
Solving the cellulose I polymorphic structural riddle: disorder in hydrogen bond networks activates diagnostic terahertz dynamics†
Emily A. Verhaeg a,
Hiromichi Hoshina*b,
Jun Kikuchic,
Luca Catalano*de and
Michael T. Ruggiero*ef
a,
Hiromichi Hoshina*b,
Jun Kikuchic,
Luca Catalano*de and
Michael T. Ruggiero*ef
aDepartment of Chemistry, University of Vermont, 82 University Place, Burlington, Vermont 05405, USA
bRIKEN, Center for Advanced Photonics, 519-1399 Aramaki-Aoba, Aoba-ku, Sendai, Miyagi 9800845, Japan. E-mail: hoshina@riken.jp
cRIKEN, Center for Sustainable Resource Science, 1-7-22 Suehiro, Tsurumi, Yokohama, Kanagawa 230-0045, Japan
dDynamic Molecular Materials Laboratory, Department of Life Sciences, University of Modena and Reggio Emilia, Modena 41125, Italy. E-mail: luca.catalano@unimore.it
eDepartment of Chemistry, University of Rochester, Rochester, NY 14627, USA. E-mail: michael.ruggiero@uvm.edu
fDepartment of Chemical Engineering, University of Rochester, Rochester, NY 14627, USA
First published on 25th June 2025
Abstract
Cellulose is a common polymer found in natural sources, with the potential to be used in a wide variety of green and technologically relevant applications. Despite years of effort, the precise three-dimensional structures of two crystalline polymorphs of cellulose, Iα and Iβ, are currently still unknown due to the presence of disorder in the intermolecular hydrogen bond networks, hampering the in-depth understanding of the structure–property relationship of this crystalline material. Disorder in the hydrogen bond networks of cellulose I polymorphs was investigated using terahertz spectroscopy, powder X-ray diffraction, and solid-state density functional theory in order to reveal previously undiscovered atomic-level details about the crystal structures. We show that the nature of the (dis)order in the hydrogen-bonded layers has a direct effect on the terahertz vibrational spectra, providing contrast that allows differentiating between various structures spectroscopically. Comparison between theoretical and experimental data indicates that these two static networks coexist spatially throughout cellulose I polymorphs.
Introduction
Cellulose is the most abundant natural polymeric material on earth.1 It is found in the cell walls of many plants, and is produced by a variety of organisms including bacteria, algae, and fungi.2 It is considered as one of the most promising building blocks for next-generation sustainable (bio)materials, thanks to its low cost, renewability, biodegradability, exceptional mechanical properties, and outstanding versatility. Indeed, it has a plethora of applications – from conventional, such as paper and textile production, to innovative, such as organic electronics and optoelectronics, drug delivery, green packaging, membranes for separation technologies and water treatment, and sensing.3–5 Because it has the potential to be used in a carbon neutral fuel cycle, it is of great interest in the bioethanol industry.6 Due to its ubiquitous nature and potential for applications, cellulose is one of the most studied materials in polymer science, and initial investigations into the molecular and bulk structures date back to the early-twentieth century.7 However, despite its importance, there are still many outstanding questions related to the precise, atomic-level structures of cellulose, potentially limiting further innovative applications.Cellulose is known to exist as many different bulk structures, including amorphous and crystalline forms. A large number of crystalline polymorphs have been reported, including celluloses I, II, III, and IV.1,8–10 These all contain polymeric chains with the cellobiose repeat unit, but differ in the way the chains pack in three dimensions. The rich structural diversity originates from the various combinations of intermolecular hydrogen bonds that are possible, which affect the bulk structures of the materials.11 Cellulose I, known as native cellulose, is the most common polymorph, and can be isolated from natural sources, including bacteria, algae, and plants.12
Determining the atomic-level crystal structure of native cellulose has been the subject of ongoing debate due to the ambiguous data collected from various cellulose I samples.1 While it was originally believed that all native celluloses crystallize in the same structure, recent studies identified two discrete crystal phases of cellulose I, commonly referred to as cellulose Iα and cellulose Iβ. While cellulose Iα is primarily found in bacteria and cellulose Iβ is primarily found in plants, they are often both present in the same sample.2,13,14 The crystal structures of cellulose Iα and cellulose Iβ were determined by Nishiyama et al. in 2003 and 2002, respectively.15,16 The difference between cellulose Iα and cellulose Iβ stems from a difference in the offset between layers. This slight difference is what led to the initial confusion when the cellulose I crystal structure was originally investigated.
The structure studies revealed that both polymorphs of cellulose I have disordered hydrogen bond networks that lead to two possible supramolecular motifs for each polymorph, referred to as A and B.15,16 Thus, despite years of work, the exact structures of cellulose I polymorphs remain unknown. While diffraction methods are the gold standard for structural characterization, alternative methods for determining crystal structures exist. One example is low-frequency vibrational spectroscopy, which excites large-amplitude vibrations of entire molecules at terahertz frequencies, effectively probing weak intermolecular forces in crystals.17,18 Due to the dependence of these weak forces on bulk structures,19 low-frequency vibrational spectroscopy has been used in numerous occasions to identify structures,20–25 specifically including the structure of biopolymers.26,27 Here, both the Iα and Iβ polymorphs of native cellulose were studied using terahertz spectroscopy and powder X-ray diffraction (PXRD). Modeling possible crystal structures and comparing the simulated data to experimental data provide insight into the accuracy of the proposed structures. This reveals previously unrevealed details about the atomic level organization of cellulose I.
Methods
Experimental
Theoretical calculations
Solid-state (fully-periodic) DFT calculations were performed using the CRYSTAL17 software package.34 The PBE-D335,36 functional and the VTZP37 basis set were used for all calculations. Geometry optimizations were initiated using the experimentally determined structures,15,16 and both the atomic positions and lattice parameters were allowed to fully relax with no constraints. It is important to note that the experimental structures exhibited disorder in the hydrogen bonding network. In order to model the various combinations of the hydrogen bond networks, structures with various combinations of the hydrogen networks were generated; two possible networks were identified, labeled as the A and B networks. The structures were simulated with either all A or all B networks, as well as a combination of the two. In the case of the IαAB structure, the unit cell symmetry did not enable using both networks, since there is only one layer per unit cell, and therefore the unit cell was redefined to be double the size in the b and c dimensions to allow for alternating layers of A and B networks. The convergence of the crystalline energy was set to a threshold of ΔE ≤ 10−8 hartree. Additionally, the convergence tolerances for the maximum root mean square (RMS) on the gradient and displacement were set to 10−5 hartree and 4.0 × 10−5 Å, respectively.Frequency calculations were performed on each optimized structure using the same level of theory, with the convergence set to a more strict threshold of ΔE ≤ 10−11 hartree. Vibrational modes (eigenvectors) and their frequencies (eigenvalues) were predicted numerically using the harmonic approximation,38,39 and IR intensities were determined through the Berry phase approach.40,41
The calculated frequency and intensity of each low-frequency (0–7.5 THz) IR active mode were used to generate a theoretical terahertz spectrum for each structure. Each vibrational mode was convolved using a Gaussian function, with the full width at half maximum (FWHM) of the bands set to 2.43 cm−1 for Iα and 2.64 cm−1 for Iβ, which were determined empirically based on the experimental peak widths. For the cellulose Iα frequency calculations, the experimental use of carbon-13 isotopes was taken into account by utilizing the larger mass of carbon when calculating the vibrational frequencies. We emphasize that the simulated terahertz spectra presented herein have not been subjected to any empirical frequency scaling factors.
