene moieties.
Siemeling et al. also reported reactions between acyclic di(amino)carbenes and CO.41 They were able to isolate the oxyallyl cation 40 and the trimeric oxyallyl–lithium chloride complex 41. To date, the X-ray structure of the latter is certainly the closest available picture of a genuine amino-substituted oxyallyl. Note that, here again, the scope of the reaction is limited to few small amino groups. Indeed, the authors showed that the formation of β-lactams, such as 42, is favoured by more bulky substituents (Scheme 18).
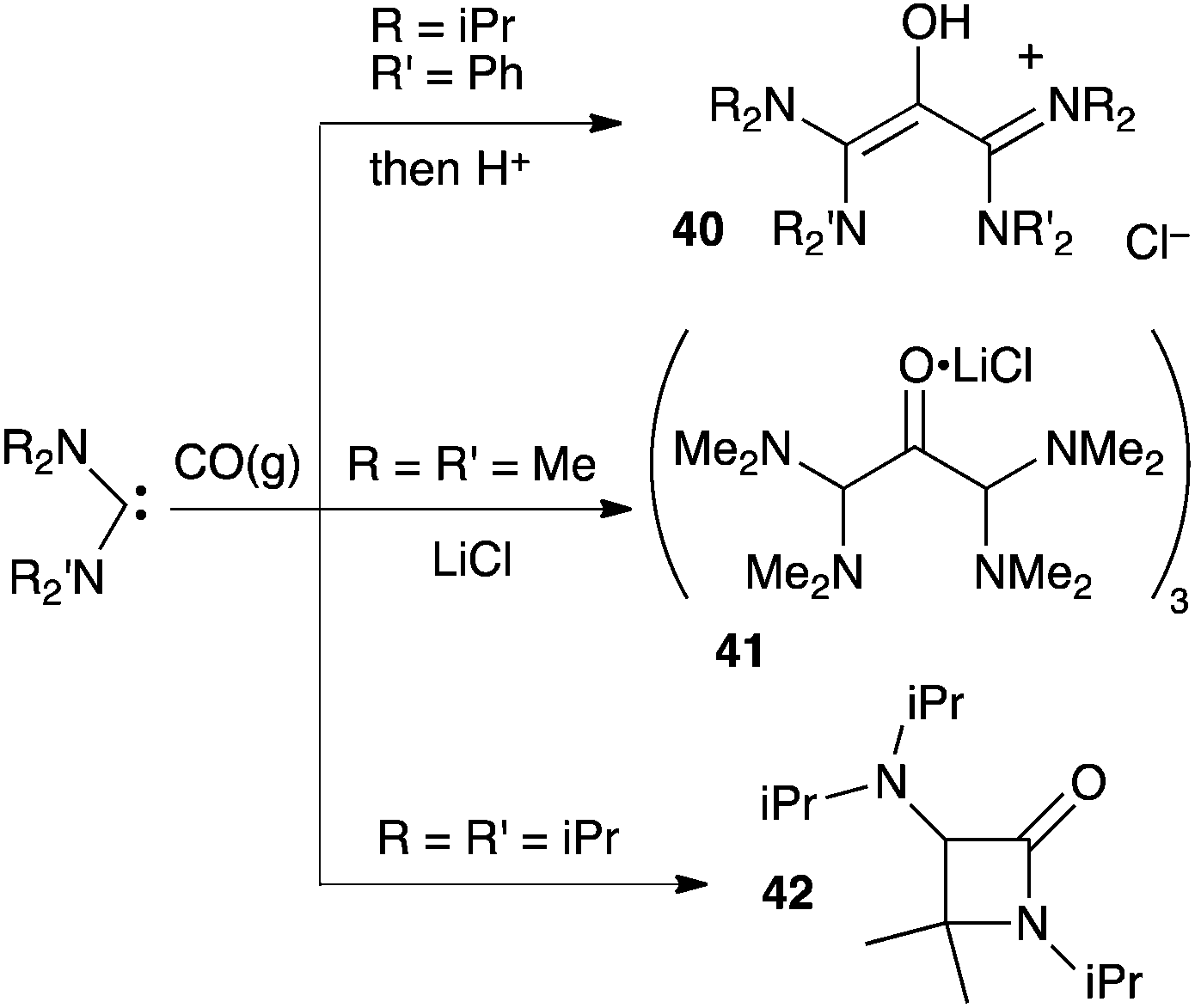 | ||
| Scheme 18 Reaction of stable acyclic di(amino)carbenes with carbon monoxide. | ||
4.2 Non-Kekulé aromatic compounds with a formal oxyallyl pattern
Several cyclic conjugated non-Kekulé compounds contain a formal oxyallyl pattern. As shown in Scheme 19, squaraines 43, croconaines 44 or dithiolane derivatives 45 can be depicted as zwitterionic aromatic rings, but also feature oxyallyl-type diradical and zwitterionic resonance forms. Compounds 43 and 44 exhibit intense π–π* transitions and have attracted considerable attention as promising near-IR dyes.44 For long, their oxyallyl character had been disregarded, their absorption being attributed to a classical donor–acceptor