
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation and testing of phosphate, oxalate and uric acid matrix-matched standards for accurate quantification of 2D elemental distribution in kidney stone sections using 213 nm nanosecond laser ablation inductively coupled plasma mass spectrometry
M. Vašinová
Galiová
ab,
K.
Štěpánková
a,
R.
Čopjaková
c,
J.
Kuta
d,
L.
Prokeš
ae,
J.
Kynický
f and
V.
Kanický
*ab
aDepartment of Chemistry, Faculty of Science, Masaryk University, Kotlářská 2, 611 37 Brno, Czech Republic. E-mail: viktork@chemi.muni.cz
bCentral European Institute of Technology (CEITEC), Masaryk University, Kamenice 5, 625 00 Brno, Czech Republic
cDepartment of Geological Sciences, Faculty of Science, Masaryk University, Kotlářská 2, 611 37 Brno, Czech Republic
dResearch Centre for Toxic Compounds in the Environment (RECETOX), Masaryk University, Kamenice 126/3, 625 00 Brno, Czech Republic
eDepartment of Physical Electronics, Faculty of Science, Masaryk University, Kotlářská 2, 611 37 Brno, Czech Republic
fDepartment of Pedology and Geology, Faculty of Forestry and Wood Technology, Mendel University in Brno, Zemědělská 3, 613 00 Brno, Czech Republic
First published on 9th March 2015
Abstract
Matrix-matched calibration for quantitative elemental mapping of kidney stones by laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS) was developed with a 213 nm Nd:YAG laser ablation device and a quadrupole mass spectrometer. The method was applied to the imaging of P, Na, Sr, Zn, Ba and Pb distributions over a section of the kidney stone specimen containing phosphate and oxalate phases. Eighteen kidney stone specimens consisting of phosphate, oxalate and urate phases in various proportions were cut into halves for both preparation of calibration pellets and bulk analysis. Homogeneity of calibration pellets was examined by scanning electron microscopy (SEM) and LA-ICP-MS, concluding that areas of individual biominerals were thoroughly mixed and their size in units of micrometers was well below the size of the used laser spot. Calcium was employed as the internal reference element, being present in sufficient contents in the studied kidney stones. Mean values of calcium contents in oxalate and phosphate phases separately were determined using an electron microprobe (EMP) in the kidney stone section further subjected to the mapping. The actual (time- and space-resolved) Ca sensitivity was computed for each 44Ca+ signal and used as the internal reference for LA-ICP-MS isotopic signals of P, Na, Sr, Zn, Ba and Pb when mapping. Dependences of particular isotopic signal intensity/Ca sensitivity ratios vs. average elemental contents by solution analysis were processed by ordinary least squares linear regression. Despite variable matrices the regression yielded calibration lines with insignificant intercepts, coefficients of determination R2 > 0.9955, and relatively narrow prediction and confidence bands. However, in addition, the applicability of four-point calibration and four single-point calibrations as less time-consuming options was examined on the basis of the NIST SRM 1486 bone meal pellet analysis. Best fit was obtained for the four-point calibration and single-point calibration with the phosphate pellet. Quantitative elemental maps of the kidney stone section were recorded and computed for P, Na, Sr, Zn, Ba and Pb. The feasibility of quantification by matrix-matched single-point calibration was verified by determination of the median elemental contents in phosphate and oxalate phases by LA-ICP-MS and their arithmetic comparison mean values obtained over the same section area using EMP.
Introduction
Laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS) has already been employed in various fields of scientific research such as biology,1–4 medicine,5–9 geology,10,11 archaeology,12–17etc. The ability to focus the laser beam onto a sample area with size from units to thousands of square micrometers together with limits of detection down to units of milligrams per kilogram makes this method interesting for elemental mapping at the “microscale”. For example, elemental distribution was studied in archaeological findings, such as tooth and bone tissues, in order to reveal the diet and migration/mobility15–17 of ancient population. Results of analyses of recent samples are used in biomedicine and archaeology.18–21 Similarly, fish scales and otoliths as indicators of migration of fish, environmental pollution, water salinity and temperature were analyzed.22,23 LA-ICP-MS is a suitable technique to investigate phytoremediation where the knowledge about deposition of toxic elements is required.1–4 Considerable interest of the scientific community is focused on the monitoring of elemental distribution in soft tissues such as brain, muscle, kidney tissue, tumors etc., or on a single cell analysis.24–27 Where elements of interest are stored, how their presence affects the distribution of other elements and how they relate to specific disease – are the questions that might be answered by LA-ICP-MS analysis.The chemical and mineralogical composition of a kidney stone is related to a particular disease which is associated with a specific kidney stone formation, depends on the medical treatment and diet of a patient, metabolic conditions, and is influenced by environmental pollution in the patient's domicile and workplace. Chemical analysis might be helpful in understanding kidney stone formation, growth, and medical treatment. In general, kidney stone (urolith) has miscellaneous mineralogical composition, and phosphate, oxalate, carbonate, uric acid, cysteine, and xanthine are the main matrices. Correspondingly, several variants of minerals are present, e.g., in the case of phosphate we find struvite MgNH4PO4·6H2O, brushite CaHPO4·2H2O, whitlockite Ca9(MgFe)(PO4)6PO3OH, and apatite (Ca5(PO4)3(OH,F,Cl)) with variable portions of OH− (hydroxylapatite), F− (fluorapatite) and CO32− (carbonate-hydroxylapatite) anions. Oxalate occurs as whewellite Ca(C2O4)·H2O and weddellite Ca(C2O4)·2H2O. The urate matrix can be present as uric acid C5H4N4O3, or uric acid dihydrate C5H4N4O3·2H2O and its salts, such as ammonium urate NH4C5H3N4O3, and sodium urate monohydrate NaC5H3N4O3·H2O. Usually, a urolith contains more than one mineral and therefore, direct analysis of solid samples is preferred in order to preserve spatial information.28–30
Various techniques have been routinely employed in mineralogical and surface analysis of kidney stones, such as infrared spectroscopy,31,32 Raman spectroscopy,32,33 X-ray diffraction,34,35 tomography,36 polarization optical crystallography, chemical microscopy, ultraviolet-visible spectroscopy and photomicroscopy, and light microscopy for macroscopic and microscopic examination. Besides, scanning electron microscopy (SEM/EDX)37 or laser induced breakdown spectrometry (LIBS)38,39 has been used for surface analysis. An exploratory study focused on the possibilities of calibration using pressed pellets prepared from uroliths for bulk analysis and microanalysis by LIBS, laser ablation-laser induced breakdown spectrometry (LA-LIBS) and LA-ICP-MS/OES brought out a comparison of calibration curves in terms of linearity and coefficients of determination. However, no elemental mapping of uroliths was attempted.40
However, when information on the spatial distribution of trace and minor elements is required, some of the above methods do not meet the requirements on limits of detection (LOD). LA-ICP-MS combines an appropriate resolution in tens of micrometers of the laser spot diameter with values of LOD in units of mg kg−1 for most elements. So far only two papers have been published on qualitative surface analysis of kidney stones by LA-ICP-MS.41,42 A qualitative elemental mapping of uroliths by LA-ICP-MS and electron microprobe (EMP) imaging by means of backscattered electrons (BSE) were applied to distinguish particular mineral phases. Bulk analysis of selected elements in oxalate and phosphate phases by LA-ICP-MS was based on calibration with NIST SRM 1486 (bone meal) and confirmed by EMP analysis, however no quantitative elemental maps were created.42
The major problem with LA-ICP-MS quantification consists in the lack of a reliable, commercially available calibration material. In such a case, effort is devoted to the preparation of laboratory-made standards. As examples, spiked gelatine,43 agarose44 or tissue freezing medium1 is presented for calibration of the LA-ICP-MS determination of metals/metalloids in soft tissues. The calibration can be based on commercially available standards with a similar matrix, which are spiked with the required analytes.2 If a suitable reference material is not available, and gelatine or agarose does not match the properties of the analyzed samples, real samples with a similar matrix, previously analyzed by independent methods, may be used with or without spiking.5,45,46 If material properties allow, the sample is crushed, milled, homogenized and the resulting powdered material is pressed into a pellet47 or mixed with the matrix prepared by the sol–gel technique with subsequent homogenization and pelletization.48–50 Another approach consists in the pressing of the powdered sample with a suitable binder.17,50
Uroliths are usually constituted by several mineral phases that are present in various proportions and differ in physical and physico-chemical properties. Consequently, different ablation rates exist in particular mineral zones and make quantitative elemental mapping by LA-ICP-MS difficult unless signal correction by a suitable internal reference is employed. Similarly, such a calibration is necessary which covers appropriately wide ranges of elemental contents and therefore, particular calibration standards would contain various proportions of minerals, and trace elements on various content levels. It is obvious that no compact calibration material is available and calibration has to be based on pressed powdered pellets.
The preparation of powdered calibration standards has been known for decades as it was being used for emission spectrography with electric arc excitation and X-ray fluorescence spectrometry. Calibration standards were typically prepared by mixing solid powdered reagents or natural previously analyzed materials with subsequent homogenization of the resulting mixtures. As both the spectral analytical methods exhibit non-spectral interferences, matrix-matched calibration with an internal reference was indispensable for quantitative analysis.
An easier way to prepare matrix-matched calibration standards for LA-ICP-MS elemental mapping of urolith sections is the use of already analyzed urolith samples, properly crushed, milled, homogenized and pressed, instead of mixing of individual chemical reagents. In comparison with spectrography or X-ray fluorescence spectrometry of powdered materials, quantitative elemental mapping of uroliths by LA-ICP-MS comprises another factor, namely the difference between the properties of calibration standards and analyzed samples. Different ablation rates would exist not only between minerals, but also between pressed pellets and the surface of urolith sections. Therefore, a reliable internal reference is necessary to compensate for these multiple effects.
