
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA glucose/O2 fuel cell-based self-powered biosensor for probing a drug delivery model with self-diagnosis and self-evaluation†
Linlin
Wang
a,
Haohua
Shao
a,
Xuanzhao
Lu
a,
Wenjing
Wang
a,
Jian-Rong
Zhang
 *ab,
Rong-Bin
Song
*a and
Jun-Jie
Zhu
*a
*ab,
Rong-Bin
Song
*a and
Jun-Jie
Zhu
*a
aState Key Laboratory of Analytical Chemistry for Life Science, Collaborative Innovation Center of Chemistry for Life Sciences, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China. E-mail: jrzhang@nju.edu.cn; rbsong@nju.edu.cn; jjzhu@nju.edu.cn
bSchool of Chemistry and Life Science, Nanjing University, Jinling College, Nanjing 210093, China
First published on 16th October 2018
Abstract
Extending the application of self-powered biosensors (SPB) into the drug delivery field is highly desirable. Herein, a robust glucose/O2 fuel cell-based biosensor is successfully integrated with a targeted drug delivery system to create a self-sustained and highly compact drug delivery model with self-diagnosis and self-evaluation (DDM-SDSE). The glucose/O2 fuel cell-based biosensor firstly performs its diagnostic function by detecting the biomarkers of cancer. The drug delivery system attached on the anode of the glucose/O2 fuel cell can be released during the diagnostic operation to guarantee the occurrence of a therapy process. Accompanied by the therapy process, the glucose/O2 fuel cell-based biosensor can also act as an evaluation component to dynamically monitor the therapy efficacy by analyzing drug-induced apoptotic cells. In addition, the use of an abiotic catalyst largely improves the stability of the glucose/O2 fuel cell without sacrificing the output performance, further ensuring long-time dynamic evaluation as well as highly sensitive diagnosis and evaluation in this DDM-SDSE. Therefore, the present study not only expands the application of SPBs but also offers a promising in vitro “diagnosis-therapy-evaluation” platform to acquire valuable information for clinical cancer therapy.
Introduction
Biofuel cell-based self-powered biosensors (BFC-SPBs) are a type of novel bioelectrochemical biosensor, which are capable of continuously providing specific quantitative information of various analytes according to the change in the output of BFCs.1–4 Compared to traditional electrochemical biosensors, BFC-SPBs possess a vital feature of obviating external power sources owing to the integration of energy transformation with target analysis, offering the potential for miniaturization and on-site analysis.1,5–7 Therefore, some studies have been devoted to developing BFC-SPBs as basic analytical tools for detecting features in various biological analysis fields, such as immunoassays, aptamer sensing, and cytosensing.7–10 More interestingly, two novel application scenarios of BFC-SPB have been proposed recently. One scenario is to extend the ability of BFC-SPBs to multiple target detection with the assistance of logic control or dual-fuel-driven strategies.1,11 The other scenario is to devise BFC-SPBs that cover not only target detection but also a detection-triggered cascade event.9,12,13 As an example, a logic controlled BFC-SPB that manifests the capability to sense and treat hyperlactacidemia has been developed, where the release of a model therapeutic agent for treatment is controlled by logic detection.12 These two scenarios reveal that the application potential of BFC-SPBs is not confined to single target detection, which motivated us to probe its more advanced applications in some unreached areas like drug delivery systems (DDSs).DDSs that combine targeting and controllable drug release are attracting more and more attention as they can overcome the poor therapy efficacy of conventional chemotherapy.14–17 In addition to major research relating to the improvement of therapy efficacy, introducing diagnosis and evaluation components into a DDS is another research hotspot, as these components would provide effective cancer risk assessments and real-time monitoring of therapeutic effects, and even clear medication dosing instructions, thus promoting the realization of future personalized medicine.14,15,18 At present, massive research efforts have been made to combine a DDS with fluorescence or nuclear magnetic imaging technologies for diagnosis and evaluation.17–20 However, the high requirements of the imaging equipment has a negative effect on the promotion and popularization of these combinations. Moreover, due to inherent characteristics of imaging technologies like low convenience and a need for imaging reagents, real-time therapy information capture is difficult and time-consuming. To address these issues, the application of BFC-SPBs as diagnosis and evaluation components in DDSs seems to be a promising strategy because this arrangement allows realization of rapid, convenient, self-sustained and on-site diagnosis and evaluation. Thus, with the intention of realizing an in vitro integrated system for cancer risk assessment, cancer therapy and real-time monitoring of therapy responses, we were interested to see whether there was a suitable BFC-SPB, which can be used to design a drug delivery model with self-diagnostic and self-evaluation functions (DDM-SDSE) by combination with a DDS.
As we know well, some biochemical substances, such as circulating miRNAs,21–23 specific proteins24,25 and circulating tumor cells,7,26 in tumor microenvironments can be recognized as noninvasive biomarkers for the diagnosis of cancer and therapy-induced apoptotic cells are identified as an indicator of therapeutic effects.27–29 Therefore, in this study, we firstly devise a glucose/O2 fuel cell-based self-powered biosensor (SPB) by designing the anode and the cathode differently with the intention of detecting such two indicators independently. Additionally, to prove the possibility of integrating the SPB with a DDS, we prefix the DDS onto the anode of the SPB we devised via a well-designed fixing strategy which can ensure that the diagnosis process is able to stimulate the DDS release synchronously; the release of DDS further assures the occurrence of targeted therapy and therapy efficacy assessments. Specifically, k562 leukemia cells are selected as model tumor cells, and circulating miRNA-125a (miR-125a) is employed as a corresponding biomarker.30,31 The detailed design and working principles of the DDM-SDSE for leukemia are depicted in Scheme 1. The glucose/O2 fuel cell-based SPB consists of a partial complementary DNA double strand-decorated porous gold nanobowl/glassy carbon anode (PDS/pAuNB/GC), and a hollow mesoporous N-doped carbon sphere/glassy carbon cathode (PSp/hmNCS/GC) modified with a phosphatidylserine-binding peptide. The DDS (HA@Dox@hmSiO2), which is fabricated by encapsulating doxorubicin (Dox, anticancer drug)-loaded hollow mesoporous silica (hmSiO2) with hyaluronic acid (HA), is linked to the 3′ end of PDS on the anode of the glucose/O2 fuel cell-based SPB by EDC/NHS chemistry (Scheme 1A).32 Initially, the pAuNB can catalyze the oxidation of glucose at the anode to produce electrons, and hmNCS can catalyze the reduction of oxygen on the cathode to accept the electrons transferred from the anode. But the maximum power density (Pmax) of DDM-SDSE is at a low level on account of the blocking effect of HA@Dox@hmSiO2 on the anode. Once the DDM-SDSE is exposed to a k562 cell-containing system, the Pmax increases to a high level as the miR-125a can competitively hybridize with PDS and simultaneously actuate the release of HA@Dox@hmSiO2 from the anode, alleviating the blocking effect. Such an increase in the Pmax works as a diagnosis signal for cancer risk assessment. Synchronously, the released HA@Dox@hmSiO2 can internalize into K562 leukemia cells by HA-mediated recognition and cellular endocytosis. With the enzyme-responsive degradation of HA in cells,33 Dox can be released from HA@Dox@hmSiO2 to induce the apoptosis of K562 cells. During the therapy process, no change occurs in the Pmax of the DDM-SDSE until the appearance of apoptotic cells. Due to the stable exposure of phosphatidylserine on the extracellular leaflet of the plasma membrane,28,29,34 the apoptotic K562 cells can be captured by the PSp modified cathode. Therefore, the Pmax of the DDM-SDSE eventually displays a dramatic time-dependent decrease due to the gradually increased blocking effect on the reduction of oxygen caused by the continuous capture of apoptotic K562 cells from the given system. Such a time-dependent decrease in Pmax serves as a signal for real-time evaluation of the therapeutic effects of HA@Dox@hmSiO2 and to further build an unbiased apoptotic kinetic model (Scheme 1B). Overall, on the basis of the above proposed strategies, an SPB has been successfully integrated with a DDS to realize a DDM-SDSE, which can serve as an in vitro “diagnosis-therapy-evaluation” research platform to provide valuable information for the clinical treatment of cancer to some extent.
