
Catalysis by electrons and holes: formal potential scales and preparative organic electrochemistry
Oana R.
Luca
*a,
Jeffrey L.
Gustafson
*b,
Sean M.
Maddox
b,
Aidan Q.
Fenwick
a and
Daryl C.
Smith
c
aJoint Center for Artificial Photosynthesis, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. E-mail: oana.luca@caltech.edu
bDepartment of Chemistry and Biochemistry, San Diego State University, San Diego, California 92182-1030, USA
cDepartment of Chemistry, Yale University, 225 Prospect Street, New Haven, CT 06520, USA
First published on 7th May 2015
Abstract
The present review surveys current chemical understanding of catalysis by addition and removal of an electron. As an overarching theme of this type of catalysis, we introduce the role of redox scales in oxidation and reduction reactions as a direct analogue of pKa scales in acid/base catalysis. Each scale is helpful in determining the type of reactivity to be expected. In addition, we describe several means of generating electrons and holes via chemical reactions, plasmonic resonance, radiolytic, photochemical and electrochemical methods. We specifically draw parallels between the now well-established fields of photoredox catalysis and chemical opportunities made available by electrochemical methods. We highlight accessible potential ranges for a series of electrochemical solvents and provide a discussion on experimental design, pitfalls and some remaining challenges in preparative organic electrochemistry.
 Oana R. Luca | Oana Luca is Project Scientist for Molecular Catalysis with the Joint Center for Artificial Photosynthesis (JCAP) at Caltech. She is a graduate of Worcester Polytechnic Institute (BS '08) and Yale University (MS '10, PhD '13) where she worked with Professor Robert H. Crabtree. Her work in inorganic and physical organic chemistry at Caltech and beyond addresses applications of electrochemical methods for the synthesis and interconversion of chemical fuels in battery, fuel cell and solar device designs. Her scientific interests span a wide range of research fields from the engineering of devices for energy production and storage, to molecular orbital interactions in surface science, to electrochemical applications in organic synthesis and in operando assessments of product distributions. |
 Jeffrey L. Gustafson | Jeffrey Gustafson received his B.S. in chemistry from San Diego State University in 2005, and he received his Ph.D. from Yale University in 2011 under the supervision of Prof. Scott Miller. In 2013, after postdoctoral work at Yale University with Prof. Craig Crews in the department of Molecular, Cellular, and Developmental Biology, he joined the faculty at San Diego State University in the department of chemistry and biochemistry as an assistant professor. |
 Sean M. Maddox | Sean Maddox received his B.S. in Chemistry from the University of California, Davis in 2013. He is currently pursuing a Ph.D. under Prof. Jeffrey Gustafson at San Diego State University in conjunction with the University of California, San Diego via the Joint Doctoral Program (JDP). Sean's research is focused on the rational design of various catalysts to effect the late-stage functionalization of aromatic systems. |
 Aidan Q. Fenwick | Aidan Fenwick B.S. ('15) in chemistry and physics from University of California, Santa Barbara, worked under Professor Peter Ford on the photochemical release of nitric oxide via chromium(III) dinitrito complexes. In 2014 he began working as a staff scientist at Caltech with Dr Oana Luca at the Joint Center for Artificial Photosynthesis. His research there focuses on the electrochemical reduction of CO2 and is the lead author of a review on challenges in establishing a standalone device that converts CO2 into a chemical fuel by means of solar irradiation. His interests are centered on the selective conversion of CO2 to high-value products. |
 Daryl C. Smith | Daryl Smith received his Associates in Applied Science in Scientific Glass Technology from Salem Community College. He completed his apprenticeship in 1989 and continued to work in manufacturing, producing catalog and custom glassware. In 2000, Daryl accepted the position of Instructional Chair of Scientific Glass at his Alma Mater, Salem Community College. For 5 years he developed and improved the curriculum to educate students in modern manufacturing processes. In 2005, Daryl left to accept his current position as Scientific Glassblower at Yale University. There he works directly with researchers in producing glassware to meet their desired needs. |
1. Introduction
The concept of chemical potential is introduced in early chemistry training and it is used to derive general assessments of reactivity based on thermodynamic considerations.1 A central factor in any such interpretation is the concept of equilibrium states2 and any chemical reactivity can be defined as a system reaching equilibrium, from a thermodynamic standpoint. Equilibria and kinetic processes involving electron transfer have been extensively studied over the past decades and several models may be used in theoretical assessments.3,4The present review describes some of these processes from the perspective of formal potential. Under this paradigm, reactions may be viewed as catalyzed by electrons and holes. These descriptions are based on the Curran analysis of such processes.5 From this viewpoint we make an analogy between protons and electrons as elementary entities being used to catalyze chemical reactions with their addition or removal from a substrate as the first step in a cycle. A common denominator in all of these processes is the concept of redox potential which describes the oxidizing or reducing ability of the relevant chemical entity.
Potential (E°) is an intrinsically thermodynamic measure of oxidizing and reducing power as depicted in the electrochemical series. Kinetic factors may complicate the picture when attempting to make an experimental assessment of such potential. A direct measure of the formal potential of a redox couple can be obtained via electrochemical measurements under a certain set of experimental parameters. To aid in the discussion, we provide a set of solvent-dependent formal potential redox scales involving known chemical oxidizing and reducing agents, akin to the medium-dependent pKa scales that often inform the choice of suitable reagents in acid/base chemistry (section 2.1.1, Fig. 1 and 2). To support this description, we touch on several means of generation and abstraction of electrons for chemical reactions, as they have been discussed in several spectroscopic and preparative studies in the fields of applied physics, inorganic, photo-, radio- and organic chemistry.
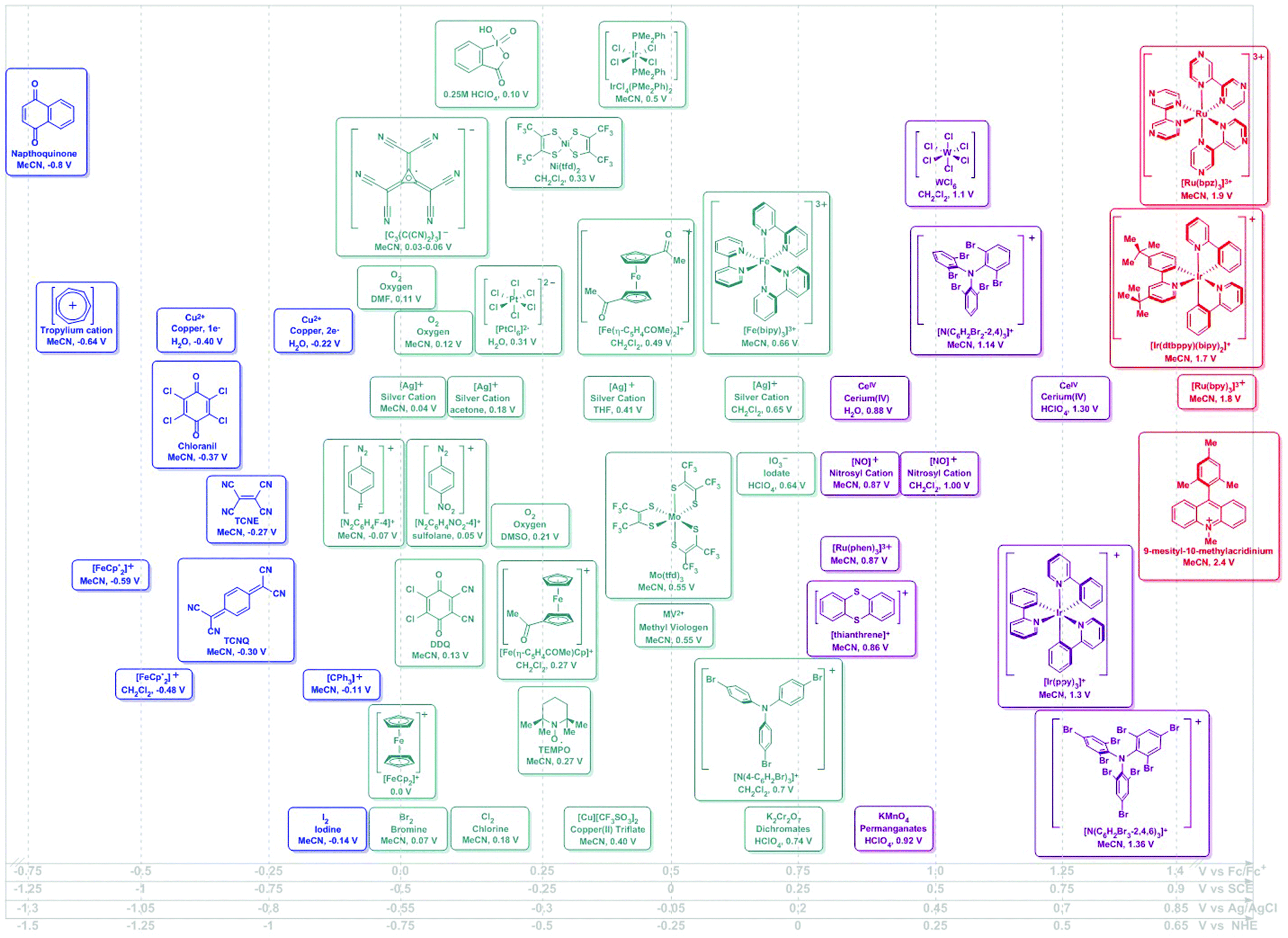 | ||
| Fig. 1 Potential ranges for known chemical oxidants. Potentials shown as vs. Fc/Fc+ using the Geiger conversions and reported potentials.6 Potentials for photo-oxidants originally reported vs. SCE (Referenced vs. Fc/Fc+ by −0.5 V). Secondary scales shown for convenience.19 | ||
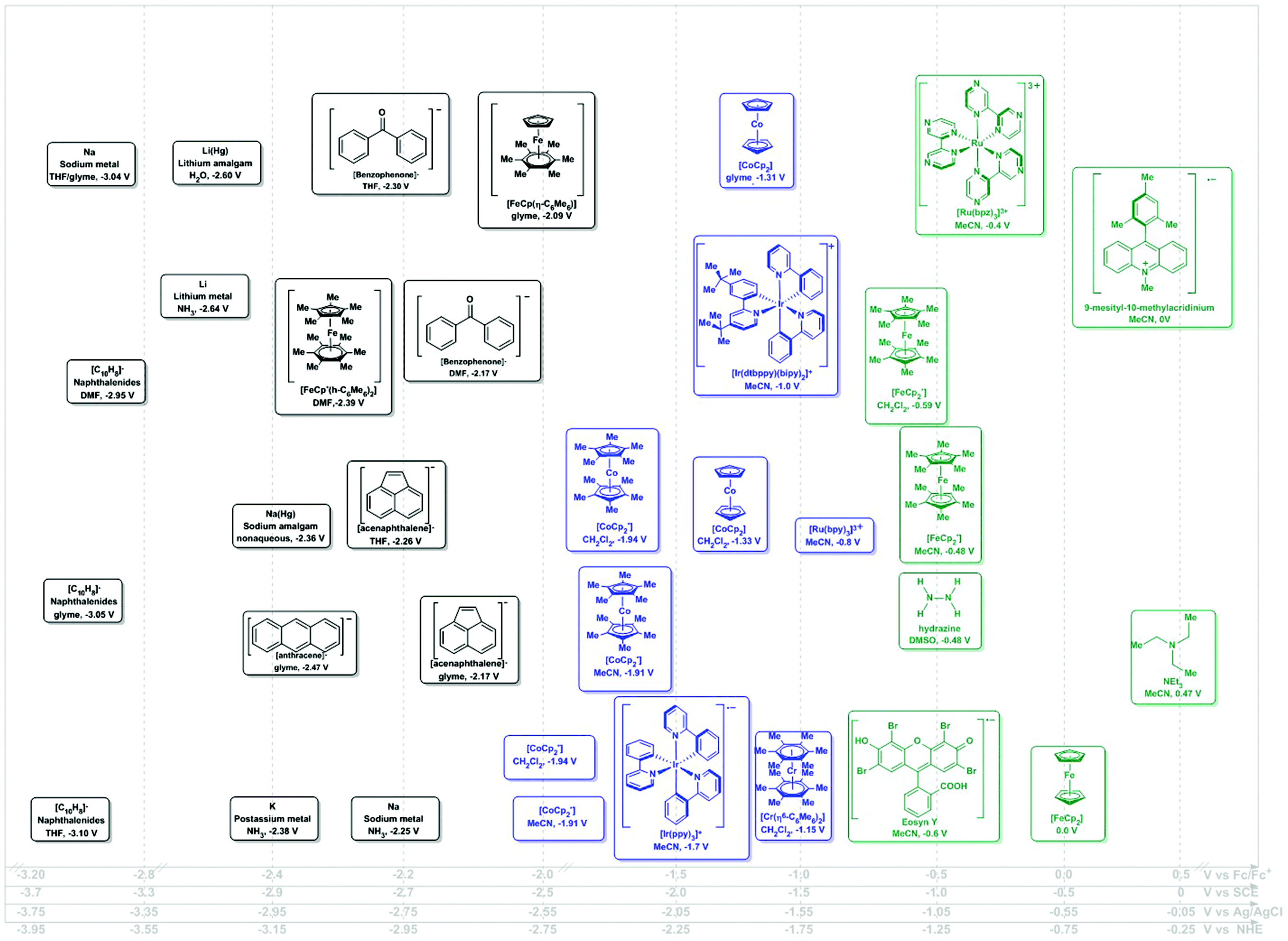 | ||
| Fig. 2 Potential ranges for known chemical reductants. Potentials shown as vs. Fc/Fc+ using the Geiger conversions and reported potentials.6 Potentials for photo-reductants originally reported vs. SCE (Referenced vs. Fc/Fc+ by −0.5 V). Secondary scales shown for convenience.19 | ||
Although a well-established field in its own, electrochemistry has not yet been widely adopted as a synthetic tool. This is somewhat surprising, given that routine methods can be used to experimentally determine the potential at which a certain electron transfer event may take place in a given molecule. Geiger makes the important distinction between measured and thermodynamic electrochemical potential and uses this concept to compare the experimentally-determined oxidizing and reducing strength of a series of organometallic species.6 Theoretical assessments of experimental observables in simple electrochemical measurements remain challenging. Appropriate referencing of such quantities has been shown as crucial to limiting systematic uncertainties in interpretations of such calculations.7,8
From a different perspective, reactions involving an electron or a hole are not a novel topic of interest and radical reactions have been extensively reviewed.9,10 Electrochemistry can provide a direct measure of the strength of oxidizing or reducing agents, given that the comparisons remain internally consistent and that the compared formal potentials are assessed under similar conditions. In the field of photoredox catalysis, the concept of formal potential has now been harnessed in numerous instances. In these reactions, the interaction between light and light-absorbing molecule generates a relatively long-lived excited state that can be quenched by a sacrificial oxidant or reductant to generate high-potential electrons or holes (their potentials being set by the properties of the absorber), allowing these dyes to act as either efficient electron donors or acceptors in a photocatalytic cycle. We discuss the details of this work in sections 2.1.4 and 3. Other means of generating electrons and holes are discussed in sections 2.1.2 and 2.1.3.
Direct access to an oxidizing or reducing agent of a desired potential is usually attainable by electrochemical means, thereby circumventing the limitations of chemical and photochemical agents, which operate at a pre-set potential and may react in unproductive ways. We then follow up these descriptions by highlighting recent advances in photoredox catalysis. We draw parallels between this emerging field of organic chemistry and chemical opportunities made available by electrochemical methods.
Lastly, we discuss the challenge of choosing experimental parameters in preparative electrolysis. We provide an experimental decision tree to aid researchers in their own future electrochemical endeavors. We also briefly describe available analytical diagnostic techniques and associated pitfalls of experimental design in macroscopic preparative instances.
