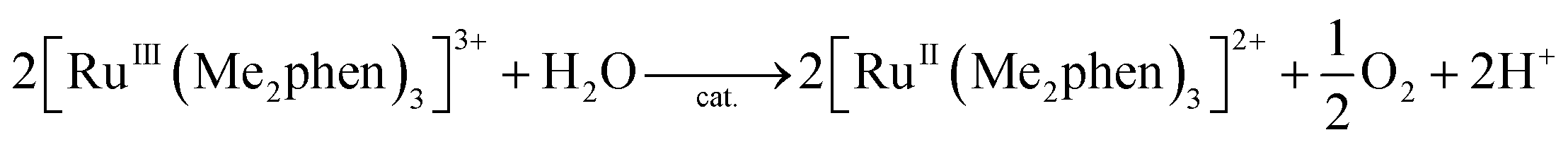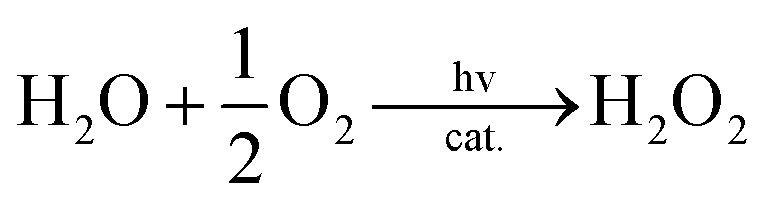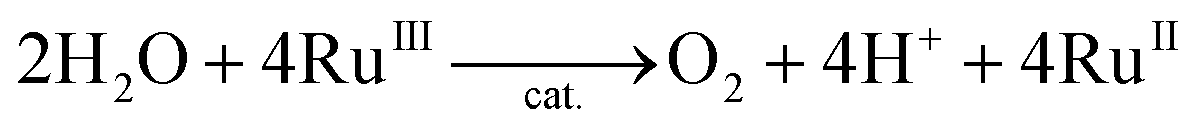DOI:
10.1039/C7SC02495A
(Edge Article)
Chem. Sci., 2017,
8, 7119-7125
Photocatalytic oxidation of benzene to phenol using dioxygen as an oxygen source and water as an electron source in the presence of a cobalt catalyst†
Received
4th June 2017
, Accepted 21st August 2017
First published on 21st August 2017
Abstract
Photocatalytic hydroxylation of benzene to phenol by dioxygen (O2) occurs under visible light irradiation of an O2-saturated acetonitrile solution containing [RuII(Me2phen)3]2+ as a photocatalyst, [CoIII(Cp*)(bpy)(H2O)]2+ as an efficient catalyst for both the water oxidation and benzene hydroxylation reactions, and water as an electron source in the presence of Sc(NO3)3. The present study reports the first example of photocatalytic hydroxylation of benzene with O2 and H2O, both of which are the most green reagents, under visible light irradiation to afford a high turnover number (e.g., >500). Mechanistic studies revealed that the photocatalytic reduction of O2 to H2O2 is the rate-determining step, followed by efficient catalytic hydroxylation of benzene to phenol with H2O2, paving a new way for the photocatalytic oxygenation of substrates by O2 and water.
Introduction
Phenol, which is an important precursor for many chemicals and industrial products (e.g., dyes, polymers, etc.), is currently produced from benzene by a three step cumene process.1,2 Since the efficiency of the cumene process is low (∼5% yield of phenol) under severe conditions (e.g., high temperature, high pressure, and strong acidic conditions), it is highly desired to develop a one-step synthesis of phenol from benzene using homogeneous and heterogeneous inorganic catalysts.3–5 Among various oxidants, hydrogen peroxide (H2O2) is frequently used as a green oxidant, which produces water or dioxygen (O2) as products, in catalytic benzene hydroxylation.6–13 O2 is a more ideal oxidant than H2O2 because of its abundance in nature, low cost, and environmental benignity.4,14,15 However, catalytic aerobic oxidation of benzene to phenol has required sacrificial reducing agents such as H2, ascorbic acid, and NADH analogs.16 In addition, the catalytic aerobic oxidation of benzene without sacrificial reducing agents has so far required harsh conditions, such as high temperatures or UV light photoirradiation.14,15,17,18 Moreover, although the photocatalytic hydroxylation of benzene to phenol using organic photocatalysts without overoxidation has been demonstrated,19–22 the turnover numbers (TONs) of the catalytic hydroxylation of benzene to phenol are still low (e.g., 13).20
We report herein an efficient photocatalytic hydroxylation of benzene to phenol (PhOH) using O2 as an oxidant in the presence of Sc(NO3)3 and utilizing [RuII(Me2phen)3]2+ (Me2phen = 4,7-dimethyl-1,10-phenanthroline) as a photocatalyst, water as an electron source, and [CoIII(Cp*)(bpy)(H2O)]2+ (Cp* = η5-pentamethylcyclopentadienyl and bpy = 2,2-bipyridine) as an efficient catalyst for water oxidation as well as benzene hydroxylation to attain a TON of greater than 500 (Scheme 1). Mechanistic aspects of the benzene hydroxylation reaction under the visible light irradiation conditions have been discussed as well.
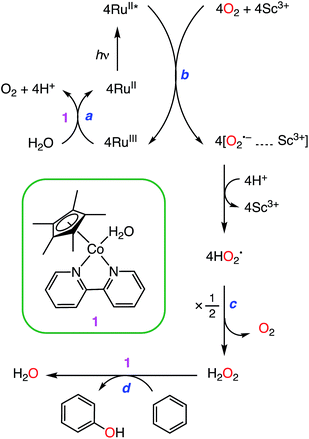 |
| | Scheme 1 Mechanism of the photocatalytic hydroxylation of benzene. | |
Results and discussion
[CoIII(Cp*)(bpy)(H2O)]2+ (1) and [RuII(Me2phen)3]2+ were synthesised according to the literature methods.23,24 Visible light irradiation of an O2-saturated solution of CH3CN (MeCN) and H2O (v/v = 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) containing a catalytic amount of 1 (1.0 μM), [RuII(Me2phen)3]2+ (1.0 mM), Sc(NO3)3 (100 mM), and benzene (1.0 M) at 298 K resulted in the formation of PhOH [eqn (1)],
:2) containing a catalytic amount of 1 (1.0 μM), [RuII(Me2phen)3]2+ (1.0 mM), Sc(NO3)3 (100 mM), and benzene (1.0 M) at 298 K resulted in the formation of PhOH [eqn (1)],| | 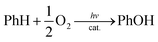 | (1) |
as shown in Fig. 1. PhOH is produced via the catalytic oxygenation of hydrogen peroxide (H2O2) with 1, which acts as not only a benzene oxygenation catalyst but also a water oxidation catalyst with [RuIII(Me2phen)3]3+ produced by photoinduced electron transfer from the excited state of [RuII(Me2phen)3]2+ to O2 in the presence of Sc3+, as shown in Scheme 1. It should be noted that the catalyst 1 acts as an efficient catalyst for water oxidation in the photocatalytic production of H2O2 in the presence of Sc(NO3)3, as reported previously.23,25 [RuII(Me2phen)3]2+ acts as a more efficient photocatalyst compared to [RuII(bpy)3]2+ for O2 reduction by the excited state, because the one-electron oxidation potential of [RuII(Me2phen)3]2+* (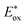 = −1.01 V vs. NHE) is more negative than that of [RuII(bpy)3]2+* (
= −1.01 V vs. NHE) is more negative than that of [RuII(bpy)3]2+* (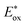 = −0.84 V vs. NHE).23 The UV-vis spectrum of the photocatalyst, [RuII(Me2phen)3]2+, during the photocatalytic oxidation remained almost identical to that of the initial stage at 445 nm, indicating that the overall reactivity decreased with time due to oxidative degradation of the catalyst 1 (see Experimental section; Fig. S1, ESI†).