Finally, PXRD patterns were predicted for each optimized geometry of cellulose I. This was done using the Mercury software.42 A wavelength of 1.54 Å (Cu Kα) was used to generate the theoretical patterns, and each peak was convolved using a symmetric pseudo–Voight peak shape, with the FWHM set to 0.7° for all Iα and Iβ structures.
Results and discussion
Cellulose Iα and cellulose Iβ are two different polymorphs of native cellulose.43 The crystal structures of these polymers have previously been solved using a combination of neutron and X-ray diffraction.15,16 Both comprise β-linked D-glucose polysaccharide chains that form an infinite polymeric network, with the polymers forming two-dimensional hydrogen bonded sheets. The triclinic (space group P1) unit cell (a = 10.4000 Å, b = 6.717 Å, c = 5.962 Å, α = 80.37°, β = 118.08°, and γ = 114.80°)15 of cellulose Iα contains polysaccharide chains that lie along the a axis. The monoclinic (space group P21) unit cell (a = 8.201 Å, b = 10.38 Å, c = 7.784 Å, and β = 96.55°)16 of cellulose Iβ contains polysaccharide chains that lie along the b axis.For both polymorphs, the parallel polymer chains form hydrogen-bonded layers, with each disaccharide building unit, called cellobiose, participating in seven intralayer hydrogen bonds.15 This makes the attraction between co-planar chains notably strong, while the lack of interlayer hydrogen bonds in the stacking direction results in sheets that are weakly bound to adjacent layers, primarily through van der Waals forces. Fig. 1 presents the structures of two single layers extracted from cellulose Iβ. The organization of how these layers stack is what differentiates between the two crystal structures, with both shown in Fig. 1. Layers within cellulose Iα lie in the (0 1 1) Miller plane. Adjacent layers are offset by about a/4 along the direction of the chain. Layers within cellulose Iβ lie in the (0 0 1) and (0 0 2) planes. Alternating layers are offset by about b/4 along the direction of the chain.
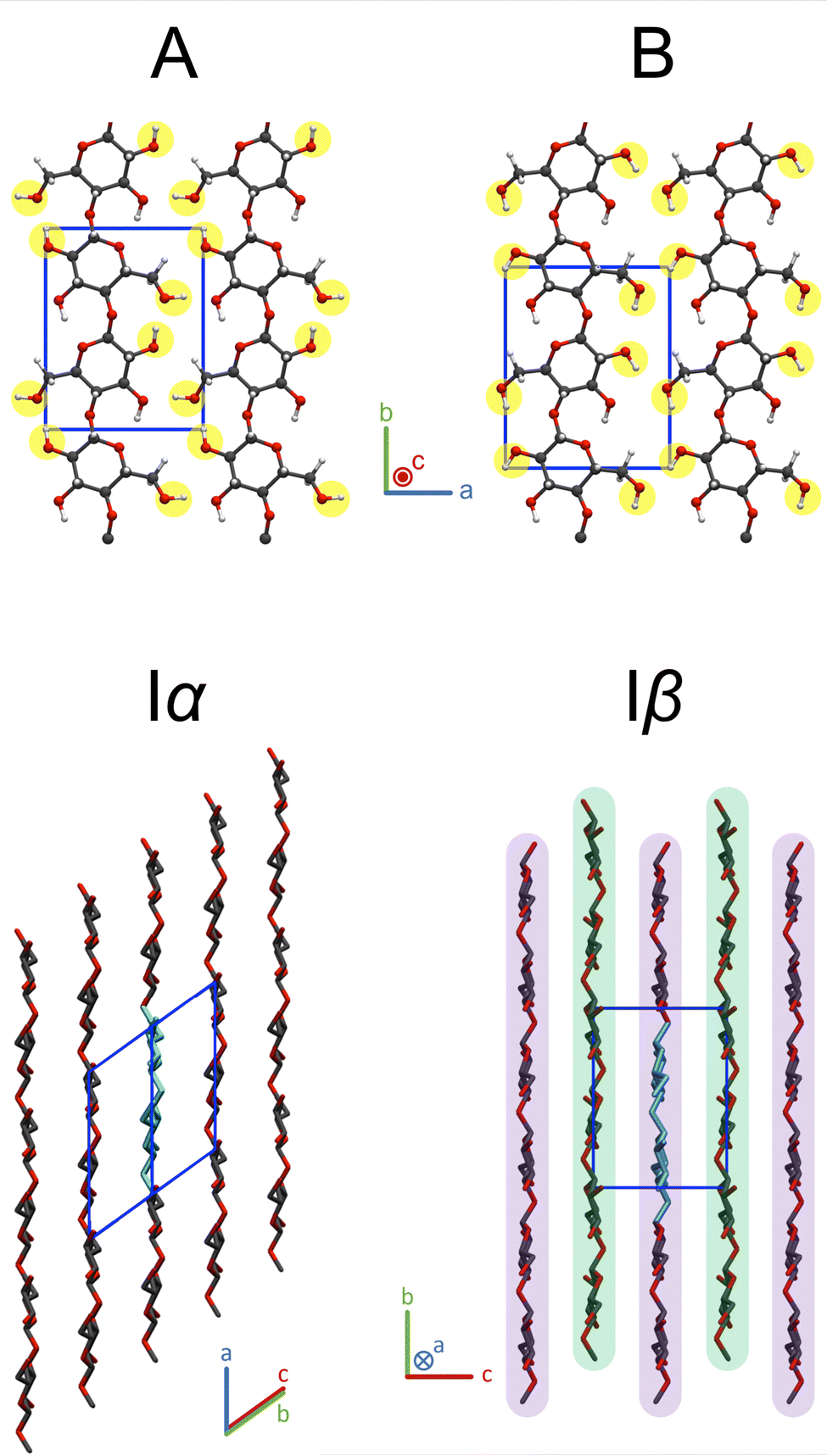 | ||
| Fig. 1 Top: Two layers of cellulose Iβ, highlighting the differences between the two hydrogen bond networks that are present in cellulose I polymorphs, labelled as A and B, respectively. Bottom left: Cellulose Iα, with hydrogen atoms removed for clarity. Bottom right: Cellulose Iβ, with hydrogen atoms removed for clarity and with center and origin sheets highlighted in purple and green, respectively. In the bottom structures, one cellobiose unit is shown in light blue. In all figures, the unit cell is outlined in dark blue. Color code: C, grey; O, red; and H, white. | ||
In cellulose Iβ, the organization of the layers gives rise to an origin sheet and a center sheet. They lie along the origin ((0 0 1) plane) and center ((0 0 2) plane) of the unit cell, respectively, as shown in Fig. 1. While there is not a significant difference between the two sheets, there are slight differences that arise from the packing of the layers. The layers can pack together more tightly due to the strained conformation of the center chains.16 Additionally, cellulose Iβ contains more interlayer C–H⋯O bonds than cellulose Iα. In the experimentally determined Iα crystal,15 the interlayer distance is 3.95 Å, while in Iβ,16 the interlayer distance is 3.89 Å, indicating that the layers in Iβ are more strongly bound. This helps to account for the increased stability of the Iβ form compared to that of the Iα form.15
The X-ray and neutron diffraction determined structures indicated that some of the hydroxyl groups were disordered in both polymorphs.15,16 Because hydrogen bonds occur between each hydroxyl group and a nearby oxygen, the disorder in hydroxyl group orientations leads to disorder in the hydrogen bond network. Nishiyama et al. concluded that there must be two possibilities for the orientation of each hydroxyl group that exhibits disorder, corresponding to two possible networks of hydrogen bonds for each polymorph.15,16 The hydrogen bonds branch between adjacent polysaccharide chains, which means that the orientation of each hydroxyl group is dependent on the others in the same layer. It is important to note that the disorder is not dynamic, but arises from the presence of two individual conformations. The two networks are at such deep energetic minima that an interconversion between the two would require a large activation barrier to be overcome.44 Additionally, because the hydrogen bond networks extend through the entire layer, a conversion from one network to the other would require all hydroxyl groups to switch orientations simultaneously, which is extremely unlikely at room temperature.44 This leads to the conclusion that there are two hydrogen-bonded layers of cellulose I: an A layer and a B layer. The two sets of hydroxyl group orientations that are present in both polymorphs are shown in Fig. 1.