No quantitative LA-ICP-MS elemental maps of uroliths have been published so far. In a study,40 calibration capabilities of laser ablation based techniques were studied for the purpose of bulk analysis and microanalysis of kidney stones. Intentionally the coarse-grained pellets were prepared from kidney stones instead of finely ground ones, thus respecting the original heterogeneity and multiphase matrix structure of uroliths consisting of several biominerals. This approach has been used because especially LIBS and LA-LIBS are more prone to matrix effects than LA-ICP-OES/MS. Thus, the urolith structure partially preserved in coarse-grained pellets might alleviate the differences between such pellets and kidney stone sections in the case of possible future elemental mapping. To eliminate coarse-grained pellet heterogeneity, a large ablated area and signal averaging were employed. As a result, a linear relationship between the content and signal was obtained, however, only loose correlation was observed, especially for LIBS. Moreover, the mineralogical composition influenced the preparation and mechanical properties of coarse-grained pellets. Pellet cohesion and strength depended on the prevailing phase (phosphate, oxalates, urates), which significantly influenced target material removal using laser beam. The study40 focused only on the comparison of laser ablation based methods in terms of calibration and did not address elemental mapping. Hole drilling, from the viewpoint of mapping a time-consuming ablation mode, was employed for this calibration study.
In another study42 qualitative elemental distributions over urolith sections were obtained by LA-ICP-MS and particular biomineral zones were identified using EMP/BSE. The influence of different ablation rates on LA-ICP-MS signal magnitude in various mineral phases was compensated by applying the sensitivity of a calcium isotope as the internal reference, for which the average content of calcium was determined in individual phases by EMP analysis. Elemental associations with mineral phases distinguished by the Ca/P signal ratio were derived based on LA-ICP-MS signal distributions compensated for different ablation rates. Calibration with a single pressed pellet prepared from NIST SRM 1486 bone meal was employed for bulk analysis of oxalate and phosphate mineral phases and confirmed by the analysis of average composition by EMP analysis.
As already stated, only qualitative 2D distribution of isotope signals has been published41,42 or calibration based on pressed coarse-grained urolith pellets suited rather for LIBS has been developed but not tested for elemental mapping.40 Single standard calibration with NIST SRM 1486 has been used for the determination of the average composition of mineral phases but not for quantification of elemental maps.42 Although it appeared that NIST SRM 1486 can be used to quantify these distributions,42 there is still scope for increasing the reliability of the calibration.
Several reasons exist to substitute real matrix-matched calibration samples for NIST SRM 1486. Contents of some elements are significantly higher in uroliths than in NIST SRM 1486 which results in inappropriate extrapolation with respect to the calibrated range. This applies to most analytes considered in this work (SRM vs. sample: 0.47%m/m Mg vs. 10%m/m Mg, 0.50%m/m Na vs. 1.50%m/m Na, 264 mg kg−1 Sr vs. 500 mg kg−1 Sr, 147 mg kg−1 Zn vs. 1000 mg kg−1 Zn, 1.3 mg kg−1 Pb vs. 20 mg kg−1 Pb). In NIST SRM 1486 only contents of Ca, P, Mg, Fe, Pb, K, Sr and Zn are certified, while contents of Si, Na, C, Al, As, Cd, Cu, F, Mn and Se are provided as additional information. Therefore, especially in the case of traces (Al, As, Cd, Cu, Mn, and Se) it would be advisable to verify their contents in a particular SRM package by solution analysis. Available indicative values of elemental contents in the SRM are too low (mg kg−1: <1 Al, 0.006 As, 0.003 Cd, 0.8 Cu, 1 Mn, 0.13 Se). Data on the level of contents of other important (potentially toxic) trace elements (Ni, Cr, Hg) are missing in the SRM certificate. Finally, NIST SRM 1486 represents only the calcium phosphate matrix, while kidney stones contain more minerals.
The aim of this study consists in developing a new, flexible matrix-matched calibration for LA-ICP-MS quantification of 2D elemental distributions in urolith sections. Fine-grained urolith pellets are considered for calibration to minimize heterogeneity and improve LA-ICP-MS signal stability. Eighteen urolith samples comprising concrements which consist of phosphate, oxalate and urate phases are included in the calibration study in order to encompass the major types of analyzed kidney stones and ranges of possible elemental contents. The maximum number of calibration samples is employed for the construction of calibration curves at first, and linearity as well as the presence of outliers due to the possible influence of the matrix is statistically tested. For practical reasons, one four-point calibration and four single-point calibrations are tested, too. The procedure consists in: (i) making of a calibration kit of pressed pellets prepared from uroliths, (ii) study of homogeneity and signal stability during laser ablation, (iii) construction of calibration curves of P, Na, Sr, Zn, Ba and Pb, (iv) verification of matrix-matched calibrations by the analysis of NIST SRM 1486, and finally (v) quantification of 2D elemental distributions and verification of results using EMP.
Experimental
Mineralogical composition of uroliths
A set of laboratory-made calibration standards and solid samples used for the testing of prepared standards consists of surgically removed human kidney stones obtained from a vast collection (more than eleven thousand uroliths) assembled by geologist Prof. Petr Martinec (Institute of Geonics, Academy of Sciences of the Czech Republic, Ostrava, Czech Republic). These uroliths have been obtained after the treatment of patients in the University Hospital Ostrava. The selection of uroliths appropriate for matrix-matched calibration was based on information about their mineralogical composition which was determined by infrared spectrometry. The calibration should include prevailing urolith matrices, which are phosphates, oxalates and uric acid, and therefore uroliths containing mainly phosphates (9 samples), uroliths consisting essentially of oxalates (5 samples) and uroliths composed of a mixture of uric acid and oxalates (4 samples) were selected for the preparation of pressed powder pellets (Table 1). Sample no. 11605 was subjected to elemental quantification of 2D maps using matrix-matched pellets.| Sample no. | Main matrix | Content of mineral (%m/m) | ||||
|---|---|---|---|---|---|---|
| Apatite | Struvite | Whewellite | Weddellite | Uric acid | ||
| a Ph = phosphate matrix, Ox = oxalate matrix, UA = uric acid. | ||||||
| 5056 | Ph | 50 | 40 | ∼ | 10 | ∼ |
| 5255 | 70 | 20 | ∼ | 10 | ∼ | |
| 5397 | 5 | 95 | ∼ | ∼ | ∼ | |
| 5996 | 35 | 65 | ∼ | ∼ | ∼ | |
| 6489 | 50 | 50 | ∼ | ∼ | ∼ | |
| 6671 | 70 | 20 | ∼ | 10 | ∼ | |
| 6686 | 60 | 40 | ∼ | ∼ | ∼ | |
| 8393 | 55 | 55 | ∼ | ∼ | ∼ | |
| 9130 | 50 | 50 | ∼ | ∼ | ∼ | |
| 5166 | Ox | 25 | ∼ | 75 | 1 | ∼ |
| 6275 | 10 | ∼ | 80 | 10 | ∼ | |
| 7851 | 5 | ∼ | 95 | ∼ | ∼ | |
| 8365 | 15 | 0 | 60 | 25 | ∼ | |
| 9081 | 5 | ∼ | 95 | ∼ | ∼ | |
| 11605 | 30 | ∼ | 40 | 30 | ∼ | |
| 6432 | UA | ∼ | ∼ | 40 | ∼ | 60 |
| 6585 | ∼ | ∼ | 5 | 5 | 90 | |
| 7301 | ∼ | ∼ | ∼ | 10 | 90 | |
| 8500 | ∼ | ∼ | ∼ | 20 | 80 | |
The phosphate matrix consists predominantly of apatite Ca5(PO4)3(OH,F,Cl) and struvite NH4MgPO4·6H2O in various proportions. The oxalate matrix comprises whewellite CaC2O4·H2O and weddellite CaC2O4·2H2O. A separate group includes mixed kidney stones containing a minor portion of oxalates (either whewellite or weddellite, exceptionally both) and uric acid (C5H4N4O3).