charge transfer between the central ring and electron-donating substituents. However, in the last decade, several theoretical studies demonstrated that the excitation was predominantly localized at the central ring, the low-energy transitions reflecting a significant diradicaloid character of the molecules.45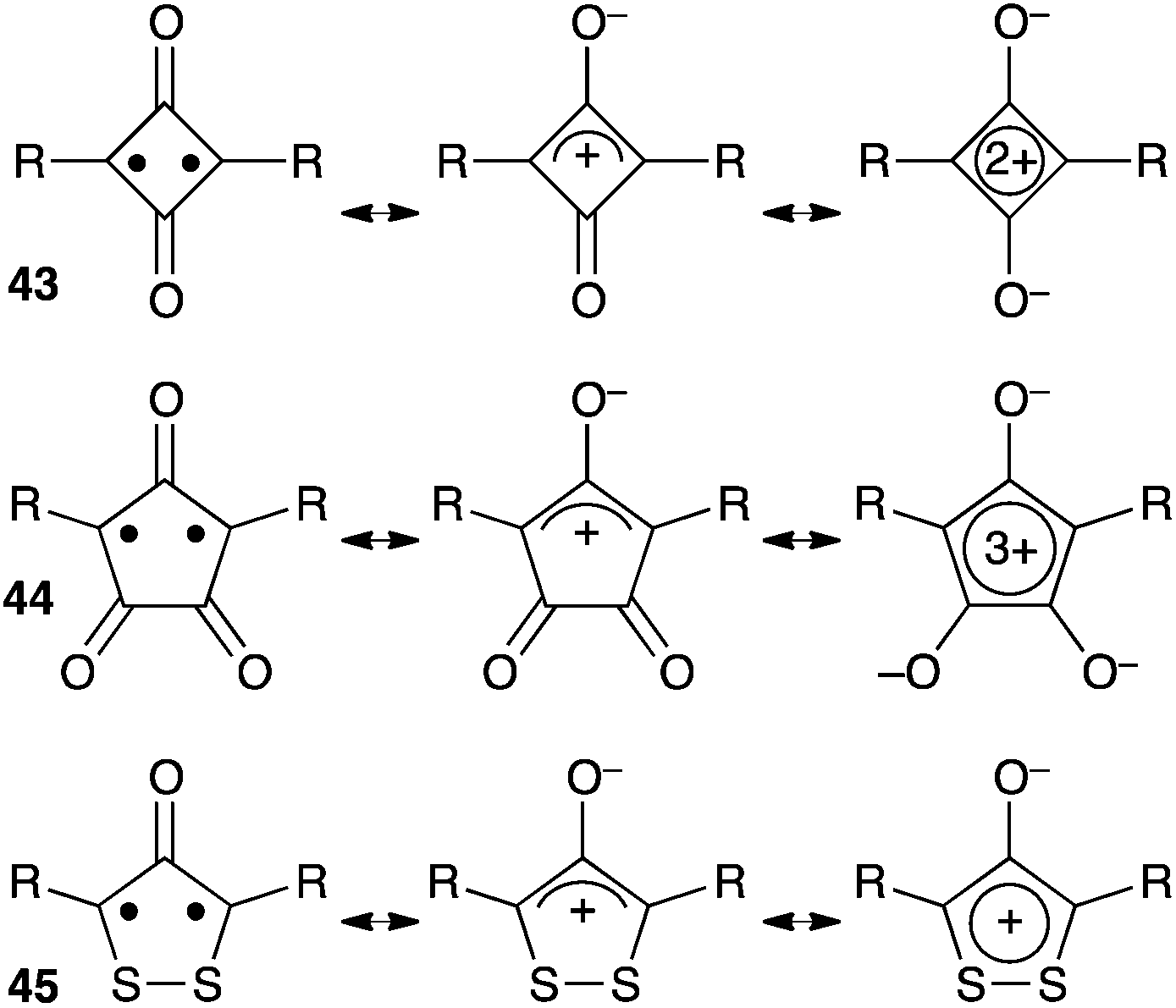 | ||
| Scheme 19 Non-Kekulé dyes with an oxyallyl pattern (R is an electron-donating group). | ||
An oxyallyl pattern can also be identified in the structure of diketohexaphyrin 46 (Scheme 20). This compound has a strong 26 π-electron aromatic character and is remarkably stable towards air and moisture. It is a non-Kekulé molecule with a singlet diradical ground state. The triplet state is very close in energy (+2.5 kcal mol−1), as shown by EPR and magnetic susceptibility measurements.46
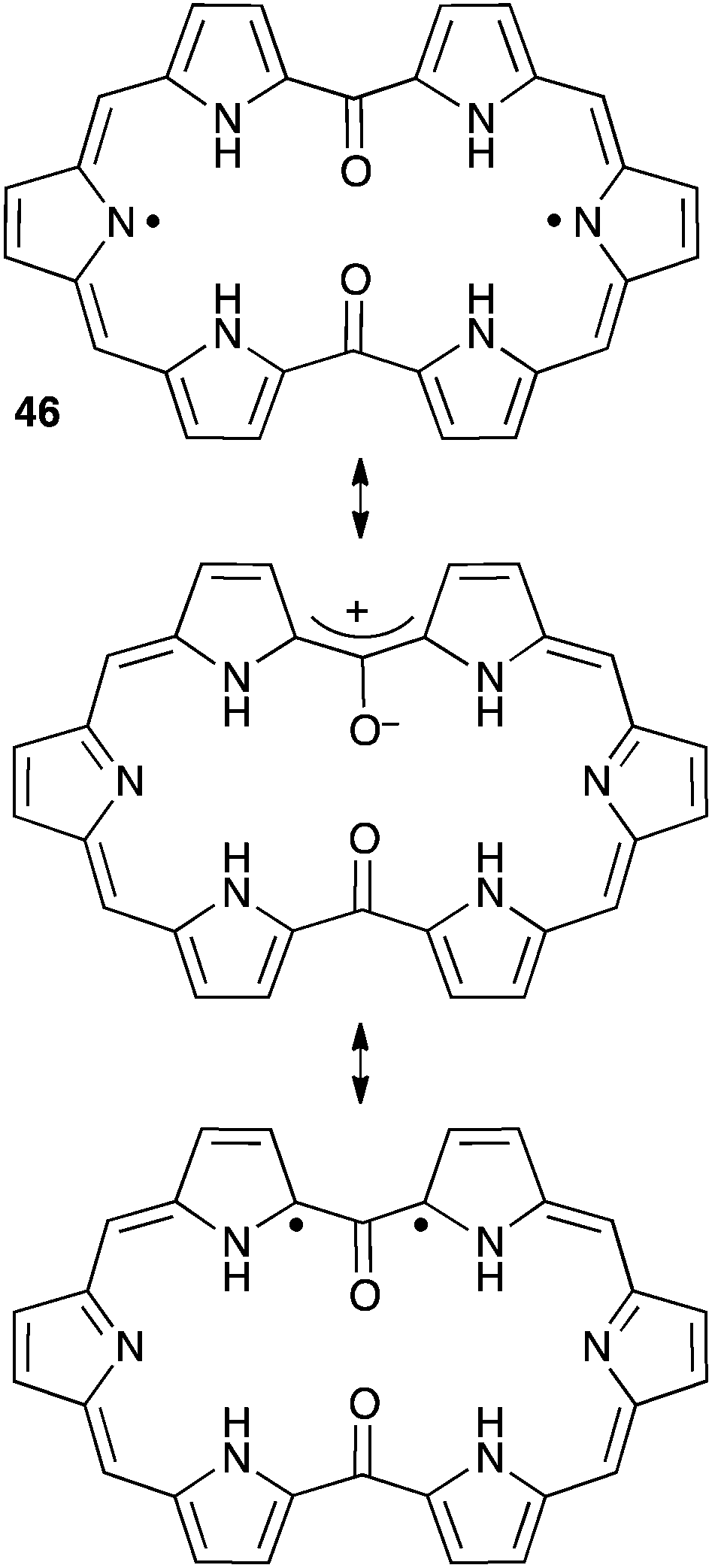 | ||
| Scheme 20 Resonance forms of non-Kekulé compound 46. | ||
4.3 Silicon-based oxyallyls
The recent progress in the quest for isolable oxyallyls has also inspired few synthetic studies in the silicon series. Importantly, although carbon and silicon are both group 14 elements, they strongly differ in terms of electronegativity, atomic radii, capacity for