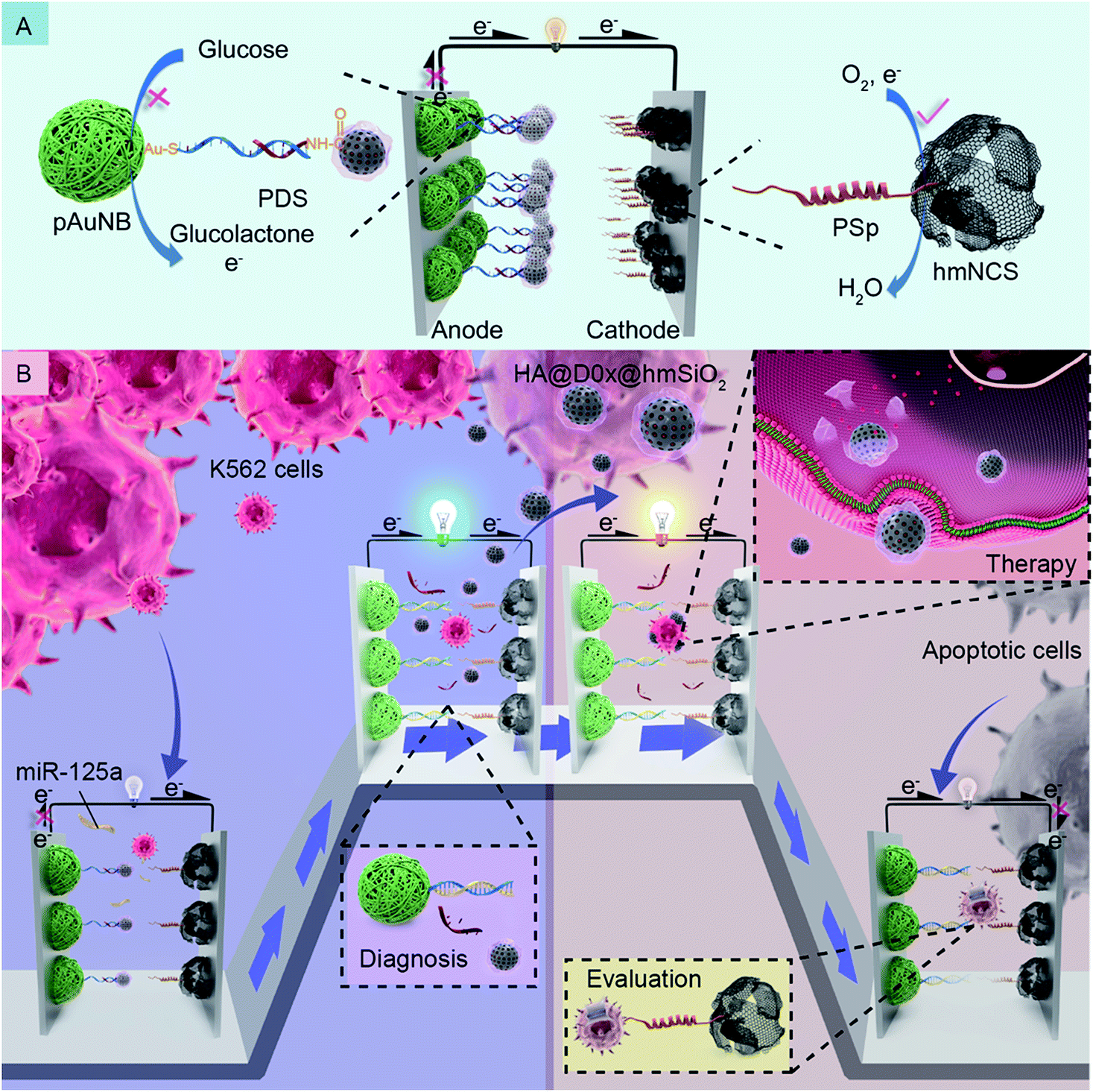 | ||
| Scheme 1 Schematic of the composition (A) and the working principles (B) of the DDM-SDSE in the presence of miR-125a and K562 leukemia cells. | ||
Results and discussion
Design and characterization of abiotic catalysts for the BFC
Because one of the designed functions of the SPB is to evaluate the therapy response of the DDS and this event will last for some time, the demand for the long-term stability of the glucose/O2 fuel cell becomes greater. However, durability has been considered to be an inherent deficiency of commonly used biocatalysts (enzymes) in BFCs.35,36 In this case, two high activity abiotic catalysts, pAuNB and hmNCS, were selected and fabricated for catalyzing the oxidization of glucose and the reduction of oxygen (see experimental procedures). The morphology of as-prepared pAuNB was first investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. S1,† the prepared pAuNB possessed a bowl-shaped structure with a well-defined cavity. The TEM image revealed that the pAuNB had a diameter range of 200–250 nm and consisted of many irregular Au particles. Careful inspection disclosed a large amount of small pores on the surface of pAuNB, which could facilitate the diffusion of electrolytes and substrates (Fig. 1A). In the high resolution TEM image, the d-spacing of the lattice was measured to be 0.24 nm, illustrating that the most exposed crystal plane was the Au (111) facet (Fig. 1B). In addition, some surface defects like dislocations and steps were observed, endowing the pAuNB with a higher surface energy and more efficient exposed catalytic active sites to improve the catalytic capability of pAuNB towards glucose.36,37 Further analysis of the chemical structure of pAuNB was carried out by X-ray diffraction (XRD). All of the peaks shown in Fig. 1C can be indexed as the pure face-centered cubic Au, which matched well with the standard data of Au (JCPDS no. 04-0784). Moreover, the intensity of the peak corresponding to Au (111) was the highest, further confirming that this facet is the most exposed crystal plane.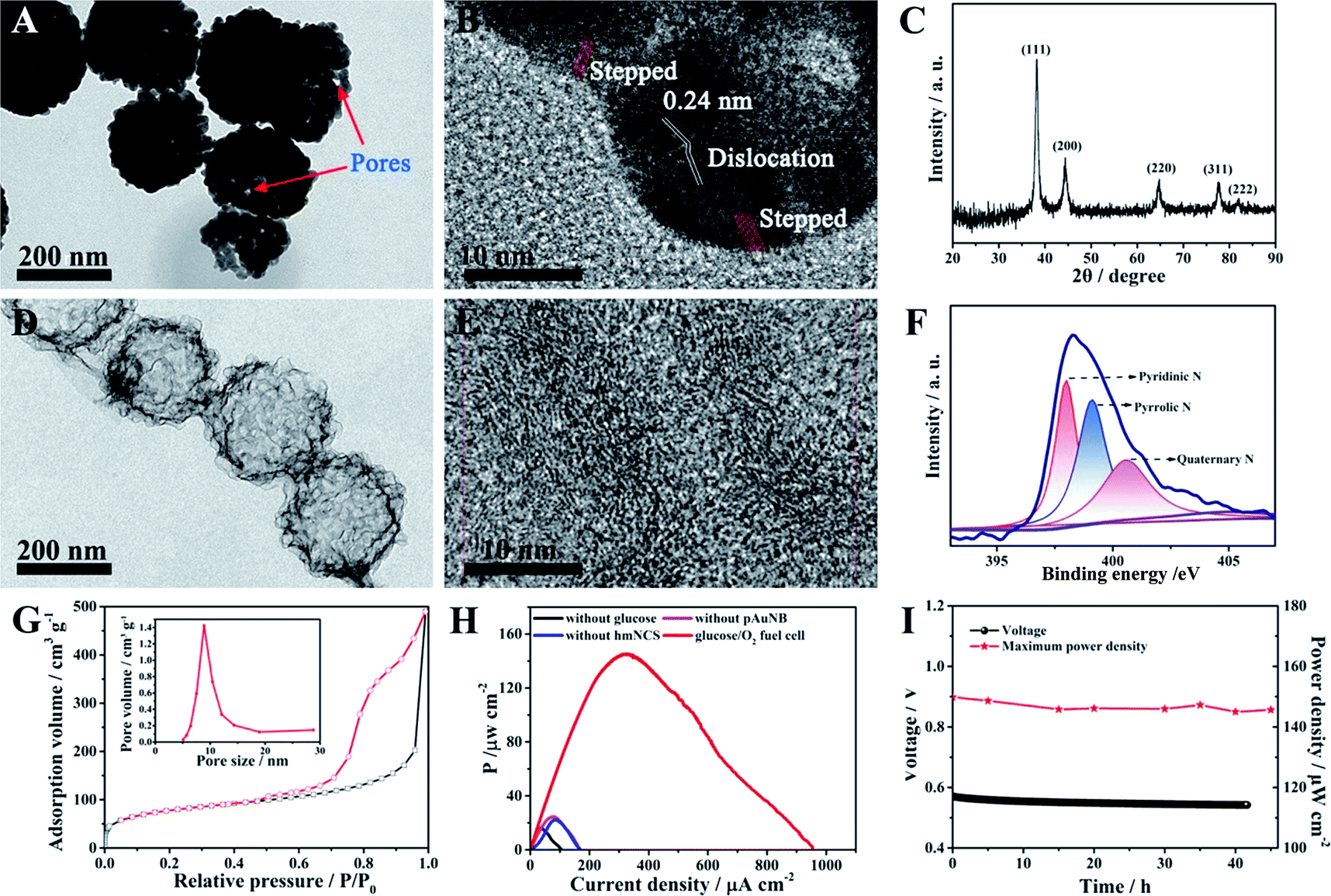 | ||
| Fig. 1 A transmission electron micrograph (TEM) image (A), a high resolution transmission electron micrograph (HRTEM) image (B) and the X-ray diffraction (XRD) pattern (C) of PAuNB. A TEM image (D), a HRTEM image (E), the X-ray photoelectron (XPS) spectra (F) and nitrogen adsorption–desorption isotherms (G) of hmNCS. Inset: the Barrett–Joyner–Halenda (BJH) pore size distributions of hmNCS. The power density curve of glucose/O2 fuel cells and control cells without fuel or catalysts (H) and the time courses of the open circuit voltage (OCV) and maximum power density (Pmax) generation (I) of the fuel cell using pAuNB and hmNCS as the anodic and cathodic catalyst, respectively. Scan rate: 1 mV s−1. Triplicate experiments were performed. | ||
The morphology and chemical features of the as-prepared hmNCS were also investigated. As shown in Fig. 1D, the hmNCS was hollow spherical-shaped with ultra-thin carbon layers and possessed a diameter of about 250 nm. Further analysis by high resolution TEM revealed that these carbon layers were partially graphitized (Fig. 1E). Besides, X-ray photoelectron spectroscopy (XPS) was performed to further probe the chemical composition of the hmNCS. The C 1s, O 1s and N 1s bands were all detected in the wide XPS spectrum (Fig. S2†), suggesting the presence of C, O and N elements in the hmNCS. The high-resolution XPS for N 1 could be separated into three peaks at 398 eV, 399 eV and 400.5 eV, which were assigned to pyridinic, pyrrolic and quaternary N (Fig. 1F). By comparing the intensities of these three peaks, we could conclude that the hmNCS was furnished with high content of pyridinic N. Such a high content of pyridinic N would significantly enhance ORR activity.35,38,39 Moreover, we recorded the N2 adsorption–desorption isotherms and pore size distribution plots for the hmNCS (Fig. 1G and the inset). Based on these isotherms, the Brunauer–Emmett–Teller (BET) surface area was calculated to be 267.8 m2 g−1, and the pore size was centered at 8.9 nm (inset). Such a high surface area and porous structure would provide many active sites to enhance their catalysis toward ORR.