2. Addition and removal of elementary entities: redox scales in the context of acid/base catalysis
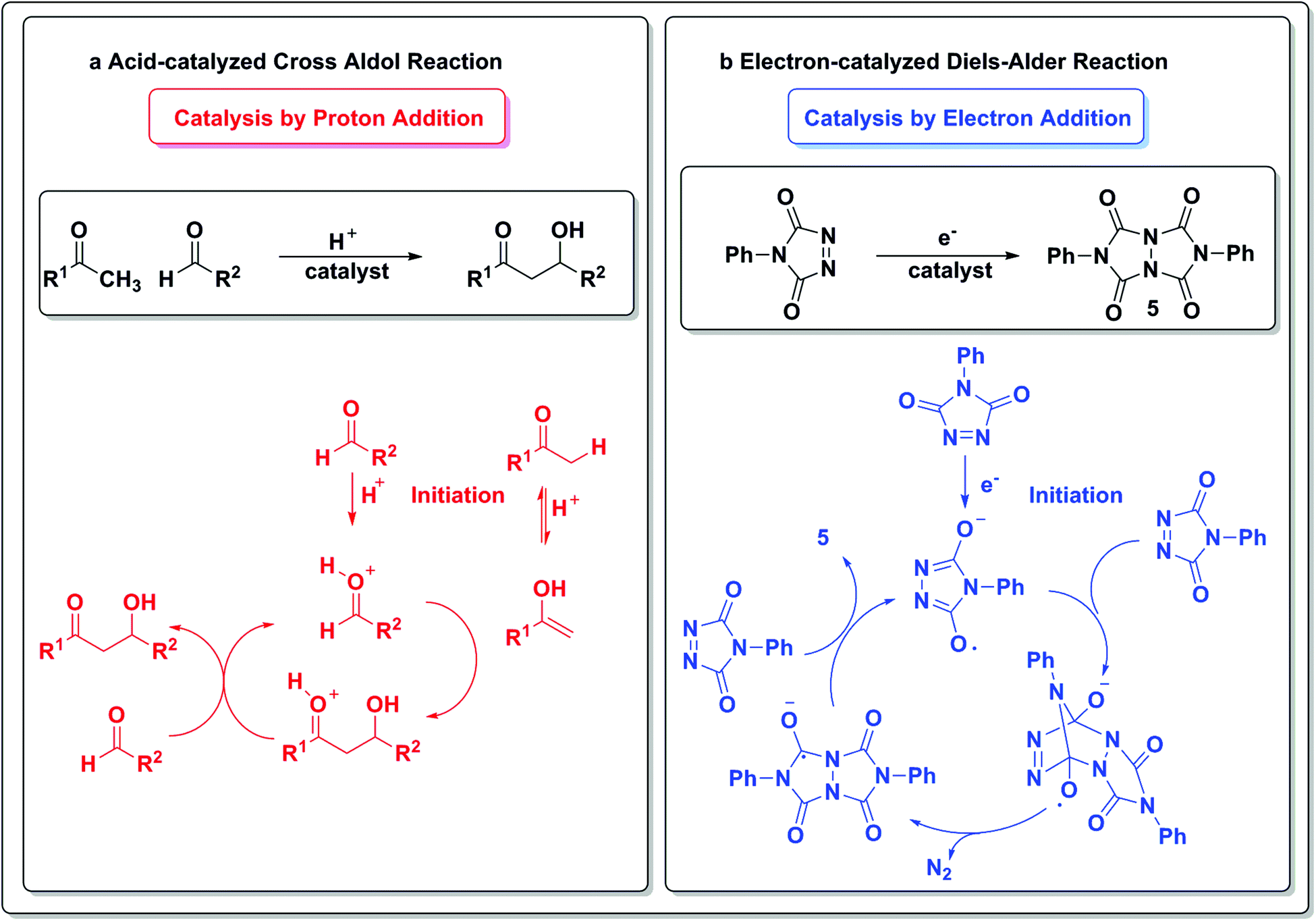
Electrons are common currency in chemical reactions and the addition or removal of an electron from a closed-shell species is normally achieved via stoichiometric radicals as initiators. This will result in the formation of reactive radical anions or radical cations, respectively. Curran has recently described how electrons can be viewed as catalysts in many chemical processes, as radical ions are powerful reactive intermediates.5While reactions between radical ions and closed-shell molecules have fast kinetics and are often uncontrolled, recent work, particularly in the photoredox catalysis field, has elegantly demonstrated that controlled radical formation can result in synthetically useful selectivities.
To draw an analogy with a more traditional branch of catalysis, acids and bases are common protagonists in catalytic reactions. In an acid-catalyzed reaction, a proton adds to the substrate (Scheme 1a) and generates a cationic species, which then can undergo further reactions. Analogously, in a reaction catalyzed by an electron (Scheme 1b), a similar sequence of events occurs. An electron is added to the substrate to generate an anion radical, which can participate in further reactions and enter a catalytic cycle. In the analogous set of reactions in acid/base chemistry, a base first abstracts a proton from a substrate (Scheme 1c), thus generating an anion poised for subsequent chemistry. Similarly, an oxidant abstracts an electron from a substrate, leaving a “hole” behind and generating a cation radical, which can then participate in further reactions (Scheme 1d).
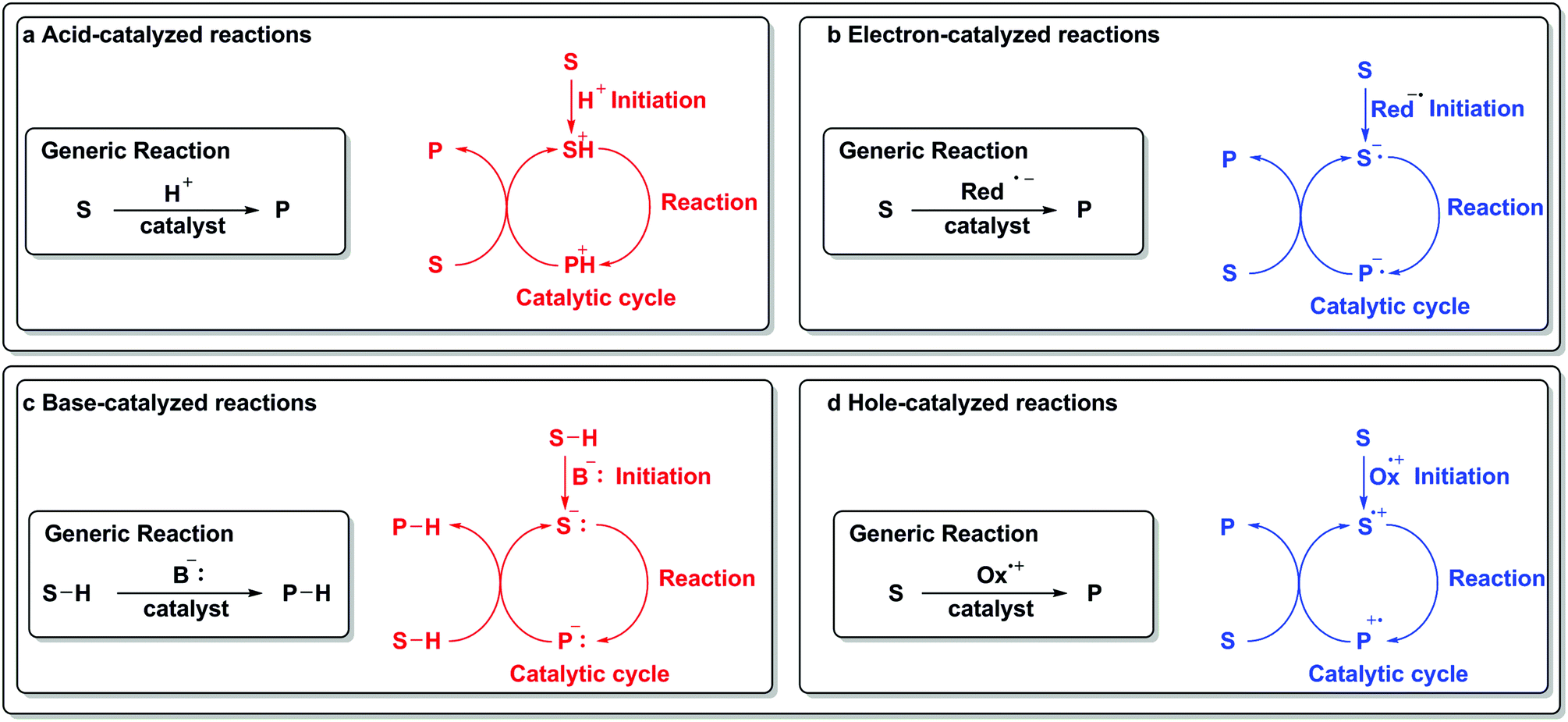 | ||
| Scheme 1 Addition or removal of an electron in redox catalysis as a direct analogy to addition or removal of a proton in acid/base chemistry.5 | ||
The ability of a proton source to donate a proton is generally assessed by means of its pKa.11 Experimental assessments of pKa often rely on proton activity given a certain medium.12 Ironically, pKa and proton activity determinations in non-aqueous media may be directly assessed via electrochemical means.12 From a redox scale standpoint, the generation of electrons and holes of set potential is relatively trivial. The formal potential of a redox couple may be measured by routine voltammetry techniques, even in cases where the electron transfer is irreversible.6 Unfortunately, the issue of lifetime and transport of electrons and holes can pose a challenge. Where an acidic or a homogeneous solution of oxidant or reductant generally has reactant molecules of set chemical potential available uniformly throughout, there is a maximal amount of reactive sites in solution (i.e., a maximal amount of efficient collisions). A reduction by photochemical, electrochemical, radiolytic or other means has both lifetime and interfacial issues to address. Charge recombination is a common problem related to using photosensitizers on solid semiconductor surfaces.13 The equivalent of this issue in the photoredox world is the lifetime of an excited state,14–17 which may simply be too short for any productive reductive or oxidative quenching to occur. Even if a reactive intermediate is generated, its availability for productive chemistry may be compromised by its decay to an unreactive ground state. Photochemical experiments also suffer from possible issues of uniform illumination areas and flux and in the case of ambient sunlight-driven processes, the day/night cycles may be detrimental to maintaining a sustained photochemical process.18
2.1. Methods to generate electrons and holes
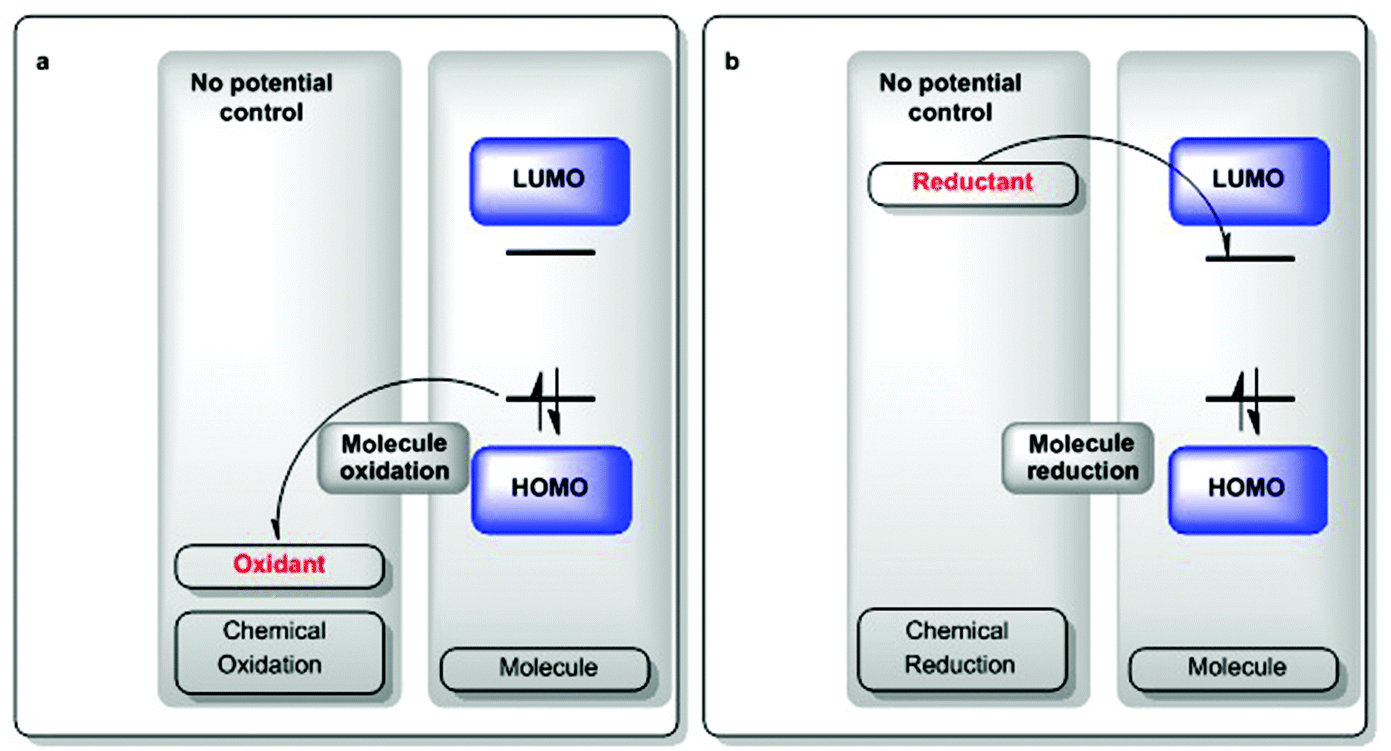 | ||
| Scheme 2 Chemical oxidations and reductions are limited by the intrinsic chemical makeup of the species.22 | ||
The oxidizing or reducing power of any species is defined by potentials intrinsic to its chemical structure, which defines the accessible energy levels of the oxidation and reduction reactions (Scheme 2). The use of some oxidants or reductants may also be limited by the nature of side-reactions or the presence of spent oxidant or reductant. However, potential scales, such as Fig. 1 and 2, may be assembled and used in rationally selecting a reagent for a specific redox transformation, given appropriate potential conversions. Since the molecules were originally measured in different media, with different electrolyte/reference electrodes, we report formal potentials vs. the Fc/Fc+ scale, as Geiger has previously reported for a similar series after application of the appropriate corrections. For convenience, we also provide alternate scales (x-axes of Fig. 1 and 2) for conversions vs. SCE, NHE and Ag/AgCl.
In the assessment of a research problem at hand, it is optimal to perform the measurements in the researcher's own setup to eliminate any artefactual conversion issues between scales. In this way, measured potentials remain internally consistent. The rankings in Fig. 1 and 2 are meant to serve as a guidance of relative redox power. A truly quantitative comparison remains elusive at this time, as all reagents would need to be interrogated under identical conditions and reported against the same reference electrode.
In a light-driven experiment, the oxidant or reductant may be generated by excitation of electrons in the HOMO of a sensitizer through the absorption of a photon of sufficient energy. The photophysical processes involved in using an excited state to effect chemical processes require a series of lifetime and quenching diagnostics. It is important to note that as this field is partially limited by the intrinsic potential of the excited state light absorbers (or catalysts under the electron/hole paradigm), tuning of the potential by structural modification of the light absorber is possible and may increase the scope of accessible chemical transformations. The oxidants and reductants produced by photoredox reactions are very strong and likely to give overoxidation/overreduction products. Fig. 1 and 2 show examples of photooxidants and reductants commonly employed in light-driven chemical transformations. Scheme 4 describes some of these processes and related potentials.
In an electrochemical experiment, an oxidant or reductant is generated at an electrode surface. One can directly effect a redox process by dialing in an appropriate potential. These types of methods have applications in battery chemistry,20,21 solar fuel generation,22 and lastly, for electrolysis in organic transformations, representing a major opportunity in synthetic chemistry, which we touch on in sections 2.1.5, 3 and 4.
The collective oscillations of the delocalized electron clouds in a metal give rise to plasmonic effects and are the cause of several optical properties of metals such as color and luster. The field of plasmonics is strongly rooted in the field of photonics and the potentials achieved by local photoexcitation of such materials may lead to the generation of an excited state. Similar to the photoexcitation of molecules followed by reductive or oxidative quenching, a plasmonic material may generate high-energy “hot” electrons and hot holes as well as local heating effects generated during their decay. Any photo-excited electron with energy higher than the Fermi energy will be thermodynamically compelled to quickly decay back to the Fermi energy. The utility of these species is still under theoretical consideration, as these parameters are important for applications of standalone catalysis.24
Such hot electrons can be utilized for chemical reactions, provided they are transferred to a molecule during their brief moment of excitation. An important report by Linic and coworkers has now shown that hydrogen dissociation is possible with the aid of hot electrons at Au surfaces.25 This is the first instance of a direct bond-breaking event by means of a hot electron. This work sets the stage for the development of further processes involving hot carriers and the principles of preparative-scale electrochemical and photochemical processes may provide valuable insights in carrying out chemical reactions via hot electrons and hot holes. Progress has been made showing that plasmonic effects are efficacious in enhancing catalytic activities at plasmonic surfaces.26–29
The field remains in its infancy as far as applications in catalysis for organic transformations. Given the advent of photoredox catalysis for organic applications, some initial observations of plasmonic effects provide encouraging precedent in the development of coupled chemical processes.
These chemical processes are also analogous to photolysis14 methods, where a light source, usually a laser, is used to increase the population of non-ground state energy levels of an analyte and then follow their decay behavior in conjunction with a spectroscopic technique.31 The sequence of techniques has uses in the study of light-induced changes in a variety of materials, from organic molecules to biological systems.32
A combined photochemical radiolytic study of the production of an NADH analog ligand on a Ruthenium center33 illustrates the versatility of radiolysis methods in generating and characterization of transient intermediates. The reaction under investigation was the reduction of acetone to isopropanol and the observed active intermediate in the reductive chemistry was shown to be an organic hydride. In the study of CO2 reduction to chemical fuels, radiolysis was used to generate and follow the decay of radical intermediates, specifically Ni–CO2 adducts supported by macrocyclic ligands.31
These types of studies require highly specialized equipment and handling capabilities for radioactive materials in some cases.33 Since these types of capabilities require advanced training and precautions, they are unlikely to become commonplace in benchtop organic chemistry. However, we would like to emphasize their existence due to their utility as mechanistic and spectroscopic probes.