= −0.84 V vs. NHE).23 The UV-vis spectrum of the photocatalyst, [RuII(Me2phen)3]2+, during the photocatalytic oxidation remained almost identical to that of the initial stage at 445 nm, indicating that the overall reactivity decreased with time due to oxidative degradation of the catalyst 1 (see Experimental section; Fig. S1, ESI†).
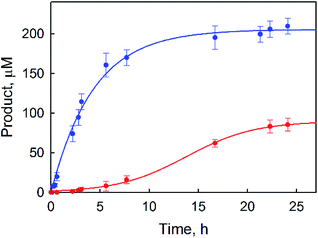 |
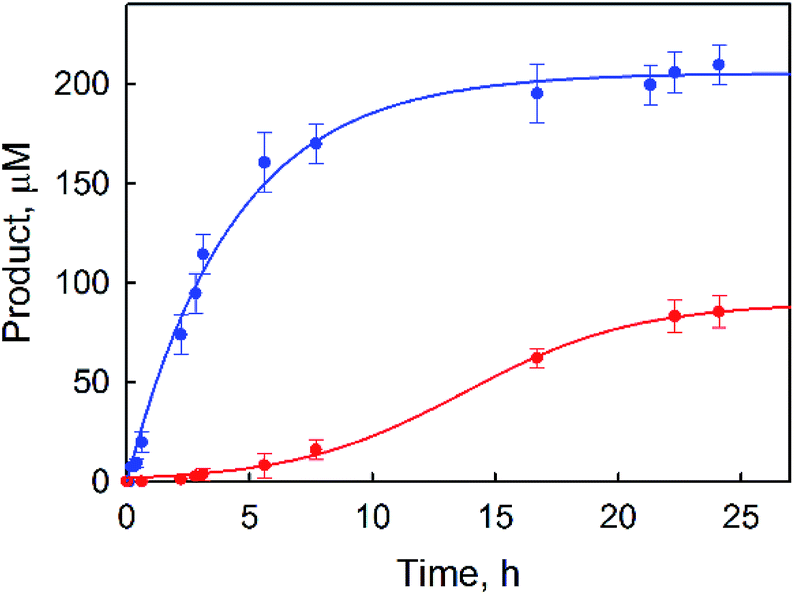
| | Fig. 1 Time courses of the products [PhOH (blue) and p-benzoquinone (red)] obtained in the photocatalytic oxidation of benzene by O2 with [RuII(Me2phen)3]2+ (1.0 mM) in the presence of a catalytic amount of 1 (1.0 μM) in an O2-saturated solvent mixture of MeCN and H2O (v/v = 23:2) containing Sc(NO3)3 (100 mM) and benzene (1.0 M) under photoirradiation (white light) at 298 K. | |
p-Benzoquinone was also produced during the photocatalytic oxidation of benzene by O2, and the yield of p-benzoquinone increased as the reaction solution was irradiated for a longer time but the yield of PhOH remained constant (Fig. 1). The observation of the induction period for the formation of p-benzoquinone in Fig. 1 suggests that p-benzoquinone is produced by the further oxidation of PhOH. The production of p-benzoquinone from PhOH was independently confirmed (Fig. S2, ESI†). The TON for the production of both phenol and benzoquinone based on 1 was determined to be 500(20), where the TON of p-benzoquinone is counted three times because p-benzoquinone is the six-electron oxidized product whereas phenol is the two-electron oxidized product. When the reaction conditions were changed by increasing the concentration of the catalyst and decreasing the concentration of the benzene substrate, a much higher product yield of phenol (∼30%) based on benzene was obtained (Fig. 2) than that in Fig. 1. The quantum yield (QY) was estimated by eqn (2),
where
R (mol s
−1) is the PhOH production rate and
I (einstein s
−1) is the rate of the number of incident photons. The quantum yield was determined to be 1.7(2)% from the amount of PhOH produced during the photocatalytic reaction under photoirradiation (
λ = 440 nm) for 1 h (Fig. S3, ESI
†). The photocatalytic reactivity increased on increasing the concentration of benzene as well as the concentration of [Ru
II(Me
2phen)
3]
2+ (Fig. S4, ESI
†). A negligible amount of PhOH was produced in the absence of
1, indicating that
1 is an essential component in the benzene hydroxylation reaction.