To probe the potential application of pAuNB and hmNCS, the glucose/O2 fuel cell with pAuNB and hmNCS as the anodic and cathodic catalyst was fabricated using cell culture medium as an electrolyte. Comparative studies of the performance of control fuel cells without glucose, pAuNB or hmNCS were also examined. The power density curve of the glucose/O2 fuel cell is displayed in Fig. 1H. Clearly, the glucose/O2 fuel cell produced a relative high maximum power density (Pmax, 145 ± 1.2 μW cm−2), suggesting good catalytic activity of pAuNB and hmNCS towards the oxidization of glucose and the reduction of oxygen, respectively. In contrast, in the cases of the fuel cells without glucose (black trace), pAuNB (pink trace) or hmNCS (blue trace), the measured power densities were all below 25 μW cm−2, suggesting that the contribution from corrosion currents is almost negligible. Moreover, the glucose/O2 fuel cell was able to operate in cell culture medium for at least 42 h with almost no decay in the output voltage and Pmax (Fig. 1I), demonstrating the good chemical stability of these two abiotic catalysts. These above results confirmed that the fabricated nanomaterials are suitable candidates for the development of robust and high-efficiency fuel cells.
Fabrication and characterization of the targeted drug delivery system
In the prepared DDS, hmSiO2 was applied as a hollow drug carrier; Dox was encapsulated as a chemotherapeutic agent. Moreover, HA, which could specifically interact with the overexpressed transmembrane glycoprotein (CD44) on the surface of K562 leukemia cells and would be degraded by hyaluronidase (HAase),33,40 was employed to cover the surface of hmSiO2 to prevent the premature release of Dox, endowing the nanocarriers with a targeting ability and allowing HAase-responsive release of drug within the cells. The hmSiO2 was first prepared using a traditional template method (see experimental procedure).41 As shown in Fig. 2A, the hmSiO2 displayed a regular hollow spherical structure with a loose shell (∼20 nm). To obtain its surface area and pore information, nitrogen absorption–desorption analysis was also conducted. Based on the nitrogen absorption–desorption curve (Fig. S3A†), the BET specific surface area was 524.7 m2 g−1. The pore size distribution revealed 4.2 nm mesopores (Fig. S3B†), which correspond to the pores formed in the loose shell. Benefiting from such a high surface area and suitable pore size, the hmSiO2 possessed a loading amount of Dox as high as 325 μg mg−1. The zeta potential experiments displayed an obvious surface charge inversion after the loading of Dox. Furthermore, with the HA coating, the zeta potential recovered to a negative value (Fig. 2B). These alterations were consistent with what was found in our previous work, confirming the successful assembly of HA@Dox@hmSiO2.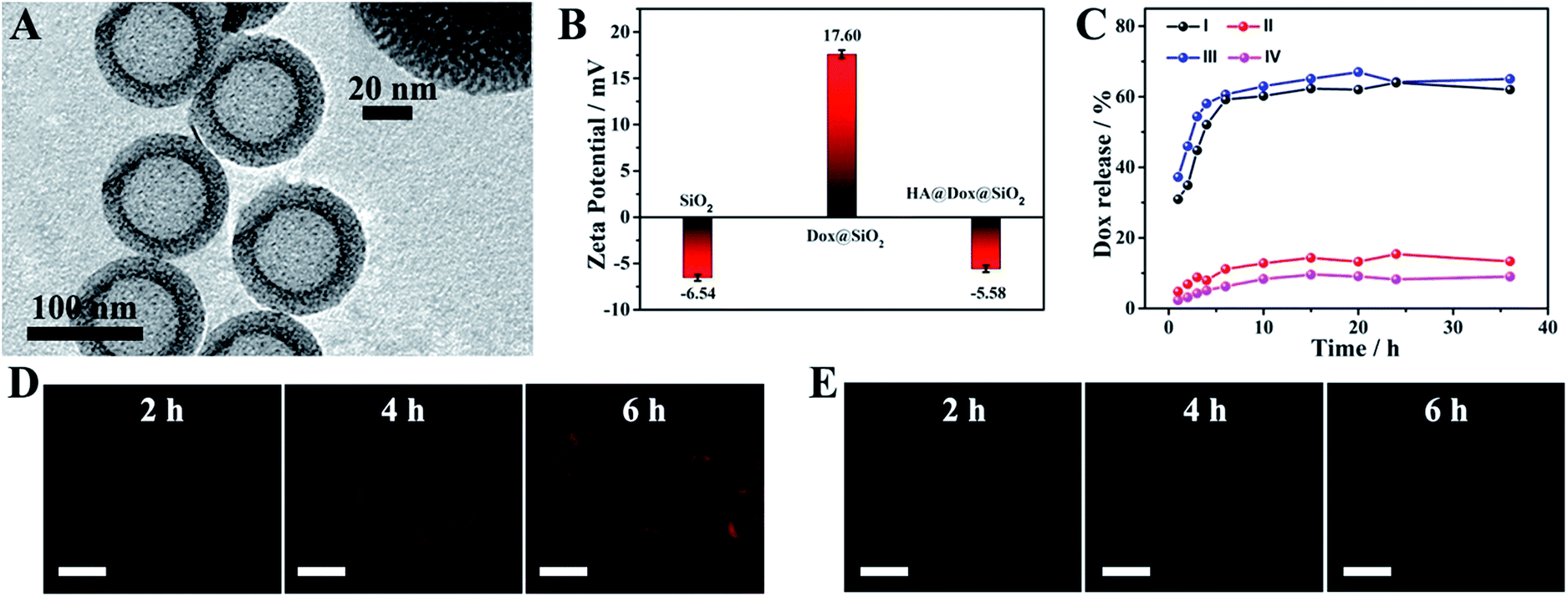 | ||
| Fig. 2 (A) A TEM image of hmSiO2, where the inset is a high-magnification image. (B) The zeta potential of hmSiO2, Dox@hmSiO2 and HA@Dox@hmSiO2. (C) The release tests of Dox from Dox@hmSiO2 (I) and HA@Dox@hmSiO2 (II–IV) under different conditions: (I) acetate buffer (pH 5.0) without HAase; (II) acetate buffer (pH 5.0) without HAase; (III) acetate buffer (pH 5.0) with HAase; and (IV) PBS buffer (pH 7.4) without HAase. Confocal laser scanning microscopy (CLSM) images of the K562 cells after incubation with (D) HA@Dox@SiO2 and (E) Dox@hmSiO2 for different times. Scale bars: 15 μm. Triplicate samples were used. | ||
To verify the role of HA in the devised targeted drug delivery system, we investigated the in vitro release profile of Dox from Dox@hmSiO2 and HA@Dox@hmSiO2 under different conditions. As shown in Fig. 2C, 62% of Dox in Dox@hmSiO2 leaked into the acetate buffer solution within 20 h (curve I), while only 13.2% of DOX in HA@Dox@hmSiO2 leaked under the same test conditions (curve II), suggesting the protective effect of the HA shell against premature release. After the addition of HAase into the acetate buffer, the release proportion of the Dox from HA@Dox@hmSiO2 increased to 67% at the same time interval (curve III). As almost the same proportion of the Dox in HA@Dox@hmSiO2 leaked into the PBS (pH = 7.4, 9%, curve IV) and acetate buffer solution (pH = 5.0, 13.2%, curve II) in the absence of HAase, the above-mentioned increase in the release proportion could be attributed mainly to the degradation of HA by HAase instead of the acidic environment. Thus, the coating of HA allowed the HAase-responsive release of drug within the cells. Moreover, we probed the targeting ability of HA by confocal laser scanning microscopy (CLSM) and flow cytometry analysis. After the cells were respectively incubated