hypervalence, etc. Therefore silaoxyallyls offer a distinct problem from their carbon analogues. Whereas carbonyls are common organic functionalities, silacarbonyl compounds remain elusive, being isolated only as adducts with Lewis bases. Kato et al. reported the first silanone (a “sila” ketone) Lewis-base adduct 47 in 2013 only.47 Interestingly the authors demonstrated that 47, which is a donor-stabilized silacyclopropanone, has a hybrid structure with a significant silaoxyallyl character (Scheme 21).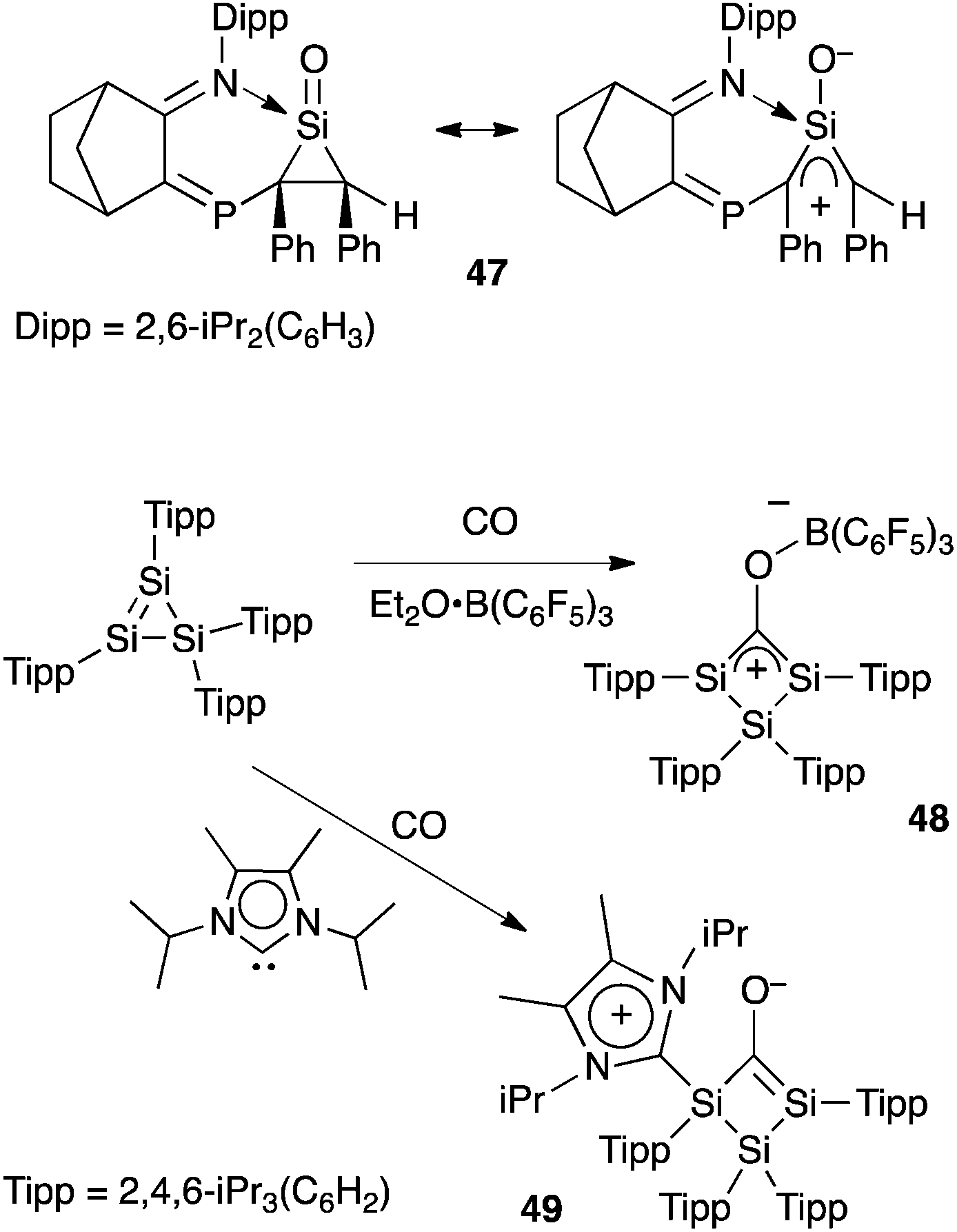 | ||
| Scheme 21 Stabilized silacyclopropanone 47 and silaoxyallyl derivatives 48–49. | ||
A year later, Scheschkewitz et al. reported the isolation of the Lewis acid adduct of sila-oxyallyl 48 from the reaction of carbon monoxide with a cyclotrisilene, in the presence of B(C6F5)3. Conversely, when performing the reaction in the presence of a stable N-heterocyclic carbene, the donor-stabilized sila-oxyallyl 49 was isolated.48 Here again, the situation contrasts with a carbon-based compound, the carbon analogues of 48 and 49 being unknown and unlikely to be isolated.
5. Conclusion