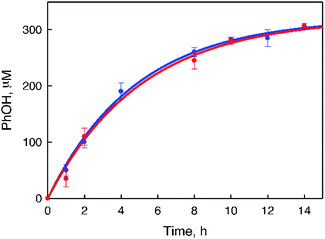 |
| | Fig. 2 Time courses of PhOH concentration produced in the photocatalytic oxidation of benzene under photoirradiation (white light) of an O2-saturated solvent mixture of MeCN and H2O (v/v = 23:2) containing 1 (0.10 mM), [RuII(Me2phen)3]2+ (1.0 mM), and benzene (1.0 mM, blue circles) or benzene-d6 (1.0 mM, red circles) in the presence of Sc(NO3)3 (100 mM) at 298 K. No p-benzoquinone was produced under the present experimental conditions. | |
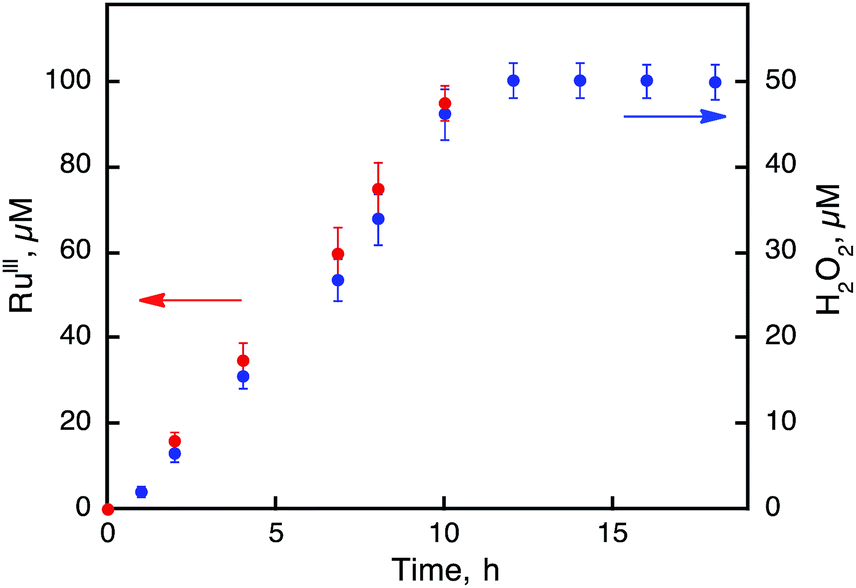
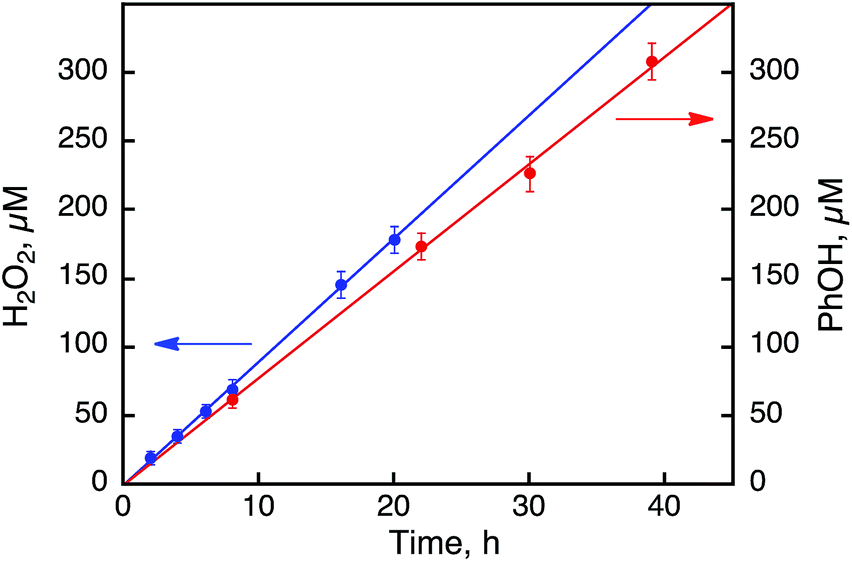
The photocatalytic hydroxylation reaction proceeds as shown in Scheme 1. Visible light irradiation of [RuII(Me2phen)3]2+ resulted in the formation of H2O2 by oxidative quenching of the photoexcited state [RuII(Me2phen)3]2+* (* denotes the excited state) with O2 in the presence of Sc(NO3)3 (Scheme 1, reaction pathways b and c), as reported for the case of [RuII(bpy)3]2+.26 The time courses of [RuIII(Me2phen)3]3+ generation and H2O2 production were obtained to give the stoichiometry of the photochemical reaction, as shown in eqn (3) (Fig. 3).
| | 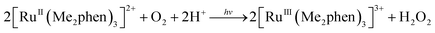 | (3) |
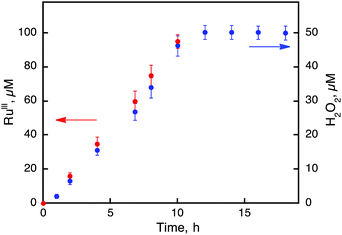 |
| | Fig. 3 Time courses of the concentrations of [RuIII(Me2phen)3]3+ (red) and H2O2 (blue) produced from H2O and O2 in the photocatalytic oxidation of [RuII(Me2phen)3]2+ by O2 under photoirradiation (white light) of an air-saturated solvent mixture of MeCN and H2O (v/v = 23:2) containing [RuII(Me2phen)3]2+ (100 μM) in the presence of Sc(NO3)3 (100 mM) at 298 K. | |
The lifetime of [RuII(Me2phen)3]2+* and emission quenching have been determined to obtain the rate constants (ket) of photoinduced electron transfer in the absence and presence of Sc(NO3)3 (Fig. S5 and Table S1, ESI†). The ket values in both the absence and presence of Sc(NO3)3 are close to the diffusion-limited value, showing that Sc(NO3)3 does not affect the oxidative quenching of [RuII(Me2phen)3]2+* by O2. The emission spectra of [RuII(Me2phen)3]2+ in the absence and presence of Sc(NO3)3 taken in a solvent mixture of MeCN and H2O (v/v = 23:2) containing different concentrations of O2 indicate that the amount of O2 in air is large enough for efficient H2O2 photogeneration (Fig. S5d, ESI†).
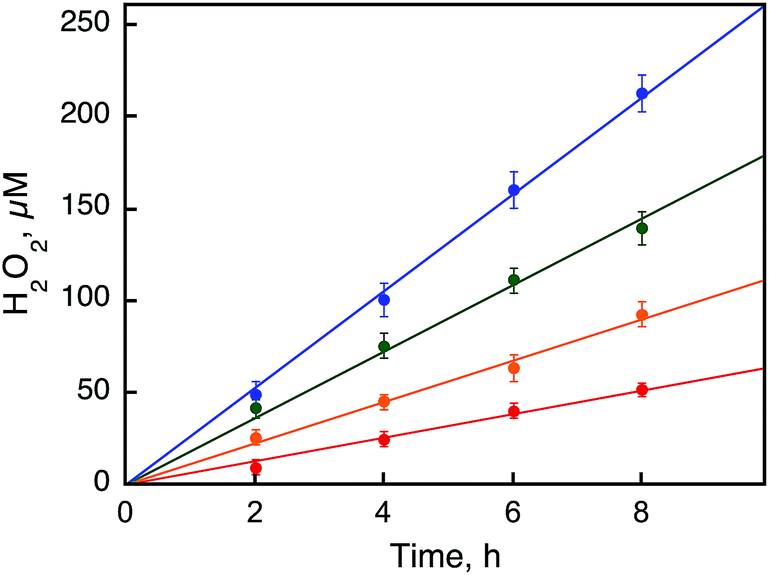
Visible light irradiation of the reaction solution without benzene results in the formation of H2O2, as shown in Fig. 4 (Scheme 1, reaction pathways a–c), and the production of H2O2 is obtained from the photooxidation of [RuII(Me2phen)3]2+ by O2 [eqn (3)] and the catalytic water oxidation by [RuIII(Me2phen)3]3+ in the presence of 1 [eqn (4)].23 Thus, the overall reaction
| | 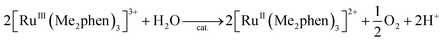 | (4) |
is the photocatalytic oxidation of H
2O by O
2 to produce H
2O
2, as given by
eqn (5).
| | 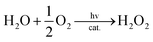 | (5) |
The rates of photocatalytic oxidation of H
2O by O
2 to produce H
2O
2 in the presence of
1 were also investigated with various concentrations of
1 at 298 K (Fig. S6, ESI
†). The zeroth-order rate constants (
kobs) were obtained from the initial slopes of the plots of the amount of H
2O
2 produced
vs. time, being proportional to the concentration of
1 (
Fig. 4 and S6, ESI
†).