Elemental composition of uroliths
The average content of the elements of interest in the uroliths was determined using a wet digestion procedure followed by an ICP-MS measurement. The carbon content was determined by elemental analysis using a LiquiToc II device with Solids Module (Elementar Analysensysteme GmbH, Hanau, Germany). The elemental composition (Ca, Mg, C, P, Na, Sr, Zn, Ba and Pb) is summarized in Table 2. According to ref. 53 the examined urolith samples contain also trace contents of Al, K, Cr, Mn, Fe, Co, Ni, Cu, Se, Rb, Zr, Mo, Cd, Sn and Hg. The ICP-MS and carbon analyses were performed at the laboratory of Research Centre for Toxic Compounds in the Environment, Faculty of Science, Masaryk University, Brno, Czech Republic. Both analytical procedures have already been reported elsewhere.51,53 The results of solution ICP-MS analysis slightly differ in some cases from those of IR screening, which probably results from the fact that each of the halves of a particular kidney stone was crushed and homogenized separately with the assumption that a kidney stone is approximately radially symmetric.| Sample no. | Main matrix | Elemental content | ||||
|---|---|---|---|---|---|---|
| %m/m | ||||||
| Ca | Mg | C | P | Na | ||
| a Ph = phosphate matrix, Ox = oxalate matrix, UA = uric acid. | ||||||
| 5056 | Ph | 23.6 ± 0.8 | 4.28 ± 0.09 | 3.29 ± 0.09 | 17.7 ± 0.4 | 0.63 ± 0.01 |
| 5255 | 23.8 ± 0.8 | 4.07 ± 0.09 | 3.02 ± 0.08 | 17.5 ± 0.4 | 0.96 ± 0.02 | |
| 5397 | 0.35 ± 0.01 | 10.1 ± 0.2 | 1.06 ± 0.03 | 14.3 ± 0.3 | 0.0223 ± 0.0004 | |
| 5996 | 12.8 ± 0.4 | 6.5 ± 0.1 | 2.84 ± 0.08 | 15.2 ± 0.3 | 0.69 ± 0.01 | |
| 6489 | 18.3 ± 0.6 | 4.8 ± 0.1 | 2.92 ± 0.08 | 16.4 ± 0.3 | 0.91 ± 0.01 | |
| 6671 | 27.5 ± 0.9 | 2.60 ± 0.05 | 3.6 ± 0.1 | 18.3 ± 0.4 | 1.52 ± 0.02 | |
| 6686 | 21.1 ± 0.7 | 4.7 ± 0.1 | 3.52 ± 0.09 | 17.5 ± 0.4 | 0.81 ± 0.01 | |
| 8393 | 20.1 ± 0.6 | 5.1 ± 0.1 | 2.29 ± 0.06 | 16.7 ± 0.4 | 0.67 ± 0.01 | |
| 9130 | 23.2 ± 0.7 | 3.90 ± 0.08 | 2.82 ± 0.08 | 16.8 ± 0.4 | 0.62 ± 0.01 | |
| 5166 | Ox | 26.5 ± 0.8 | 0.061 ± 0.001 | 11.5 ± 0.3 | 2.05 ± 0.04 | 0.223 ± 0.004 |
| 6275 | 26.4 ± 0.8 | 0.0404 ± 0.0008 | 11.4 ± 0.3 | 1.50 ± 0.03 | 0.151 ± 0.002 | |
| 7851 | 24.5 ± 0.8 | 0.0280 ± 0.0006 | 11.9 ± 0.3 | 0.339 ± 0.007 | 0.135 ± 0.002 | |
| 8365 | 25.9 ± 0.8 | 0.115 ± 0.002 | 8.6 ± 0.2 | 2.63 ± 0.06 | 0.111 ± 0.002 | |
| 9081 | 24.5 ± 0.8 | 0.0231 ± 0.0005 | 10.9 ± 0.3 | 0.227 ± 0.005 | 0.086 ± 0.001 | |
| 11605 | 27.3 ± 0.9 | 0.160 ± 0.003 | 9.8 ± 0.3 | 5.0 ± 0.1 | 0.258 ± 0.004 | |
| 6432 | UA | 1.33 ± 0.04 | 0.00288 ± 0.00006 | 26.6 ± 0.7 | 0.0360 ± 0.0008 | 0.0214 ± 0.0003 |
| 6585 | 0.076 ± 0.002 | 0.00080 ± 0.00002 | 27.2 ± 0.7 | 0.0138 ± 0.0003 | 0.0133 ± 0.0002 | |
| 7301 | 0.31 ± 0.01 | 0.00134 ± 0.00003 | 23.6 ± 0.7 | 0.0191 ± 0.0004 | 0.0428 ± 0.0007 | |
| 8500 | 0.97 ± 0.03 | 0.00240 ± 0.00005 | 24.9 ± 0.7 | 0.0212 ± 0.0004 | 0.014 ± 0.0002 | |
| Sample no. | Main matrix | Elemental content | |||
|---|---|---|---|---|---|
| mg kg−1 | |||||
| Sr | Zn | Ba | Pb | ||
| 5056 | Ph | 314 ± 16 | 251 ± 5 | 10.1 ± 0.3 | 10.4 ± 0.3 |
| 5255 | 309 ± 15 | 939 ± 17 | 14.4 ± 0.4 | 67 ± 2 | |
| 5397 | 4.4 ± 0.2 | 29.0 ± 0.5 | 0.72 ± 0.02 | 0.324 ± 0.008 | |
| 5996 | 122 ± 6 | 373 ± 7 | 3.3 ± 0.1 | 9.7 ± 0.2 | |
| 6489 | 180 ± 9 | 458 ± 8 | 4.1 ± 0.1 | 3.02 ± 0.08 | |
| 6671 | 415 ± 21 | 711 ± 13 | 24.3 ± 0.8 | 13.8 ± 0.3 | |
| 6686 | 329 ± 16 | 945 ± 17 | 12.3 ± 0.4 | 51 ± 1 | |
| 8393 | 181 ± 9 | 477 ± 9 | 11.3 ± 0.4 | 19.0 ± 0.5 | |
| 9130 | 517 ± 26 | 590 ± 11 | 22.4 ± 0.7 | 5.9 ± 0.1 | |
| 5166 | Ox | 63 ± 3 | 137 ± 2 | 2.05 ± 0.06 | 9.6 ± 0.2 |
| 6275 | 38 ± 2 | 180 ± 3 | 0.84 ± 0.03 | 19.6 ± 0.5 | |
| 7851 | 33 ± 2 | 31.0 ± 0.6 | 0.54 ± 0.02 | 5.1 ± 0.1 | |
| 8365 | 102 ± 5 | 280 ± 5 | 9.7 ± 0.3 | 7.8 ± 0.2 | |
| 9081 | 31 ± 2 | 45.4 ± 0.8 | 1.04 ± 0.03 | 5.4 ± 0.2 | |
| 11605 | 116 ± 6 | 534 ± 10 | 5.0 ± 0.2 | 27.9 ± 0.7 | |
| 6432 | UA | 1.72 ± 0.09 | 9.7 ± 0.2 | 0.17 ± 0.01 | 0.349 ± 0.009 |
| 6585 | 0.081 ± 0.004 | 7.0 ± 0.1 | 0.066 ± 0.002 | 0.58 ± 0.01 | |
| 7301 | 1.17 ± 0.06 | 3.05 ± 0.05 | 0.052 ± 0.002 | 0.179 ± 0.004 | |
| 8500 | 4.3 ± 0.2 | 6.7 ± 0.1 | 0.199 ± 0.006 | 0.83 ± 0.02 | |
Making of pellets
Eighteen uroliths were employed for the preparation of matrix-matched calibration standards (Table 1, samples no. 5056–9130). The preparation of pellets presents an essential critical step in terms of precision and accuracy of LA-ICP-MS measurement. In particular, the homogeneity of the pellets and the grain size are crucial prerequisites to achieve a stable LA-ICP-MS signal. Besides, sufficient pellet cohesion is necessary to minimize cracking and crumbling of materials at ablation.The use of pellets prepared from finely ground kidney stones was aimed at homogenizing the multiphase composition of uroliths to reduce the possible spread of calibration points. Kidney stones considered for pellet preparation were crushed in an agate bowl and subsequently milled and homogenized in a ball mill (Planetary Micro Mill Pulverisette 7, FRITSCH; Germany). One portion of each pulverized sample was pressed without adding any binder into pellets with a diameter of 12 mm and 2 mm thickness. Pelletization was performed for 30 s at the pressure of 1.3 GPa using a manual hydraulic press (Mobiko Company, Czech Republic). The remaining powdered sample was dissolved for subsequent ICP-MS analysis.
In contrast to coarse-grained pellets, which were applied to the study of calibration capabilities in LIBS, LA-LIBS, and LA-ICP-OES/MS methods,40 the fine grained ones eliminated the lack of pellet cohesion. The difference in particle size was significant. While scanning electron microscope (SEM) exposures of intact and ablated coarse-grained pellets in ref. 40 show particle size between 10 and 50 μm and in some cases even above 100 μm, SEM images in this work (Fig. 1) show particles around 1 μm, i.e., about one or two orders of magnitude smaller.
 | ||
| Fig. 1 BSE images of 7851 pressed pellet surface: (a) homogeneous character of the pellet in a large scale view, length of the scale 500 μm, (b) fine scale with evenly distributed phosphate (bright grains) within the fine-grained oxalate matrix (dark color), length of the scale 20 μm, while the square outline defines the size of the laser beam spot (65 × 65 μm). | ||
Preparation of the urolith section for elemental mapping
For LA-ICP-MS elemental mapping of the urolith section surface, sample no. 11605 was cut into two parts using a diamond saw and one part of the urolith was mounted onto polymethylmethacrylate (PMM) and polished. The other part was pulverized and left for the determination of the average composition.Laser ablation-inductively coupled plasma-mass spectrometry procedures
Laser ablation experiments were performed at the Laboratory of Atomic Spectrochemistry, Department of Chemistry, Faculty of Science, Masaryk University. A pulsed Nd:YAG laser system UP 213 (New Wave Research, Inc., Fremont, CA, USA) working at 213 nm and a pulse duration of 4.2 ns was used. The ablation system is equipped with a SuperCell (New Wave Research) designed to enable rapid elution of the ablation-generated aerosol in a large format cell. Helium was used as the carrier gas with a flow rate of 1 L min−1. Aerosol was transported from the ablation cell using a 1 m long polyurethane transport tube (i.d. of 4 mm) to the ICP (ICP-MS Agilent 7500ce, Agilent Technologies, Santa Clara, CA, USA). The instrument was operated with a collision cell in He-mode for minimization of possible polyatomic interferences.The urolith section (no. 11605) and calibration pellets were rastered with the square-shaped laser beam (65 μm) in line scanning mode at a scan speed of 40 μm s−1 with a fluence of 7.5 J cm−2 and laser repetition rate of 5 Hz. Rasters composed of an appropriate number of parallel lines and spaced apart 100 μm were employed. The operating parameters are summarized in Table 3. Calibration sets were designed and statistically tested for P, Na, Sr, Zn, Ba and Pb, while Ca was employed as the internal reference. LA-ICP-MS data were collected also for carbon as the matrix element, for Mg as a medium/minor element, for K as a minor/trace element and for Al, Mn, Fe, Cu, Rb, and Sn as trace elements. Significantly different contents of various types of uroliths were also found for Fe, Cu, and Sn by ICP-MS solution analysis53 which corresponded to a range of LA-ICP-MS signals measured in this work.