For long, the quest for the observation of oxyallyl derivatives had faced seemingly intractable issues. An abundant literature bears witness of unsuccessful attempts by several research groups. We would like to stress the importance of these publications, which have clearly paved the way to the recent achievements. This is certainly a teachable lesson and one has to wonder whether such apparent “failures” would be published and made available to the community today.Pivotal advances have marked the last decade, among which are the establishment of the transition-state nature of the parent compound 1 and the observation of alkyl-substituted oxyallyl 32. Theoretical calculations have played a key role in ascertaining the identification of the new compounds. They also unveiled the unexpected oxyallyl character of bicyclo[1.1.0]butanones and compounds 43–45, which had been mistakenly considered for long as classical cyclopropanones or donor–acceptor-type dyes respectively.
The scene is set for new synthetic challenges. We have already shown that the design of highly persistent alkyl-substituted oxyallyls is within the reach, and we have even discussed a few possible strategies to achieve this goal. Another type of stable oxyallyl derivative relies on the introduction of strong +M donating 1,3-substituents, favouring singlet zwitterionic limit forms. The small singlet–triplet gap of these species allows for considering potential applications in the field of functional near-IR dyes. Furthermore, the preliminary results also revealed the original redox properties. For example, the radical-cation of oxyallyls 37 and 39 proved to be remarkably air-stable. However, very few models of oxyallyls featuring such “push–push” substitution are available. Their synthesis relies on reactions with narrow scopes: the low yield rearrangement of some croconaines and the carbonylation of rare non-bulky electrophilic stable carbenes. It is clear that further development in this field will require more general and versatile synthetic routes to be designed in the future.
Acknowledgements
The authors acknowledge Agence Nationale de la Recherche (ANR-14-CE06-0013-01) for fundings.References
- (a) N. J. Turro and W. B. Hammond, J. Am. Chem. Soc., 1966, 88, 3672 CrossRef CAS; (b) S. E. Schaafsma, H. Steinberg and T. J. De Boer, Recl. Trav. Chim. Pays-Bas, 1966, 85, 1170 CrossRef CAS PubMed.
- R. Hoffmann, J. Am. Chem. Soc., 1968, 90, 1475 CrossRef CAS.
- J. M. Pochan, J. E. Baldwin and W. H. Flygare, J. Am. Chem. Soc., 1969, 91, 1896 CrossRef CAS.
- For a recent review on diradicals, see: M. Abe, Chem. Rev., 2013, 113, 7011 CrossRef CAS PubMed.