A dye, also referred to as a photoabsorber, is a molecule that absorbs light. The photophysical properties of the state generated by absorption of a photon in a potential scale may be used in light-driven chemical processes. The lifetime of an excited state generally limits the types of processes that can be effected.
In the parlance of solar fuels, the dye absorbs a photon to reach an excited state, which can then drive water splitting to either oxygen or hydrogen, depending on the nature of catalysts and accessible energy levels and quenchers in the medium. Organic and metal-based photoabsorbers are commonly employed, however each is likely to undergo different types of charge transfers. In the case of an organic light absorber, interaction with light may cause an excitation from a bonding (HOMO) to an antibonding LUMO orbital (π → π*). Metal-to-ligand charge transfer (MLCT, d → π*) comes into play in the case of the canonical Ru and Ir photosensitizers.13
In order to generate a long-lived excited state, the light-absorbing unit may be interfaced with a semiconductor material with appropriate conduction band levels in a potential landscape (Scheme 3a).17 Light absorption of the dye generates an excited state (1D*) which can either directly interact with the conduction band of the semiconductor (shown as a fast ps-scale e− injection in Scheme 3) or can relax down to a triplet state 3D*. In Scheme 3a, the excitation generates a hole in the dye (Scheme 3a, center) and the semiconductor funnels the electrons away from the system (Scheme 3a, left). If the potential of the hole is appropriately large, it may effect oxidation in a catalyst present at the 3-level interface (Scheme 3a, right). However, the lifetime and fate of that excited state dictate the kind of chemistry it can drive and fast recombination may compromise the entire scheme. In this scheme, oxidative quenching of the 1D* can produce oxidizing equivalents in the dye, which can then act as oxidants of their own. The scheme in reverse would effect an electron injection into the dye, which can then act as a reductant, while the same issues related to the lifetime of the charge-separated state apply.
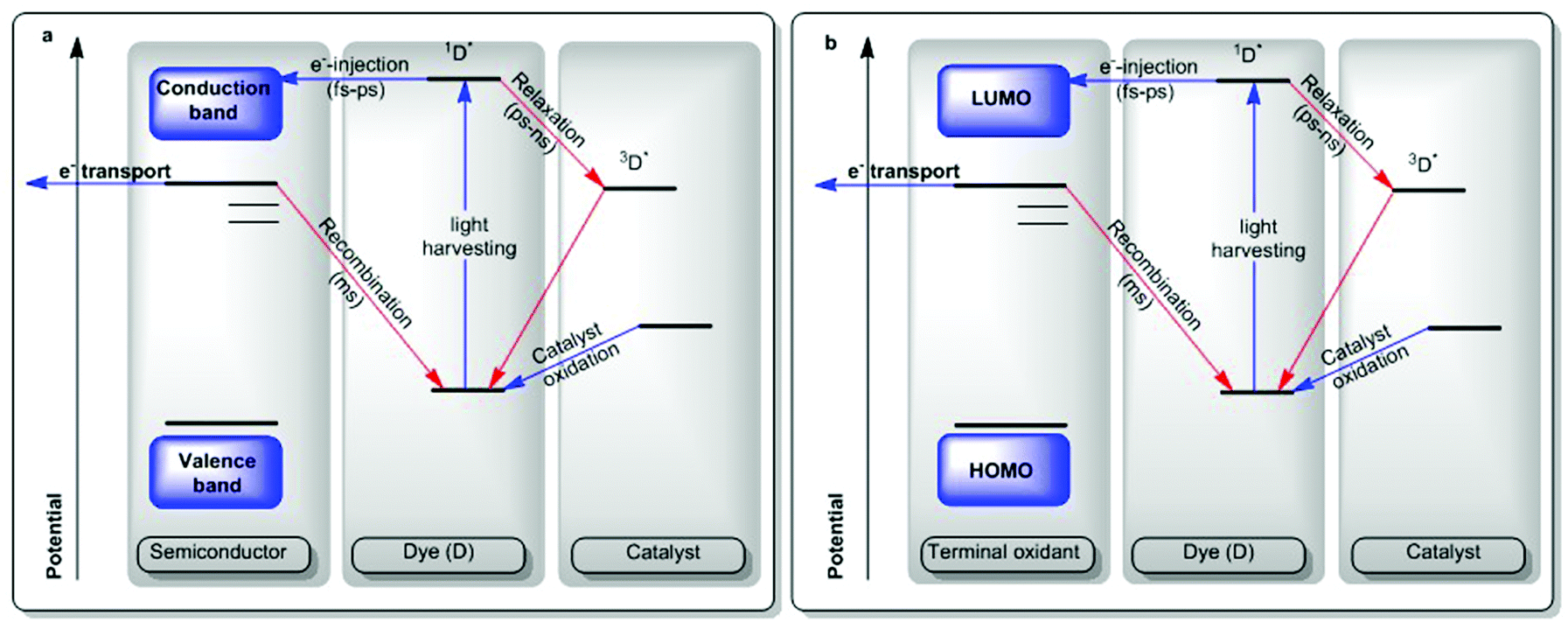 | ||
| Scheme 3 Light-driven catalysis: physical processes involved in generation of an oxidized catalyst. Catalysis by holes as shown.13 | ||
In molecular parlance, given the case of a homogeneous molecular photochemical transformation, a similar set of components come into play, but instead of a conduction band of a semiconductor, a molecular terminal oxidant of appropriate potential may be utilized in an almost identical scheme (Scheme 3b).
Although largely applied in the solar fuels field, photon-induced electron-transfer (PET) has remained a long-standing contributor to the field of organic chemistry.39,40 The design principles used in the development of photosensitizers for solar-fuel applications may be applied to drive transformations involving the generation and subsequent reactivity of organic radicals (Scheme 4).13
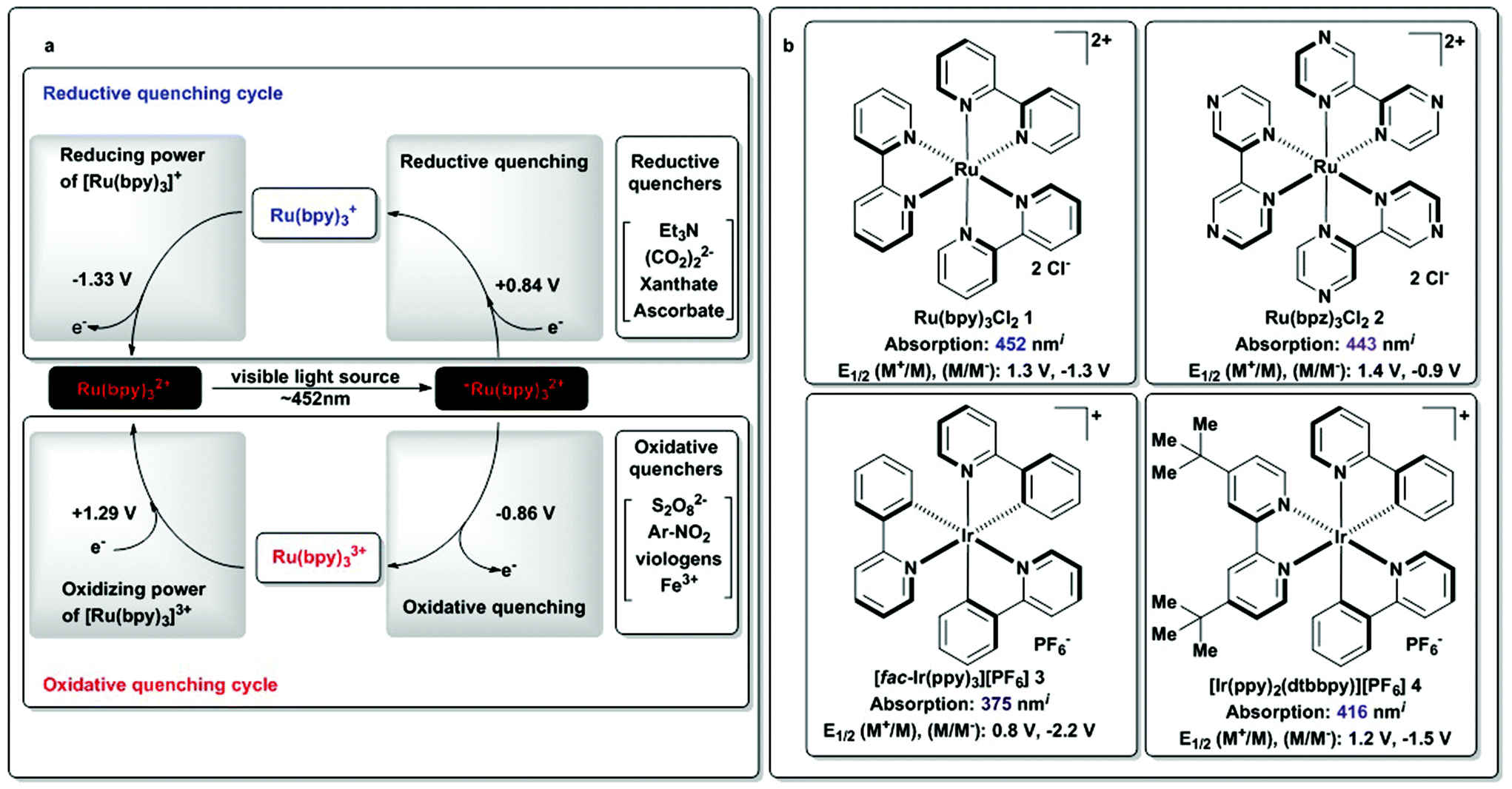 | ||
| Scheme 4 (a) Photoredox cycle for [Ru(bpy)3]2+ (1) and (b) alternate photosensitizers. iDetermined in MeCN; redox potentials in V vs. SCE. SCE: saturated calomel electrode.13 | ||
Scheme 4 illustrates both reductive and oxidative quenching cycles involving [Ru(bpy)3]2+ as photosensitizer, with potentials vs. SCE of each of the species involved.41–43 Upon light excitation of [Ru(bpy)3]2+, the excited state can act as either a reductant or oxidant, depending on the nature of the quencher available at the time of the reaction. [Ru(bpy)3]+ has been shown to be a −1.33 V (vs. SCE) reducing agent, while [Ru(bpy)3]3+ is a 1.29 V (vs. SCE) oxidant. For convenience and ease of comparison, we provide some examples of such species in Scheme 4a.
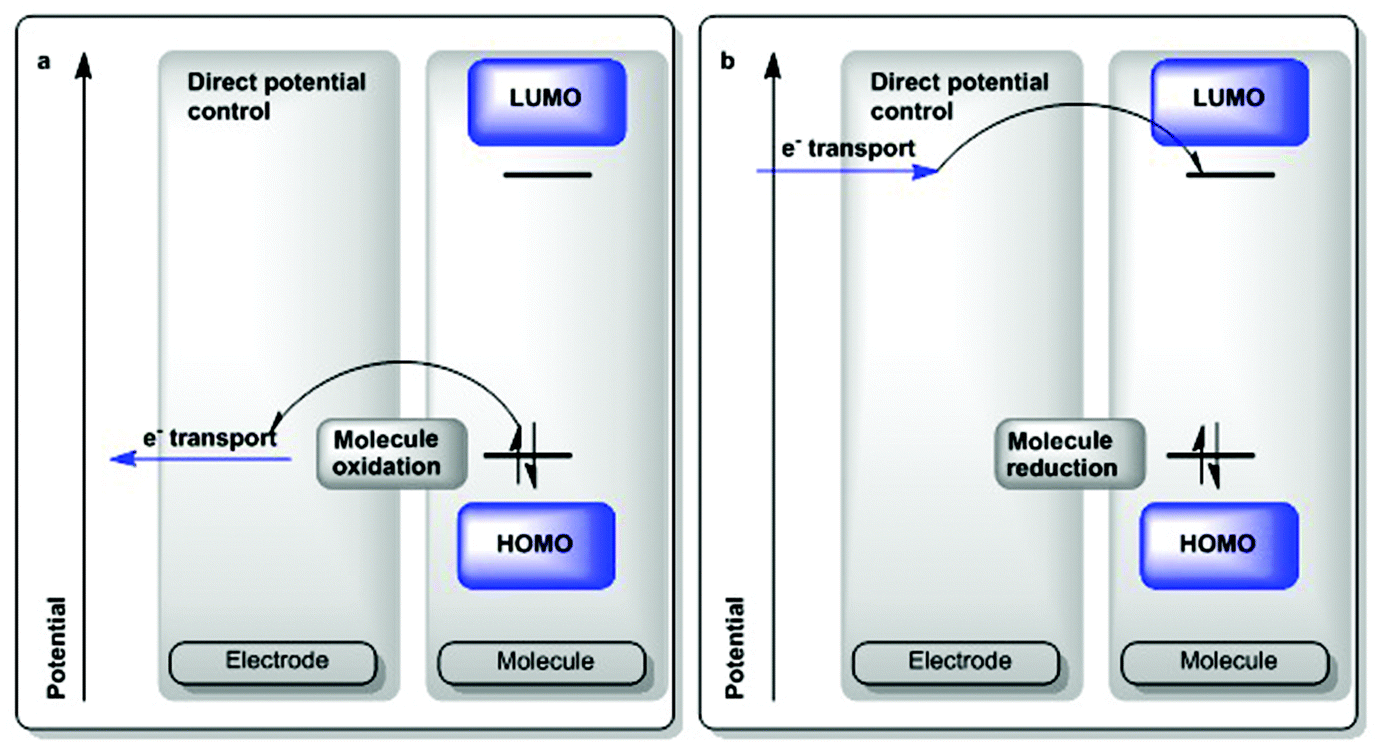 | ||
| Scheme 5 Electrochemical oxidation and reduction.45 | ||
To return to the analogy between redox scales and pKa scales, pKa scales are also highly solvent dependent and are used as a general reference to assess the relative ability of a certain molecule to donate or accept a proton. To give a concrete example of this phenomenon, there is a distinct difference between pKa values measured in water versus dimethyl sulfoxide (DMSO) of several orders of magnitude. Bordwell notes that proton availability in organic solvents is orders of magnitude lower than in aqueous conditions.44 For organic transformations under non-aqueous conditions, DMSO pKa values are usually considered as a general guide in the choice of a suitable reagent, even when the organic reaction occurs in a different solvent and the pKa values are likely different.