One question that remains unanswered is whether the two networks coexist throughout cellulose I crystals or if they exist exclusively within distinct domains. In order to investigate the precise atomic structure and hydrogen-bond ordering in cellulose I polymorphs, DFT simulations were initiated. Three variations of cellulose Iα were used – a structure with all hydroxyl groups in the A conformation (IαA), a structure with all hydroxyl groups in the B conformation (IαB), and a structure with alternating layers of A and B (IαAB). Because of the difference between the origin and center sheets in cellulose Iβ, there are four variations of the structure – one with all A layers (IβA), one with all B layers (IβB), one with A origin sheets and B center sheets (IβAB), and one with B origin sheets and A center sheets (IβBA). All layers in cellulose Iα are symmetrically equivalent, so IαBA is identical to IαAB, and therefore IαBA does not need to be considered. Initially, geometry optimizations for all seven crystals were performed. All unit cell side lengths and angles, as well as the errors between the PBE/VTZP optimized and experimentally determined parameters, are reported in Table 1. In order to directly compare the cellulose IαAB structure to the others, the lengths of the optimized a and b unit cell vectors were halved, as these dimensions were doubled prior to the optimization to allow for altering A and B layers. It is clear that the IαA, IβA, and IβAB structures show the best agreement with the experimentally determined unit cell parameters, with the errors in the unit cell dimensions averaging to less than one percent. This indicates that the B network results in an optimized unit cell that disagrees with the experimentally determined unit cell.
| Dimension | Experiment | A | B | AB | BA | |
|---|---|---|---|---|---|---|
| Iα | a (Å) | 10.400 | 10.403 | 10.435 | 10.388 | |
| b (Å) | 6.717 | 6.605 | 6.496 | 6.940 | ||
| c (Å) | 5.962 | 5.911 | 6.099 | 5.791 | ||
| α (°) | 80.37 | 80.99 | 85.13 | 81.28 | ||
| β (°) | 118.08 | 117.45 | 121.66 | 115.74 | ||
| γ (°) | 114.80 | 114.63 | 105.21 | 117.70 | ||
| Average error | 0.665% | 3.869% | 1.994% | |||
| Relative energy (kJ mol−1) | +2.078 | +42.749 | +21.154 | |||
| Iβ | a (Å) | 8.201 | 8.149 | 8.562 | 8.117 | 8.155 |
| b (Å) | 10.380 | 10.404 | 10.297 | 10.379 | 10.430 | |
| c (Å) | 7.784 | 7.728 | 7.811 | 7.912 | 7.952 | |
| β (°) | 96.55 | 96.34 | 100.24 | 96.14 | 89.13 | |
| Average error | 0.447% | 2.341% | 0.776% | 2.720% | ||
| Relative energy (kJ mol−1) | 0 | +50.907 | +28.053 | +60.134 | ||
In addition to comparing the unit cell parameters of the seven optimized structures, the bulk energy of each crystal is reported in Table 1. For each structure, the energy of one unit cell was divided by the number of cellobiose units contained in the unit cell. Because the energy of IβA was the lowest, its value was set to zero and the rest of the energies are reported relative to IβA. It can be seen that the A and AB layers of both Iα and Iβ are significantly lower in energy than the B and BA layers. For cellulose Iα, the structure including IαAB is more stable than consecutive layers of A and B at distinct locations. This is also true for cellulose Iβ, as long as the layers that take the B form are the center sheets of the crystal. The fact that the A hydrogen bond network is more energetically favorable than the B hydrogen bond network is consistent with previous results determined using other computational methods.44
To further evaluate the proposed structures of each polymorph, theoretical PXRD patterns of the optimized structures were generated and compared to experimental data. Fig. 2 presents the theoretical and experimental patterns of both polymorphs. By looking at the peaks around 2θ = 15°, it can be observed that the IαB, IβB, and IβBA patterns differ the most from the experimentally determined patterns, with those same structures being the least stable energetically of all seven studied structures. To quantify the accuracy of each predicted pattern, the three major reflections for each of the data sets are listed in Table 2 as well as the average error between the reflections from each proposed structure and the experiment. The structures that produce PXRD reflections with errors that average to be about one percent or less are IαA, IαAB, IβA, and IβAB. Overall, it is clear that the orientation of the hydroxyl groups has a drastic effect on the intermolecular forces – and by extension, the bulk structures.