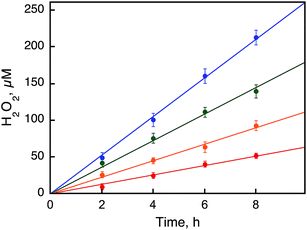 |
| | Fig. 4 Time courses of the photocatalytic production of H2O2 from H2O and O2 with [RuII(Me2phen)3]2+ (0.10 mM) in the presence of a catalytic amount of 1 [0.50 mM (red), 1.0 mM (orange), 1.5 mM (green), and 2.0 mM (blue)] in an air-saturated solvent mixture of MeCN and H2O (v/v = 23:2) containing Sc(NO3)3 (100 mM) under photoirradiation (white light) at 298 K. | |
The catalytic water oxidation by [RuIII(Me2phen)3]3+ with 1 to evolve O2 [eqn (6); Scheme 1, reaction pathway a]
| | 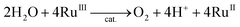 | (6) |
was independently confirmed, where O
2 was evolved, accompanied by the formation of [Ru
II(Me
2phen)
3]
2+. The evolved O
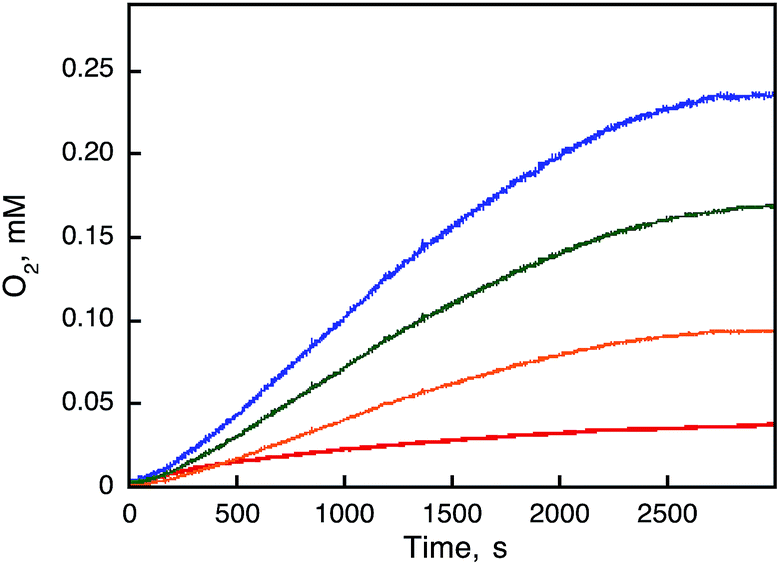
2 concentrations were investigated by a Clark oxygen electrode, showing an increase in the amount of O
2 evolved after addition of [Ru
III(Me
2phen)
3]
3+ (1.0 mM) to a solvent mixture of MeCN and H
2O (1.0 mL; v/v = 9
:
1) containing Sc(NO
3)
3 (100 mM) and various concentrations of
1 (
Fig. 5), where the O
2 yield increased with an increasing concentration of
1 to approach 100% (1/4 of the initial concentration of [Ru
III(Me
2phen)
3]
3+). The reaction rate was accelerated with an increasing concentration of
1 to account for oxidation of water by [Ru
III(Me
2phen)
3]
3+ with
1 under acidic conditions due to the presence of Sc(NO
3)
3. Formation of [Ru
II(Me
2phen)
3]
2+ accompanied by water oxidation was also confirmed by monitoring the UV-vis spectral changes (Fig. S7a, ESI
†). The time courses of the oxidation of
1 by [Ru
III(Me
2phen)
3]
3+ were obtained by spectral changes at
λ = 445 nm due to [Ru
II(Me
2phen)
3]
2+, showing that the rate of [Ru
II(Me
2phen)
3]
2+ formation from [Ru
III(Me
2phen)
3]
3+ increased with the increase of the concentration of
1 (Fig. S7b, ESI
†).
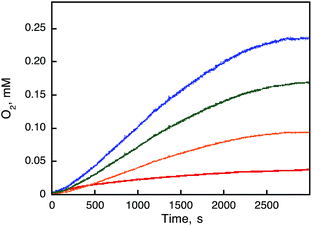 |
| | Fig. 5 Time courses of O2-evolution observed in the catalytic oxidation of H2O by [RuIII(Me2phen)3]3+ (1.0 mM) with 1 [10 μM (red), 50 μM (orange), 100 μM (green), and 200 μM (blue)] in the presence of Sc(NO3)3 (100 mM) in a deaerated solvent mixture (1.0 mL) of MeCN and H2O (v/v = 9:1) at 298 K. The induction periods were observed due to the reaction time to oxidize 1 by [RuIII(Me2phen)3]3+ to produce the catalytically active species. | |
Catalytic oxidation of benzene to phenol with H2O2 [eqn (7)]
| |  | (7) |
has also been confirmed in a solvent mixture of MeCN and H
2O (23
:
2) containing Sc(NO
3)
3 in order to stabilize H
2O
2 with the same conditions for the overall photocatalytic reaction as shown in
Fig. 6 (
Scheme 1, reaction pathway d). The rate of benzene hydroxylation increased with increasing concentrations of
1 (
Fig. 6) and H
2O
2 (Fig. S8, ESI
†). A combination of the photocatalytic oxidation of H
2O by O
2 to produce H
2O
2 [
eqn (5)] and the catalytic hydroxylation of benzene to phenol by H
2O
2 [
eqn (7)] affords the overall photocatalytic oxidation of benzene to phenol by O
2 [
eqn (1)]. The photocatalytic reactions of an O
2-saturated solution of MeCN and H
2O (v/v = 23
:
2) containing
1 (0.50 mM), [Ru
II(Me
2phen)
3]
2+ (0.10 mM), and Sc(NO
3)
3 (100 mM) under photoirradiation (white light) were examined in the absence and presence of benzene to compare their reactivities (
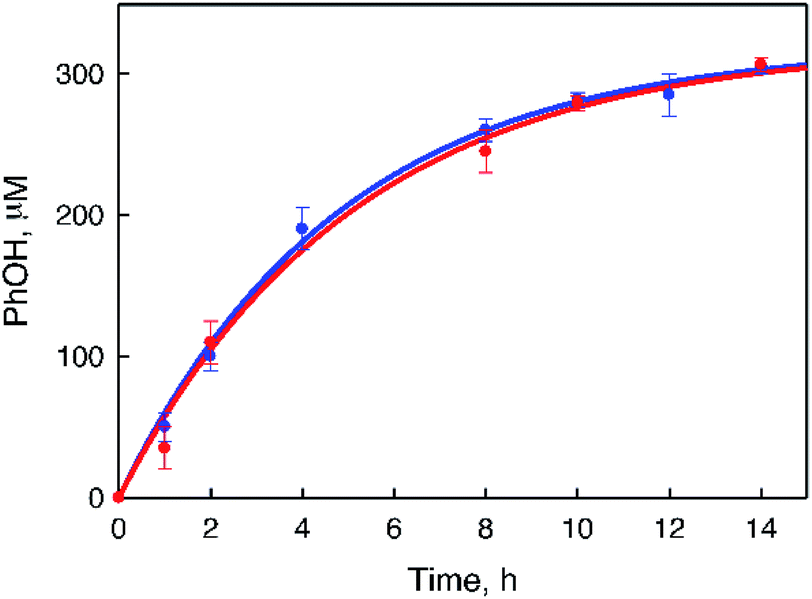
Fig. 7). The rate of H
2O
2 production from H
2O and O
2 in the absence of benzene was slightly higher than that of PhOH generation in the photocatalytic hydroxylation of benzene, indicating that H
2O
2 produced in the photocatalytic oxidation of H
2O by O
2 reacted with benzene efficiently in the presence of
1 to produce PhOH.