| Laser ablation system | New wave research UP213 |
|---|---|
| Laser | Nd:YAG |
| Ablation chamber | SuperCell |
| Wavelength | 213 nm |
| Pulse duration | 4.2 ns |
| Fluence | 7.5 J cm−2 |
| Repetition rate | 5 Hz |
| Carrier gas flow rate | He 1.0 L min−1 + Ar 0.6 L min−1 |
| Ablation mode | Line scan |
| Ablation spot size | 65 μm (square) |
| Scan speed | 40 μm s−1 |
| Distance between lines | 100 μm (for mapping only) |
| ICP-MS | ICP-QMS, Agilent 7500ce |
|---|---|
| Rf power input | 1500 W |
| Plasma gas flow rate | Ar 15.0 L min−1 |
| Auxiliary gas flow rate | Ar 1.0 L min−1 |
| Sampling depth | 8 mm |
| Collision cell | He 2.5 mL min−1 |
| Acquisition time and isotopes monitored | 0.1 s: 12,13C+, 23Na+, 27Al+, 31P+, 39K+, 55Mn+, 56,57Fe+, 63Cu+, 66,68Zn+, 85Rb+, 86,88Sr+, 118Sn+, 135,137Ba+, 208Pb+ 0.05 s: 24,26Mg+ 0.01 s: 42,43,44Ca+ |
Scanning electron microscopy study and electron microprobe analysis
The samples were examined by SEM and EMP analysis prior to LA-ICP-MS experiments. The sample surface was carbon coated for the SEM and EMP study. The quality of pellets in respect to the grain size and phase homogeneity was inspected using SEM. MIRA III SEM equipped with a back-scattered electron (BSE) detector (laboratories of TESCAN, a. s., Brno, Czech Republic) was employed. Back-scattered electron images (1024 × 1024 pixels) were acquired under the following analytical conditions: an accelerating voltage of 20 kV and a beam current of 20 nA.The chemical composition (including major and trace elements) of specific phases within the kidney stone sample no. 11605 was determined using a Cameca SX100 electron microprobe (Department of Geological Sciences, Masaryk University, Brno) to check the trueness of results obtained by the quantification of 2D elemental distribution via LA-ICP-MS. The instrument was operated at an accelerating voltage of 15 kV, a beam current of 6 nA and scanning beam mode over the area 20 × 15 μm. The following calibration standards and analytical lines were used: (Kα) lines: fluorapatite (Ca, P), albite (Na), sanidine (Si, Al, K), Mg2SiO4 (Mg), NaCl (Cl), hematite (Fe), gahnite (Zn), SrSO4 (S) and topaz (F); (Lα) lines: SrSO4 (Sr), baryte (Ba). The peak counting times ranged from 10 to 20 s for major elements, and from 20 to 80 s for minor elements. The matrix effects were corrected using the PAP routine.52
Results and discussion
Matrix-matched urolith standards
The homogeneity of pellets is of utmost importance for reliable LA-ICP-MS calibration. The critical case may be urolith containing a minor portion of a mineral which substantially differs in chemical composition from the matrix mineral. Therefore, the structure of pellets was inspected using SEM. As an example, the pellet prepared from urolith no. 7851 containing 95%m/m of oxalate and 5%m/m of phosphate is discussed. The BSE large-scale image of the surface of this pellet does not indicate apparent lack of homogeneity, although bright grains consisting of phosphate and dark oxalate grains are easily distinguishable (Fig. 1a). The detailed image demonstrates almost uniform distribution of phosphate-rich grains characterized by bright spots (≪1 μm) within the dark oxalate area (Fig. 1b). As the ablation event occurs on the area of (65 × 65) μm2 (marked with a square in Fig. 1b), the inhomogeneity at a low micrometric scale is acceptable and would not result in isotopic signal instability and biased results. Indeed, as it is obvious from Fig. 2 where three line scans over the surface of pellet no. 7851 are recorded, the time-resolved signal of minor and trace elements such as Na (0.13%m/m), Sr (0.003%m/m), and Pb (0.0005%m/m) exhibits a continuous course uninterrupted by any significant spike, which would otherwise indicate a time/space resolved ablation of a grain enriched with a particular element. This confirms a good “targeted” homogeneity, although these elements are predominantly associated with minor phosphates.53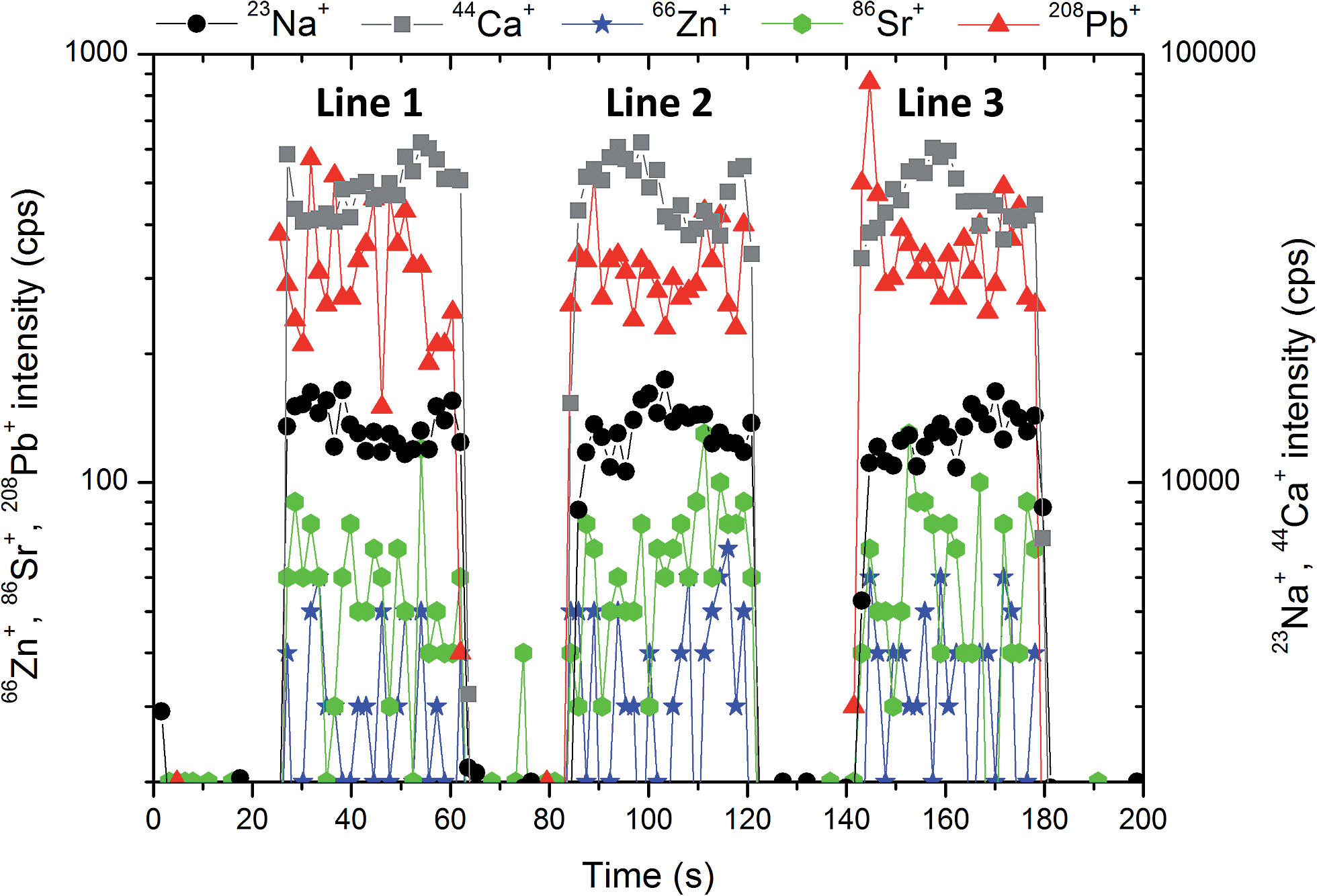 | ||
| Fig. 2 The stability of the signal of 23Na+, 44Ca+, 66Zn+, 86Sr+, 208Pb+ during the ablation of pellet no. 7851 within 3 lines with the length of 1.5 mm, approximately. | ||
Compactness of pellets, their stability during ablation and relative insignificance of microinhomogeneity in comparison with the size of the ablation spot in Fig. 1b can also be assessed on the basis of signal variability of the main constituents. The major element present in both phosphate uroliths (with the exception of urolith no. 5397, which consists primarily of struvite) and oxalate uroliths is calcium (∼12–27%m/m Ca), while one of the organic matrix elements in uric acid samples is carbon (∼25%m/m C), as it follows from Tables 1 and 2.
As phosphate pellets contain apatite and struvite in various proportions and also include ∼10% of weddelite in several cases, a fluctuation of 44Ca+ signal along the line scans over the surface of a particular pellet indicates the degree of homogeneity of such a mixture and pellet compactness (presence of cracks, crumbling by the interaction with a laser beam). The RSD of the 44Ca+ signal measured with all phosphate pellets falls within the range of 0.5–10.8% for three ablation tracks located at different areas of the pellet surface. As an example, development of the 44Ca+ signal in time along the mentioned ablation lines is shown for urolith pellet no. 7851 in Fig. 2. The oxalate pellets contain predominantly whewellite, while weddelite content ranges up to 25% and the phosphate mineral group constitutes from 5 to 30% of the total. Signal variability of 44Ca+ in oxalate pellets is characterized by RSD ranging from 0.6 to 4.9%. Calcium is a minor element (0.1–1.3%m/m) in uric acid group uroliths, and therefore 44Ca+ signal exhibits the most significant fluctuation with RSD between 4.4% and 51%. On the other hand, RSD of 12C+ signal ranging from 3.3 to 7.1% documents quite a good stability and homogeneity of uric acid pellets. Nevertheless, urate-based pellets exhibit the highest fluctuation of 12C+ signal in comparison with phosphate (0.4–2.0%) and oxalate (0.6–2.5%) groups, which is probably due to the presence of some fine cracks on the pellet surface. The internal precision, which reflects the microscale homogeneity, is expressed as the median value of %RSD of 12C+ isotopic signal fluctuation, and is equal to 4.5%, 4.1% and 9.9% for groups of uroliths with prevailing phosphate, oxalate and urate matrices, respectively. It should be generally noted that calcium and carbon signal fluctuation reflects, besides pellet compactness and homogeneity of their distributions in all kinds of pellets, also both the influence of ablation rate variability and overall instrumental precision of the LA-ICP-MS system. Obviously, Ca and C signal stability can be considered an appropriate common indicator of a particular pellet quality and suitability for calibration.
The occurrence of kidney stones with only one matrix is quite uncommon. It is not an exception that more than two matrices can be found in uroliths. Therefore, we attempted to create a single calibration which would be applicable to various types of kidney stones. The concept of matrix-matched calibration in our approach means that a single calibration would be able to cover phosphate, oxalate and urate matrices. A necessary prerequisite for application of the common calibration is the same or at least close sensitivity for these matrices.