 | ||
| Fig. 2 Theoretical and experimental PXRD patterns of cellulose I polymorphs. DFT-predicted (PBE/VTZP) patterns of each structure along with experimental data, all normalized. Dashed lines indicate the peak positions of the experimental patterns. Left: Cellulose Iα. Right: Cellulose Iβ. | ||
| Reflection | Experiment | A | B | AB | BA | |
|---|---|---|---|---|---|---|
| Iα | (0 1 0) | 14.5 | 14.7 | 14.2 | 14.4 | |
| (0 0 1) | 16.9 | 16.9 | 17.1 | 17.0 | ||
| (0 1 1) | 22.7 | 23.0 | 23.0 | 23.1 | ||
| Average error | 0.900% | 1.525% | 1.014% | |||
| Iβ | (1 0 −1) | 14.9 | 15.0 | 14.1 | 14.8 | 15.5 |
| (1 0 1) | 16.6 | 16.7 | 16.9 | 16.5 | 15.7 | |
| (0 0 2) | 22.9 | 23.1 | 23.1 | 22.6 | 22.3 | |
| Average error | 0.716% | 2.683% | 0.861% | 4.023% | ||
Due to the relatively limited number of reflections inherent to these cellulose polymorphs, full-pattern refinement methods, such as Rietveld refinements, were not employed here, as they would not significantly enhance structural assignments beyond what is already confidently established by our combined experimental and theoretical analyses. And while XRD is effective for studying static structures, low-frequency vibrational spectroscopy is a powerful tool for investigating the interatomic forces in solids. Due to the dependence of terahertz motions on weak, long-range forces, it can be used to effectively probe weak intermolecular forces, and by extension to indirectly probe the structure, as each unique structure will have a unique spectral fingerprint.17,19,45
Fig. 3 shows the simulated spectra of each of the Iα structures overlaid with the experimental spectrum. It is clear that neither the IαA nor IαB spectra alone are able to account for all of the experimentally observed transitions. There is a peak in the experimental spectrum at about 4.7 THz that is not accurately predicted by either IαA or IαB, but is present in the IαAB spectrum. While it appears that the simulated mode at 4.9 THz in the IαB spectrum could correspond to the peak at 4.7 THz, the lattice motion associated with the eigenvector matches the motion of the mode at 5.0 THz in the IαA spectrum, and given that the simulated lattice is expanded with respect to the experimental parameters, it is likely that this mode is estimated to be red-shifted. By visualizing the motion associated with the 4.7 THz peak in the IαAB spectrum, it is clear that this mode is due to asymmetry in the layers of the crystal. The A and B layers are vibrating out of phase with each other, which is only possible because of the fact that they are nonequivalent. In the IαA and IαB crystals, all layers are symmetrically equivalent, so there is no potential for asymmetric motion, and thus this mode at 4.7 THz is only activated when the two different layers are adjacent to one another.
 | ||
| Fig. 3 Simulated terahertz spectra for IαA, IαB, and IαAB. Each is overlaid with the experimental spectrum. | ||
Fig. 4 shows the simulated spectra of each cellulose Iβ structure, again with the experimental data. The peak in the experimental spectrum around 2.1 THz is not accurately predicted for the IβA or IβBA structures. While it is present for both the IβB and IβAB structures, the motion associated with this frequency is different for the two structures. Although there are different modes predicted in each structure, the concept of additional motion due to asymmetry in the layers does not apply here like it does in the Iα polymorph. Because of the alternating origin and center sheets in Iβ, adjacent layers are already asymmetric, so the concept of alternating A and B layers does not add asymmetry to the structure.
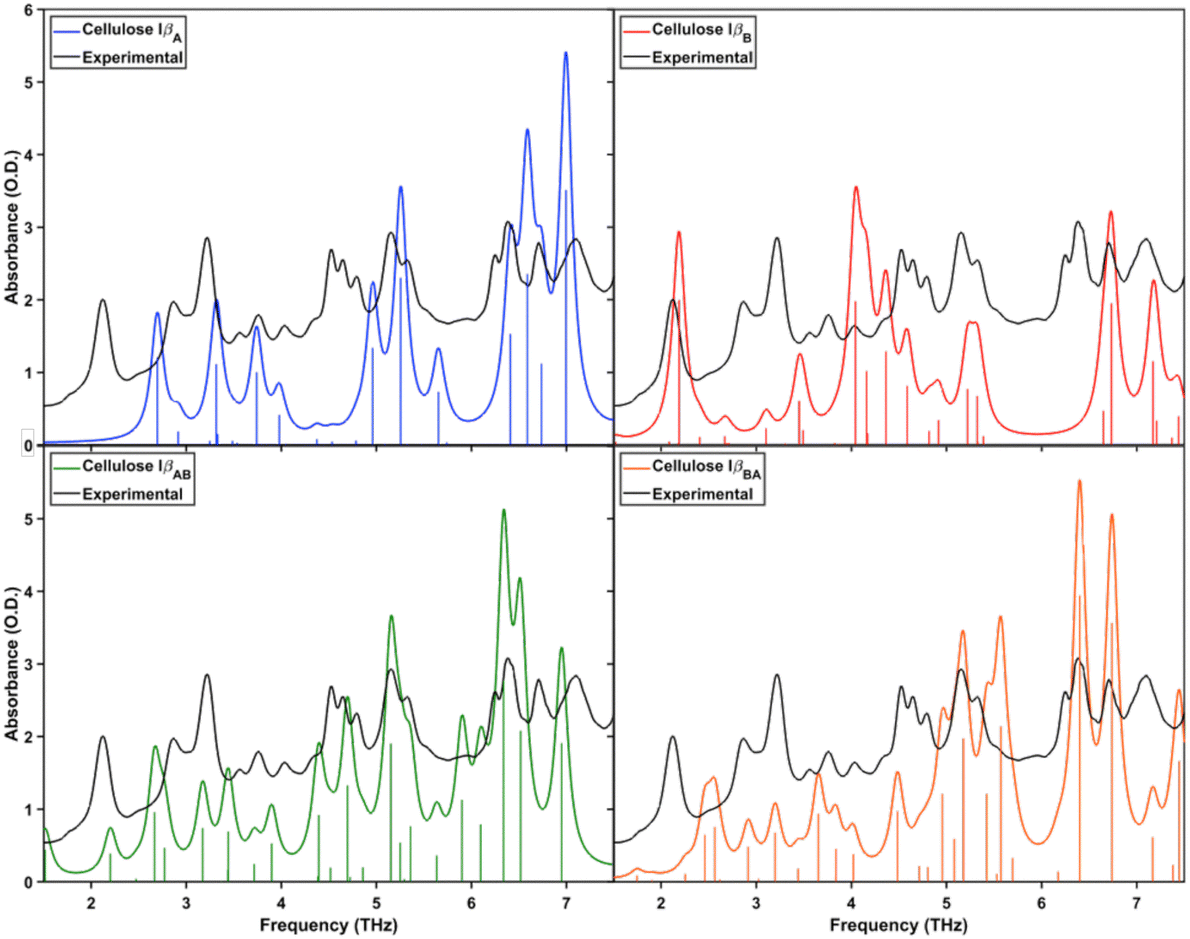 | ||
| Fig. 4 Simulated terahertz spectra for IβA, IβB, IβAB, and IβBA. Each is overlaid with the experimental spectrum. | ||
Similar to the vibrational mode at 2.1 THz, the region of 4.5–5 THz shows similarities between simulated IβB, IβAB, and IβBA spectra, but the associated motion is different due to the difference between structures. Finally, the range 5.75–6 THz only contains predicted peaks in the IβAB structure, which means that this motion is a direct result of the interaction between A origin sheets and B center sheets.