27 In fact, the rate of PhOH production (59 μM after 1 h) from H
2O
2 (1.0 mM) and benzene (1.0 M) with
1 (0.50 mM, red dots in
Fig. 6) is much faster than that of PhOH production (estimated to be 7.5 μM after 1 h) in the photocatalytic reaction (
Fig. 7), because the concentration of H
2O
2 produced in the photocatalytic oxidation of H
2O by O
2 is much smaller than 1.0 mM. Since the rate of PhOH production from H
2O
2 is proportional to the concentration of H
2O
2 (Fig. S8, ESI
†), the rate of PhOH production in the photocatalytic hydroxylation of benzene by O
2 corresponds to that of PhOH production in the catalytic oxidation of benzene with 127 μM H
2O
2, which may be the steady state concentration during the photocatalytic hydroxylation of benzene.
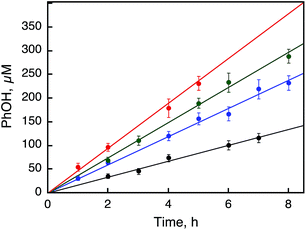 |
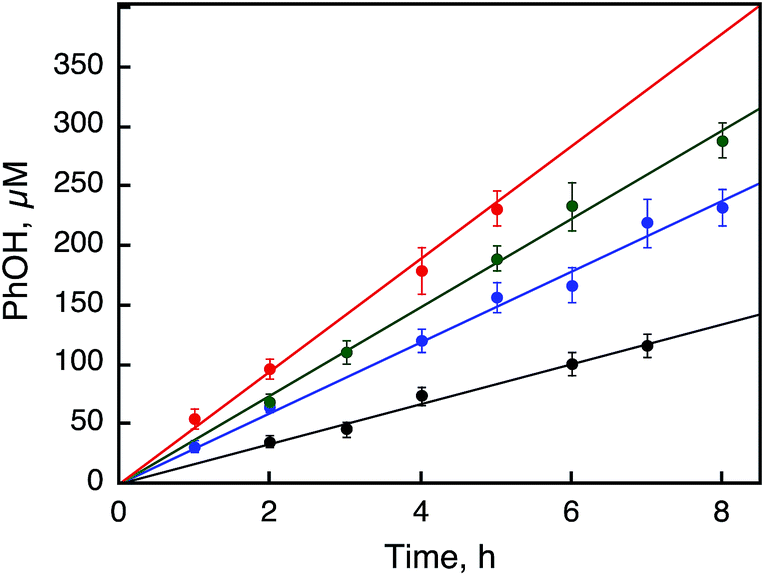
| | Fig. 6 Time courses of the catalytic hydroxylation of benzene to PhOH with H2O2 (1.0 mM) in the presence of 1 [10 μM (black), 50 μM (blue), 100 μM (green), and 500 μM (red)] in a solvent mixture of MeCN and H2O (v/v = 23:2) containing benzene (1.0 M) and Sc(NO3)3 (100 mM) at 298 K. | |
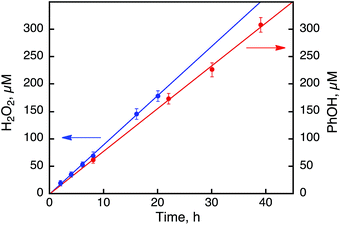 |
| | Fig. 7 Time courses of the products from the photoinduced reaction under photoirradiation (white light) in an O2-saturated solvent mixture of MeCN and H2O (v/v = 23:2) containing 1 (0.50 mM), [RuII(Me2phen)3]2+ (0.10 mM), and Sc(NO3)3 (100 mM) in the absence (blue) and presence (red) of benzene (1.0 M) at 298 K. | |
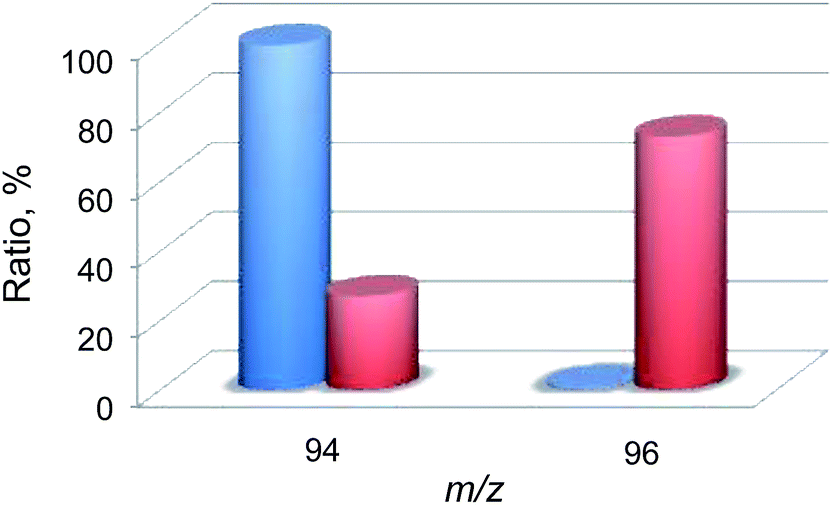
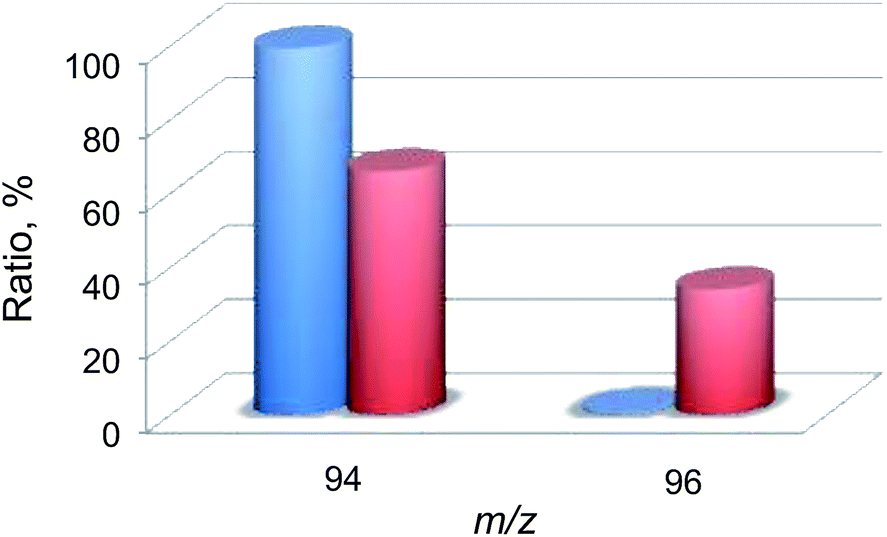
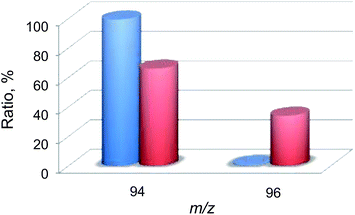
Then, 18O2-labeling experiments were performed to confirm that the oxygen atom in the phenol product in eqn (1) derives from O2 (Fig. 8). Indeed, Ph18OH (73%) was formed as the major product together with Ph16OH (27%) in the 18O2-labeling experiments, indicating that the oxygen atom in the PhOH product obtained in the photocatalytic oxidation of benzene by H218O2 derived from 18O2 (98% 18O-enriched; Fig. 8). The formation of Ph16OH (27%) indicates that 16O2 was produced by the oxidation of H216O by [RuIII(Me2phen)3]3+, leading to the production of H216O2 to oxidize benzene to produce Ph16OH instead of Ph18OH (Scheme 1, reaction pathway a). The 18O-labeling experiments were also performed by replacing H216O with H218O to support that the oxygen atom in the phenol product derives from O2 rather than H2O (Fig. 9). In this case, Ph16OH (66%) was the major product together with Ph18OH (34%), consistent with the 18O2-labeling experiments described above.
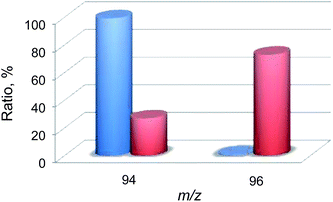 |
| | Fig. 8 A comparison of the relative abundances of authentic PhOH (blue) and the PhOH product (red) yielded in the photocatalytic hydroxylation of benzene by 1 (0.10 mM) in the presence of [RuII(Me2phen)3]2+ (1.0 mM), Sc(NO3)3 (100 mM), and benzene (2.0 M) under photoirradiation (white light) in an 18O2-saturated solvent mixture of MeCN and H2O (v/v = 23:2) at 298 K for 24 h. The peaks at m/z = 94 and 96 correspond to Ph16OH and Ph18OH, respectively. | |