with Dox@hmSiO2 and HA@Dox@hmSiO2 for different times, the CLSM images of the cells were collected. Red fluorescence from Dox was observed in the CLSM images in the group of HA@Dox@hmSiO2, indicating the endogenous HAase-triggered release of drug within the cells (Fig. 2D). More importantly, the fluorescence intensity in the CLSM image collected at any time was obviously enhanced compared to that of corresponding CLSM image in the group of Dox@hmSiO2 (Fig. 2E), illustrating the targeting ability of HA. Flow cytometry analysis further confirmed the above results, as the increased rate of fluorescence intensity over time in the HA@Dox@hmSiO2 group was higher than that of the Dox@hmSiO2 group (Fig. S4†).
Construction and electrochemical analysis of the anode and cathode
Having the above-discussed materials at hand, we then investigated the feasibility of the anode with the integrated ability of a sensing biomarker and actuating the drug delivery event by recording the polarization curves in the presence of glucose. As shown in Fig. 3A, an extremely weak oxidation current was observed for the naked GC electrode (curve I), while an obvious oxidation current at about 0.1 V was detected for the pAuNB-modified GC electrode (pAuNB/GC, curve II), which could correspond to the oxidation of glucose. The current was as high as 1.51 ± 0.06 mA cm−2, confirming the good catalysis of pAuNB towards glucose oxidation. Modifying pAuNB/GC with thiolate PDS slightly decreased the current to 0.99 ± 0.04 mA cm−2, due to the steric effects of PDS (curve III). After further incubating the above electrode with HA@Dox@hmSiO2, the current sharply decreased (curve IV), indicating the successful assembly of the HA@Dox@hmSiO2/PDS/pAuNB/GC anode. Moreover, with the addition of 1.0 × 10−15 M miR-125a, the current recovered to 0.92 ± 0.03 mA cm−2 (curve V). In contrast, nearly no change in the current was observed in presence of 1.0 × 10−14 M miR-21 (0.32 ± 0.02 mA cm−2, curve VI vs. 0.24 ± 0.04 mA cm−2, curve IV). These results suggested that the competitive formation of fully hybridized base pairs between miR-125a and the PDS specifically triggered the model drug-HA@Dox@hmSiO2 to move away from the anode to diminish the blocking effect. Therefore, the designed anode could be employed to diagnose the biomarker and to actuate the drug delivery event, simultaneously.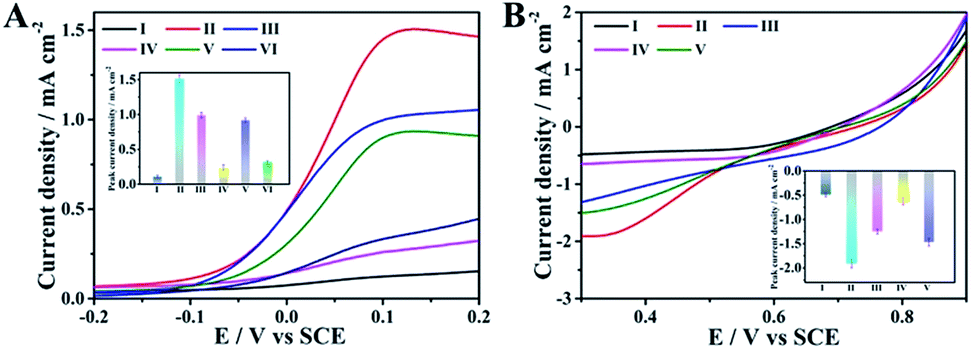 | ||
| Fig. 3 (A) Polarization curves and the current at 0.1 V (inset) of different electrodes in PBS buffer (10 mM, pH = 7.4) with 5 mM of glucose: (I) GC; (II) pAuNB/GC; (III) PDS/pAuNB/GC; (IV) HA@Dox@hmSiO2/PDS/pAuNB/GC; (V) HA@Dox@hmSiO2/PDS/pAuNB/GC after the incubation with miR-125a (1.0 × 10−15 M) and (VI) HA@Dox@hmSiO2/PDS/pAuNB/GC after the incubation with miR-21 (1.0 × 10−14 M). (B) Polarization curves and the current at 0.3 V (inset) of different electrodes in O2 saturated PBS buffer: (I) GC; (II) hmNCS/GC; (III) PSp/hmNCS/GC; (IV) PSp/hmNCS/GC after the incubation with apoptotic cells and (V) PSp/hmNCS/GC after the incubation with viable cells. The scan rate of all tests is 5 mV s−1. Triplicate experiments were performed. | ||
Similarly, polarization analysis was also performed to explore the feasibility of the cathode for evaluation of therapeutic effects. As shown in Fig. 3B, no obvious oxygen reduction current was detected for the GC electrode in the physiological solution (curve I). In contrast, an obvious oxygen reduction current of 1.91 ± 0.09 mA cm−2 was observed at 0.3 V for the hmNCS modified GC electrode under same conditions (hmNCS/GC, curve II), highlighting the high ORR activity of hmNCS. The functionalization of the hmNCS/GC electrode with PSp decreased the reduction current at 0.3 V to 1.25 ± 0.05 mA cm−2 due to steric effect of PSp (curve III), verifying the successful assembly of the PSp/hmNCS/GC cathode. As the PSp could capture the apoptotic K562 cells to produce an extra blocking effect, a further decrease in reduction current at 0.3 V was discovered after incubating the PSp/hmNCS/GC cathode with apoptotic K562 cells for 30 min (0.63 ± 0.07 mA cm−2, curve IV). But incubating the same cathode with living K562 cells generated negligible reduction of the current at 0.3 V (1.47 ± 0.08 mA cm−2, curve V vs. 1.25 ± 0.05 mA cm−2, curve III), illustrating the good selectivity of the PSp/hmNCS/GC cathode. Therefore, the designed cathode has the capability to evaluate therapeutic effects by the real-time supervision of apoptotic cells.