In order to learn more about the atomic level details of cellulose I polymorphs, observations from the data presented above are used to determine the organization of the hydrogen bond networks.
Nishiyama et al. determined that cellulose Iα contains 55% A layers and 45% B layers from neutron diffraction experiments.15 Evaluation of the three proposed structures via analysis of PXRD patterns showed that IαA and IαAB best match experiment, with errors of about one percent. The error of IαB is almost double that of IαAB. When it comes to the vibrational spectra, the mode at 4.7 THz is only predicted by IαAB, while IαB shows significant disagreement with the experiment.
In order to maintain the occupancy of the hydrogen atoms at the A positions at the determined 55%, there are multiple possibilities for the concentration of each proposed structure. Cellulose Iα could be composed of 55% IαA and 45% IαB, 10% IαA and 90% IαAB, or a number of other combinations that contain all three structures. Looking at the strong disagreement between the theoretical IαB data and experimental data – as well as the large energetic instability (see Table 1) – it is likely that this structure is actually not present on its own, which leaves the only possible combination to arrive at the reported concentration of layers determined previously to be 10% IαA and 90% IαAB.
These concentrations are used to calculate the weighted average of the simulated terahertz spectra and PXRD patterns, which are shown in Fig. 5. The averaged PXRD pattern shows good agreement with the experimental data. Almost all of the peaks in the experimental and theoretical terahertz spectrum are in agreement, with some minor disagreements in the 6–6.5 THz region. This indicates that the composition of 10% IαA and 90% IαAB is an accurate representation of cellulose Iα crystals, which implies that the A and B hydrogen bond networks coexist throughout the crystal rather than existing exclusively in distinct domains.
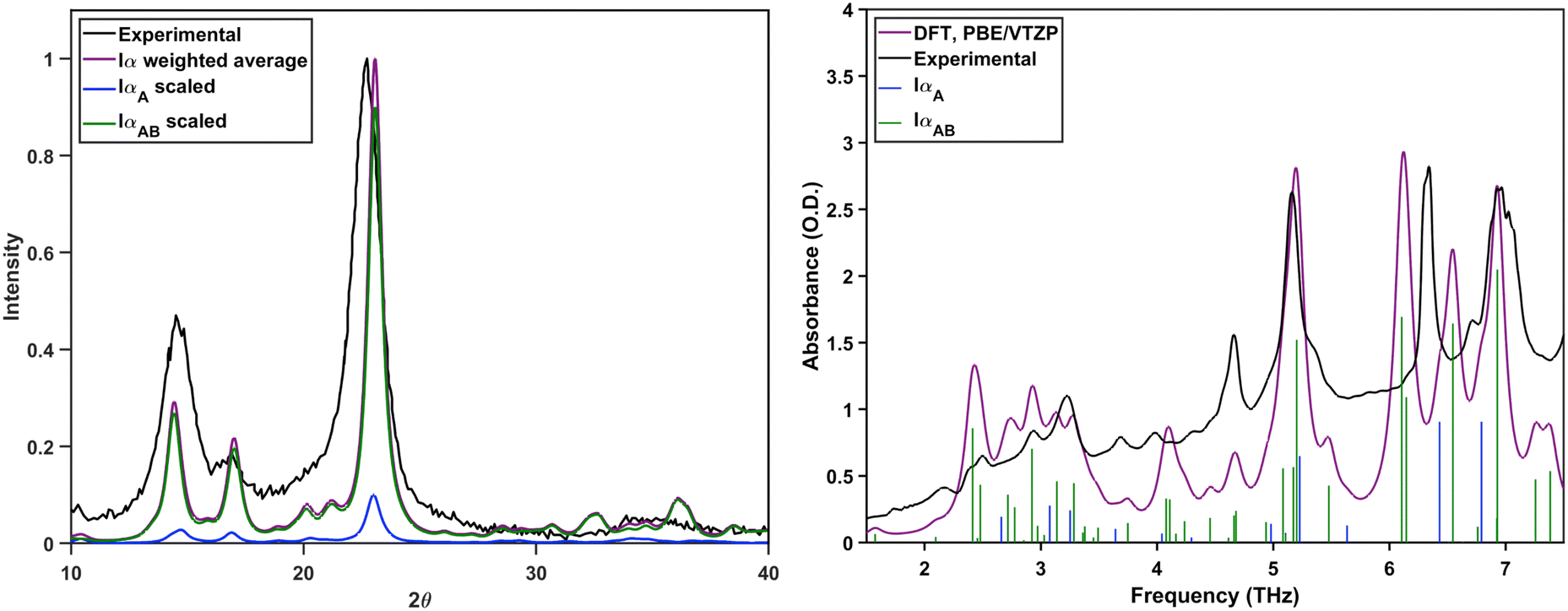 | ||
| Fig. 5 Left: Normalized experimental PXRD pattern of cellulose Iα, normalized weighted average of the theoretically generated IαA and IαAB PXRD patterns, and the scaled components of the weighted average. Right: Experimental terahertz spectrum and the weighted average of the simulated IαA and IαAB spectra. Colored sticks indicate the contribution from each structure. | ||
In cellulose Iβ, Nishiyama et al. determined that 70–80% of the layers have hydroxyl groups that are in the A orientation.16 Evaluation of the four cellulose Iβ geometries and PXRD patterns indicates that the IβA and IβAB structures are in agreement with experimentally determined values, with the error of both structures for both methods being below one percent. While the terahertz analysis is more qualitative, it can be seen that the spectrum is best predicted with the IβAB structure.
Because of the four possible structures, the possibilities for the composition are endless. In order to assist with determining the composition, the four simulated PXRD patterns were fit to the experimental pattern such that the root mean squared deviation (RMSD) between the experimental and weighted averages of simulated patterns was minimized. The significant difference between the predicted reflections in the four proposed Iβ structures made the fitting possible. This was not possible for the Iα structures because of the more similar angles of the reflections, which led to ambiguous results. The PXRD fitting resulted in concentrations of 42% IβA, 58% IβAB, and 0% IβB and IβBA. Because this leads to a composition of 71% A layers, which aligns with the previously reported occupancies,15 it was used to generate the weighted averages of both the terahertz spectra and PXRD patterns.