 |
| | Fig. 9 A comparison of the relative abundances of authentic PhOH (blue) and the produced PhOH (red) in the photocatalytic hydroxylation of benzene by O2 with 1 (0.10 mM) in the presence of [RuII(Me2phen)3]2+ (1.0 mM), Sc(NO3)3 (100 mM), and benzene (2.0 M) under photoirradiation (white light) of an 16O2-saturated solvent mixture of MeCN and H218O (v/v = 23:2) at 298 K for 24 h. The peaks at m/z = 94 and 96 correspond to Ph16OH and Ph18OH, respectively. | |
The photocatalytic mechanism of benzene hydroxylation to phenol by O2 is summarized in Scheme 1. The photoexcitation of [RuII(Me2phen)3]2+ in an O2-saturated solvent mixture of MeCN and H2O (v/v = 23:2) containing Sc(NO3)3 resulted in electron transfer from the triplet excited state of [RuII(Me2phen)3]2+ to O2 to produce [RuIII(Me2phen)3]3+ and the O2˙−–Sc3+ complex.23,28 The strong binding of Sc3+ to O2˙−, which was detected by EPR, inhibits the back electron transfer from the O2˙−–Sc3+ complex to [RuIII(Me2phen)3]3+, followed by disproportion with H2O to produce H2O2.23 H2O2 has attracted considerable attention as an ideal solar fuel for a one-compartment H2O2 fuel cell with a theoretical maximum output potential of 1.09 V, which is comparable to that of a hydrogen fuel cell (1.23 V).23 [RuIII(Me2phen)3]3+ can oxidize H2O to O2 with catalysis by 1. Benzene hydroxylation to PhOH was also catalysed by 1. As reported previously, the NMR peaks assignable to the bpy and Cp* of 1 remained the same after the photocatalytic production of H2O2, indicating that 1 acted as a homogeneous catalyst during the reaction.23 However, the catalytically active intermediates such as the Co(IV)-oxo species29 have yet to be detected in the catalytic benzene hydroxylation with H2O2. There was no deuterium kinetic isotope effect (KIE) for benzene (Fig. 2), indicating that C–H bond cleavage is not involved in the rate-determining step of the photocatalytic hydroxylation reaction.
Conclusions
In conclusion, we have shown that benzene is oxidized to phenol by dioxygen with a high TON (e.g., 500) under visible light irradiation of a reaction solution containing [RuII(Me2phen)3]2+ as a photocatalyst, [CoIII(Cp*)(bpy)(H2O)]2+ as an efficient catalyst for both water oxidation and benzene hydroxylation, and water as an electron source in the presence of Sc(NO3)3. The combination of the photocatalytic H2O2 production by H2O oxidation with O2 and the catalytic hydroxylation of benzene to phenol with H2O2 demonstrated in this study has paved a new road towards direct oxygenation of substrates by O2 as the most environmentally benign oxidant as well as oxygen source without any electron source except H2O, which is also the most environmentally benign hydrogen source.