Evaluation and visual verification of the DDM-SDSE
Given the above feasibility analysis, integration of the fabricated anode and cathode is expected to form a DDM-SDSE system which is capable of realizing the cascade functions of diagnosis, therapy and evaluation. To confirm this, the DDM-SDSE was incubated in K562 cell culture medium and the Pmax of the DDM-SDSE was collected over time. As shown in Fig. 4A, upon the emergence of miR-125a, the Pmax increased quickly in the initial 2 h, followed by a plateau for about 4 h, and then started to decrease. Finally, the Pmax no longer decreased after operating for 14 h (square, black line). In contrast, we operated the similar DDM-SDSE without miR-125a, and there was nearly no change in the Pmax observed during the whole operation time (Fig. S5†). This result suggested that the change of the Pmax in the presence of miR-125a could be attributed to the occurrence of certain events instead of system disturbances from operation conditions, the degradation of catalyst activity and so on. At certain time points, CLSM analysis of K562 cells from the DDM-SDSE was performed to make clear the aforementioned changes in the Pmax. In these experiments, Dox (red fluorescence) could be employed as an endogenous dye to reflect the internalization of the targeted drug delivery system into cells, while an extrinsic dye (Hoechst 33258, brilliant blue fluorescence) was used to differentiate the apoptotic/living cells.33 As shown in Fig. 4B and S6,† red fluorescence appeared in the second hour, indicating the presence of Dox in the cells. However, in the absence of miR-125a, no obvious red fluorescence was observed in the CLSM images of the cells collected at any time (Fig. S7†). Considering that the drug-loaded nanocarriers were prefixed on the anode, we can draw conclusions that the diagnosis of miR-125a and the liberation of the targeted drug delivery system have been successfully realized in the devised DDM-SDSE. The release of drug-loaded nanocarriers diminished the blocking effect on the anode, thus the rising tendency of Pmax in the initial 2 h clearly demonstrated the diagnosis process. Subsequently, a common therapy-lag phenomenon has been reflected in the Pmax of DDM-SDSE.42,43 Specifically, the intensity of the intracellular red fluorescence increased significantly in the following 4 h and then remained stable, suggesting that the internalization of the liberated drug-loaded nanocarrier into cells and the release of Dox within cells had both been completed within 6 h. However, at this time, no Dox-induced apoptotic cells existed, as no brilliant blue fluorescence was observed. Therefore, the unchanged Pmax during this period presented well the delayed result of the Dox therapy. With extending time, the apoptotic characteristic (blue chromatin condensation) was progressively enhanced, indicating the increased proportion of apoptotic cells in the medium. In contrast, for the control group, no apoptotic characteristics were detected at the same time interval (Fig. S7†), demonstrating that the increased proportion of apoptotic cells is caused by the miR-125a triggered drug nanocarrier release from the anode. Since apoptotic cells could be captured to block the ORR on the cathode, the continuous descending trend of Pmax in this period perfectly provided precise information of cell apoptosis and the therapy effect. Moreover, an MTT (3-[4,4-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay was also carried out to screen cell activity. In the presence of miR-125a, the variation tendency of the cell activity coincided well with that of CLSM analysis, and the cell activity in the control group showed no distinct change (Fig. S8†). These results further confirmed that the fabricated DDM-SDSE can work as an in vitro “diagnosis-therapy-evaluation” research platform.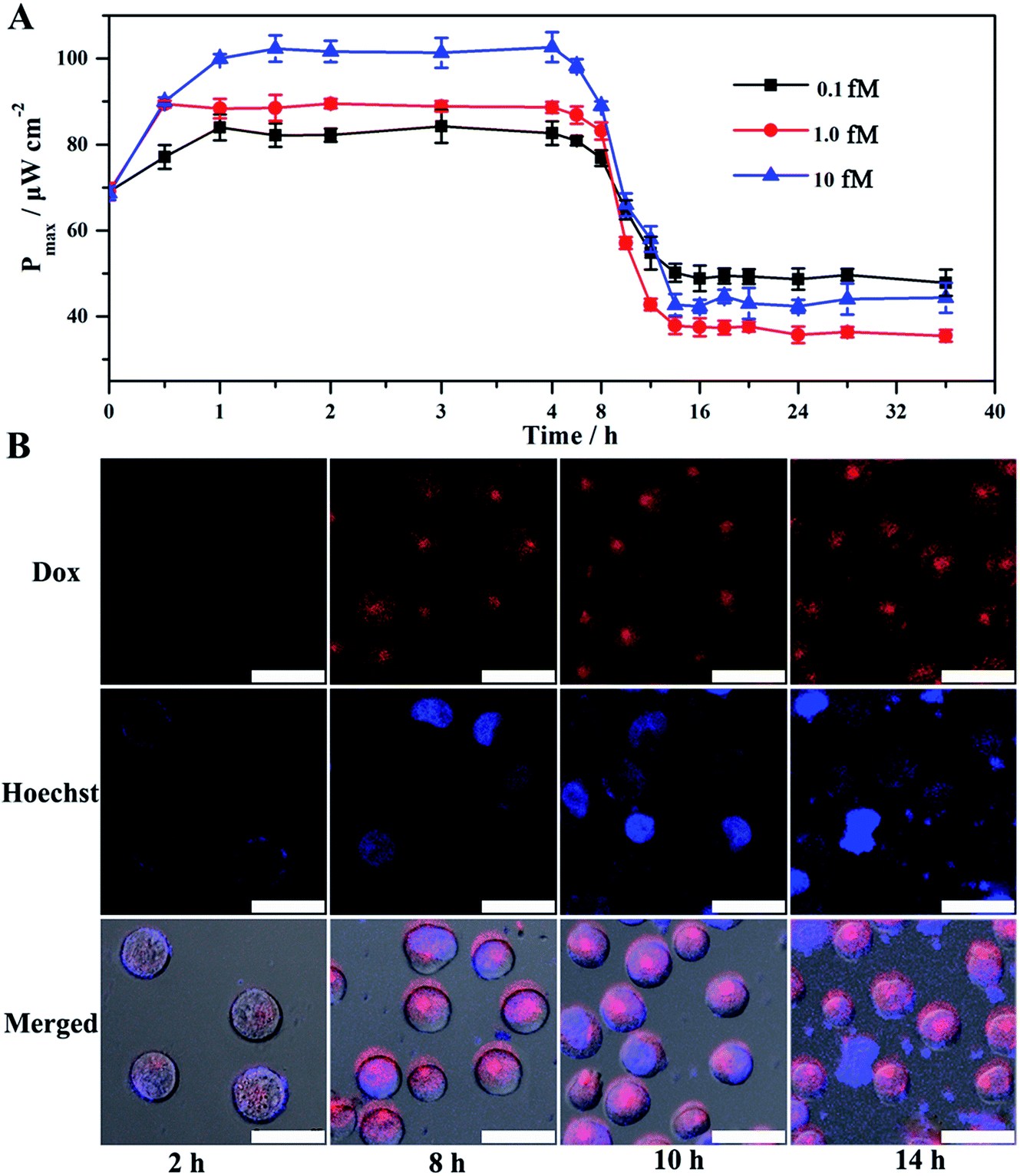 | ||
| Fig. 4 (A) The time course of maximum power density generation in the DDM-SDSE. The RPMI 1640 medium containing 1.0 × 104 K562 cells and different concentrations of miR-125a (0.1 fM, black line, square; 1.0 fM, red line, circle; and 10 fM, blue line, triangle) is used as the electrolyte for the DDM-SDSE, respectively. Scan rate: 1 mV s−1. (B) CLSM images of K562 cells collected from the DDM-SDSE after operation in 1.0 × 104 K562 cell-containing RPMI 1640 medium for different times in the presence of 0.1 fM miR-125a. The cells were further stained with Hoechst (blue fluorescence) before imaging, and the red fluorescence derived from Dox. Scale bar: 25 μm. Triplicate experiments were performed. | ||
In this DDM-SDSE, miR-125a serves not only as the biomarker of cancer for its diagnosis, but also as the initiator for the cascade “drug nanocarrier release-therapy-evaluation”. As a result of this design, we speculated that the DDM-SDSE may possess miR-125a-dependent drug delivery capacity. To verify this hypothesis, we recorded the Pmax of DDM-SDSE with varying amounts of miR-125a (Fig. 4A). It is obvious that the Pmax of all of the DDM-SDSEs generally displayed a similar variation in trend. But the Pmax at the plateaus gradually increased with an increase in miR-125a concentration, because more drug nanocarriers were ousted from the anode, decreasing the blocking effect on the anode. Furthermore, the plateau stage had the tendency to shorten due to the facilitated therapy caused by the increased amount of the liberated drug-loaded nanocarrier. From the above results, we concluded that the