The averaged terahertz spectrum and PXRD pattern are shown in Fig. 6. Both are in agreement with experimental data. For the vibrational spectra, the weighted average of the simulated modes is in agreement with the experiment. The intensities of some of the modes, particularly those at 2.1, 4.6, and 7.0 THz, do not agree, but this minimal disagreement is not enough to cause major concern. These subtle intensity differences likely originate from minor structural variations or slight residual disorder uniquely affecting cellulose Iβ, but they do not significantly alter the overall conclusions. This indicates that the composition of 42% IβA and 58% IβAB accurately represents cellulose Iβ crystals, which means that the origin sheets take the A layer, while center sheets can take either the A hydrogen-bonded network or the B one. Similar to cellulose Iα, the two hydrogen bond networks coexist throughout Iβ crystals instead of existing solely at distinct locations.
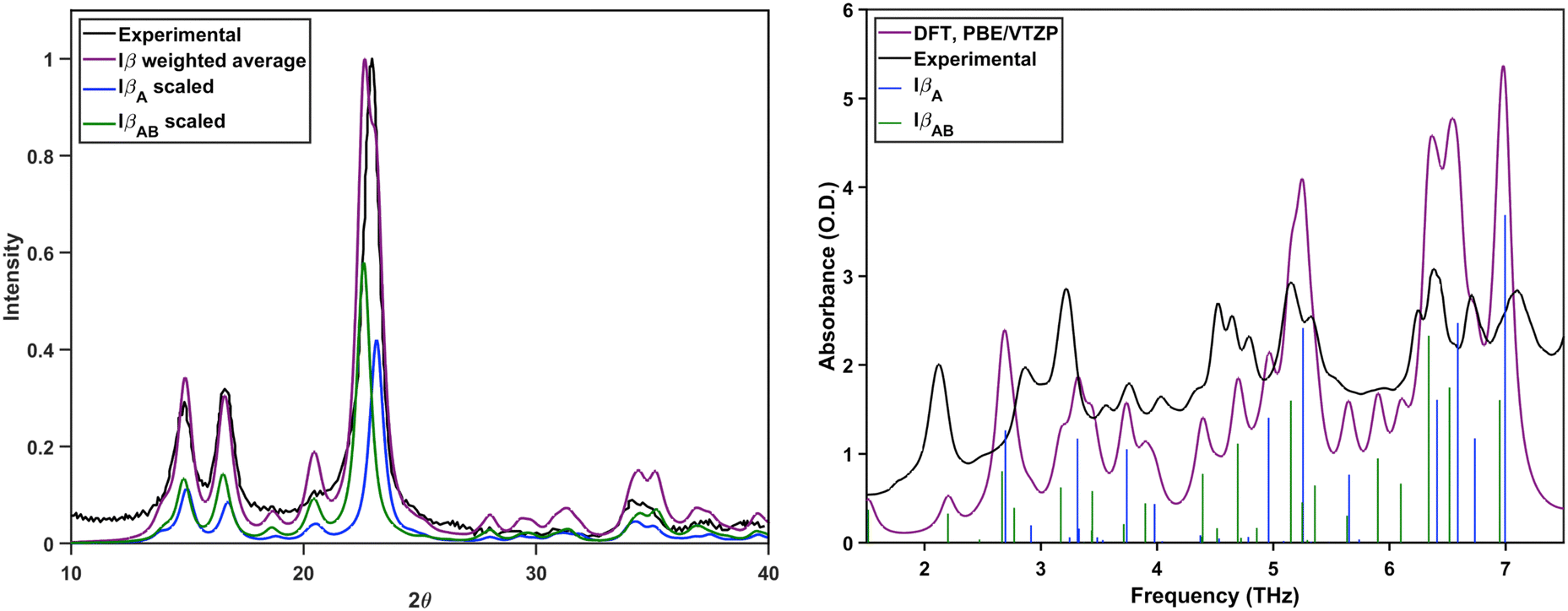 | ||
| Fig. 6 Left: Normalized experimental PXRD pattern of cellulose Iβ, normalized weighted average of the theoretically generated IβA and IβAB PXRD patterns, and the scaled components of the weighted average. Right: Experimental terahertz spectrum and the weighted average of the simulated IβA and IβAB spectra. Colored sticks indicate the contribution from each structure. | ||
With the combination of experimental and theoretical PXRD patterns and terahertz spectra, it has been shown that in both polymorphs of cellulose I, the A and B networks coexist throughout the crystal. The mismatch between layers results in the activation of additional vibrational modes, which appear in the simulated vibrational spectra. It can be concluded that the asymmetry that arises from nonequivalent hydrogen bonding in adjacent layers gives rise to these additional modes. When adjacent layers have the same hydrogen bonding network, these vibrational modes are symmetrical and therefore there is no change in the dipole moment, making them IR inactive. The asymmetry in the layers translates to asymmetry in the vibrational modes, making them IR active. These modes are present in the experimental spectra, so it can be concluded that in cellulose I crystals, both networks coexist throughout the sample. It is important to note that due to the relatively large particle size and high crystallinity of the cellulose samples studied here, the contributions from surfaces, grain boundaries, or other short-range defects to the observed spectroscopic signals are negligible, ensuring that the spectra predominantly reflect the bulk crystalline properties.
One explanation for the difference in abundance of each hydrogen bond network between polymorphs could stem from the Iα to Iβ phase transition. It has been shown that upon heating, cellulose Iα transitions to cellulose Iβ by translation of the layers along the direction of the chain axis.14,46 During this transition, the center sheets of cellulose Iβ adopt a slightly strained conformation, which allows the layers of the Iβ crystal to pack together more tightly, resulting in an overall stabilization of the crystal.16 This transition is shown in Fig. 7. It is possible that in order to further stabilize the crystal, the origin sheets of cellulose Iβ transition to the A hydrogen-bonded network. Since the center sheets are already more strained, the supramolecular motif of the hydrogen bonding network does not have as much of an effect on the bulk energy of the structure. This proposed conversion mechanism within Iβ from IβBA to IβA is also shown in Fig. 7. Our observations of the coexistence and specific ratios of hydrogen bond networks in cellulose suggest that previously reported energetic barriers for hydrogen bond interconversion might be influenced by local structural disorder. This represents an interesting avenue for future research.