Experimental section
Materials
All solvents and chemicals were of reagent-grade quality, obtained commercially and used without further purification, unless otherwise noted. Ruthenium(III) chloride hydrate, ammonium hexafluorophosphate, n-butyllithium solution (2.7 M in heptane), n-pentane, and tetrahydrofuran were purchased from Aldrich Chemicals. The chemicals, such as 4,7-dimethyl-1,10-phenanthroline (Me2phen), silver sulphate, lead dioxide, and cobalt(II) chloride, were purchased from Alfa Aesar. 2,2′-Bipyridine, 1,2,3,4,5-pentamethylcyclopentadiene, and TiIV(O)(tpyp) (tpyp = 5,10,15,20-tetra(4-pyridyl)porphyrinato anion) were obtained from Tokyo Chemical Industry Co., Ltd. Sc(NO3)3·4H2O was supplied by Mitsuwa Chemicals Co., Ltd. 18O2 gas (98% 18O-enriched) was purchased from ICON Services Inc. (Summit, NJ. USA). The purification of water (18.2 MΩ cm) was performed with a Milli-Q system (Millipore, Direct-Q 3 UV). Acetonitrile was dried according to published procedures and distilled prior to use.30 The cobalt(III) starting complex, [CoIII(Cp*)(bpy)(H2O)]2+ (1, Cp* = η5-pentamethylcyclopenta-dienyl and bpy = 2,2-bipyridine), and the tris(4,7-dimethyl-1,10-phenanthroline)ruthenium(II) complex, [RuII(Me2phen)3]2+, were prepared according to the published methods.25,31
Instrumentation
UV-vis spectra were recorded on a Hewlett Packard 8453 diode array spectrophotometer equipped with a UNISOKU Scientific Instruments Cryostat USP-203A. Product analysis for the oxidation reactions was performed with an Agilent Technologies 6890N gas chromatograph (GC) and a Thermo Finnigan (Austin, Texas, U.S.A.) FOCUS DSQ (dual stage quadrupole) mass spectrometer interfaced with a Finnigan FOCUS gas chromatograph (GC-MS). The amount of evolved oxygen was recorded by a Clark-type oxygen electrode made by Hansatech Ltd.
Product analysis
The products formed in the oxidation of benzene by 1 and O2 in the presence of [RuII(Me2phen)3]2+ and Sc(NO3)3 in a solvent mixture of MeCN and H2O (v/v = 23:2) at 298 K were identified by GC and GC-MS by a comparison of the mass peaks and retention time of the products with respect to the authentic samples, and the product yields were determined by comparing the responsive peak areas of the sample products against standard curves prepared with known authentic compounds using the internal standard decane. The quantum yield (QY) of the photocatalytic hydroxylation of benzene has been determined under visible light irradiation of monochromatized light using a Compact Xenon Light Source (MAX-302; Asahi Spectra Co., Ltd). The amount of hydrogen peroxide produced was determined by spectroscopic titration with an acidic solution of a [TiIVO(tpypH4)]4+ complex.31 [TiIVO(tpypH4)]4+ (50 μM) was prepared by dissolving 3.4 mg of the TiO(tpyp) complex into water (100 mL) containing hydrochloric acid (50 mM). A small portion (100 μL) of the photocatalytic reaction solution was taken and diluted up to 1.0 mL with water. To 0.25 mL of the diluted sample, 0.25 mL of perchloric acid (4.8 M) and 0.25 mL of [TiIVO(tpypH4)]4+ (50 μM) were added. The mixed solution was then allowed to stand for 5 min at room temperature. This sample solution was diluted up to 2.5 mL with water and used for the spectroscopic measurement. The absorbance at λ = 434 nm (AS) due to [TiIVO(tpypH4)]4+ was measured using a Hewlett Packard 8453 diode array spectrophotometer. In a similar manner, a blank solution was prepared by adding distilled water instead of the sample solution in the same volume with its absorbance designated as AB. The difference in absorbance at 434 nm was determined as follows: ΔA434 = AB − AS. Based on ΔA434 and the volume of the solution, the amount of hydrogen peroxide was determined according to the literature.23 A Clark-type oxygen electrode was used to obtain oxygen evolution data, and calibrated daily using Ar deoxygenated and oxygen saturated atmospheric solutions. Clark electrode experiments were performed by adding 10 μL of [RuIII(Me2phen)3]3+ (100 mM) to an O2-saturated solvent mixture (1.0 mL) of MeCN and H2O (v/v = 9:1) containing 1 (10–200 μM) and monitoring the O2 evolved.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by SENTAN projects from the Japan Science and Technology Agency (JST), JSPS KAKENHI (No. 16H02268) to S. F. and by the CRI (NRF-2012R1A3A2048842 to W. N.), GRL (NRF-2010-00353 to W. N.), and Basic Science Research Program (2017R1D1A1B03029982 to Y. M. L. and 2017R1D1A1B03032615 to S. F.) through the NRF of Korea.
Notes and references
-
M. Weber, M. Weber and M. Kleine-Boymann, Phenol. Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2004, vol. 26, p. 503 Search PubMed.
-
(a) R. Molinari and T. Poerio, Asia–Pac. J. Chem. Eng., 2010, 5, 191 CrossRef CAS;
(b) R. J. Schmidt, Appl. Catal., A, 2005, 280, 89 CrossRef CAS.
-
(a) L. Balducci, D. Bianchi, R. Bortolo, R. D’Aloisio, M. Ricci, R. Tassinari and R. Ungarelli, Angew. Chem., Int. Ed., 2003, 42, 4937 CrossRef CAS PubMed;
(b) J.-H. Yang, G. Sun, Y. Gao, H. Zhao, P. Tang, J. Tan, A.-H. Lu and D. Ma, Energy Environ. Sci., 2013, 6, 793 RSC.
- S. Fukuzumi and K. Ohkubo, Asian J. Org. Chem., 2015, 4, 836 CrossRef CAS.
-
(a) X. Cai, Q. Wang, Y. Liu, J. Xie, Z. Long, Y. Zhou and J. Wang, ACS Sustainable Chem. Eng., 2016, 4, 4986 CrossRef CAS;
(b) W. Wang, L. Shi, N. Li and Y. Ma, RSC Adv., 2017, 7, 12738 RSC.
-
(a) Y. Morimoto, S. Bunno, N. Fujieda, H. Sugimoto and S. Itoh, J. Am. Chem. Soc., 2015, 137, 5867 CrossRef CAS PubMed;
(b) Y. Aratani, Y. Yamada and S. Fukuzumi, Chem. Commun., 2015, 51, 4662 RSC.