- (a) P. J. Chenier, J. Chem. Educ., 1978, 55, 286 CrossRef CAS; (b) F. G. Bordwell, R. G. Scamehorn and W. R. Springer, J. Am. Chem. Soc., 1969, 91, 2087 CrossRef CAS; (c) F. G. Bordwell and J. G. Strong, J. Org. Chem., 1973, 38, 579 CrossRef CAS; (d) N. Tsuchida, S. Yamazaki and S. Yamabe, Org. Biomol. Chem., 2008, 6, 3109 RSC.
- (a) E. J. Corey, S. P. T. Matsuda, R. Nagata and M. B. Cleaver, Tetrahedron Lett., 1988, 29, 2555 CrossRef CAS; (b) R. Koljak, O. Boutaud, B.-H. Shieh, N. Samel and A. R. Brash, Science, 1997, 277, 1994 CrossRef CAS; (c) B. A. Hess Jr., L. Smentek, A. R. Brash and J. K. Cha, J. Am. Chem. Soc., 1999, 121, 5603 CrossRef.
- For reviews, see: (a) A. G. Lohse and R. P. Hsung, Chem. – Eur. J., 2011, 17, 3812 CrossRef CAS PubMed; (b) M. Harmata, Chem. Commun., 2010, 46, 8904 RSC; (c) M. Harmata, Chem. Commun., 2010, 46, 8886 RSC; (d) H. F. Bettinger, Angew. Chem., Int. Ed., 2010, 49, 670 CrossRef CAS PubMed; (e) M. Harmata, Adv. Synth. Catal., 2006, 348, 2297 CrossRef CAS PubMed; (f) M. A. Battiste, P. M. Pelphrey and D. L. Wright, Chem. – Eur. J., 2006, 12, 3438 CrossRef CAS PubMed; (g) I. V. Hartung and H. M. R. Hoffmann, Angew. Chem., Int. Ed., 2004, 43, 1934 CrossRef CAS PubMed; (h) M. Harmata and P. Rashatasakhon, Tetrahedron, 2003, 59, 2371 CrossRef CAS; (i) M. Harmata, Acc. Chem. Res., 2001, 34, 595 CrossRef CAS PubMed; (j) J. H. Rigby and F. C. Pigge, Org. React., 1997, 51, 351 CAS; (k) M. Harmata, Tetrahedron, 1997, 53, 6235 CrossRef CAS; (l) A. R. Katritzky and N. Dennis, Chem. Rev., 1989, 89, 827 CrossRef CAS.
- P. Dowd, J. Am. Chem. Soc., 1966, 88, 2587 CrossRef CAS.
- (a) L. J. Shaad, B. A. Hess Jr. and R. Zahradnik, J. Org. Chem., 1981, 46, 1909 CrossRef; (b) Y. Osamura, W. T. Borden and K. Morokuma, J. Am. Chem. Soc., 1984, 106, 5112 CrossRef CAS; (c) M. B. Coolidge, K. Yamashita, K. Morokuma and W. T. Borden, J. Am. Chem. Soc., 1990, 112, 1751 CrossRef CAS; (d) B. A. Hess Jr., U. Eckart and J. Fabian, J. Am. Chem. Soc., 1998, 120, 12310 CrossRef.
- (a) P. G. Wenthold, J. Hu, R. R. Squires and W. C. Lineberger, J. Am. Chem. Soc., 1996, 118, 475 CrossRef CAS; (b) P. G. Wenthold, J. Hu, R. R. Squires and W. C. Lineberger, J. Am. Soc. Mass Spectrom., 1999, 10, 800 CrossRef CAS.
- W. T. Borden and E. R. Davidson, J. Am. Chem. Soc., 1977, 99, 4587 CrossRef CAS.
- C. A. Schalley, S. Blanksby, J. N. Harvey, D. Schröder, W. Zummack, J. H. Bowie and H. Schwarz, Eur. J Org. Chem., 1998, 987 CrossRef CAS.
- See also: F. Turecek, D. E. Drinkwater and F. W. McLafferty, J. Am. Chem. Soc., 1991, 113, 5950 CrossRef CAS.
- (a) T. Ichino, S. M. Villano, A. J. Gianola, D. J. Goebbert, L. Velarde, A. Sanov, S. J. Blanksby, X. Zhou, D. A. Hrovat, W. T. Borden and W. C. Lineberger, Angew. Chem., Int. Ed., 2009, 48, 8509 CrossRef CAS PubMed; (b) T. Ichino, S. M. Villano, A. J. Gianola, D. J. Goebbert, L. Velarde, A. Sanov, S. J. Blanksby, X. Zhou, D. A. Hrovat, W. T. Borden and W. C. Lineberger, J. Phys. Chem. A, 2011, 115, 1634 CrossRef CAS PubMed.