The pellets were divided into three groups according to their mineralogical composition: (i) 9 pellets with a predominant phosphate matrix; (ii) 5 pellets with the prevailing oxalate matrix; and (iii) 4 pellets with the main content being uric acid (Table 1). The response in these three groups of uroliths was evaluated using the 44Ca+ signal. Sensitivity was calculated for each pellet as the ratio of the 44Ca+ net signal to the Ca content in the pellet, the values in each group were averaged and standard deviations were calculated and plotted (Fig. 3), while sensitivities in oxalate and uric acid matrices were normalized to the sensitivity in the phosphate group. Thus, the normalized sensitivity in the phosphate group is equal to one. The Ca contents in pellets with phosphate and oxalate as the main constituents are both high and close, while the pellets with uric acid/oxalates contain only 0.1–1.3%m/m Ca. As observed, the highest sensitivity was obtained for the group of phosphate-based pellets while the sensitivity in oxalate and uric acid pellets reaches 81% and 96% of the maximum value (Fig. 3). However, non-parametric Kruskal–Wallis test indicated no statistically significant difference between sensitivities.
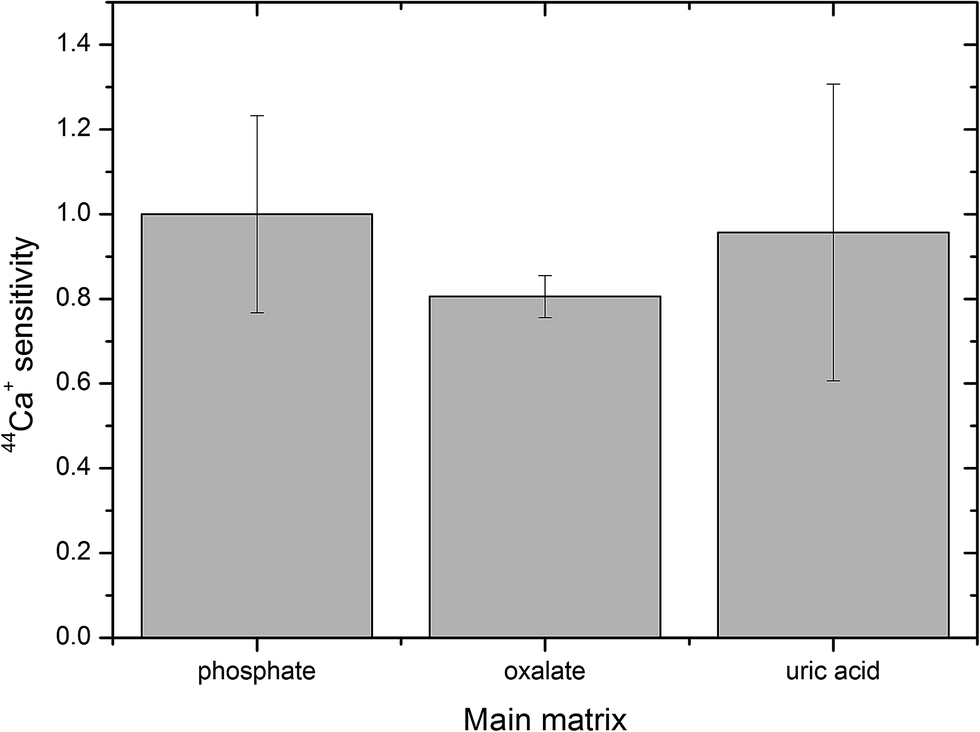 | ||
| Fig. 3 Sensitivity of the calcium isotopic signal in oxalate and uric acid matrices normalized to the sensitivity in phosphate, error bars represent standard deviation. | ||
Development of matrix-matched calibration kits
Eighteen pellets prepared from urolith specimens listed in Table 1 were used for the construction of calibration plots for P, Na, Sr, Zn, Ba and Pb. Calibration kits for particular analytes are listed in Table 4. Calibration for Mg of the same quality was obtained too (not presented in this work). The LA-ICP-MS measurement was repeated three times on each pellet using three parallel lines with the length of 1.2 mm under conditions specified in the Experimental part. Background intensity was recorded prior to ablation and then subtracted from the signal measured during the ablation period. The sensitivity of the 44Ca+ signal was applied as an internal reference for the correction of matrix effects.| Element | Calibration standards Ph/Ox/UAa | R 2 outliers included | R 2 outliers left out | Regression range %m/m | LOD %m/m |
|---|---|---|---|---|---|
| a Ph = phosphate matrix, Ox = oxalate matrix, UA = uric acid matrix. b LOD in mg kg−1 calculated as the triple standard error of regression divided by the slope of the regression line, 3sy/x/b, considering the zero value intercept. For completeness of information, regression ranges are presented here while the actual calibrations are limited to values above LODs. | |||||
| P | 0/5/4 | 0.9991 | 0.9991 | 0.014–2.6 | <0.1 |
| Na | 6/5/4 | 0.9936 | 0.9955 | 0.013–1.5 | <0.1 |
| Sr | 6/4/4 | 0.9896 | 0.9960 | 0.0001–0.052 | <0.004 |
| Zn | 6/4/4 | 0.9868 | 0.9966 | 0.0003–0.094 | <0.007 |
| Ba | 7/4/4 | 0.9777 | 0.9957 | 0.05–24b | <2b |
| Pb | 9/1/3 | 0.9861 | 0.9974 | 0.18–67b | <4b |
Calibration points were plotted as analyte signals divided by 44Ca+ sensitivities vs. elemental contents. Calibration curves were fitted using a computer program Origin. Linear regression models that resulted from the tests and two sets of coefficients of determination (R2) were computed; one set with all data points included, and the other after outliers had been eliminated (Table 4). In Fig. 4, calibration curves of P, Na, Sr, Zn, Ba and Pb are plotted together with confidence and prediction bands without outliers. Ordinary least squares linear regression was applied and the regression line was fitted for calibration points representing arithmetic means of 3 replicates for each pellet. Each replicate represents the average value of all MS readings obtained during the line scan ablation measurement. Vertical error bars represent the combined uncertainty of replicates calculated based on the standard deviation of the signal of isotope (analyte) and standard deviation of calcium isotope signal (internal reference). Horizontal error bars represent uncertainty of ICP-MS solution analysis. Regression lines computed without outliers show tighter correlation, which is obvious from values of R2 > 0.99, namely in the range 0.9955–0.9991 (Table 4). At this point it should be emphasized that calibrations for Sr, Mg, Ba, and Na obtained with coarse-grained pellets40 exhibit worse values of R2 (0.75–0.90). All calibration plots are described by the equation y = bx with a zero value of the intercept at the level of confidence (1 − α) = 0.975. The intercepts are not statistically significant regardless of whether the outliers are retained in the regression sets or excluded on the basis of statistical testing. The absence of intercepts together with the proved validity of linear models indicates that the internal reference compensates for different ablation rates associated with the three various mineral matrices, and thus eliminates systematic error. The calibration line for phosphorus was fitted only with data points for oxalate and uric acid pellets, while phosphate data points were omitted as phosphorus is the main constituent in phosphate uroliths. In Fig. 4, calibration curves of P, Na, Sr, Zn, Ba and Pb are plotted together with confidence and prediction bands without outliers. Limits of detection were calculated as the triple standard error of regression divided by the slope of the regression line, 3sy/x/b, considering the zero value intercept. It has to be emphasized that limits of detection calculated through the calibration lines generally depend on both the number and distribution of calibration standards and are in principle higher than those (instrumental) obtained simply based on the triple standard deviation of the LA-ICP-MS blank signal recorded when ablation is off. Consequently, the LOD values in Table 4 follow from regression calculations performed in the presented regression ranges, and are associated with concrete calibration lines displayed in Fig. 4, while LOD values at a particular mapping may differ.