Molecules with stable, accessible electronic states typically show well-behaved, reversible, 1e− redox responses by cyclic voltammetry.45,46 Ferrocene is such a molecule and given its stability and general inertness, it is regarded as an appropriate internal standard and reference couple.47 The locus of such redox behavior in certain coordination and organometallic species remains the topic of discussion and intensive studies, as organic radical fragments on metals have been shown to act as electron or hole reservoirs and may impart important catalytic properties due to the attainment of unexpected electronic states.48 Several formal redox potentials measured in ionic liquid media have also been reported.49
2.2. Reactivity of radical ions
One of the purposes of this review is to highlight formal potential scales as analogues to pKa scales and as such, we limit the discussion to potential ranges and means of generation for the initial step. However, Curran notes an interesting point:5 free protons or free bases are usually not explicitly written out in acid or base catalysis aside from the initial step. They are usually shown as first interacting with a substrate and then carried forward in subsequent steps as part of a different chemical entity. Similarly, catalysis by electrons and holes normally involves bound electrons or holes, often generated and transported by some chemical means. The mechanism of transport is therefore an intriguing aspect to consider, as radical lifetimes are highly medium- and substrate-dependent. Interestingly, organic fragments have now been shown to be able to store and accumulate electrons or holes in order to allow access to multi-electron processes that were previously thought to be inaccessible.48 Given this description, the field of catalysis by redox-active ligands becomes a subset of catalysis by electrons and holes.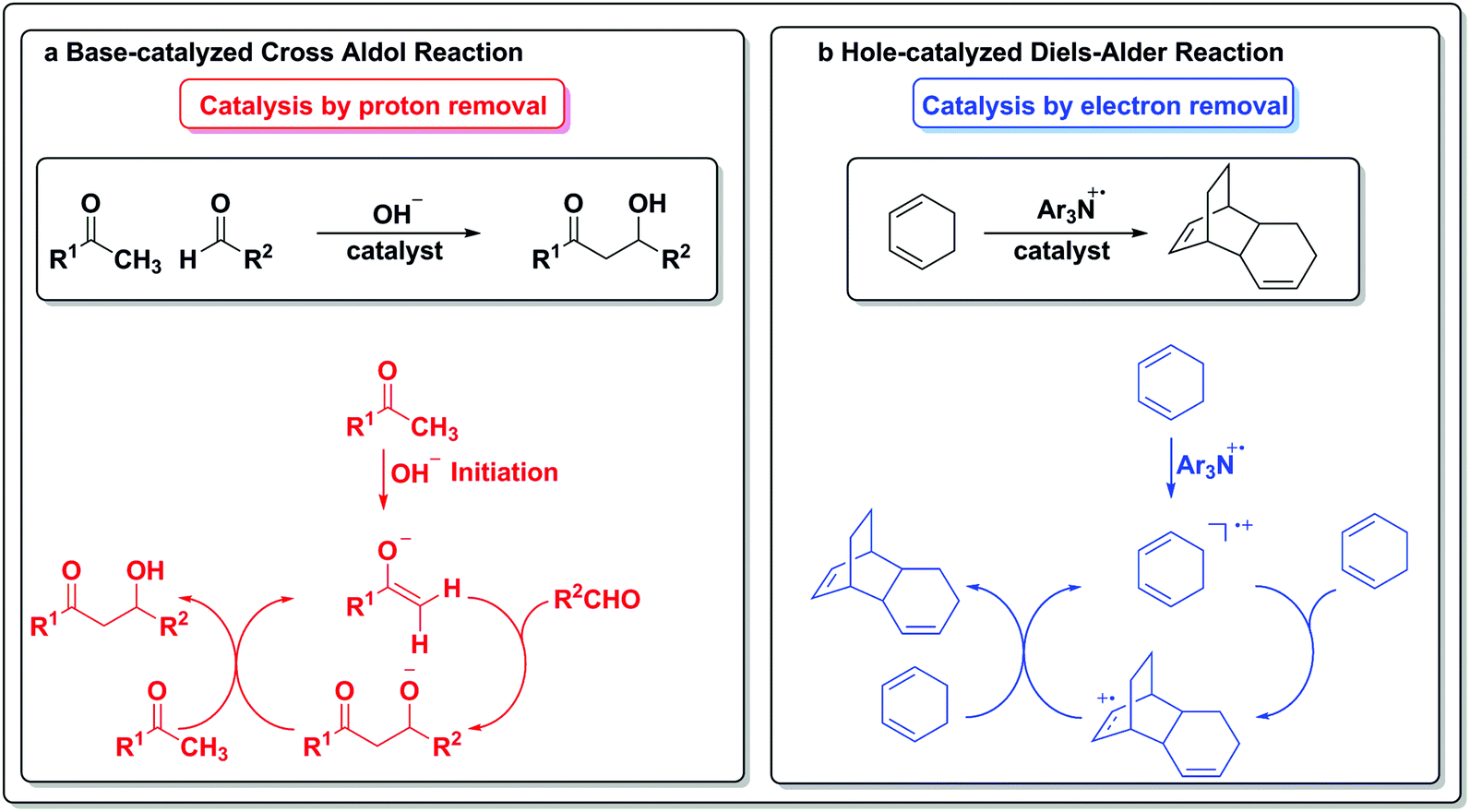 | ||
| Scheme 6 Catalysis by removal of an elementary unit.5 | ||
The electron in the redox scale paradigm is an elementary particle that can be added or removed from a particular substrate and also act as a catalyst for a transformation (Scheme 6b). In the case of the hole-catalyzed Diels–Alder reaction, a triarylamine radical cation can act as an electron scavenger and generate an activated radical cation diene, which can then react stepwise with another equivalent of itself to form an activated adduct that can propagate the hole to another equivalent of substrate and close the cycle.
 | ||
| Scheme 7 Catalysis by addition of an elementary unit.5 | ||
3. Parallels between photoredox catalysis and electrochemistry
3.1. Photoredox catalysis – synthesis of organic molecules using sunlight53
Photoredox catalysis has captured the imagination of the synthetic chemistry community over the last decade, culminating in many novel radical-based transformations that were previously intractable or inherently unselective using more traditional chemistry.54–57 The breadth of transformations achieved thus far via photoredox catalysis is indicative of the potential of synthetic electrochemistry. Thus, the discussion of key examples herein is intended to suggest opportunities for the field of synthetic electrochemistry. The examples of photoredox catalysis in this review are by no means comprehensive and we refer the reader to several excellent recent reviews over the last few years.18,56–59 We introduce potentials in the context of modes of reactivity, as they can also be achieved electrochemically and highlight possible advantages and drawbacks of an electrochemical approach.Applications of photoactive catalysts such as [Ru(bpy)3]Cl2 (1) that are capable of converting light to electrochemical potential, have been studied for decades.60–62 These catalysts absorb a photon of UV or visible light, depending on the metal and the electronic properties of the ligands (see Scheme 4 for illustrative examples) to give several high-energy excited states. These states often quickly relax to the lowest-energy singlet excited state, which can then either fluoresce to return to the ground state or can undergo intersystem crossing to give a triplet excited state. The triplet state has a relatively long lifetime (on the order of 1100 ns for 1), as its relaxation to the ground state is forbidden. This long lifetime provides sufficient time for other relaxation pathways, including electron transfer with an organic substrate (Schemes 3 and 4) to occur. One of the key aspects of the triplet excited state is that it is both a stronger oxidant and reductant than the ground state, as its highest-energy electron is in an exited energy orbital, allowing it to be more easily donated (reduction) while a low-energy orbital (the HOMO in the ground state) is only singly occupied, rendering it available for an external electron. Because of the possibility of competing reactivities, “sacrificial” reductants (e.g., tertiary amines) or “sacrificial” oxidants (e.g., dioxygen) are often added to influence the reaction pathway. Finally, as photoredox catalysis has gained popularity there has been extensive catalyst tuning to optimize the reductive and oxidative properties towards a particular set of reactions. A depiction of this analysis is available in Schemes 3 and 4.
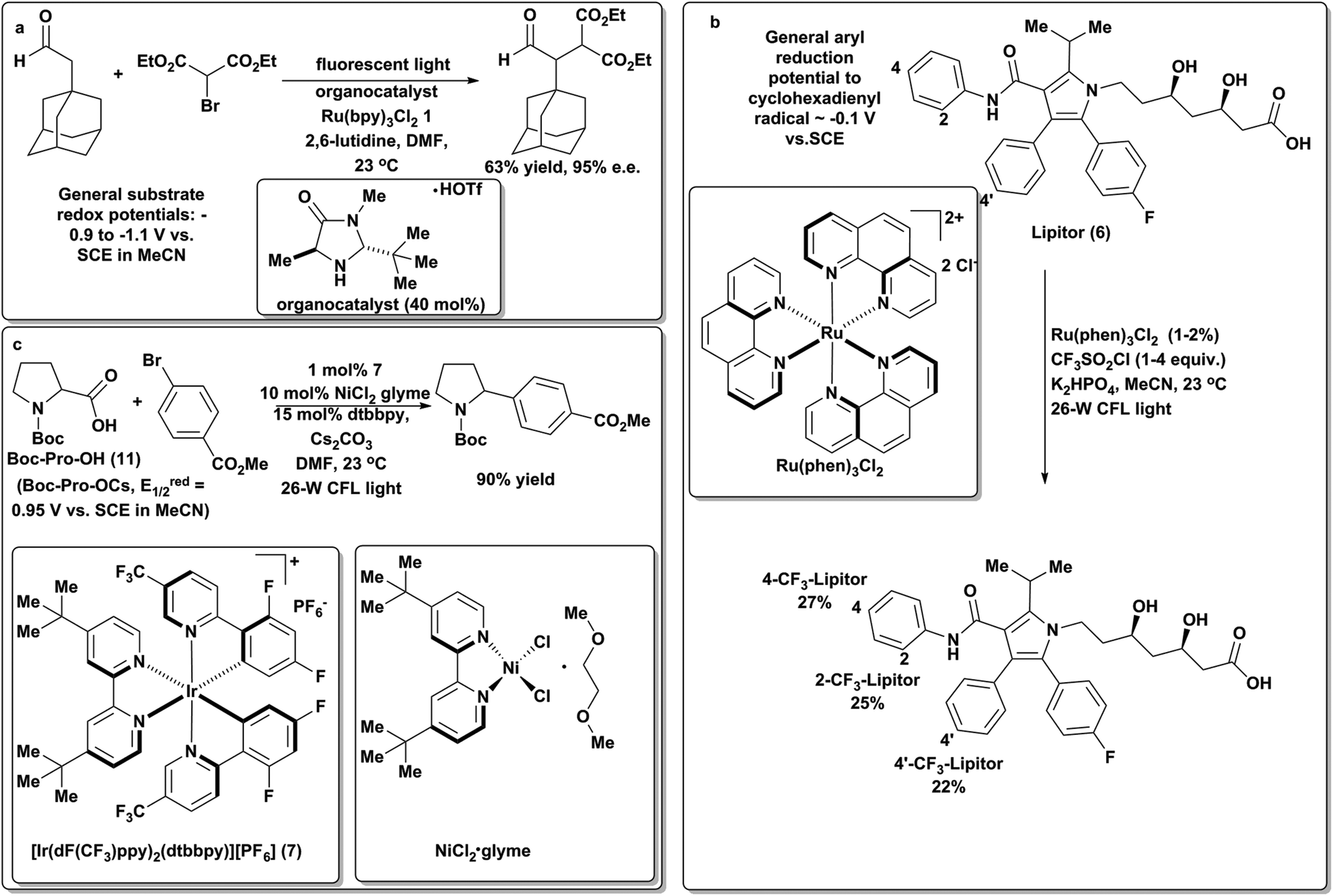
While the use of photocatalysis to generate organic radicals had been known for some time, it gained attention in synthetic chemistry in 2008 when MacMillan utilized 1 and visible light to generate electrophilic α-ketoradicals which could react with a chiral “SOMOphilic” enamine to forge the key C–C bond in the resulting radical intermediate. This intermediate is then oxidized by a ruthenium(II)* species to give an iminium ion. Following hydrolysis, enantioenriched α-alkylated aldehydes are generated62 with little sensitivity to steric properties of the coupling partners (Scheme 8a). Overall, the successful realization of this sequence represents a remarkable advance in the field. One of the important aspects of this work was the demonstration that photoredox catalysis allowed for facile and mild access to organic radicals that could be leveraged in diverse chemical transformations. Moreover, this approach obviated the need for stoichiometric, toxic reagents (e.g., organotin compounds) that are a hallmark of traditional radical-based transformations. Since these seminal results, MacMillan and coworkers have applied photoredox catalysis to effect myriad diverse transformations including arene and heterocycle trifluoromethylation63via the generation of highly electrophilic trifluoromethyl radicals. The potential utility of this chemistry was highlighted by the mild trifluoromethylation of several complex pharmaceuticals, including Lipitor (6, Scheme 8b). More recently, they have leveraged iridium-based photoredox catalysts (e.g., 7) to generate radicals α to amines via single-electron oxidation of the corresponding carboxylate, followed by a rapid decarboxylation (a common reaction in electrochemistry). These intermediates have been combined with Ni(II) catalysts to effect arylation64 and vinylation, representing a novel and operationally simple approach to C(sp3)–C(sp2) cross-coupling (Scheme 8c).
 | ||
| Scheme 8 Representative transformations using photoredox catalysis.62–64 | ||
3.2. Reductive dehalogenations and related reactions
As protagonists of traditional radical-induced transformations, organic halides are ideal candidates for electrochemical transformations due to their propensity for forming radicals. In particular, radical dehalogenation reactions are commonly performed by reactions with tributyltin hydride.65 These species are highly toxic; thus, methods to perform these transformations with less toxic materials have long been sought. Moreover, electrochemically-induced reductive cyclizations of organic halides represent powerful synthetic strategies in complex molecule synthesis and have recently been the subject of a comprehensive review.66 In these chemistries a tertiary amine was used as a sacrificial one-electron reductant which led to iminium byproducts upon a second one-electron oxidation. Using this as inspiration Stephenson and coworkers then utilized the iridium based photoredox catalyst Ir(ppy)2(dtbbpy)PF6 as a means to generate iminiums directly in situ from the corresponding unactivated amine to act as electrophiles in an aza-Henry (Scheme 9c) reaction with nitromethane.67 Conversely nucleophilic α-amino radicals can be generated via deprotonation of the initial (one-electron) oxidized amine. Stephenson has recently leveraged this reactivity to add N-methyl morpholine to complex unfunctionalized pyradazines in the context of the direct synthesis of JAK2 inhibitor LY2784544 and related analogues (Scheme 9d).68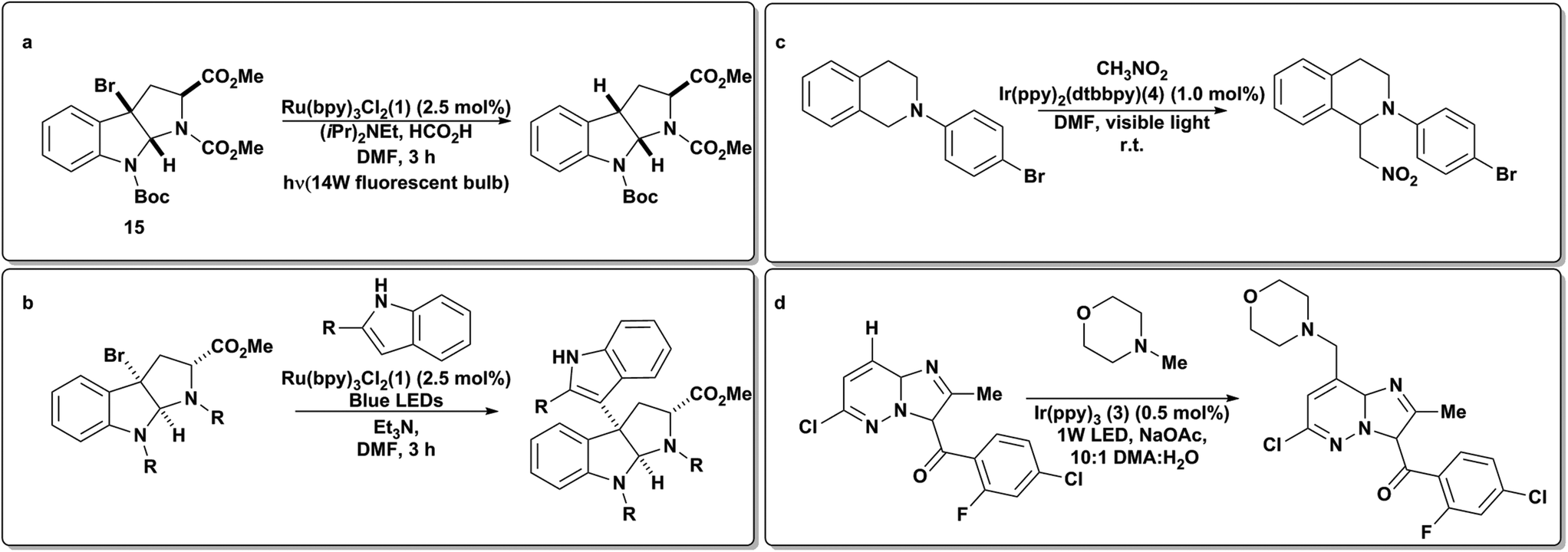 | ||
| Scheme 9 Reductive dehalogenation by photoredox catalysis.67 | ||
Stephenson and coworkers have studied applications of photoredox catalysis as a green and chemoselective entry into organic radical chemistry. Early on, the application of visible light to 1 was exploited to effect a tin-free reductive dehalogenation of alkyl bromides with exceptional chemoselectivity and functional group tolerance (Scheme 9a).69 Since then, this strategy has been applied to generate electrophilic radicals for intra-70 and intermolecular addition to indoles, including in the synthesis of Glicladin C (Scheme 9b).71 More recently, they have applied fac-Ir(ppy)3 (Scheme 4, 3) to generate radicals from unactivated aryl, alkyl and alkenyl iodides.72
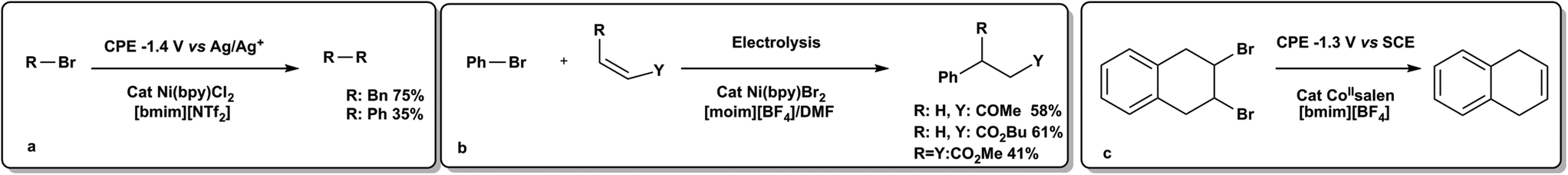
Alternatively, organic halides can be irreversibly reduced directly by the electrode surface, by either one or two electrons, or indirectly, through reaction with the anion radical of a mediator or catalyst. Pd(II), Ni(II) or Co(II) coordination complexes have been reported as mediators of this process (Scheme 10).66,73 The reduced form of these complexes can be produced as the first step in the reaction. These species were shown to react further with organic halides to form reactive organometallic species that lead to productive chemistry. A review of reactivity of electro-generated reduced species of Ni and Pd is available.73 The potential at which the mediator is reduced is generally more positive than the potential necessary to directly reduce the organic halide. The reduced mediator species is then able to transfer reducing equivalents to the organic fragment to afford a radical species, R˙, which can, in turn, become further reduced to an anion or react as a radical. This is similar to the transformations developed by Stephenson in Scheme 9a.69
 | ||
| Scheme 10 (a) Reductive dehalogenation of aryl and benzyl bromides.66 (b) Reductive coupling of activated olefins with aryl bromides.74 (c) Electroreductive dehalogenation of vicinal dihalides.75 | ||
Since ionic liquids have been shown to provide a promising medium for electrochemical transformations, several reductive dehalogenations examples have been reported in such media. As discussed in section 4, the ease of product separation (due to special miscibility properties), the absence of supporting electrolytes (due to suitable conductivity properties), the large electrochemical windows and their physical properties (recyclability, low flammability and low vapor pressure), render them seemingly ideal candidates as solvents for electrochemical transformations. However, the water content of these materials can be highly variable and the electrochemical window of these solvent media may be altered by adventitious water (solvent windows are depicted in Fig. 4). Adventitious water and traces of dissolved oxygen may also interfere with observed chemical yields. Given this issue and the relatively negative potentials, drying precautions and degassing are commonly necessary in the execution of such an electrochemical experiment.