- A. M. Oertel, V. Ritleng, A. Busiah, L. F. Veiros and M. J. Chetcuti, Organometallics, 2011, 30, 6495 CrossRef CAS.
- R. M. Bullock, R. T. Hembre and J. R. Norton, J. Am. Chem. Soc., 1988, 110, 7868 CrossRef CAS.
- (a) M. Frey, T. A. Jenny and H. Stoeckli-Evans, Organometallics, 1990, 9, 1806 CrossRef CAS; (b) K. Ehrlich and G. F. Emerson, J. Am. Chem. Soc., 1972, 94, 2464 CrossRef CAS.
- A. Ohsuka, T. Hirao, H. Kurosawa and I. Ikeda, Organometallics, 1995, 14, 2538 CrossRef CAS.
- (a) D. B. Sclove, J. F. Pazos, R. L. Camp and F. D. Greene, J. Am. Chem. Soc., 1970, 92, 7488 Search PubMed; (b) J. F. Pazos, J. G. Pacifici, G. O. Pierson, D. B. Sclove and F. D. Greene, J. Org. Chem., 1974, 39, 1990 CrossRef CAS.
- (a) M. H. J. Cordes and J. A. Berson, J. Am. Chem. Soc., 1996, 118, 6241 CrossRef CAS; (b) M. H. J. Cordes and J. A. Berson, J. Am. Chem. Soc., 1992, 114, 11010 CrossRef CAS.
- T. S. Sorensen and F. Sun, J. Chem. Soc., Perkin Trans. 2, 1998, 1053 RSC.
- A. S. Ichimura, P. M. Lahti and A. R. Matlin, J. Am. Chem. Soc., 1990, 112, 2868 CrossRef CAS.
- T. Hirano, T. Kumagai, T. Miyashi, K. Akiyama and Y. Ikegami, J. Org. Chem., 1991, 56, 1907 CrossRef CAS.
- A. P. Masters, M. Parvez, T. S. Sorensen and F. Sun, J. Am. Chem. Soc., 1994, 116, 2804 CrossRef CAS.
- J. K. Crandall and R. P. Haseltine, J. Am. Chem. Soc., 1968, 90, 6251 CrossRef CAS.
- See also: R. Noyori and M. Katô, Tetrahedron Lett., 1968, 9, 5075 CrossRef.
- A. R. Matlin, P. M. Lahti, D. Appella, A. Straumanis, S. Lin, H. Patel, K. Jin, K. P. Schrieber, J. Pauls and P. Raulerson, J. Am. Chem. Soc., 1999, 121, 2164 CrossRef CAS.
- S. Bhargava, J. Hou, M. Parvez and T. S. Sorensen, J. Am. Chem. Soc., 2005, 127, 3704 CrossRef CAS PubMed.
- See also: A. Rauk, T. S. Sorensen and F. Sun, J. Am. Chem. Soc., 1995, 117, 4506 CrossRef CAS.
- G. Kuzmanich, F. Spänig, C.-K. Tsai, J. M. Um, R. M. Hoekstra, K. N. Houk, D. M. Guldi and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2011, 133, 2342 CrossRef CAS PubMed.
- M. Abe, H. Furunaga, D. Ma, L. Gagliardi and G. J. Bodwell, J. Org. Chem., 2012, 77, 7612 CrossRef CAS PubMed.
- B. A. Hess Jr., J. Am. Chem. Soc., 2002, 124, 920 CrossRef PubMed.
- M. Tian, S. Tatsuura, M. Furuki, Y. Sato, I. Iwasa and L. S. Pu, J. Am. Chem. Soc., 2003, 125, 348 CrossRef CAS PubMed.
- (a) S. Tatsuura, M. Tian, M. Furuki, Y. Sato, I. Iwasa and H. Mitsu, Appl. Phys. Lett., 2004, 84, 1450 CrossRef CAS PubMed; (b) S. Tatsuura, T. Matsubara, M. Tian, H. Mitsu, I. Iwasa, Y. Sato and M. Furuki, Appl. Phys. Lett., 2004, 85, 540 CrossRef CAS PubMed.