 | ||
| Fig. 7 The transition from cellulose Iα to Iβ. First, the layers translate along the direction of the chain. Second, in order to further stabilize the crystal, IβBA transitions to IβA. Layers with the A network are shown in blue, and layers with the B network are shown in red. In the Iβ crystals, center sheets are shown in a lighter shade. | ||
Conclusion
Through a combination of terahertz spectroscopy, powder X-ray diffraction, and solid-state DFT simulations, we have elucidated the structural organization of hydrogen bond networks within the cellulose I polymorphs. Our results demonstrate that neither cellulose Iα nor Iβ can be accurately described by structures comprising purely A or B hydrogen-bonded layers. Instead, the best agreement with experimental PXRD patterns and terahertz vibrational spectra is achieved by models in which the A and B networks coexist within the same crystal. The presence of mixed layers activates vibrational modes that are otherwise IR-inactive in fully symmetric structures, serving as a unique spectral fingerprint of the layer-level disorder. For cellulose Iα, the optimal match is achieved with a composition of approximately 10% IαA and 90% IαAB, while for cellulose Iβ, a composition of 42% IβA and 58% IβAB provides the best agreement. These ratios not only align with previously reported occupancies from neutron diffraction studies but also minimize energetic and structural discrepancies in theoretical models. Finally, we propose that the differences in hydrogen-bond network abundance between the two polymorphs may arise from the thermally induced Iα to Iβ phase transition, during which structural rearrangements promote stabilization via an increased A-layer content. This study underscores the importance of incorporating both static and dynamic structural information when investigating complex biopolymeric materials and highlights the utility of terahertz spectroscopy to sensitively probe supramolecular organization.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
MTR, EA, and LC thank the National Science Foundation (award numbers CHE-2346689 and DMR-2348765), the American Chemical Society Petroleum Research Fund (61794-DNI10), and the University of Rochester for support. This work was supported by Industry-Academia Collaborative R&D from the Japan Science and Technology Agency, JST. The authors are grateful to Shigeharu Moriya (RIKEN) for providing holo-cellulose, Yuuri Tsuboi (RIKEN) for sample preparation, and Chiko Otani (RIKEN) and Hiroaki Minamide (RIKEN) for their help in the spectral measurements.References
- K. H. Gardner and J. Blackwell, The Structure of Native Cellulose, Biopolymers, 1974, 13, 1975–2001 CrossRef CAS.
- J. Sugiyama, T. Okano, H. Yamamoto and F. Horii, Transformation of Valonia Cellulose Crystals by an Alkaline Hydrothermal Treatment, Macromolecules, 1990, 23, 3196–3198 CrossRef CAS.
- J. Verma, M. Petru and S. Goel, Cellulose based materials to accelerate the transition towards sustainability, Ind. Crops Prod., 2024, 210, 118078 CrossRef CAS.
- J. Poisson and K. Zhang, Unique Optical Properties of Cellulosic Materials, Acc. Mater. Res., 2024, 5, 920–932 CrossRef CAS.
- Y. Cho, P. T. T. Ninh, S. Hwang, S. Choe and J. Myung, Sustainability Meets Functionality: Green Design Approaches to Cellulose-Based Materials, ACS Mater. Lett., 2025, 1563–1592 CrossRef CAS.
- I. N. Pulidindi, B. B. Kimchi and A. Gedanken, Can Cellulose be a Sustainable Feedstock for Bioethanol Production?, Renewable Energy, 2014, 71, 77–80 CrossRef CAS.
- K. H. Meyer and H. Mark, Über Den Bau Des Krystallisierten Anteils Der Cellulose, Ber. Dtsch. Chem. Ges. A, 1928, 61, 593–614 CrossRef.
- F. J. Kolpak and J. Blackwell, Determination of the Structure of Cellulose II, Macromolecules, 1976, 9, 273–278 CrossRef CAS PubMed.
- M. Wada, H. Chanzy, Y. Nishiyama and P. Langan, Cellulose IIII Crystal Structure and Hydrogen Bonding by Synchrotron X-ray and Neutron Fiber Diffraction, Macromolecules, 2004, 37, 8548–8555 CrossRef CAS.
- M. Wada, L. Heux and J. Sugiyama, Polymorphism of Cellulose I Family: Reinvestigation of Cellulose IVI, Biomacromolecules, 2004, 5, 1385–1391 CrossRef CAS PubMed.
- A. Ishikawa, T. Okano and J. Sugiyama, Fine Structure and Tensile Properties of Ramie Fibres in the Crystalline Form of Cellulose I, II, IIII and IVI, Polymer, 1997, 38, 463–468 CrossRef CAS.
- I. Nieduszynski and R. D. Preston, Crystallite Size in Natural Cellulose, Nature, 1970, 225, 273–274 CrossRef CAS.
- J. Sugiyama, R. Vuong and H. Chanzy, Electron Diffraction Study on the Two Crystalline Phases Occurring in Native Cellulose from an Algal Cell Wall, Macromolecules, 1991, 24, 4168–4175 CrossRef CAS.
- F. Horii, H. Yamamoto, R. Kitamaru, M. Tanahashi and T. Higuchi, Transformation of Native Cellulose Crystals Induced by Saturated Steam at High Temperatures, Macromolecules, 1987, 20, 2946–2949 CrossRef CAS.
- Y. Nishiyama, J. Sugiyama, H. Chanzy and P. Langan, Crystal Structure and Hydrogen Bonding System in Cellulose Iα from Synchrotron X-ray and Neutron Fiber Diffraction, J. Am. Chem. Soc., 2003, 125, 14300–14306 CrossRef CAS PubMed.
- Y. Nishiyama, P. Langan and H. Chanzy, Crystal Structure and Hydrogen-Bonding System in Cellulose Iβ from Synchrotron X-ray and Neutron Fiber Diffraction, J. Am. Chem. Soc., 2002, 124, 9074–9082 CrossRef CAS PubMed.
- P. A. Banks, E. M. Kleist and M. T. Ruggiero, Investigating the Function and Design of Molecular Materials through Terahertz Vibrational Spectroscopy, Nat. Rev. Chem., 2023, 7, 480–495 CrossRef PubMed.
- E. M. Kleist and M. T. Ruggiero, Advances in Low-Frequency Vibrational Spectroscopy and Applications in Crystal Engineering, Cryst. Growth Des., 2021, 22, 939–953 CrossRef.
- P. A. Banks, L. Burgess and M. T. Ruggiero, The Necessity of Periodic Boundary Conditions for the Accurate Calculation of Crystalline Terahertz Spectra, Phys. Chem. Chem. Phys., 2021, 23, 20038–20051 RSC.
- S. A. Ajibade, L. Catalano, J. Kölbel, D. M. Mittleman and M. T. Ruggiero, Terahertz Spectroscopy Unambiguously Determines the Orientation of Guest Water Molecules in a Structurally Elusive Metal–Organic Framework, J. Phys. Chem. Lett., 2024, 15, 5549–5555 CrossRef CAS PubMed.
- R. Yang, X. Dong, G. Chen, F. Lin, Z. Huang, M. Manzo and H. Mao, Novel Terahertz Spectroscopy Technology for Crystallinity and Crystal Structure Analysis of Cellulose, Polymers, 2021, 13(1), 6 CrossRef CAS.