3.3 Photocatalysis for cycloadditions
Yoon and coworkers have applied visible light photoredox catalysis to effect cycloaddition chemistry. In 2008, they were one of the early groups to apply 1 in the context of synthetic organic chemistry. They reported an intramolecular [2 + 2] enone cyclization (Scheme 11a),76 where a photo-generated catalytic intermediate promotes a one-electron reduction of an aryl enone that can then undergo an intramolecular radical cycloaddition with any suitable Michael acceptor to give bicyclic cyclobutanes in excellent yield. Notably this chemistry proceeded with ambient sunlight through a window as the sole light source.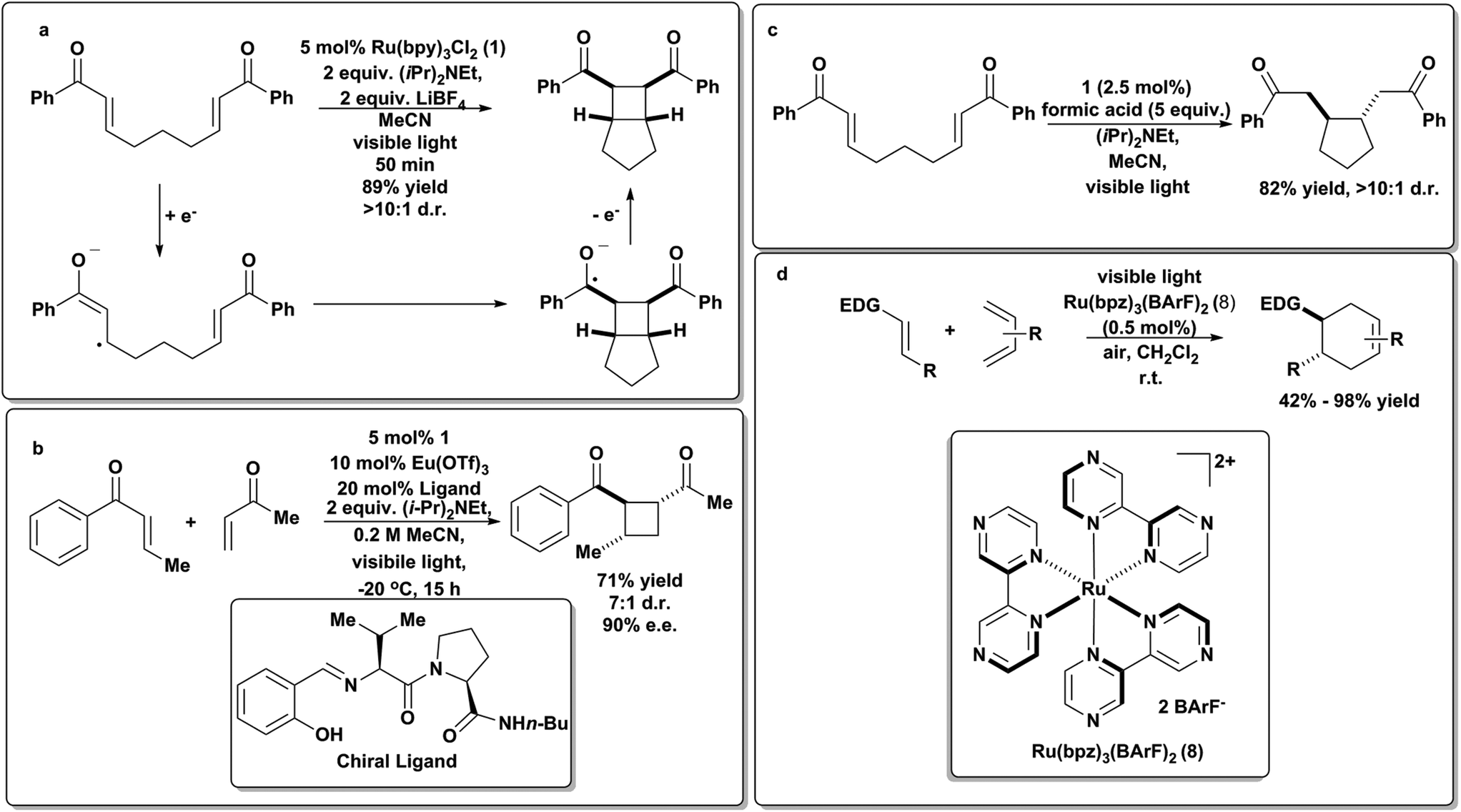 | ||
| Scheme 11 Photoredox cycloadditions.78,79 | ||
This group then found that they could leverage the lower reduction potentials of aryl enones to effect efficient cross intermolecular [2 + 2] cycloadditions under similar conditions.77,78 They later found that the presence of chiral Lewis acids resulted in enantioselective cross [2 + 2] cycloadditions (Scheme 11b), representing a breakthrough in the synthesis of enantioenriched, chiral cyclobutanes. These reactions required the presence of Lewis acidic Li+, presumably to stabilize the radical anion formed from the enone upon one-electron reduction. They also found that neutral radicals could be formed by protonation of the initially formed radical anion in the presence of a Brønsted acid (formic acid) and that this neutral radical possessed markedly different reactivity, proceeding via a 5-exo-trig cyclization, rather than a [2 + 2] enone cyclization (Scheme 11c).79 The Yoon group has also studied other cycloadditions, most notably the radical cation Diels–Alder reaction in which the photoredox catalyst [Ru(bpz)3](BArF)2 (8) was used to generate radical cations from electron-rich olefins that then underwent facile [4 + 2] cycloadditions with electron-rich dienes, to give “electronically mismatched” Diels–Alder adducts (Scheme 11d) that were intractable using traditional thermal conditions.80 Key to this work was the availability of bipyridine ligand analogs that allowed for systematic tuning of the ruthenium photocatalyst to possess a large enough excited state oxidation potential to directly oxidize olefins.
3.4. Hole-catalyzed Diels–Alder cycloadditions: a case for equivalent chemical, electrochemical and photoredox manifolds
In addition to proceeding under photoredox catalysis (Scheme 11d), the hole-catalyzed Diels–Alder process can also be effected via chemical or electrochemical means. A stable cation radical Ar3N+·SbCl6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 81 can be used to initiate and catalyze the transformation (Scheme 12). As discussed in the previous section, 8 (Schemes 4 and 11) was also used to generate reducing equivalents in solution to drive the same type of reaction.18,82 Surprisingly, the reaction occurred in a halogenated solvent and under aerobic conditions, where reductions are likely to be hindered by side-reactivity and possibly excited-state quenching due to oxygen reduction pathways. Most excitingly, photoredox chemistry in flow systems have now been reported.58
81 can be used to initiate and catalyze the transformation (Scheme 12). As discussed in the previous section, 8 (Schemes 4 and 11) was also used to generate reducing equivalents in solution to drive the same type of reaction.18,82 Surprisingly, the reaction occurred in a halogenated solvent and under aerobic conditions, where reductions are likely to be hindered by side-reactivity and possibly excited-state quenching due to oxygen reduction pathways. Most excitingly, photoredox chemistry in flow systems have now been reported.58
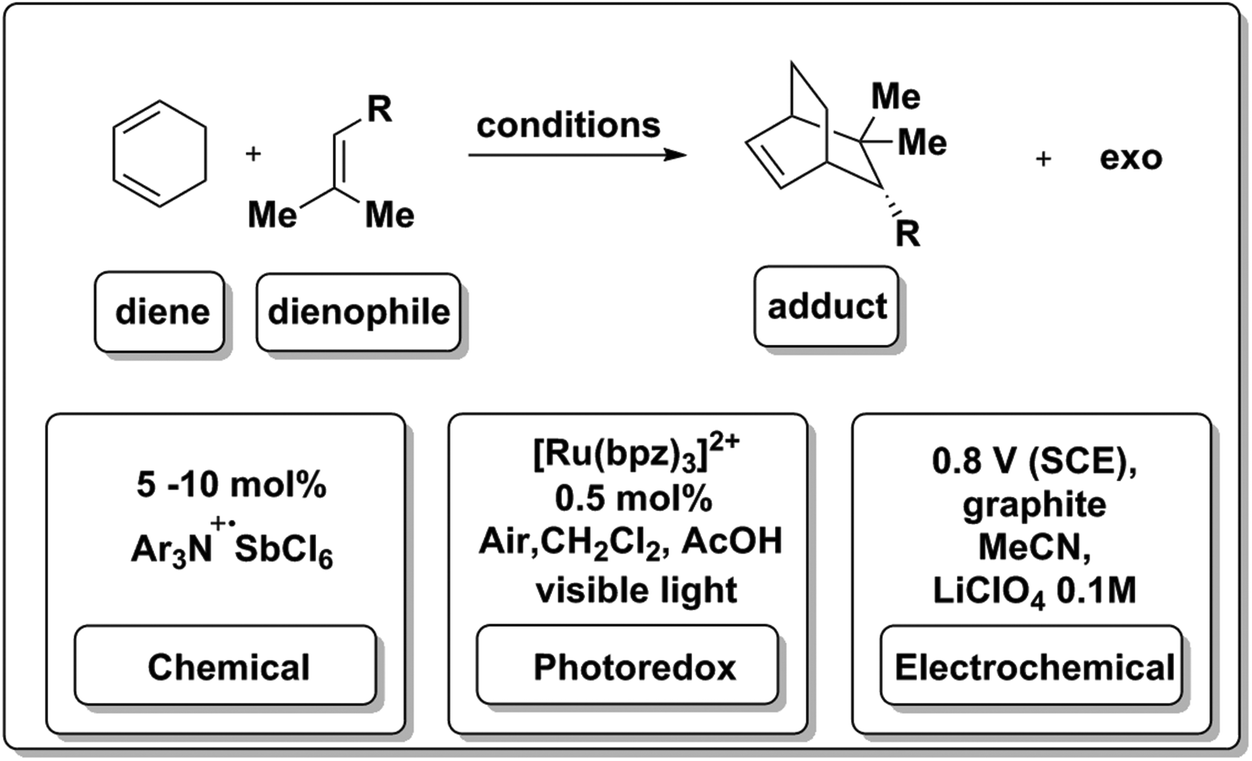
The electrochemical Diels–Alder reaction83 was also achieved at a graphite electrolyte at modest potentials (0.8 V vs. SCE) in acetonitrile. A hetero-Diels–Alder variant of this transformation has also been reported.84 This type of Diels–Alder reaction is a good example of an electron catalyzing the a reaction, as it is generated by three independent means at the appropriate potential (Scheme 12).
3.5. Organic molecules as light-absorbers and redox catalysts
In addition to metal-based photoredox catalysts there has been an emerging number of examples employing organic dyes such as methylene blue (9), Eosin Y (10) and acridinium salts (Scheme 13).59 In 2011, Zeitler and coworkers disclosed metal-free reductive dehalogenations and enamine alkylations utilizing Eosin Y and 530 nm light. Notably, only slightly attenuated yields were obtained using a 23 W fluorescent bulb or ambient sunlight.85 König then used similar conditions to generate aryl radicals from diazonium salts to effect the direct arylation of heteroarenes.86 Shortly thereafter, Xiao and coworkers87 utilized 1 to effect the oxidative hydroxylation of aryl boronic acids (Scheme 13a), noting that Eosin Y could also perform this reaction. This was followed by an elegant mechanistic study by Scaiano88 which shed light on the excited state of this process, leading to the realization that Methylene Blue, a dye known for its ability to generate singlet oxygen, was an optimal photocatalyst for this transformation. Fukuzumi89 has rationally designed an acridinium-based organic salt (11) that functions in the visible region of light (λ = 450 nm) with intriguing excited-state properties, including an extended excited-state lifetime and excellent oxidizing capabilities. Nicewicz has applied this catalyst to the anti-Markovnikov functionalization of olefins (Scheme 13c) and has made progress towards a long-standing goal of the synthetic community.59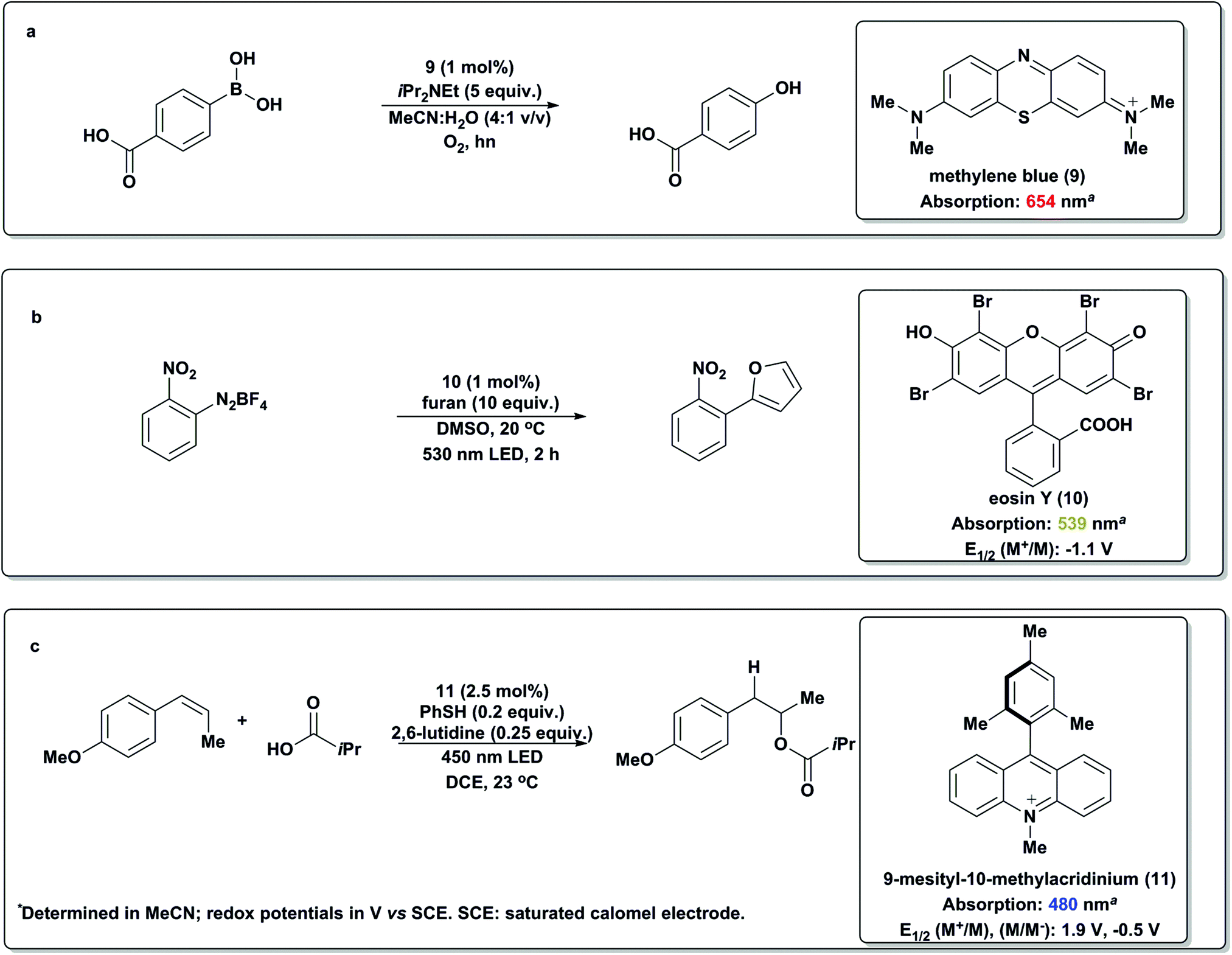 | ||
| Scheme 13 Organic dyes as photoredox mediators.60,84–86 | ||
Organic electrochemical redox mediators are chemical compounds that catalyze transformations by means of an electrochemically generated anion or cation radical storing electrons or holes within the bonding framework of an organic molecule.90,91 Several inorganic mediators are known.92 An example of an electrochemical reaction involving an organic mediator is the oxidation of primary benzylic amines to the corresponding imines by using catalytic DDQ, a known oxidant for dehydrogenation and regenerating it at an inert electrode surface.93
4. Analytical and preparative aspects of the electrochemical experiment
4.1. Experimental decision tree
The advantage of electrochemical methods is that they also offer a suite of diagnostic measurements that can be used to identify suitable conditions for the transformation. Generally, the experiment setup requires three actors: the working electrode, the counter electrode and a reference. All of the data observed in an electrochemical experiment convey only what happens at the working electrode surface, while the reaction at the counter electrode does not get recorded. Cyclic Voltammetry (CV)46 is a diagnostic technique for a type of electrolysis at a fixed potential, also known as controlled potential electrolysis (CPE) which is one of the two types of bulk electrolysis. Currents observed during a bulk electrolysis are in the mA range for 10–100 mM concentrations of analytes. Both techniques require three electrodes. In a CV experiment, a potential range is scanned while current is recorded, as the solution moves between the two potential points in a cyclic fashion. Reversible and irreversible redox events have very distinct electrochemical responses, but detailed analyses of those measurements lie outside of the scope of this survey discussion.45,46,95,96 Once a potential of suitable current density is identified, a bulk electrolysis at controlled potential can be set up.