- K. Yesudas and K. Bhanuprakash, J. Phys. Chem. A, 2007, 111, 1943 CrossRef CAS PubMed.
- T. S. Sorensen and F. Sun, J. Am. Chem. Soc., 1997, 119, 11327 CrossRef CAS . See also references cited herein.
- (a) V. Lavallo, Y. Canac, B. Donnadieu, W. W. Shoeller and G. Bertrand, Angew. Chem., Int. Ed., 2006, 45, 3488 CrossRef CAS PubMed; (b) T. W. Hudnall and C. W. Bielawski, J. Am. Chem. Soc., 2009, 131, 16039 CrossRef CAS PubMed.
- (a) U. Siemeling, C. Färber, C. Bruhn, M. Leibold, D. Selent, W. Baumann, M. von Hopffgarten, C. Goedecke and G. Frenking, Chem. Sci., 2010, 1, 697 RSC; (b) C. Goedecke, M. Leibold, U. Siemeling and G. Frenking, J. Am. Chem. Soc., 2011, 133, 3557 CrossRef CAS PubMed.
- D. Martin, C. E. Moore, A. L. Rheingold and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7014 CrossRef CAS PubMed.
- J. K. Mahoney, D. Martin, F. Thomas, C. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2015, 137, 7519 CrossRef CAS PubMed.
- T. Schulz, C. Färber, M. Leibold, C. Bruhn, W. Baumann, D. Selent, T. Porsch, M. C. Holthausen and U. Siemeling, Chem. Commun., 2013, 49, 6834 RSC.
- D. A. Dixon, A. J. Arduengo III, K. D. Dobbs and D. V. Khasnis, Tetrahedron Lett., 1995, 36, 645 CrossRef CAS.
- For earlier observation of transient oxyallyl radical cations, see: (a) L. Eberson and P. Kubacek, Acta Chem. Scand., 1990, 44, 384 CrossRef CAS; (b) H. Ikeda, F. Tanaka, K. Akiyama, S. Tero-Kubota and T. Miyashi, J. Am. Chem. Soc., 2004, 126, 414 CrossRef CAS PubMed.
- (a) L. Hu, Z. Yan and H. Xu, RSC Adv., 2013, 3, 7667 RSC; (b) G. Qian and Z. Y. Wang, Chem. – Asian J., 2010, 5, 1006 CrossRef CAS PubMed; (c) J. Fabian, H. Nakazumi and M. Matsuoka, Chem. Rev., 1992, 92, 1197 CrossRef CAS.
- (a) G. Krishna Chaitanya, L. P. Avinash and K. Bhanuprakash, RSC Adv., 2015, 5, 18813 RSC; (b) J. Fabian and R. Peichert, J. Phys. Org. Chem., 2010, 23, 1137 CrossRef CAS PubMed; (c) K. Srinivas, C. Prabhakar, C. L. Devi, K. Yesudas, K. Bhanuprakash and V. J. Rao, J. Phys. Chem. A, 2007, 111, 3378 CrossRef CAS PubMed; (d) C. Prabhakar, K. Yesudas, G. Krishna Chaitanya, S. Sitha, K. Bhanuprakash and V. J. Rao, J. Phys. Chem. A, 2005, 109, 8604 CrossRef CAS PubMed.
- (a) T. Koide, K. Furukawa, H. Shinokubo, J.-Y. Shin, K. S. Kim, D. Kim and A. Osuka, J. Am. Chem. Soc., 2010, 132, 7246 CrossRef CAS PubMed; (b) M. Ishida, J.-Y. Shin, J. M. Lim, B. S. Lee, M.-C. Yoon, T. Koide, J. L. Sessler, A. Osuka and D. Kim, J. Am. Chem. Soc., 2011, 133, 15533 CrossRef CAS PubMed.
- R. Rodriguez, T. Troadec, D. Gau, N. Saffon-Merceron, D. Hashizume, K. Miqueu, J.-M. Sotiropoulos, A. Baceiredo and T. Kato, Angew. Chem., Int. Ed., 2013, 52, 4426 CrossRef CAS PubMed.
- M. J. Cowley, V. Huch and D. Scheschkewitz, Chem. – Eur. J., 2014, 20, 9221 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2015 |