designed DDM-SDSE can possess the ability of on-demand drug delivery because the liberated amount of drug-loaded nanocarrier is determined by the expression levels of miR-125a. We further evaluated the quantitative analysis capability of self-diagnosis in the DDM-SDSE. With an increasing concentration of miR-125a , the value of stable Pmax progressively increased, and had a linear relationship with the concentration of miR-125a over the range of 0.10–25 fM (Fig. 5A–C). The limit of detection for miR-125a was estimated to be 0.1 fM, which was better or at least comparable to those of other miRNA analyses on the basis of fluorescence or electrocatalytic amplification.44 Such ideal sensitivity endows the designed DDM-SDSE with the potential for the early-stage diagnosis of tumors. Next, we explored whether the self-evaluation component in the DDM-SDSE could be used to quantitatively detect the Dox-induced apoptotic cells. The result illustrated that the Pmax gradually decreased with an increase of the cell numbers. The plot of the reduction of Pmax against the cell number gave a linear line in the range of 5–2000 apoptotic cells, with a limit of detection of 5 cells (Fig. 5D and E). This sensitivity is comparable to that of the most sensitive cytosensing approaches.7,32 More importantly, on the basis of the linear line, we can calculate detailed amounts of Dox-induced apoptotic K562 cells in the DDM-SDSE at any given time (Fig. 5F). As a result, the designed DDM-SDSE system can precisely assess the therapy response of the involved DDS.45
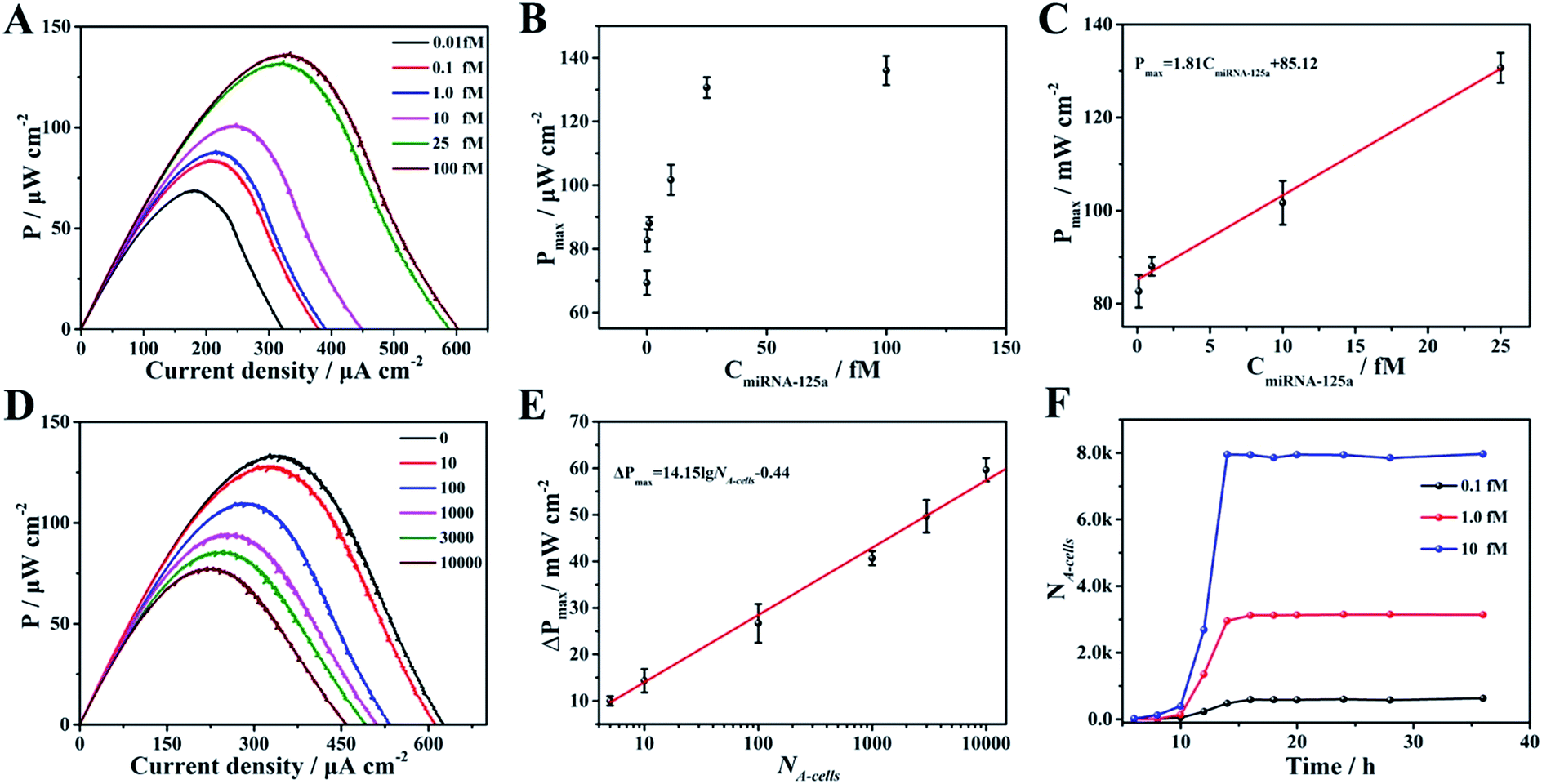 | ||
| Fig. 5 (A) The power density curves of the DDM-SDSE operating in 1.0 × 104 K562 cell-containing RPMI 1640 medium in the presence of different concentrations of miR-125a. (B) The plot of Pmaxvs. the concentration of miR-125a. (C) The plot of Pmaxvs. the concentration of miR-125a from 0.01 fM to 10 fM. (D) The power density curves of the DDM-SDSE operating in 25 fM miR-125a-containing RPMI 1640 medium in the presence of different numbers of apoptotic cell. (E) The reduction of Pmaxvs. the logarithm of the apoptotic cell numbers (NA-cells). (F) Time curves of apoptotic cell number generation in the DDM-SDSE in the presence of different concentrations of miR-125a. | ||
Conclusions
In summary, we have developed a self-sustained and highly compact drug delivery model with self-diagnosis and self-evaluation for realization of an in vitro “diagnosis-therapy-evaluation” research platform by integration of a targeted drug delivery system with a robust glucose/O2 fuel cell-based self-powered biosensor. The comprehensive design that covers the diagnostic event-triggered drug delivery on the anode, competitive reaction, blocking effect and the establishment of the evaluation model on the cathode ensures the successful fabrication of this in vitro integrated platform. In addition, due to the use of pAuNB and hmNCS as the anodic and cathodic catalyst, the stability and output performances of the fabricated glucose/O2 fuel cell-based self-powered biosensor are good enough to achieve long-term dynamic evaluation as well as highly sensitive diagnosis and evaluation in this in vitro integrated platform. Clearly, this study hits a sustainable cross-link between SPBs and drug delivery systems. It not only expands the application of SPBs into triple cascade events, but also adds a new promising in vitro “diagnosis-therapy-evaluation” platform to provide valuable information for clinical cancer therapy.Experimental
Fabrication of HA@Dox@hmSiO2
We firstly fabricated hmSiO2 by a template method, where polystyrene spheres (PS) were used as the template.41 Specifically, 250 mg of AETAC (80 wt%), 90 mL of ultrapure water and 10 mL of styrene were mixed to form a uniform solution. After heating the solution up to 90 °C with mechanical agitation and N2 agitation, 2.5 mL of V-50 (100 mg mL−1) was added, and then kept at 90 °C for 24 h. Subsequently, the PS was obtained using a sedimentation method in which iced ethanol was used as the solvent. To prepare hmSiO2 NPs, 0.8 g CTAB was firstly dissolved in a mixed solvent (80 mL of ultrapure water, 60 mL of ethanol and 1.5 mL of NH4OH (28%)), then the as-synthesized PS (10 mL, 93 mg mL−1) was added dropwise into the above mixture under vigorous stirring conditions, followed by the addition of 4.0 g of TEOS. After stirring for another 12 h, the black precipitate was collected by centrifugation. The black precipitate was then washed thoroughly with water and dried at room temperature. Finally, the precipitate was calcined under an air atmosphere (600 °C, 6 h) to remove PS and CTAB to obtain the hmSiO2 NPs.48The HA@Dox@hmSiO2 NPs were prepared in two steps: (1) embedding Dox into hmSiO2 NPs for the formation of Dox@hmSiO2 NPs, and (2) coating the Dox@hmSiO2 NPs with HA to prevent undesired drug leakage and to target the overexpressed transmembrane glycoprotein (CD44) on the surface of K562 leukemia cells.49 In detail, 1 mg hmSiO2 was incubated in the solution of Dox (1 mL, 1 mg mL−1) for 24 h while stirring at room temperature, and then the solution was centrifuged to remove the residual Dox. The obtained Dox@hmSiO2 NPs were redispersed in 1 mL of water. Subsequently, 1 mg of HA was added to the suspension of Dox@hmSiO2. After stirring for 24 h, the suspension was centrifuged, and then repeatedly washed with PBS buffer (10 mM, pH = 7.4). The loading amount of HA in the obtained HA@Dox@hmSiO2 NPs was calculated to be 325 μg mg−1.