A less utilized technique is the reverse of cyclic voltammetry, cyclic chronopotentiometry (CC).97,98 In this procedure, a certain current (current density if reported against electrode surface area) range is scanned, while measuring the voltage response. This technique is a direct diagnostic tool for electrolysis performed under controlled current conditions: given passage of a certain number of electron equivalents per unit time, Faraday's constant allows for calculation of the amount of time electrolysis may take. More commonly, CV may be used as a diagnostic in this instance, although the measurements are not directly analogous. CC identifies what kind of voltage response to expect at a chosen current density. Generally, a controlled current electrolysis (CCE) can be run at current densities as low as 1 mA cm−2, especially in the optimization stages, but is usually run between 50–100 mA cm−2 at concentrations up to 0.5 M of analyte. 50–100 mM analyte concentrations are generally sufficient.94 At such high concentrations of a likely organic substrate, however, measurements and possible abatement strategies of resistance may be necessary (e.g., increasing electrolyte concentration or switching to a more intrinsically conductive solvent). At the end of a CPE or CCE experiment, the initial diagnostic may be used again to assess the magnitude of conversion. CC may be performed using only a working and a counter electrode, but the use of a reference is helpful in recording a voltage response against a stable voltage point set by the reference throughout the experiment.
Both CV and CC techniques are conducted under diffusion-limited conditions (without stirring) and they do not commonly produce macroscopic quantities of product. Their interpretation does not provide any information regarding the explicit chemical nature of the observed responses, although inferences can be made. In a sense, the level of information obtained from a CV is analogous to the level of information obtained from Thin Layer Chromatography (TLC).39 Monitoring of reaction progress during a preparative-scale bulk electrolysis may provide insight into the mechanism of the overall reaction.99
22,105 to solar fuels and oxidative dehydrogenation of heterocycles,93,106,107 transformations relevant to hybrid-fuel cell flow batteries. When deciding whether to consider electrochemistry for organic synthesis, the existence of a radical species in a known and related mechanistic pathway is usually a favorable indication. When a radical mechanism for a related mechanism has not been previously proposed, it does not necessarily mean that a radical mechanism is impossible, but one must proceed with cautious optimism. An interesting case of unexpected radical chemistry in the recent literature was reported by Herzon108et al., in their work on Mn-mediated oxidative dimerization a precursor to (−)-Lomaiviticin aglycon.
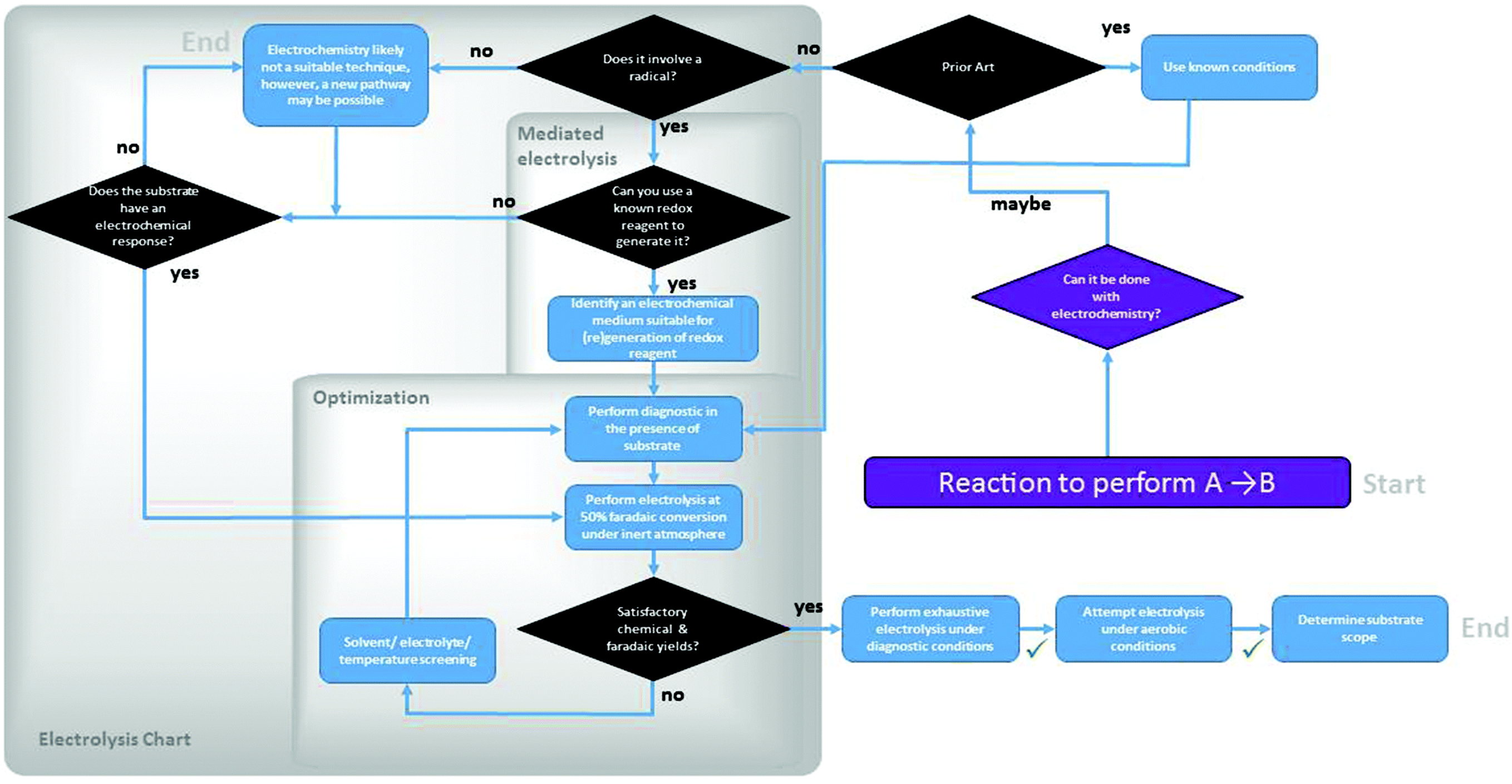 | ||
| Fig. 3 Experimental decision tree in the optimization of an organic transformation using electrochemical methods. | ||
A Faradaic yield is one of the metrics of assessment of an electrolysis experiment. From the amount of charge passed through the solution, a theoretical amount of product to be expected can be determined using Faraday's laws (96487 C mol−1 e− passed). A quantitative chemical analysis then provides a measure of experimental chemical yield. The ratio of the two yields provides the Faradaic yield. Once the optimal conditions are identified in the sequence, testing under aerobic conditions may reveal that the process is tolerant of air.
These general guidelines and the accompanying flowchart are meant to aid a synthetic electrochemical optimization processes. Assessment and amendment on a case-by-case basis is of course at the researcher's discretion.
4.2. Solvent windows in protic and aprotic media94,109
When the chemical transformation requires a proton source, the electrochemical solvent window is a relevant consideration. The solvent window is the separation between the accessible cathodic and anodic potentials of a solvent/electrolyte system. Any productive electrochemical transformation will need to occur at potentials located between the solvent/electrolyte limits. The conductivity of an organic solvent is generally poor and addition of salt is necessary to allow current to flow through the solution. Although several references covering the topic are now available, for convenience, we provide a series of electrochemical solvent/supporting electrolyte accessible solvent windows in Fig. 4.94,109 The anodically-limiting step is generally the oxidation of the anionic fragment of the supporting electrolyte. For example, I− anions are easily oxidized and their salts have rather small potential windows on the oxidation side of the electrochemical spectrum. A recent study has now shown direct participation of I− oxidation in productive aziridination chemistry,101 and we would like to highlight the cathodic I−/I3− redox couple used in solar cell technology as an example of I− participation in reductive chemistry.92 Conversely, cathodically-limiting reactions usually result from the reduction of the cationic species of the electrolyte. Tetrabutylammonium salts, for example, have been shown to be more reductively stable than their lower alkane congeners and as such, they are often preferred.110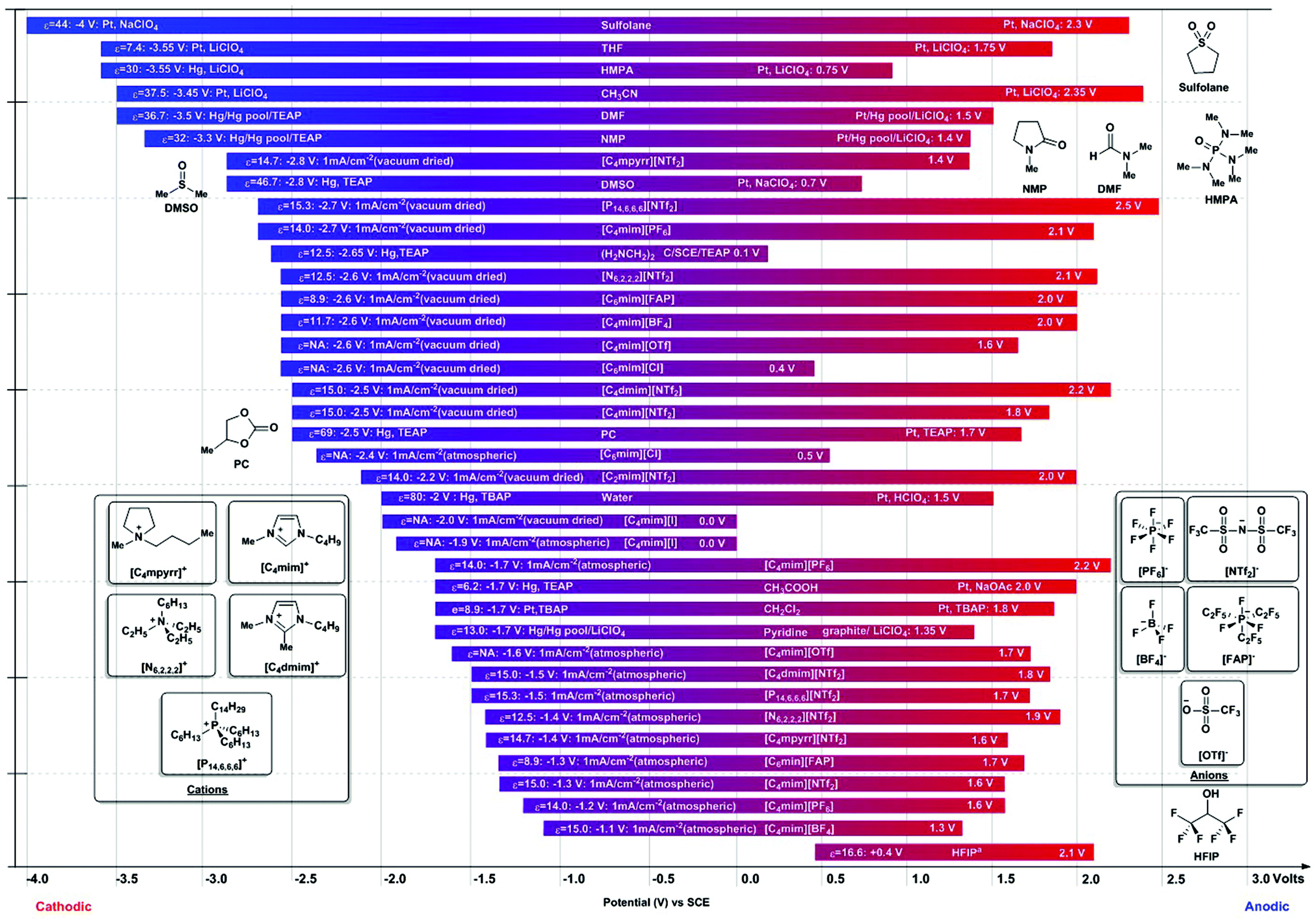 | ||
| Fig. 4 Potential solvent windows.94,109 a HPF: ε = 16.6: Anodic limit: +0.4 V: Pt, TBAPF6: Cathodic Limit: Pt, TBAPF6: 2.1 V Sulfolane: tetramethylene sulfone, THF: tetrahydrofuran, HMPA: hexamethylphosphotriamide, CH3CN: acetonitrile, DMF: N,N-dimethylformamide, NMP: N-methylpyrrolidone, [C4mpyrr][NTf2]: N-butyl-N-methyl-pyrrolidinium bis(trifluoromethylsulfonyl)imide, DMSO: dimethyl sulfoxide, [P14,6,6,6][NTf2]: tris(P-hexyl)tetradecylphosphonium trifluorotris(pentafluoroethyl)phosphate, [C4mim][PF6]: 1-butyl-3-methylimidazolium hexafluorophosphate, [N6,2,2,2][NTf2]: N-hexyltriethylammonium bis(trifluoromethylsulfonyl)imide, [C6mim][FAP]: 1-hexyl-3-methylimidazolium tris(perfluoroethyl)trifluorophosphate, [C4mim][BF4]: 1-butyl-3-methylimidazolium tetrafluoroborate, [C4mim][OTf]: 1-butyl-3-methylimidazolium trifluoromethylsulfonate, [C6mim][Cl]: 1-hexyl-3-methylimidazolium chloride, [C4dmim][NTf2]: 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide, [C4mim][NTf2]: 1-butyl-3-methylimidazolium bis(trifluoromethylsolfonyl)imide, PC: propylene carbonate, [C2mim][NTf2]: 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide, [C4mim][I]: 1-hexyl-3-methylimidazolium iodide, CH3COOH: acetic acid, CH2Cl2: dichloromethane, HFIP: hexafluoro-2-propanol. | ||
Some other issues come into play in the choice of an appropriate medium for an electrochemical reaction. The lifetime of hydrocarbon radicals may be compromised by protic media. Anion radicals are easily protonated in such media (ethanol, isopropanol and methanol). Protic solvents may also act as nucleophilic scavengers for cation radicals. In aprotic media, the presence of adventitious protic impurities may compromise the observed chemical process as well as limit accessible solvent windows.94 Acetonitrile is often used due to its relative inertness to anion and cation radicals, but this solvent may require additional drying. Propionitrile, butyronitrile and mixtures thereof are applicable solvents for the generation of both anion and cation radicals, whereas dimethylformamide (DMF), also a common electrochemical solvent, is known to react with cation radicals but is generally appropriate for anion radicals. Methylene chloride is used for the generation of cation radicals, however, the solutions have high resistance, which limits the applications of such media. High salt concentrations (>0.2 M)93 may be used to minimize such problems.
Ionic liquids have emerged as an important class of solvents, as their intrinsic conductivity allows them to operate as electrochemical solvents without additional supporting electrolyte;111 many have wide electrochemical windows.112,113 Their low vapor pressure and unique miscibility profiles renders them useful in separation processes and they can be easily reused.114 Adventitious water present in the ionic liquid medium has been shown to narrow the accessible electrochemical window. This effect is evidenced by the examples in Fig. 4, which illustrates the potential window difference between dried ionic liquids and those interrogated under atmospheric conditions. An extreme example of such behavior is [C4mim]BF4. Under dry conditions the ionic solvent has a window spanning from −2.6 V to 2 V, a total of 4.6 V, whereas under “wet” conditions in open atmosphere, that window becomes only 2.4 V wide (from −1.1 V and 1.3 V on the anodic side). Experimental aspects of electrolyses performed in ionic liquids have been recently reviewed.115 For convenience, several other solvent/electrolyte systems are shown in Fig. 4.
4.3. Electrodes
The choice of electrode materials is crucial in the design of an electrochemical experiment. The difference in kinetics of electron transfer at surface/liquid interfaces may require consideration in the development of a preparative application. Analytical techniques such as cyclic voltammetry are available to probe these differences. Monographs related to the theoretical basis of cyclic voltammetry and bulk electrolysis are available.95,96,116Not all surfaces are created equal. The observed electrochemical behavior of a certain analyte of interest occurs at the interface of the liquid medium and the surface of the working electrode. Given this fact, commercially available electrodes of a specific type of metal or carbonaceous materials are a convenient option for electrode choice in current state-of-the-art experiments. However, if the purpose of the electrochemical work is ultimately preparative-scale electrolysis, a large surface area electrode of the same material may be necessary. Such electrodes may be available from the same source. This allows for internally consistent voltage measurements.