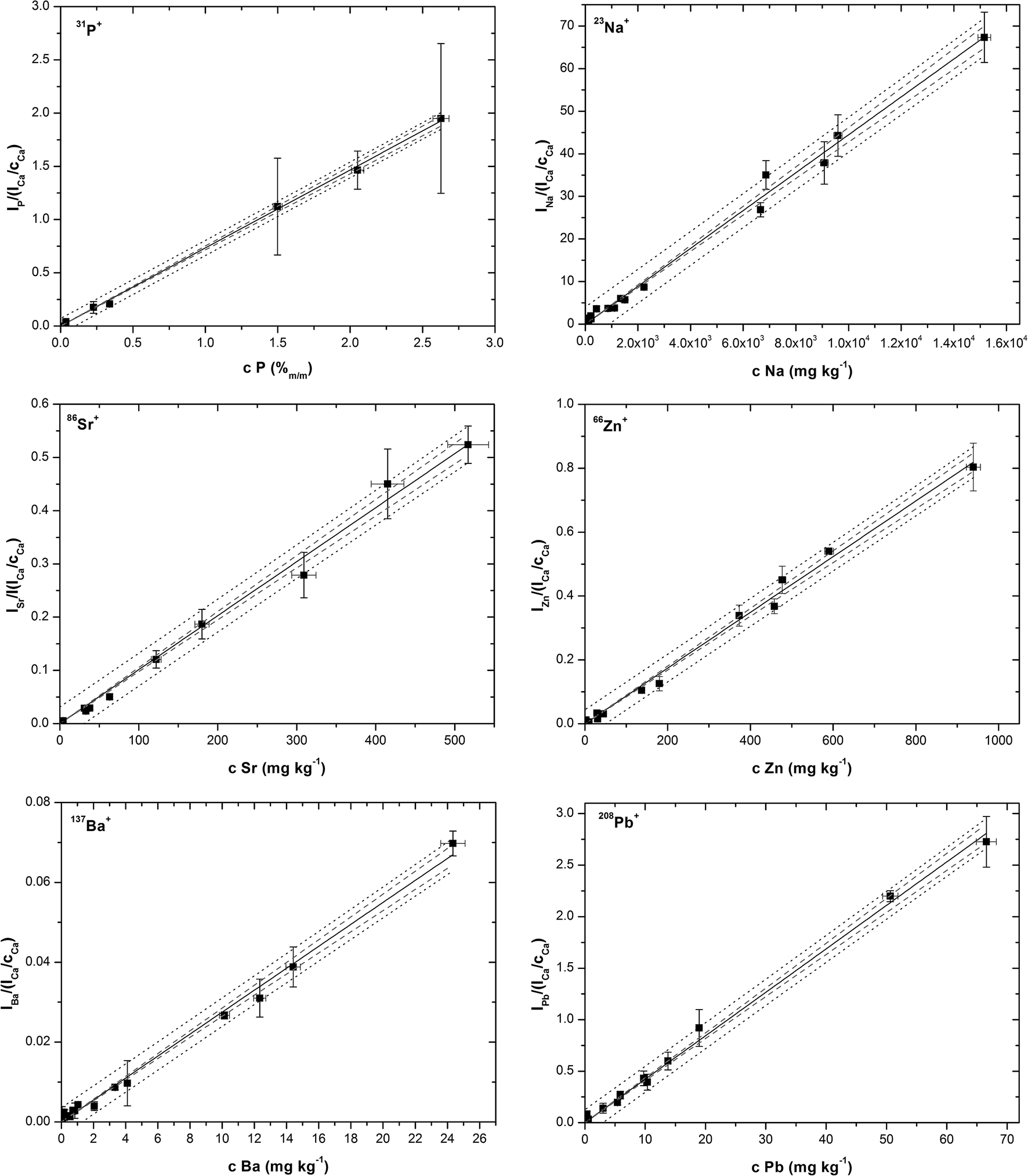 | ||
| Fig. 4 Calibration lines for P, Na, Sr, Zn, Ba and Pb after statistical analysis and outliers exclusion. | ||
Verification of matrix-matched calibrations by the analysis of NIST SRM 1486
Practical application of the presented calibration kits is limited by the size of the ablation cell (SuperCell), which holds only five pellets because the PMM block with the urolith section occupies a relatively large space (∼5 cm2). To minimize both the analysis time and the effect of handling the ablation cell on measurement stability, the calibration pellets and the urolith section should be inserted into the ablation cell at the same time. Based on the satisfactory results of the calibration dependence studies, we decided to explore the possibility of reducing the number of calibration points to four. In the extreme case, calibration with a single pellet would bring, besides reducing the time of analysis, in particular the possibility to place more samples for mapping into the ablation cell. Therefore, four calibration pellets were selected to represent both the phosphate (3 pellets, 6686, 8393 and 9130) and the oxalate (1 pellet, 6275) matrices. The fifth pellet was NIST SRM 1486, the bone meal, which was employed to prove whether the calibration kit is really matrix-matched, in the first approximation as regards the analysis of the powdered material with the phosphate matrix.The ablation pattern applied to pellets consisted of ten parallel lines 2.8 mm in length. Measurement was performed under conditions specified in the Experimental part. Two calibration procedures were examined: (i) the four-point calibration comprising three phosphate and one oxalate pellets; (ii) four single-point calibrations based on particular phosphate and oxalate pellets. Relative intensities representing ratios of signals of individual isotopes and the 44Ca+ sensitivity were processed. Isotope 44Ca+ sensitivity correlates reasonably with signals of analytes. This means that the internal reference follows analyte signal variations, which are due to both matrix-dependent ablation rate, and the fluctuations of instrumental operating conditions of the LA-ICP-MS system.
The NIST SRM 1486 elemental contents resulting from calibrations (i) and (ii) are shown together with the certified and published uncertified values in Table 5. Besides the four-point calibration with the coefficient of determination in the range of 0.990–0.999, best fit is obtained with single-point calibrations using phosphate pellets, while oxalate matrix calibration leads to an underestimation of Ba content due to the Ba content in pellet no. 6275 being below LOD (Table 4). Similarly, the Pb content in NIST 1486 is three times below LOD and cannot be determined under the method conditions used. Finally, the calibration with phosphate pellet no. 9130 was selected for mapping of the urolith section.
| Element | Certified values | Four-point calibration | 6686 Phosphate | 8393 Phosphate | 9130 Phosphate | 6275 Oxalate |
|---|---|---|---|---|---|---|
| mg kg−1 | ||||||
| a In %m/m. b Non-certified content. c The value is not included in the NIST SRM 1486 certificate; Ba content presented in Table 5 is taken from Georem.54 | ||||||
| P | 12.30 ± 0.19a | 12.07 ± 0.68a | 12.06 ± 0.69a | 11.66 ± 0.66a | 12.19 ± 0.69a | 12.94 ± 0.74a |
| Na | (0.50)a,b | 0.48 ± 0.03a | 0.45 ± 0.02a | 0.34 ± 0.02a | 0.45 ± 0.02a | 0.32 ± 0.02a |
| Sr | 264 ± 7 | 273 ± 17 | 285 ± 18 | 274 ± 17 | 265 ± 16 | 269 ± 17 |
| Zn | 147 ± 16 | 148 ± 18 | 210 ± 17 | 153 ± 12 | 151 ± 12 | 128 ± 10 |
| Ba | 189–314c | 249 ± 12 | 235 ± 17 | 196 ± 14 | 227 ± 16 | 52.7 ± 3.8 |
| Pb | 1.335 ± 0.014 | 3.64 ± 0.48 | 2.68 ± 0.35 | 0.511 ± 0.068 | 0.99 ± 0.13 | 0.445 ± 0.059 |
Quantification of 2D elemental distributions
Besides the sensitivities of particular analytes measured in calibration pellets, the sensitivities of analytes obtained for sections of uroliths play an important role in quantification. The section of kidney stone exhibits different physical properties than the pressed pellet. Ablation rate depends on the mineral phase and its density and hardness. Consequently, different ablation rates in various minerals can be expected in a given urolith section in comparison with the pressed pellet.The elemental map of the part of the urolith no. 11605 section surface (1.4 × 1.9) mm2 was quantified. The photograph of the whole sample surface with the analyzed area marked with a rectangle is presented in Fig. 5a. Sample no. 11605 contains 30%m/m of phosphate and 70%m/m of oxalate of which 40%m/m is whewellite and 30%m/m weddellite.
 | ||
| Fig. 5 (a) Photograph of no. 11605 kidney stone surface with the marked area analyzed via LA-ICP-MS and EMP imaging and (b) BSE image marked surface with the description of phases (Ph – phosphate and Ox – oxalate). | ||
As shown in Fig. 5b, the analyzed area is composed mainly of oxalate (whewellite > weddellite) and fluorapatite according to EMP analysis. Fluorapatite predominates in the central part of the kidney stone. Calculation of the structural formula from the results of EMP analysis indicates a rather fine-grained mixture of predominant fluorapatite with a minor amount of Mg-rich phosphate (probably struvite) compared to pure fluorapatite. The chemical heterogeneity of fluorapatite observed in the BSE image reflects a varying admixture of Mg-rich phosphate (a higher amount of Mg-rich phosphate decreases the brightness of the BSE image). Fluorapatite is overgrown by fine-grained, colomorph oxalate, possibly weddellite. Coarse-grained compact oxalate, probably whewellite, predominates in the kidney stone. The outermost part of the kidney stone is composed of intergrowths of both types of oxalate (whewellite and weddellite). Contents of minor elements in whewellite and weddellite are similar and generally decrease from the inner part of the kidney stone (close to the fluorapatite) to the rim.
A single-point calibration was used with respect to the low content of some elements in the oxalate matrix and with the aim to reduce the time of analysis. For this purpose, phosphate pellet no. 9130 was used as the calibration standard.
Elemental 2D maps are shown in Fig. 6, where Ca distribution is presented on the level of intensities (Fig. 6a), while P, Na, Sr, Zn, Ba and Pb (Fig. 6b–g) are quantified. It should be noted right at the beginning that the quantification of the inhomogeneous surface is hampered by the presence of defects (cracks) in phases. Besides, spatial resolution in the direction of x-axis is deteriorated by using the line scanning mode which induces overlapping of signals on the boundary of the phases. The spatial resolution of the elemental maps in the scanned area is also affected by the fact that lines are spaced 100 μm. As a result, the image of the boundary is not sharp.