Fabrication of the HA@Dox@hmSiO2/pAuNB/GC anode
Before anode fabrication, the pAuNB that served as the anodic catalyst was synthesized as follows:46,47 420 mg of AgNO3 and 400 mg of PVP were dissolved in 50 mL of ethylene glycol, followed by dropwise addition of 1.25 mL of HCl (37%) while stirring. The mixture was heated to 150 °C in the darkness, maintained at this temperature for 20 min, and finally cooled to room temperature. After centrifugation (9000 rpm, 10 min), the product (AgCl nanocubes) was washed with ethanol and dried under vacuum at 40 °C. Next, a 100 μL suspension of the as-prepared AgCl nanocubes (0.04 g mL−1), and 130 μL of hydroquinone (28 mM) were dispersed in a PVP aqueous solution (4.5 mL, 65 mM), followed by the addition of 55 μL of HAuCl4 solution (48.6 mM) under gentle agitation conditions. When the color of the solution turned to blue grey, stirring was stopped and the solution was left to stand for 2 h. The solution was then centrifuged, and the precipitation was incubated in 0.5 mL of NH4OH (25%) for 5 min to remove the AgCl nanocubes. Finally, the pAuNB was obtained through centrifugation (5500 rpm) and washed thoroughly.For the construction of the HA@Dox@hmSiO2/pAuNB/GC anode, 10 μL of the pAuNB solution (1 mg mL−1) was firstly dropped onto the surface of the GC electrode with a diameter of 3.0 mm. After drying, 10 μL PDS (1.0 × 10−5 M) was dropped onto the surface of the pAuNB/GC electrode and incubated at 4 °C overnight to form the PDS/pAuNB/GC electrode. The PDS and the pAuNB/GC electrode were bound by Au–S bonding.44 After the PDS/pAuNB/GC electrode was rinsed by water, the PDS/pAuNB/GC electrode was incubated with 10 μL of BSA (1 wt%) for 1 h to block the nonspecific binding sites. Finally, the electrode was incubated with 10 μL of the HA@Dox@hmSiO2 solution (0.1 mg mL−1) at 4 °C overnight. The formation of the HA@Dox@hmSiO2/pAuNB/GC anode was due to the amidation reaction between the –NH2 at the 3′ end of PDS and the carboxyl of HA. The HA@Dox@hmSiO2/pAuNB/GC anode was rinsed thoroughly to remove unbonded HA@Dox@hmSiO2 NPs.
Fabrication of PSp/hmNCS/GC cathode
The hmNCS was firstly prepared according to the previous literature.50 Briefly, 60 mL of ethanol, 11.36 mL of ultrapure water and 1.30 mL of ammonia solution were mixed together and stirred for 10 min, followed by the addition of 2.38 mL of TEOS. After stirring for 7 h at room temperature, 183.1 mL of H2O and 37.2 mL of ethanol were added into the mixture, and then the mixture was kept stirring for an additional 15 min. At this point, 6 g of CTAC (25%, wt%) was added dropwise under vigorous stirring, and the mixture was stirred for another 30 min. Subsequently, 0.41 g of resorcinol, 0.84 mL of formaldehyde and 2.94 mL of TEOS were successively added with vigorous stirring while the time interval of addition was 30 min. Finally, this mixture was stirred overnight and then transferred into autoclaves, and maintained at 150 °C for 12 h. After centrifugation, the precipitate was washed and dried under vacuum. The product was calcined at 350 °C for 3 h and at 950 °C for 4 h, and then etched by 10% HF to remove SiO2. After washing several times, we obtained the hmNCS.For the construction of the PSp/hmNCS/GC cathode, 10 mg of hmNCS was first dispersed in 10 mL of carboxymethyl chitosan solution (0.1 mg mL−1) by ultrasound, and then the solution was stirred for 12 h, leading to the introduction of carboxyl onto the surface of hmNCS. Then, 10 μL of the as-prepared carboxylated hmNCS solution was dropped onto the surface of the GC electrode and dried at 37 °C for 2 h. The above obtained electrode was further incubated with 10 μL PSp (FNFRLKAGAKIRFGRGC, 1 mg mL−1) for 12 h and then rinsed with water to remove unbonded PSp. Because of the amidation reaction between –NH2 on the peptide and –COOH on the hmNCS, the PSp/hmNCS/GC cathode was obtained.
Construction of DDM-SDSE
The above prepared anode and cathode were placed in the same electrolytic cell containing 200 μL of cell culture medium and K562 cells. After the addition of glucose, we recorded the polarization curves of the DDM-SDSE at each interval by LSV (onset voltage: the open-circuit voltage; scan rate: 1 mV s−1), and the power output (P) was calculated according to the formula P = UI. The results were normalized to the projective surface area of the GC electrode (0.07 cm2).Cell culture and cell cytotoxicity assay
The leukemia cells (K562 cell) bought from Nanjing Key Gen Biotech Co., Ltd. were cultured in RPMI 1640 medium (Gibco, Grand Island, NY) containing 10% fetal calf serum (FCS, Sigma), penicillin (100 μg mL−1) and streptomycin (100 μg mL−1) in an incubator (5% CO2, 37 °C). For MTT analysis, the cells were removed from the DDM-SDSE system at different times and washed with PBS, and then incubated in 10 μL of MTT solution (0.5 mg mL−1). After 4 h, the stained cells were lysed by DMSO. Finally, we recorded the absorbance (abs) at 630 nm in a Full Wavelength Scanning Multi-function Reader (Varioskan Flash, ThermoFisher Scientific) and calculated the cell viability according to the equation: cell viability (%) = (mean abs. value of sample/mean abs. value of control) × 100%.Drug targeted delivery and in vitro cytotoxicity
In order to observe the cellular fluorescence in K562 cells, the cells were removed from the DDM-SDSE system at different times and then transferred to confocal microscopy dishes, and were finally stained by Hoechst 33258 at 37 °C for 15 min. After thoroughly washing with PBS, we observed fluorescence immediately using CLSM (TCS SP5, Leica).Dox release experiments
In order to explore the protective effect of HA, the HAase-mediated drug release and the rates of drug efflux in vitro, the HA@Dox@hmSiO2 and Dox@hmSiO2 were incubated in 20 mL of PBS (10 mM, pH = 7.4) with or without HAase for different times. After centrifugation, the supernatant was used as a sample. The absorbance of these samples at 480 nm was then measured using a UV-vis spectrophotometer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully appreciate support from the National Natural Science Foundation (21775067, 21335504), the International Cooperation Foundation from Ministry of Science and Technology (2016YFE0130100), the China Postdoctoral Science Foundation (2018T110475) and the Fundamental Research Funds for the Central Universities (020514380161).Notes and references
- M. Zhou, Electroanalysis, 2015, 27, 1786–1810 CrossRef CAS.
- M. Zhou, Y. Du, C. Chen, B. Li, D. Wen, S. Dong and E. Wang, J. Am. Chem. Soc., 2010, 132, 2172–2174 CrossRef CAS PubMed.
- S. Xu, Y. Qin, C. Xu, Y. Wei, R. Yang and Z. L. Wang, Nat. Nanotechnol., 2010, 5, 366–373 CrossRef CAS PubMed.
- H. Sakai, T. Nakagawa, Y. Tokita, T. Hatazawa, T. Ikeda, S. Tsujimura and K. Kano, Energy Environ. Sci., 2009, 2, 133–138 RSC.
- Y. Yu, J. Zhai, Y. Xia and S. Dong, Nanoscale, 2017, 9, 11846–11850 RSC.
- D. P. Hickey, R. C. Reid, R. D. Milton and S. D. Minteer, Biosens. Bioelectron., 2016, 77, 26–31 CrossRef CAS PubMed.
- P.-P. Gai, Y.-S. Ji, W.-J. Wang, R.-B. Song, C. Zhu, Y. Chen, J.-R. Zhang and J.-J. Zhu, Nano Energy, 2016, 19, 541–549 CrossRef CAS.