For cyclic voltammetry, an electrode of well-defined, dimensionally stable surface area is necessary for a rigorous analytical measurement. Under certain conditions, given comparative diffusion coefficients and an internal standard, such electrodes allow for quantitation of the number of electrons associated with a CV wave of an analyte, as has been reported.103 For oxidations, possible working electrodes could be: graphite, glassy C and platinum in solvents like methanol, MeCN, CH2Cl2 and CF3COOH. Working electrodes for reductions could be: graphite, GC, mercury pool, graphite and Pt with anodes of possibly the same materials. Carbonaceous materials are highly versatile, as they can be handled rather easily and are generally cost-efficient compared to the noble metals. High-surface area carbon cloth anodes and cathodes have been reported in electrochemical devices.117,118 Another useful type of carbon material for both anodes and cathodes is a type of high-surface area glassy carbon called reticulated vitreous carbon.119,120
Under high-current conditions, SCE and Ag/AgCl electrodes do not remain stable under non-aqueous conditions in the long term.6Reference electrodes should be checked against a suitable redox couple. Even though a reference may not be necessary for a controlled current electrolysis, a reference electrode provides a potential anchoring point and allows for a measure of reproducibility between experiments. The ferrocene (Fc/Fc+) redox couple is a common reference point used in electrochemistry. It may, however, shift at different electrodes and under different electrolyte conditions.47 In order to determine whether the reference potential changed during a preparative electrolysis, an ex situ determination of a redox couple against the reference in use before and after the experiment is necessary. Ferrocene has also been measured in a series of ionic liquids123,124 as well as cobaltocene.124
Errors in reported potentials may be caused by re-referencing against already unreliable reference points. We recommend providing multiple reference scales if a conversion is necessary. The reference scale that was used in the direct measurement, in particular, is the most useful. Reporting potentials in non-aqueous electrochemistry is most suitable against ferrocene or even cobaltocene (for reductive chemistry). Computational studies of systematic uncertainties in referencing from an in silico standpoint have been made available.7
4.4. Temperature125,126
If a temperature other than ambient conditions is chosen, several considerations need to be taken into account. Systems with sluggish kinetics benefit from rate enhancement effects at elevated temperatures and current enhancements may allow for higher detection limits in electroanalytical experiments.127 High-temperature electrolysis methods have been reviewed.128,129 Conversely, low-temperature electrochemistry may allow for observation of electro-generated intermediates without complicating subsequent chemical reactions.130 For this purpose, butyronitrile 0.1 M NBu4PF6 has been shown to be an ideal solvent for electrochemical transformations as low as −130 °C. For experiments at −60 °C and above, a cell jacket with a circulating bath is an appropriate choice; however, a slush bath could also be a more convenient solution with enough equilibration time.The conductivity of the solvent medium is temperature-dependent. An extensive study of conductivity in aqueous media in the presence of varying amounts and types of solvent131 revealed that conductivity may decrease at higher temperature. Having a higher salt content may counteract this effect. Electrochemistry at high temperature has now been the topic of a review.129 A report on high-temperature electrolysis of water vapor as a means for H2 production has also been made available.127 These studies have shown that at higher temperatures, the extent of solubility of certain salts and analytes may change and thermal chemical reactions may take place.132,133
4.5. Cell design and pitfalls
An appropriate apparatus is generally required to perform preparative electrolyses. A series of general principles in cell design can help to identify suitable reaction vessels for different reactions. A minimal distance between electrodes needs to be maintained and the materials that are utilized for the electrodes, connectors and air tight seals need to be compatible with and stable to the starting materials and products. The cell should be amenable to temperature control to allow for optimization. Most importantly, uniform current, mass transport and heat distribution in the cell are necessary to ensure reproducibility of results.Materials that are used in electrochemical experiments will ideally operate over a wide temperature range, show good dimensional and chemical stability and be easy to fabricate and modify. Glass is the common material used for chemical transformations. It is normally etched by HF, particularly highly basic solutions and basic fused salts, but is generally impervious to most other reagents. Borosilicate glass is typically used in laboratory applications. Borosilicate glass frits are also available; often they are used as electrode separators. In these frits, porosity varies—the finer the frit, the higher the resistance introduced in the system. Fine frits are suitable for anode-electrode separators, while medium frits allow ion migration. Modular assemblies, such as joints, frits and stopcocks are available in different types of glass. Soda-Lime glass, for example, consists of 71–73% SiO2, 14–17% Na2O, 0–1.5% K2O, 5–6% CaO2, 3.5–4.5 MgO and up to 0.1% Fe2O3. If etching does occur, metal ion impurities in the glass walls could be released into the reaction chamber.
Impurities catalyzing reactions and substrate decomposition. Precautions should be taken to avoid introducing impurities. This includes taking care not to handle the electrodes with contaminated gloves and using pure solvents and electrolytes. To ensure reproducibility of measurements, commercial reagents that are not of guaranteed electrochemical grade should be purified. Impurities can arise from the solid components of the experiment: dissolution of rubber gaskets is one possible case where such adventitious contamination may occur. When homemade electrodes are prepared, solder or Ag-conducting epoxide may be used to assemble the setup. Under oxidizing conditions, such materials could lead to contamination of the solvent with Sn or Ag species. Ag contaminants may also arise from Ag-based reference electrodes leaching into solution. For both reductive and oxidative purposes, pre-electrolysis of the solvent medium with a dummy electrodes has been used to remove such artefactual contributions.134,135
“Homeopathic” levels of metallic impurities and their implications in augmenting the efficacy of catalytic processes are the subject of a recent review.136 Pre-electrolysis is a possible means to remove transition metal impurities present in the medium. Metallic electrodeposition can remove ions from a liquid medium,137 a method that has been used for water purification,138 specifically in the removal of heavy metal ions from waste water.139
Changes in the phase of the analyte material during the electrochemical experiment often arise in the case of electrocatalytic production of solar fuels from abundant sources like water.136 For these experiments, Crabtree has introduced the term “operationally homogeneous” to describe a catalyst or mediator that retains, or often improves, its catalytic activity even after a phase change and electrodeposition (which may not be obvious events to characterize without further complex experiments). Products formed at the counter electrode and migration to the working chamber may cause complications in interpreting the data obtained during an electrochemical experiment.
An electrolysis cell design for benchtop electrochemistry is shown in Fig. 5. The 14/20-sized ports allow for a Schlenk adapter to convert the cell into a Schlenk electrochemical flask amenable to common air-free techniques. Supporting electrolyte and non-sensitive solids can be introduced directly into the cell. The modularity of the cell design, particularly the provision of a central O-ring seal, allows for introduction of large-surface area electrodes without the size limitation of narrow port-sizes in the upper part of the cell. The jacketed cell also allows for temperature control during the experiment. If high or low temperature is required, a reflux condenser or cold finger may be introduced via one of the upper ports.
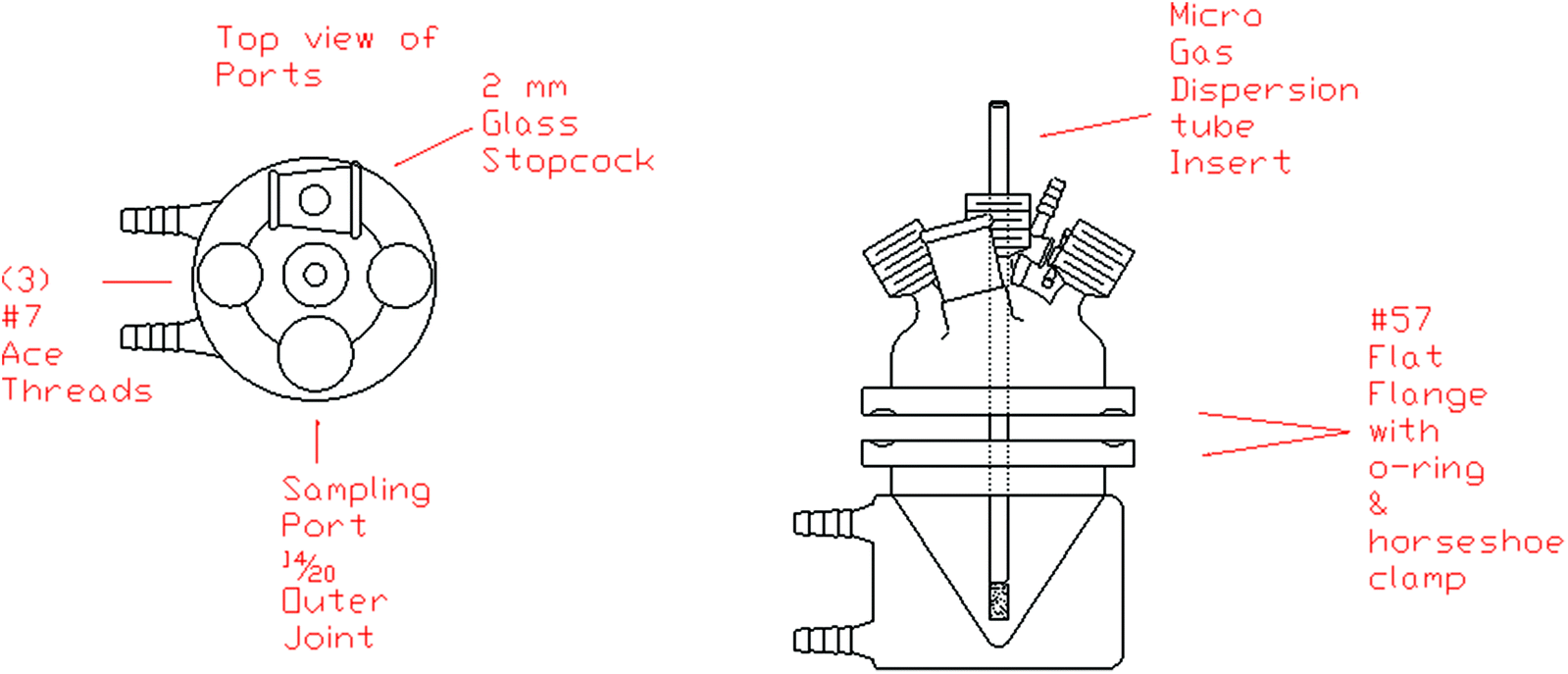 | ||
| Fig. 5 Schlenk CV and electrolysis cell design for small-volume, large-surface area electrodes. | ||
Two-chamber, concentrically-arranged cells may be set up with a glass dispersion tube as a fritted separator. These are also easily interfaced with one of the access ports. If a gas is evolved during electrolysis, a common headspace may be achieved by adding a hole to the dispersion tube in the central insert, above the immersion level. This is a suitable alternative to the traditional H-cell, where field effect irregularities may cause problems.
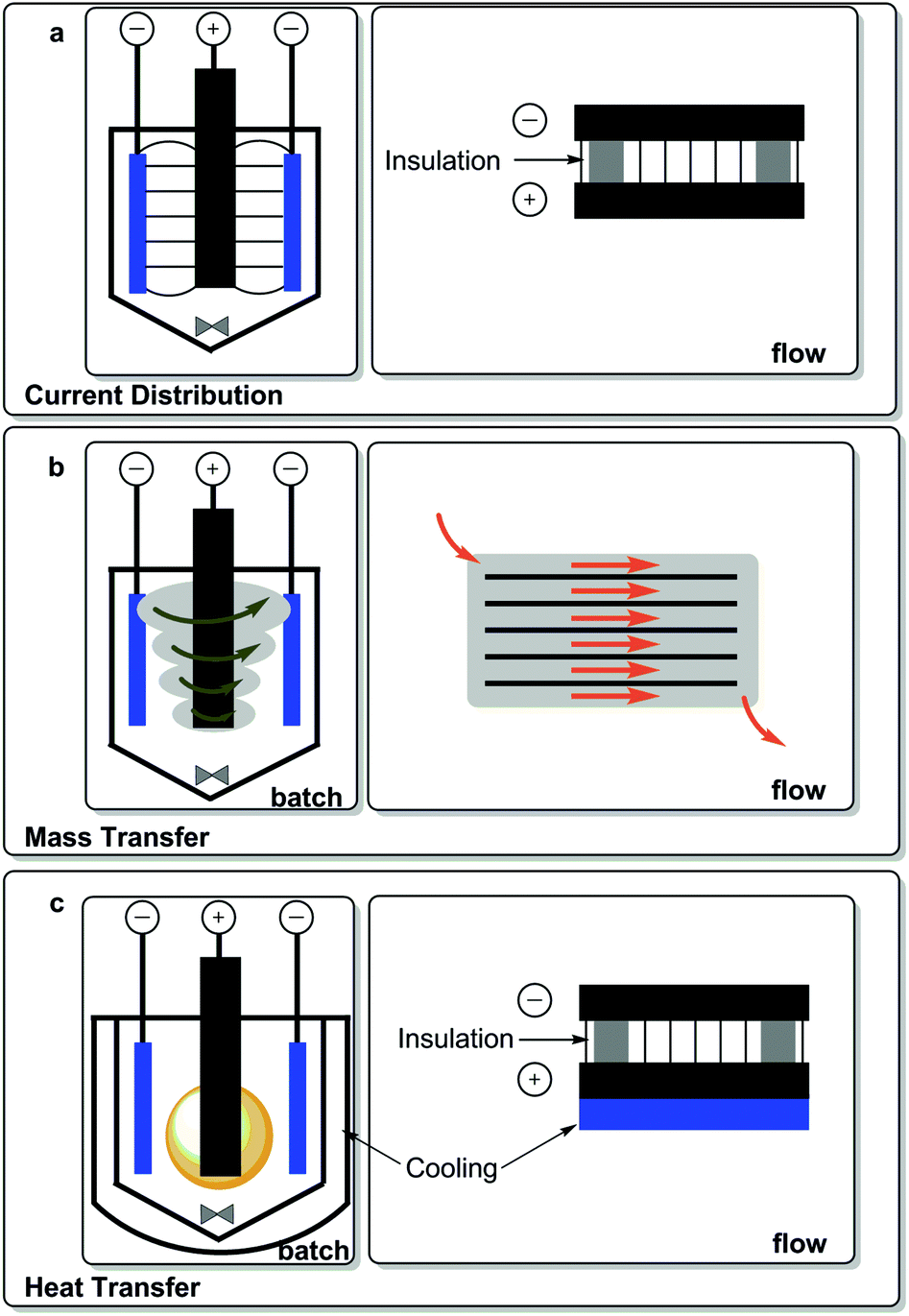 | ||
| Fig. 6 Current distribution, mass and heat transfer in a concentrical electrolysis cell and flow system.140 | ||
An interesting recent report described the advantages of maintaining uniform fields in thin-film flow electrolysis cells and reported electrolyses reactions being possible in the absence of supporting electrolyte, thereby alleviating some of the product separation issues related to salt removal.140 The field remains constant throughout the experiment in this optimal experimental setup Fig. 6. Flow chemistry has garnered interest in pharmaceutical applications.141 Microfluidic electrolysis for organic electrolysis has now been the subject of a report by Pletcher and coworkers.142
Reticulated vitreous carbon electrodes are of interest due to their low cost, high surface area and favorable electrochemical properties. These have now been implemented in flow electrolysis.143 Comparisons between chemical processes and their kinetics in batch vs. flow have been described.144 Reactions in batch (in a traditional flask), coupled with in situ observation of rates product formation, can provide a wealth of information regarding the kinetics of the reactions.
5. Conclusions
Redox processes are often investigated in reference to the oxidation and reduction potentials of the relevant chemical species. As these redox processes may be carried out by a variety of different methods, electrochemical means of reduction and oxidation may soon become commonplace in the laboratory. Even though contributions from thermal and environmental effects are still under study in these types of reactions,145 recent advances speak to the potential widespread utility of reactions catalyzed by electrons and holes in organic synthesis. The concept of formal redox potential now sets the stage for further applications of electrochemical methods in synthetic chemistry and we remain forward-looking to developments in the field.Acknowledgements
This material is based upon work performed by the Joint Center for Artificial Photosynthesis, a DOE Energy Innovation Hub, supported through the Office of Science of the U.S. Department of Energy under Award Number DE-SC0004993. JLG would like to thank SDSU for startup funds. We thank Dr Charles McCrory, Dr John Gregoire, Dr James Blakemore, Dr Kevin Oberg, Mr Chris Nalbandian, Dr Corinne Allen and Professors Donna Blackmond, Carl Koval, Keary Engle, Phil Baran, Robert Crabtree, Seth Herzon, Jonas Peters, T. Don Tilley, Clifford Kubiak and Nathan Lewis for insightful discussions.Notes and references
- M. P. Tarazona and E. Saiz, J. Chem. Educ., 1995, 72, 882 CrossRef CAS.
- O. R. Luca, Equilibrium as a Conceptual Framework for an Integrated, Synergistic Introductory Course in Chemistry and Physics, 2014, DOI:10.7907/S4WC74.
- L. Tender, M. T. Carter and R. W. Murray, Anal. Chem., 1994, 66, 3173–3181 CrossRef CAS.