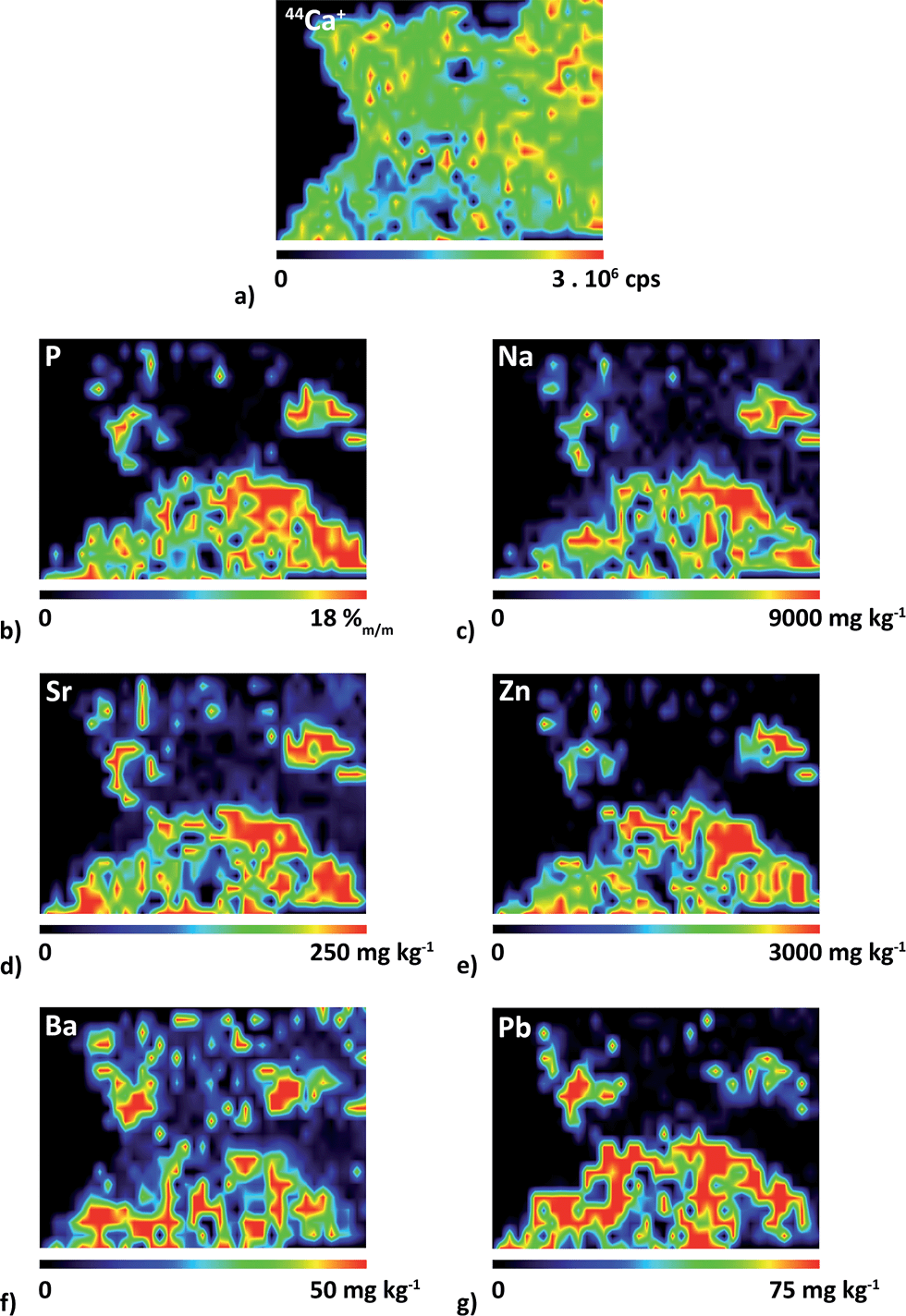 | ||
| Fig. 6 Distributions of (a) 44Ca+ and of elemental contents of (b) P, (c) Na, (d) Sr, (e) Zn, (f) Ba and (g) Pb which were quantified using the single-point calibration with pellet no. 9130. | ||
The quantification procedure is as follows. Prior to quantification, the signal corresponding to PMM resin was removed and thus filtered values were divided into two groups characterizing section areas. The area comprising 31P+ signal corresponds exclusively to the phosphate matrix whereas the area covered by the 44Ca+ signal defines the whole sample section surface as calcium is common for both oxalate and phosphate phases. This distinction allows quantifying elemental contents separately in particular phases.
A correction for different ablation rates in oxalate and phosphate matrices of the urolith section and in calibration pellets is of utmost importance for obtaining quantitative elemental distributions. The average ablation rate in the phases of the sample section and in the pellet was determined on the basis of a large number of measurements. For this purpose, 50 and 80 values of 44Ca+ signal in phosphate and oxalate phases were employed, respectively. Considering the Ca content in both phases obtained via EMP analysis and median intensity values of 44Ca+ for each of the phases, the value of 1.3 was found which indicates that the Ca sensitivity in the phosphate phase is higher than that in the oxalate phase. In other words, the ablation rate in the oxalate phase reaches 80% of that in the phosphate phase. This is in good agreement with a comparison of Ca sensitivities for pellets (Fig. 3). As a result, 44Ca+ distribution (Fig. 6a) corrected for the difference in the average ablation rate exhibits slightly higher intensity values in the phosphate-rich domain, and the intensity drops in small areas with material defects (cracks). The comparison of Ca (Fig. 6a) and P (Fig. 6b) distributions reveals that 42% of the examined part of the section surface corresponds to the phosphate matrix. It is apparent that the found elemental distributions of Na, Sr, Zn, Ba and Pb (Fig. 6c–g) are concentrated to the area corresponding to the phosphate-based matrix (Fig. 6b).
Results of analyses of phosphate and oxalate phases in urolith no. 11605 are presented in Table 6. To verify that the quantification of 2D maps (Fig. 6), which is based on pellet no. 9130, is not distorted by this single-point calibration, median values of elemental contents in both phases were also determined using three other single-point calibrations with pellets no. 6686, 8393 and 6275, employed for the analysis of NIST SRM 1486. The range of thus obtained four median values is presented in Table 6 together with their arithmetic mean and corresponding standard deviation for each analyte. It is obvious from these low SD values that all the used single-pellet calibrations provide close results. Content variability over the phase area is expressed as the median absolute deviation (MAD), which is the median of the absolute deviations from the content's median. The median was selected in order to minimize the influence of the presence of cracks and distortion of signal by line scanning mode. For comparison, average values (arithmetic mean) with standard deviations are presented for EMP analyses of the studied phases. However, contents of Ba and Pb in phosphate and Sr, Zn, Ba and Pb in oxalate are below LOD of EMP analysis. The agreement between LA-ICP-MS and EMP analysis results is satisfactory when considering the fact that the variability in elemental contents within phosphate and oxalate area phases reaches at least 32% (P), 51% (Na) and 44% (Zn), respectively. As the quantification relies on the single-point calibration instead of the multi-point one, LODs of LA-ICP-MS measurement calculated as the triple standard deviation of background intensity divided by sensitivity were as follows: P 200 mg kg−1, Na 20 mg kg−1, Sr 4 mg kg−1, Zn 0.7 mg kg−1, Ba 0.05 mg kg−1 and Pb 0.03 mg kg−1.
| Element | Median content range | Arithmetic mean of median contents | SD | Variability in content MAD | Arithmetic mean content |
|---|---|---|---|---|---|
| LA-ICP-MS | EMP analysis | ||||
| mg kg−1 | % | mg kg−1 | |||
| a %m/m. | |||||
| Phosphate area | |||||
| P | 17.5–19.4a | 18.36a | 0.80a | 32 | 17.39 ± 0.22a |
| Na | 0.56–0.79a | 0.68a | 0.12a | 74 | 0.93 ± 0.06a |
| Sr | 244–262 | 251.7 | 7.8 | 71 | <LOD |
| Zn | 2303–3791 | 2890 | 630 | 47 | 2740 ± 630 |
| Ba | 7–30 | 23 | 11 | 49 | <LOD |
| Pb | 26–59 | 39 | 18 | 76 | <LOD |
| Oxalate area | |||||
| P | 1342–1489 | 1405 | 61 | 65 | 2400 ± 1200 |
| Na | 694–973 | 840 | 148 | 51 | 870 ± 330 |
| Sr | 39–41 | 39.8 | 1.2 | 82 | <LOD |
| Zn | 26–43 | 32.4 | 7.1 | 44 | <LOD |
| Ba | 2–11 | 8.3 | 4.0 | 55 | <LOD |
| Pb | 1–3 | 2.15 | 0.98 | 63 | <LOD |
Conclusions
The kidney stone investigation plays an important role in medical treatment when the process of formation and growth of uroliths as well as the influence of environmental pollution, for instance, have to be known. In this study, a new flexible matrix-matched calibration was designed, tested and used to quantify the distribution of elements in uroliths by laser ablation-inductively coupled plasma-mass spectrometry. Two-dimensional distributions of isotopic signals obtained from the selected area of the urolith section surface were converted to maps of elemental contents using the matrix-matched calibration. Calibration kits comprising 9–15 pressed pellets (depending on a particular element) were tested for homogeneity, signal stability and linear response with satisfactory results. Pellets were prepared without any binder from powdered uroliths consisting of phosphate, oxalate and urate phases. Despite the fact that the individual calibration pellets differ significantly in proportions of mineral phases and therefore also in physical and physico-chemical properties, a linear calibration model in the range of three orders of magnitude and with zero intercept was obtained for P, Na, Mg, Zn, Ba and Pb. The low scatter of calibration points is characterized by coefficients of determination >0.9955 and narrow confidence and prediction bands. This tight correlation was achieved through internal standardization. Correction of isotopic signals for different ablation rates in phosphate, oxalate and urate matrices of calibration pellets was successfully accomplished by internal standardization with the response factor of 44Ca+. For this purpose, calcium contents in particular uroliths employed for the preparation of calibration pellets were determined by solution analysis, while the average content of calcium in oxalate and phosphate phases of the mapped urolith section surface was determined using an electron microprobe.To reduce the time of analysis, the four-point and four single-point calibrations were tested by the analysis of a pressed pellet of bone meal NIST SRM 1486. The four-point calibration with the coefficient of determination in the range of 0.990–0.999 and single-point calibrations with phosphate matrix pellets yielded better trueness than those for the oxalate pellet. This proved the feasibility of quantification of elemental maps using calibration with a single pellet. The trueness of quantitative elemental maps obtained by LA-ICP-MS was indirectly proved by comparison of averaged elemental contents in phosphate/oxalate phases with those determined by an electron microprobe. Limits of detection of minor or trace elements (Sr, Zn, Pb, and Ba) calculated from the standard error of regression (calibration) were found to be between units and hundreds of mg kg−1.
The proposed calibration exhibits advantages over using the bone meal NIST SRM 1486. Unlike this SRM, calibration samples prepared from uroliths cover an existing wide range of elemental contents in the analyzed samples without extrapolation, which is on the other hand unavoidable in the case of Na, Mg, K, Pb, Sr and Zn quantification using NIST 1486. The number of calibration standards can be flexibly adapted to elemental contents in a particular urolith. Besides P, Na, Mg, Zn, Ba and Pb, urolith pressed pellets can also encompass linear multi-point calibrations for C, K, Al, Mn, Fe, Cu, Rb and Sn, while NIST SRM 1486 covers only Ca, Mg, P, Fe, K, Pb, Sr, and Zn with certified values. However, the real content of K, Pb, Sr, and Zn in analyzed uroliths is frequently above particular certified values.
Acknowledgements
The authors acknowledge the Czech Science Foundation for a grant to support the project 203/09/1394. This work was also supported by the European Regional Development Fund project “CEITEC” (CZ.1.05/1.1.00/02.0068). R. C. acknowledges support from Specific research programme MUNI/A/1005/2013 of the Masaryk University.References
- M. Debeljak, J. T. van Elteren and K. Vogel-Mikus, Anal. Chim. Acta, 2013, 787, 155–162 CrossRef CAS PubMed.