- N. V. Fredeen, N. I. Lesack, A. Ciocoiu, A. M. Garner, W. F. Zandberg, A. Jirasek and J. F. Holzman, The Dynamic Morphology of Glucose as Expressed via Raman and Terahertz Spectroscopy, OSA Continuum, 2020, 3, 515–527 CrossRef.
- F. Zhang, H.-W. Wang, K. Tominaga, M. Hayashi, S. Lee and T. Nishino, Elucidation of Chiral Symmetry Breaking in a Racemic Polymer System with Terahertz Vibrational Spectroscopy and Crystal Orbital Density Functional Theory, J. Phys. Chem. Lett., 2016, 7, 4671–4676 CrossRef CAS PubMed.
- F. Zhang, H.-W. Wang, K. Tominaga, M. Hayashi and T. Sasaki, Terahertz Fingerprints of Short-Range Correlations of Disordered Atoms in Diflunisal, J. Phys. Chem. A, 2019, 123, 4555–4564 CrossRef CAS PubMed.
- M. T. Ruggiero, J. Kölbel, Q. Li and J. A. Zeitler, Predicting the structures and associated phase transition mechanisms in disordered crystals via a combination of experimental and theoretical methods, Faraday Discuss., 2018, 211, 425–439 RSC.
- M. T. Ruggiero, J. Sibik, R. Orlando, J. A. Zeitler and T. M. Korter, Measuring the Elasticity of Poly-L-Proline Helices with Terahertz Spectroscopy, Angew. Chem., Int. Ed., 2016, 55, 6877–6881 CrossRef CAS PubMed.
- H. Hoshina, T. Kanemura and M. T. Ruggiero, Exploring the Dynamics of Bound Water in Nylon Polymers with Terahertz Spectroscopy, J. Phys. Chem. B, 2019, 124, 422–429 CrossRef PubMed.
- S. Hestrin and M. Schramm, Synthesis of Cellulose by Acetobacter xylinum. 2. Preparation of Freeze-Dried Cells Capable of Polymerizing Glucose to Cellulose, Biochem. J., 1954, 58, 345–352 CrossRef CAS PubMed.
- K. Okushita, E. Chikayama and J. Kikuchi, Solubilization Mechanism and Characterization of the Structural Change of Bacterial Cellulose in Regenerated States through Ionic Liquid Treatment, Biomacromolecules, 2012, 13, 1323–1330 CrossRef CAS PubMed.
- K. Okushita, T. Komatsu, E. Chikayama and J. Kikuchi, Statistical Approach for Solid-State NMR Spectra of Cellulose Derived from a Series of Variable Parameters, Polym. J., 2012, 44, 895–900 CrossRef CAS.
- T. Mori, E. Chikayama, Y. Tsuboi, N. Ishida, N. Shisa, Y. Noritake, S. Moriya and J. Kikuchi, Exploring the Conformational Space of Amorphous Cellulose Using NMR Chemical Shifts, Carbohydr. Polym., 2012, 90, 1197–1203 CrossRef CAS PubMed.
- C. J. Strachan, T. Rades, D. A. Newnham, K. C. Gordon, M. Pepper and P. F. Taday, Using terahertz pulsed spectroscopy to study crystallinity of pharmaceutical materials, Chem. Phys. Lett., 2004, 390, 20–24 CrossRef CAS.
- W. B. Stoll, P. A. Banks, S. G. Dannenberg, R. Waterman, L. Catalano and M. T. Ruggiero, Interrogation of the Intermolecular Forces That Drive Bulk Properties of Molecular Crystals with Terahertz Spectroscopy and Density Functional Theory, Cryst. Growth Des., 2025, 25, 3697–3706 CrossRef CAS PubMed.
- R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich-Wilson, B. Civalleri, L. Maschio, M. Rérat, S. Casassa, J. Baima, S. Salustro and B. Kirtman, Quantum-Mechanical Condensed Matter Simulations with CRYSTAL, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2018, 8, e1360 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, Fully Optimized Contracted Gaussian Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- F. Pascale, C. M. Zicovich-Wilson, F. L. Gejo, B. Civalleri, R. Orlando and R. Dovesi, The Calculation of the Vibrational Frequencies of Crystalline Compounds and its Implementation in the CRYSTAL Code, J. Comput. Chem., 2004, 25, 888–897 CrossRef CAS PubMed.
- C. M. Zicovich-Wilson, F. Pascale, C. Roetti, V. R. Saunders, R. Orlando and R. Dovesi, Calculation of the Vibration Frequencies of α-quartz: The Effect of Hamiltonian and Basis Set, J. Comput. Chem., 2004, 25, 1873–1881 CrossRef CAS PubMed.
- Y. Noel, C. M. Zicovich-Wilson, B. Civalleri, P. D’arco and R. Dovesi, Polarization Properties of ZnO and BeO:an ab initio Study through the Berry Phase and Wannier Functions Approaches, Phys. Rev. B:Condens. Matter Mater. Phys., 2001, 65, 014111 CrossRef.
- R. Dovesi, B. Kirtman, L. Maschio, J. Maul, F. Pascale and M. Rérat, Calculation of the Infrared Intensity of Crystalline Systems. A Comparison of Three Strategies Based on Berry Phase, Wannier Function, and Coupled-Perturbed Kohn–Sham Methods, J. Phys. Chem. C, 2018, 123, 8336–8346 CrossRef.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, Mercury 4.0: From Visualization to Analysis, Design and Prediction, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- R. H. Atalla and D. L. Vanderhart, Native Cellulose: A Composite of Two Distinct Crystalline Forms, Science, 1984, 223, 283–285 CrossRef CAS PubMed.
- R. Witter, U. Sternberg, S. Hesse, T. Kondo, F.-T. Koch and A. S. Ulrich, 13C Chemical Shift Constrained Crystal Structure Refinement of Cellulose Iα and its Verification by NMR Anisotropy Experiments, Macromolecules, 2006, 39, 6125–6132 CrossRef CAS.
- M. T. Ruggiero, T. Bardon, M. Strli
![[c with combining breve]](https://www.rsc.org/images/entities/char_0063_0306.gif) , P. F. Taday and T. M. Korter, The role of terahertz polariton absorption in the characterization of crystalline iron sulfate hydrates, Phys. Chem. Chem. Phys., 2015, 17, 9326–9334 RSC.
, P. F. Taday and T. M. Korter, The role of terahertz polariton absorption in the characterization of crystalline iron sulfate hydrates, Phys. Chem. Chem. Phys., 2015, 17, 9326–9334 RSC. - M. Wada, T. Kondo and T. Okano, Thermally Induced Crystal Transformation from Cellulose Iα to Iβ, Polym. J., 2003, 35, 155–159 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc01544h |
| This journal is © The Royal Society of Chemistry 2025 |