- M. Chou, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1977, 99, 5615–5623 CrossRef CAS.
- A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765–773 CrossRef CAS PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- S. J. Konezny, M. D. Doherty, O. R. Luca, R. H. Crabtree, G. L. Soloveichik and V. S. Batista, J. Phys. Chem. C, 2012, 116, 6349–6356 CAS.
- J. A. Keith, K. A. Grice, C. P. Kubiak and E. A. Carter, J. Am. Chem. Soc., 2013, 135, 15823–15829 CrossRef CAS PubMed.
- C. P. Jasperse, D. P. Curran and T. L. Fevig, Chem. Rev., 1991, 91, 1237–1286 CrossRef CAS.
- P. Renaud and M. Gerster, Angew. Chem., Int. Ed., 1998, 37, 2562–2579 CrossRef CAS.
- D. D. Perrin, B. Dempsey and E. P. Serjeant, pKa prediction for organic acids and bases, Springer, 1981 Search PubMed.
- W. C. Barrette Jr., H. Johnson Jr. and D. T. Sawyer, Anal. Chem., 1984, 56, 1890–1898 CrossRef CAS.
- K. J. Young, L. A. Martini, R. L. Milot, R. C. Snoeberger, V. S. Batista, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2012, 256, 2503–2520 CrossRef CAS PubMed.
- R. Bensasson, E. Land and T. Truscott, Excited States and Free Radicals in Biology and Medicine, Contributions from Flash Photolysis and Pulse Radiolysis, New York, 1993 Search PubMed.
- S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455–1464 CrossRef CAS PubMed.
- M. Grätzel, Inorg. Chem., 2005, 44, 6841–6851 CrossRef PubMed.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- Y. Xi, H. Yi and A. Lei, Org. Biomol. Chem., 2013, 11, 2387–2403 CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical methods: fundamentals and applications, Wiley, New York, 1980 Search PubMed.
- O. R. Luca, Virtual Hydrogen Storage - Strategies for the Production and Storage of Hydrogen, Yale University, 2013 Search PubMed.
- P. Driscoll, E. Deunf, L. Rubin, O. Luca, R. Crabtree, C. Chidsey, J. Arnold and J. Kerr, ECS Trans., 2011, 35, 3–17 CAS.
- A. Q. Fenwick, J. M. Gregoire and O. R. Luca, J. Photochem. Photobiol., B, 2014 DOI:10.1016/j.jphotobiol.2014.12.019.
- C. Kittel, Elementary statistical physics, Courier Corporation, 2004 Search PubMed.
- R. Sundararaman, P. Narang, A. S. Jermyn, W. A. Goddard Iii and H. A. Atwater, Nat. Commun., 2014, 5 Search PubMed.
- P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CAS.
- W. Hou, Z. Liu, P. Pavaskar, W. H. Hung and S. B. Cronin, J. Catal., 2011, 277, 149–153 CrossRef CAS PubMed.
- S. Mukherjee, F. Libisch, N. Large, O. Neumann, L. V. Brown, J. Cheng, J. B. Lassiter, E. A. Carter, P. Nordlander and N. J. Halas, Nano Lett., 2012, 13, 240–247 CrossRef PubMed.
- M. Sun and H. Xu, Small, 2012, 8, 2777–2786 CrossRef CAS PubMed.
- S. Peng, M. T. Sheldon, W.-G. Liu, A. Jaramillo-Botero, W. A. Goddard and H. A. Atwater, Appl. Phys. Lett., 2015, 106, 023102 CrossRef PubMed.
- D. C. Grills, A. R. Cook, E. Fujita, M. W. George, J. M. Preses and J. F. Wishart, Appl. Spectrosc., 2010, 64, 563–570 CrossRef CAS PubMed.
- J. Schneider, H. Jia, K. Kobiro, D. E. Cabelli, J. T. Muckerman and E. Fujita, Energy Environ. Sci., 2012, 5, 9502–9510 CAS.
- R. A. Moss, Chem. Rev., 2013, 113, 6903–6904 CrossRef CAS PubMed.
- D. Polyansky, D. Cabelli, J. T. Muckerman, E. Fujita, T.-a. Koizumi, T. Fukushima, T. Wada and K. Tanaka, Angew. Chem., Int. Ed., 2007, 119, 4247–4250 CrossRef PubMed.
- C. C. Moser, in The Biophysics of Photosynthesis, Springer, 2014, pp. 121–139 Search PubMed.
- J. R. Durrant, S. A. Haque and E. Palomares, Coord. Chem. Rev., 2004, 248, 1247–1257 CrossRef CAS PubMed.
- P. Bai and M. Z. Bazant, Nat. Commun., 2014, 5, DOI:10.1038/ncomms4585.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811, 265–322 CrossRef CAS.
- S. Ašperger, in Chemical Kinetics and Inorganic Reaction Mechanisms, Springer, 2003, pp. 177–201 Search PubMed.
- D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC.
- M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS PubMed.
- J.-F. Yin, M. Velayudham, D. Bhattacharya, H.-C. Lin and K.-L. Lu, Coord. Chem. Rev., 2012, 256, 3008–3035 CrossRef CAS PubMed.
- J. P. Sauvage, J. P. Collin, J. C. Chambron, S. Guillerez, C. Coudret, V. Balzani, F. Barigelletti, L. De Cola and L. Flamigni, Chem. Rev., 1994, 94, 993–1019 CrossRef CAS.
- A. S. Polo, M. K. Itokazu and N. Y. M. Iha, Coord. Chem. Rev., 2004, 248, 1343–1361 CrossRef CAS PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- D. K. Gosser, Cyclic voltammetry: simulation and analysis of reaction mechanisms, VCH, New York, 1993 Search PubMed.
- P. T. Kissinger and W. R. Heineman, J. Chem. Educ., 1983, 60, 702–706 CrossRef CAS.
- R. R. Gagne, C. A. Koval and G. C. Lisensky, Inorg. Chem., 1980, 19, 2854–2855 CrossRef CAS.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- K. Seto, T. Nakayama and B. Uno, J. Phys. Chem. B, 2013, 117, 10834–10845 CrossRef CAS PubMed.
- B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- F. Fringuelli and A. Taticchi, Dienes in the Diels–Alder reaction, 1990, ISBN: 978-0-471-85549-1 Search PubMed.
- D. W. Borhani and F. D. Greene, J. Org. Chem., 1986, 51, 1563–1570 CrossRef CAS.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- C. K. Prier, D. a. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- J. W. Tucker, Y. Zhang, T. F. Jamison and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2012, 51, 4144–4147 CrossRef CAS PubMed.
- D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355–360 CrossRef CAS.
- T. J. Meyer, Acc. Chem. Res., 1989, 22, 163–170 CrossRef CAS.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed.
- Z. Zuo, D. Ahneman, L. Chu, J. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed.
- J. F. Bunnett, Acc. Chem. Res., 1992, 25, 2–9 CrossRef CAS.
- E. Dunach, M. J. Medeiros and S. Olivero, New J. Chem., 2006, 30, 1534–1548 RSC.
- A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS PubMed.
- J. J. Douglas, K. P. Cole and C. R. J. Stephenson, J. Org. Chem., 2014, 79, 11631–11643 CrossRef CAS PubMed.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- J. W. Tucker, J. M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Org. Lett., 2010, 12, 368–371 CrossRef CAS PubMed.
- L. Furst, J. M. R. Narayanam and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2011, 50, 9655–9659 CrossRef CAS PubMed.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- E. Duñach, D. Franco and S. Olivero, Eur. J. Org. Chem., 2003, 1605–1622 CrossRef PubMed.
- R. Barhdadi, C. Courtinard, J. Y. Nedelec and M. Troupel, Chem. Commun., 2003, 1434–1435 RSC.
- Y. Shen, T. Tajima, M. Atobe and T. Fuchigami, Electrochemistry, 2004, 72, 849 CAS.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed.
- J. Du and T. P. Yoon, J. Am. Chem. Soc., 2009, 131, 14604–14605 CrossRef CAS PubMed.
- J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392–396 CrossRef CAS PubMed.
- J. Du, L. R. Espelt, I. a. Guzei and T. P. Yoon, Chem. Sci., 2011, 2, 2115–2119 RSC.
- S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350–19353 CrossRef CAS PubMed.
- D. J. Bellville, D. W. Wirth and N. L. Bauld, J. Am. Chem. Soc., 1981, 103, 718–720 CrossRef CAS.
- S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350–19353 CrossRef CAS PubMed.
- J. Mlcoch and E. Steckhan, Tetrahedron Lett., 1987, 28, 1081–1084 CrossRef CAS.
- A. E. Hurtley, M. A. Cismesia, M. A. Ischay and T. P. Yoon, Tetrahedron, 2011, 67, 4442–4448 CrossRef CAS PubMed.
- M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed.
- D. P. Hari, P. Schroll and B. Koenig, J. Am. Chem. Soc., 2012, 43, 2958–2961 CrossRef PubMed.
- Y.-Q. Zou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Jørgensen and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784–788 CrossRef CAS PubMed.
- S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, J. Am. Chem. Soc., 2013, 135, 13286–13289 CrossRef CAS PubMed.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- Y. Oh and X. Hu, Chem. Soc. Rev., 2013, 42, 2253–2261 RSC.
- G. Boschloo and A. Hagfeldt, Acc. Chem. Res., 2009, 42, 1819–1826 CrossRef CAS PubMed.
- O. R. Luca, T. Wang, S. J. Konezny, V. S. Batista and R. H. Crabtree, New J. Chem., 2011, 35, 998–999 RSC.
- D. T. Sawyer, A. Sobkowiak and J. L. Roberts, Electrochemistry for Chemists, Wiley & Sons, New York, 2nd edn, 1995 Search PubMed.
- R. S. Nicholson, Anal. Chem., 1965, 37, 1351–1355 CrossRef CAS.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS PubMed.
- H. B. Herman and A. J. Bard, Anal. Chem., 1964, 36, 510–514 CrossRef CAS.
- J. H. Wagenknecht, J. Org. Chem., 1977, 42, 1836–1838 CrossRef CAS.
- A. G. O'Brien, A. Maruyama, Y. Inokuma, M. Fujita, P. S. Baran and D. G. Blackmond, Angew. Chem., Int. Ed., 2014, 126, 12062–12065 CrossRef PubMed.
- N. D. Schley and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 16588–16593 CrossRef CAS PubMed.
- J. Chen, W.-Q. Yan, C. M. Lam, C.-C. Zeng, L.-M. Hu and R. D. Little, Org. Lett., 2015, 17, 986–989 CrossRef CAS PubMed.
- O. Luca, S. J. Konezny, J. D. Blakemore, D. Colosi, S. Saha, G. W. Brudvig, V. S. Batista and R. H. Crabtree, New J. Chem., 2012, 36, 1149–1152 RSC.
- O. R. Luca, S. J. Konezny, G. B. Hunsinger, P. Müller, M. K. Takase and R. H. Crabtree, Polyhedron, 2014, 82, 2–6 CrossRef CAS PubMed.
- O. R. Luca, J. D. Blakemore, S. J. Konezny, J. M. Praetorius, T. J. Schmeier, G. B. Hunsinger, V. S. Batista, G. W. Brudvig, N. Hazari and R. H. Crabtree, Inorg. Chem., 2012, 51, 8704–8709 CrossRef CAS PubMed.
- D. C. Lacy, C. C. L. McCrory and J. C. Peters, Inorg. Chem., 2014, 53, 4980–4988 CrossRef CAS PubMed.
- O. R. Luca, D. L. Huang, M. K. Takase and R. H. Crabtree, New J. Chem., 2013, 37, 3402–3405 RSC.
- O. R. Luca, B. A. Thompson, M. K. Takase and R. H. Crabtree, J. Organomet. Chem., 2013, 730, 79–83 CrossRef CAS PubMed.
- C. M. Woo, S. L. Gholap, L. Lu, M. Kaneko, Z. Li, P. C. Ravikumar and S. B. Herzon, J. Am. Chem. Soc., 2012, 134, 17262–17273 CrossRef CAS PubMed.
- H. Lund and O. Hammerich, Organic electrochemistry, M. Dekker, New York, 2001 Search PubMed.
- H. O. House, E. Feng and N. P. Peet, J. Org. Chem., 1971, 36, 2371–2375 CrossRef CAS.
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- H. Ohno, Electrochemical aspects of ionic liquids, John Wiley & Sons, 2011 Search PubMed.
- M. J. Earle and K. R. Seddon, Pure Appl. Chem., 2000, 72, 1391–1398 CrossRef CAS.
- A. P. Doherty, L. Diaconu, E. Marley, P. L. Spedding, R. Barhdadi and M. Troupel, Asia–Pac. J. Chem. Eng., 2012, 7, 14–23 CrossRef CAS PubMed.
- Y.-F. Wu and C.-H. Chen, J. Electrochem. Soc., 2014, 161, E3276–E3282 CrossRef CAS PubMed.
- S. Cheng and B. E. Logan, Electrochem. Commun., 2007, 9, 492–496 CrossRef PubMed.
- R. Elazari, G. Salitra, A. Garsuch, A. Panchenko and D. Aurbach, Adv. Mater., 2011, 23, 5641–5644 CrossRef CAS PubMed.
- J. Friedrich, C. Ponce-de-León, G. Reade and F. Walsh, J. Electroanal. Chem., 2004, 561, 203–217 CrossRef CAS PubMed.
- J. Wang, Electrochim. Acta, 1981, 26, 1721–1726 CrossRef CAS.
- O. R. Luca, D. L. Huang, M. K. Takase and R. H. Crabtree, New J. Chem., 2013, 37, 3402–3405 RSC.
- O. R. Luca, T. Wang, S. J. Konezny, V. S. Batista and R. H. Crabtree, New J. Chem., 2011, 35, 998–999 RSC.
- L. Nagy, G. Gyetvai, L. Kollár and G. Nagy, J. Biochem. Biophys. Methods, 2006, 69, 121–132 CrossRef CAS PubMed.
- A. Lewandowski, L. Waligora and M. Galinski, J. Solution Chem., 2013, 42, 251–262 CrossRef CAS PubMed.
- R. P. Van Duyne and C. N. Reilley, Anal. Chem., 1972, 44, 153–158 CrossRef.
- R. P. Van Duyne and C. N. Reilley, Anal. Chem., 1972, 44, 158–169 CrossRef.
- W. Doenitz, R. Schmidberger, E. Steinheil and R. Streicher, Int. J. Hydrogen Energy, 1980, 5, 55–63 CrossRef CAS.
- S. D. Ebbesen, S. H. Jensen, A. Hauch and M. B. Mogensen, Chem. Rev., 2014, 114, 10697–10734 CrossRef CAS PubMed.
- G. G. Wildgoose, D. Giovanelli, N. S. Lawrence and R. G. Compton, Electroanalysis, 2004, 16, 421–433 CrossRef CAS PubMed.
- R. P. Van Duyne and C. N. Reilley, Anal. Chem., 1972, 44, 142–152 CrossRef.
- T. Isono, J. Chem. Eng. Data, 1984, 29, 45–52 CrossRef CAS.
- S. P. Pinho and E. A. Macedo, Fluid Phase Equilib., 1996, 116, 209–216 CrossRef CAS.
- M. Hodes, P. Griffith, K. A. Smith, W. S. Hurst, W. J. Bowers and K. Sako, AIChE J., 2004, 50, 2038–2049 CrossRef CAS PubMed.
- X.-y. Zou, H.-w. Xie, Y.-c. Zhai and X.-c. Lang, J. Mol. Sci., 2012, 3, 008 Search PubMed.
- D. Jagner and A. Graneli, Anal. Chim. Acta, 1976, 83, 19–26 CrossRef CAS.
- R. H. Crabtree, Chem. Rev., 2011, 112, 1536–1554 CrossRef PubMed.
- T. Kohyama, Entegris Applications Note, 2010 Search PubMed.
- X. Feng, Z. Wu and X. Chen, Sep. Purif. Technol., 2007, 57, 257–263 CrossRef CAS PubMed.
- Y. D. Gamburg and G. Zangari, Theory and practice of metal electrodeposition, Springer Science & Business Media, 2011 Search PubMed.
- K. Watts, A. Baker and T. Wirth, J. Flow Chem., 2014, 2014, 2–11 Search PubMed.
- L. Malet-Sanz and F. Susanne, J. Med. Chem., 2012, 55, 4062–4098 CrossRef CAS PubMed.
- R. Green, R. Brown and D. Pletcher, J. Flow Chem, 2015, 5, 31–36 CrossRef.
- W. Blaedel and J. Wang, Anal. Chem., 1979, 51, 799–802 CrossRef CAS.
- F. E. Valera, M. Quaranta, A. Moran, J. Blacker, A. Armstrong, J. T. Cabral and D. G. Blackmond, Angew. Chem., Int. Ed., 2010, 49, 2478–2485 CrossRef CAS PubMed.
- A. Q. Fenwick, J. M. Gregoire and O. R. Luca, J. Photochem. Photobiol., B, 2015 DOI:10.1016/j.jphotobiol.2015.04.003.
| This journal is © the Partner Organisations 2015 |