- A. Hanc, A. Piechalak, B. Tomaszewska and D. Baralkiewicz, Int. J. Mass Spectrom., 2014, 363, 16–22 CrossRef CAS PubMed.
- M. Galiova, J. Kaiser, K. Novotny, M. Hartl, R. Kizek and P. Babula, Microsc. Res. Tech., 2011, 74, 845–852 CAS.
- A. Kötschau, G. Büchel, J. W. Einax, C. Fischer, W. von Tümpling and D. Merten, Microchem. J., 2013, 110, 783–789 CrossRef PubMed.
- S. R. Ellis, A. L. Bruinen and R. M. A. Heeren, Anal. Bioanal. Chem., 2014, 406, 1275–1289 CrossRef CAS PubMed.
- J. Koelmel, T. Leland and H. Wang, Environ. Pollut., 2013, 174, 222–228 CrossRef CAS PubMed.
- A. Hanc, I. Komorowicz, M. Iskra, W. Majewski and D. Baralkiewicz, Anal. Bioanal. Chem., 2011, 399, 3221–3231 CrossRef CAS PubMed.
- S. Gruhl, F. Witte, J. Vogt and C. Vogt, J. Anal. At. Spectrom., 2009, 24, 181–188 RSC.
- J. Chou, C. Austin, P. Doble, B. Ben-Nissan and B. Milthorpe, J. Tissue Eng. Regener. Med., 2014, 8, 515–520 CAS.
- E. G. Grosch, J. Kosler, N. McLoughlin, K. Drost and J. Slama, Precambrian Res., 2011, 191, 85–99 CrossRef CAS PubMed.
- C. M. Fisher, H. P. Longerich, S. E. Jackson and J. M. Hanchar, J. Anal. At. Spectrom., 2010, 25, 1905–1920 RSC.
- M. Bertini, A. Izmer, F. Vanhaecke and E. M. Krupp, J. Anal. At. Spectrom., 2013, 28, 77–91 RSC.
- A. Nevin, G. Spoto and D. Anglos, Appl. Phys. A: Mater. Sci. Process., 2012, 106, 339–361 CrossRef CAS.
- C. Chataigner, M. Isikli, B. Gratuze and V. Cil, Archaeometry, 2014, 56, 351–374 CrossRef PubMed.
- C. Stadlbauer, C. Reiter, B. Patzak, G. Stingeder and T. Prohaska, Anal. Bioanal. Chem., 2007, 388, 593–602 CrossRef CAS PubMed.
- T. Prohaska, C. Latkoczy, G. Schultheis, M. Teschler-Nicola and G. Stingeder, J. Anal. At. Spectrom., 2002, 17, 887–891 RSC.
- M. Vasinova Galiova, M. Nyvltova Fisakova, J. Kynicky, L. Prokes, H. Neff, A. Z. Mason, P. Gadas, J. Kosler and V. Kanicky, Talanta, 2013, 105, 235–243 CrossRef CAS PubMed.
- A. E. Dolphin and A. H. Goodman, Am. J. Phys. Anthropol., 2009, 140, 399–409 CrossRef PubMed.
- A. E. Dolphin, A. H. Goodman and D. D. Amarasiriwardena, Am. J. Phys. Anthropol., 2005, 128, 878–888 CrossRef PubMed.
- D. Hare, C. Austin, P. Doble and M. Arora, J. Dent., 2011, 39, 397–403 CrossRef CAS PubMed.
- A. Arvidsson, B. Liedberg, K. Moller, B. Lyven, A. Sellen and A. Wennerberg, J. Dent., 2002, 30, 67–75 CrossRef CAS.
- Z. A. Doubleday, C. Izzo, S. H. Woodcock and B. M. Gillanders, Aquat. Biol., 2013, 18, 271–280 CrossRef.
- A. D. Clarke, K. H. Telmer and J. M. Shrimpton, Ecol. Freshwat. Fish, 2007, 16, 354–361 CrossRef PubMed.
- B. Wu, J. S. Becker and J. Sabine, Int. J. Mass Spectrom., 2012, 323, 34–40 CrossRef PubMed.
- S. D. Urgast, D. G. Ellingsen, B. Berlinger, E. Eilertsen, G. Friisk, V. Skaug, Y. Thomassen, J. H. Beattie, I. Kwun and J. Feldmann, Anal. Bioanal. Chem., 2012, 404, 89–99 CrossRef PubMed.
- O. Reifschneider, C. A. Wehe, I. Raj, J. Ehmcke, G. Ciarimboli, M. Sperling and U. Karst, Metallomics, 2013, 5, 1440–1447 RSC.
- D. Drescher, C. Giesen, H. Traub, U. Panne, J. Kneipp and N. Jakubowski, Anal. Chem., 2012, 84, 9684–9688 CrossRef CAS PubMed.
- L. Prien and C. Frondel, J. Urol., 1947, 57, 949–994 Search PubMed.
- M. L. Stoller and M. V. Meng, Urinary Stone Disease: the Practical Guide to Medical and Surgical Management, Humana press, New Jersey, 2007 Search PubMed.
- N. Mandel, Lab. Med., 1986, 17, 449–458 CAS.
- S. Tamosaityte, V. Hendrixson, A. Zelvys, R. Tyla, Z. A. Kucinskiene, F. Jankevicius, M. Pucetaite, V. Jablonskiene and V. Sablinskas, J. Biomed. Opt., 2013, 18, 027011 CrossRef PubMed.
- C. Conti, M. Casati, C. Colombo, M. Realini, L. Brambilla and G. Zerbi, Spectrochim. Acta, Part A, 2014, 128, 413–419 CrossRef CAS PubMed.
- R. Selvaraju, A. Raja and G. Thiruppathi, Spectrochim. Acta, Part A, 2013, 114, 650–657 CrossRef CAS PubMed.
- S. D. Blaschko, J. Miller, T. Chi, L. Flechner, S. Fakra, A. Kahn, P. Kapahi and M. L. Stoller, J. Urol., 2013, 189, 726–734 CrossRef CAS PubMed.
- K. Kaneko, Y. Matsuta, M. Moriyama, M. Yasuda, N. Chishima, N. Yamaoka, T. Fukuuchi, K. Miyazawa and K. Suzuki, Int. J. Urol., 2014, 21, 341–346 CrossRef PubMed.
- E. S. Wisenbaugh, R. G. Paden, A. C. Silva and M. R. Humphreys, Urology, 2014, 83, 1243–1247 CrossRef PubMed.
- V. Uvarov, I. Popov, N. Shapur, T. Abdin, O. N. Gofrit, D. Pode and M. Duvdevani, Environ. Geochem. Health, 2011, 33, 613–622 CrossRef CAS PubMed.
- B. G. Oztoprak, J. Gonzalez, J. Yoo, T. Gulecen, N. Mutlu, E. E. Russo, O. Gundogdu and A. Demir, Appl. Spectrosc., 2012, 66, 1353–1361 CrossRef CAS PubMed.
- J. Anzano and R. J. Lasheras, Talanta, 2009, 79, 352–360 CrossRef CAS PubMed.
- K. Stepankova, K. Novotny, M. Vasinova Galiova, V. Kanicky, J. Kaiser and D. W. Hahn, Spectrochim. Acta, Part B, 2013, 81, 43–49 CrossRef CAS PubMed.
- M. A. Chaudhri, J. Watling and F. A. Khan, J. Radioanal. Nucl. Chem., 2007, 271, 713–720 CrossRef CAS.
- M. Vasinova Galiova, R. Copjakova, R. Skoda, K. Stepankova, M. Vankova, J. Kuta, L. Prokes, J. Kynicky and V. Kanicky, Spectrochim. Acta, Part A, 2014, 100, 105–115 CrossRef CAS PubMed.
- A. Izmer, D. Gholap, K. De Houwer, F. Cuyckens and F. Vanhaecke, J. Anal. At. Spectrom., 2012, 27, 413–418 RSC.
- H. J. Stärk and R. Wennrich, Anal. Bioanal. Chem., 2011, 399, 2211–2217 CrossRef PubMed.
- E. Moreno-Gordaliza, C. Giesen, A. Lazaro, D. Esteban-Fernandez, B. Humanes, B. Canas, U. Panne, A. Tejedor, N. Jakubowski and M. M. Gomez-Gomez, Anal. Chem., 2011, 83, 7933–7940 CrossRef CAS PubMed.
- D. J. Hare, J. Lear, D. Bishop, A. Beavis and P. A. Doble, Anal. Methods, 2013, 5, 1915–1921 RSC.
- D. Tabersky, N. A. Luechinger, M. Rossier, E. Reusser, K. Hametner, B. Aeschlimann, D. A. Frick, S. C. Halim, J. Thompson, L. Danyushevsky and D. Guenther, J. Anal. At. Spectrom., 2014, 29, 955–962 RSC.
- I. Hubova, M. Hola, J. Pinkas and V. Kanicky, J. Anal. At. Spectrom., 2007, 22, 1238–1243 RSC.
- H. Sela, Z. Karpas, H. Cohen, Y. Zakon and Y. Zeiri, Int. J. Mass Spectrom., 2011, 307, 142–148 CrossRef CAS PubMed.
- J. A. T. Pugh, A. G. Cox, C. W. McLeod, J. Bunch, B. Whitby, B. Gordon, T. Kalber and E. White, J. Anal. At. Spectrom., 2011, 26, 1667–1673 RSC.
- J. Kuta, J. Machát, D. Benova, R. Červenka and T. Kořistková, Cent. Eur. J. Chem., 2012, 10, 1475–1483 CrossRef CAS PubMed.
- J. L. Pouchou and F. Pichoir, in PAP, Procedure for Improved Quantitative Microanalysis, ed. J. T. Armstrong, Microbeam Analysis Proc. San Francisco Press, San Francisco, 1985, pp. 104–106 Search PubMed.
- J. Kuta, J. Machat, D. Benova, R. Cervenka, J. Zeman and P. Martinec, Environ. Geochem. Health, 2013, 35, 511–522 CrossRef CAS PubMed.
- http://georem.mpch-mainz.gwdg.de/sample_query.asp, 16/10/2014.
| This journal is © The Royal Society of Chemistry 2015 |