- P. Gai, C. Gu, T. Hou and F. Li, ACS Appl. Mater. Interfaces, 2018, 10, 9325–9331 CrossRef CAS PubMed.
- M. Rasmussen, S. Abdellaoui and S. D. Minteer, Biosens. Bioelectron., 2016, 76, 91–102 CrossRef CAS PubMed.
- C. Hou, S. Fan, Q. Lang and A. Liu, Anal. Chem., 2015, 87, 3382–3387 CrossRef CAS PubMed.
- L.-L. Wang, H.-H. Shao, W.-J. Wang, J.-R. Zhang and J.-J. Zhu, Nano Energy, 2018, 44, 95–102 CrossRef CAS.
- M. Zhou, N. Zhou, F. Kuralay, J. R. Windmiller, S. Parkhomovsky, G. Valdés-Ramírez, E. Katz and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 2686–2689 CrossRef CAS PubMed.
- M. Zhou and J. Wang, Electroanalysis, 2012, 24, 197–209 CrossRef CAS.
- B. Yang, Y. Chen and J. Shi, Chem, 2018, 4, 1248–1313 Search PubMed.
- J. Bai, X. Jia, W. Zhen, W. Cheng and X. Jiang, J. Am. Chem. Soc., 2018, 140, 106–109 CrossRef CAS PubMed.
- D. Niu, Y. Li and J. Shi, Chem. Soc. Rev., 2017, 46, 569–585 RSC.
- D. Mao, W. Wu, S. Ji, C. Chen, F. Hu, D. Kong, D. Ding and B. Liu, Chem, 2017, 3, 991–1007 CAS.
- X. Gu and B. Z. Tang, Chem, 2017, 3, 922–924 CAS.
- D. Y. Lee, J. Y. Kim, Y. Lee, S. Lee, W. Miao, H. S. Kim, J. J. Min and S. Jon, Angew. Chem., Int. Ed., 2017, 56, 13684–13688 CrossRef CAS PubMed.
- Z. Yaari, D. Da Silva, A. Zinger, E. Goldman, A. Kajal, R. Tshuva, E. Barak, N. Dahan, D. Hershkovitz and M. Goldfeder, Nat. Commun., 2016, 7, 13325–13335 CrossRef CAS PubMed.
- L.-h. Xu, Y. Guo, X.-L. Zhang, J.-j. Chen and S.-y. Hu, Cell. Physiol. Biochem., 2016, 38, 939–949 CrossRef CAS PubMed.
- T. Kawaguchi, S. Komatsu, D. Ichikawa, M. Tsujiura, H. Takeshita, S. Hirajima, M. Miyamae, W. Okajima, T. Ohashi and T. Imamura, Int. J. Mol. Sci., 2016, 17, 1459–1474 CrossRef PubMed.
- H. Dong, J. Lei, L. Ding, Y. Wen, H. Ju and X. Zhang, Chem. Rev., 2013, 113, 6207–6233 CrossRef CAS PubMed.
- C. Liu, Z. Zhao, J. Fan, C. J. Lyon, H. J. Wu, D. Nedelkov, A. M. Zelazny, K. N. Olivier, L. H. Cazares, S. M. Holland, E. A. Graviss and Y. Hu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3969–3974 CrossRef CAS PubMed.
- R. Peri-Naor, T. Ilani, L. Motiei and D. Margulies, J. Am. Chem. Soc., 2015, 137, 9507–9510 CrossRef CAS PubMed.
- V. Plaks, C. D. Koopman and Z. Werb, Science, 2013, 341, 1186–1188 CrossRef CAS PubMed.
- Y. Yuan, R. T. Kwok, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2014, 136, 2546–2554 CrossRef CAS PubMed.
- P. Miao, J. Yin, L. Ning and X. Li, Biosens. Bioelectron., 2014, 62, 97–101 CrossRef CAS PubMed.
- J. J. Zhang, T. T. Zheng, F. F. Cheng, J. R. Zhang and J. J. Zhu, Anal. Chem., 2011, 83, 7902–7909 CrossRef CAS PubMed.
- X. Chen, W. Xiong and H. Li, Oncol. Lett., 2016, 12, 4937–4948 CrossRef CAS PubMed.
- J. Lu, G. Getz, E. A. Miska, E. Alvarez-Saavedra, J. Lamb, D. Peck, A. Sweet-Cordero, B. L. Ebert, R. H. Mak, A. A. Ferrando, J. R. Downing, T. Jacks, H. R. Horvitz and T. R. Golub, Nature, 2005, 435, 834–838 CrossRef CAS PubMed.
- P. Gai, R. Song, C. Zhu, Y. Ji, W. Wang, J.-R. Zhang and J.-J. Zhu, Chem. Commun., 2015, 51, 16763–16766 RSC.
- J. Ding, T. Liang, Y. Zhou, Z. He, Q. Min, L. Jiang and J. Zhu, Nano Res., 2017, 10, 690–703 CrossRef CAS.
- C. Xiong, K. Brewer, S. Song, R. Zhang, W. Lu, X. Wen and C. Li, J. Med. Chem., 2011, 54, 1825–1835 CrossRef CAS PubMed.
- C.-e. Zhao, P. Gai, R. Song, Y. Chen, J. Zhang and J.-J. Zhu, Chem. Soc. Rev., 2017, 46, 1545–1564 RSC.
- M. Xu, Y. Sui, G. Xiao, X. Yang, Y. Wei and B. Zou, Nanoscale, 2017, 9, 2514–2520 RSC.
- M. Chu, Y. Zhang, L. Yang, Y. Tan, W. Deng, M. Ma, X. Su, Q. Xie and S. Yao, Energy Environ. Sci., 2013, 6, 3600–3604 RSC.
- N. Mano and A. de Poulpiquet, Chem. Rev., 2018, 118, 2392–2468 CrossRef CAS PubMed.
- Y. Ito, H. J. Qiu, T. Fujita, Y. Tanabe, K. Tanigaki and M. Chen, Adv. Mater., 2014, 26, 4145–4150 CrossRef CAS PubMed.
- R. Sackstein, J. S. Merzaban, D. W. Cain, N. M. Dagia, J. A. Spencer, C. P. Lin and R. Wohlgemuth, Nat. Med., 2008, 14, 181–187 CrossRef CAS PubMed.
- S. Yang, D. Chen, N. Li, Q. Xu, H. Li, F. Gu, J. Xie and J. Lu, Small, 2016, 12, 360–370 CrossRef CAS PubMed.
- M. Simeoni, P. Magni, C. Cammia, G. De Nicolao, V. Croci, E. Pesenti, M. Germani, I. Poggesi and M. Rocchetti, Cancer Res., 2004, 64, 1094–1101 CrossRef CAS PubMed.
- R. E. Eliaz, S. Nir, C. Marty and F. C. Szoka, Cancer Res., 2004, 64, 711–718 CrossRef CAS PubMed.
- A. D. Castaneda, N. J. Brenes, A. Kondajji and R. M. Crooks, J. Am. Chem. Soc., 2017, 139, 7657–7664 CrossRef CAS PubMed.
- T. Lammers, S. Aime, W. E. Hennink, G. Storm and F. Kiessling, Acc. Chem. Res., 2011, 44, 1029–1038 CrossRef CAS PubMed.
- M. C. Ortega-Liebana, J. L. Hueso, R. Arenal and J. Santamaria, Nanoscale, 2017, 9, 1787–1792 RSC.
- S. Pedireddy, H. K. Lee, C. S. Koh, J. M. Tan, W. W. Tjiu and X. Y. Ling, Small, 2016, 12, 4531–4540 CrossRef CAS PubMed.
- K. Shin, J. W. Choi, G. Ko, S. Baik, D. Kim, O. K. Park, K. Lee, H. R. Cho, S. I. Han and S. H. Lee, Nat. Commun., 2017, 8, 15807–15819 CrossRef CAS PubMed.
- T. Jiang, R. Mo, A. Bellotti, J. Zhou and Z. Gu, Adv. Funct. Mater., 2014, 24, 2295–2304 CrossRef CAS.
- S. Mezzavilla, C. Baldizzone, K. J. Mayrhofer and F. Schüth, ACS Appl. Mater. Interfaces, 2015, 7, 12914–12922 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and instruments used in this study, characteristics of materials, comparison of flow cytometry results of cells that were incubated with HA@Dox@hmSiO2 and Dox@hmSiO2 nanoparticles for different times, output performance, CLSM and MTT results of control experiments. See DOI: 10.1039/c8sc04019b |
| This journal is © The Royal Society of Chemistry 2018 |