-
(a) D. Wang, M. Wang and Z. Li, ACS Catal., 2015, 5, 6852 CrossRef CAS;
(b) X. Ye, Y. Cui, X. Qiu and X. Wang, Appl. Catal., B, 2014, 152–153, 383 CrossRef CAS;
(c) G. Wen, S. Wu, B. Li, C. Dai and D. S. Su, Angew. Chem., Int. Ed., 2015, 54, 4105 CrossRef CAS PubMed.
-
(a) M. Yamada, K. D. Karlin and S. Fukuzumi, Chem. Sci., 2016, 7, 2856 RSC;
(b) B. Xu, W. Zhong, Z. Wei, H. Wang, J. Liu, L. Wu, Y. Feng and X. Liu, Dalton Trans., 2014, 43, 15337 RSC;
(c) L. Wu, W. Zhong, B. Xu, Z. Wei and X. Liu, Dalton Trans., 2015, 44, 8013 RSC.
-
(a) W. Ge, Z. Long, X. Cai, Q. Wang, Y. Zhou, Y. Xu and J. Wang, RSC Adv., 2014, 4, 45816 RSC;
(b) C. Wang, L. Hu, Y. Hu, Y. Ren, X. Chen, B. Yue and H. He, Catal. Commun., 2015, 68, 1 CrossRef CAS.
-
(a) P. Borah, X. Ma, K. T. Nguyen and Y. Zhao, Angew. Chem., Int. Ed., 2012, 51, 7756 CrossRef CAS PubMed;
(b) P. K. Khatri, B. Singh, S. L. Jain, B. Sain and A. K. Sinha, Chem. Commun., 2011, 47, 1610 RSC;
(c) Y. Leng, J. Liu, P. Jiang and J. Wang, Chem. Eng. J., 2014, 239, 1 CrossRef CAS.
-
(a) H. Wang, M. Zhao, Q. Zhao, Y. Yang, C. Wang and Y. Wang, Ind. Eng. Chem. Res., 2017, 56, 2711 CrossRef CAS;
(b) S. Verma, R. B. N. Baig, M. N. Nadagouda and R. S. Varma, ACS Sustainable Chem. Eng., 2017, 5, 3637 CrossRef CAS.
-
(a) L. Muniandya, F. Adama, A. R. Mohamed, A. Iqbal and N. R. A. Rahman, Appl. Surf. Sci., 2017, 398, 43 CrossRef;
(b) J. Xu, Q. Jiang, T. Chen, F. Wu and Y.-X. Li, Catal. Sci. Technol., 2015, 5, 1504 RSC;
(c) P. R. Makgwane and S. S. Raya, J. Mol. Catal. A: Chem., 2015, 398, 149 CrossRef CAS.
-
(a) L. Carneiro and A. R. Silva, Catal. Sci. Technol., 2016, 6, 8166 RSC;
(b) E. Gu, W. Zhong and X. Liu, RSC Adv., 2016, 6, 98406 RSC;
(c) E. Gu, W. Zhong, H. Ma, B. Xu, H. Wang and X. Liu, Inorg. Chim. Acta, 2016, 444, 159 CrossRef CAS;
(d) B. Xu, W. Zhong, Z. Wei, H. Wang, J. Liu, L. Wu, Y. Feng and X. Liu, Dalton Trans., 2014, 43, 15337 RSC.
-
(a) S. Niwa, M. Eswaramoorthy, J. Nair, A. Raj, N. Itoh, H. Shoji, T. Namba and F. Mizukami, Science, 2002, 295, 105 CrossRef CAS PubMed;
(b) K. Sato, T. Hanaoka, S. Niwa, C. Stefan, T. Namba and F. Mizukami, Catal. Today, 2005, 104, 260 CrossRef CAS.
-
(a) A. Okemoto, Y. Tsukano, A. Utsunomiya, K. Taniya, Y. Ichihashi and S. Nishiyama, J. Mol. Catal. A: Chem., 2016, 411, 372 CrossRef CAS;
(b) B. B. Sarma, R. Carmieli, A. Collauto, I. Efremenko, J. M. L. Martin and R. Neumann, ACS Catal., 2016, 6, 6403 CrossRef CAS.
-
(a) Z. Long, Y. Zhou, W. Ge, G. Chen, J. Xie, Q. Wang and J. Wang, ChemPlusChem, 2014, 79, 1590 CrossRef CAS;
(b) Z. Long, Y. Zhou, G. Chen, P. Zhao and J. Wang, Chem. Eng. J., 2014, 239, 19 CrossRef CAS;
(c) S. Shang, B. Chen, L. Wang, W. Dai, Y. Zhang and S. Gao, RSC Adv., 2015, 5, 31965 RSC.
- Y. Aratani, K. Oyama, T. Suenobu, Y. Yamada and S. Fukuzumi, Inorg. Chem., 2016, 55, 5780 CrossRef CAS PubMed.
-
(a) R. Hamada, Y. Shibata, S. Nishiyama and S. Tsuruya, Phys. Chem. Chem. Phys., 2003, 5, 956 RSC;
(b) Z. Long, Y. Zhou, G. Chen, W. Ge and J. Wang, Sci. Rep., 2014, 4, 3651 CrossRef CAS PubMed;
(c) Z. Long, Y. Liu, P. Zhao, Q. Wang, Y. Zhou and J. Wang, Catal. Commun., 2015, 59, 1 CrossRef CAS;
(d) S. S. Acharyya, S. Ghosh, R. Tiwari, C. Pendem, T. Sasaki and R. Bal, ACS Catal., 2015, 5, 2850 CrossRef CAS;
(e) R. Bal, M. Tada, T. Sasaki and Y. Iwasawa, Angew. Chem., Int. Ed., 2006, 45, 448 CrossRef CAS PubMed.
-
(a) T. D. Bui, A. Kimura, S. Ikeda and M. Matsumura, J. Am. Chem. Soc., 2010, 132, 8453 CrossRef CAS PubMed;
(b) Y. Ide, M. Torii and T. Sano, J. Am. Chem. Soc., 2013, 135, 11784 CrossRef CAS PubMed;
(c) Y. Ide, M. Matsuoka and M. Ogawa, J. Am. Chem. Soc., 2010, 132, 16762 CrossRef CAS PubMed.
- K. Ohkubo, T. Kobayashi and S. Fukuzumi, Angew. Chem., Int. Ed., 2011, 50, 8652 CrossRef CAS PubMed.
- K. Ohkubo, A. Fujimoto and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 5368 CrossRef CAS PubMed.
- K. Ohkubo, K. Hirose and S. Fukuzumi, Chem.–Eur. J., 2015, 21, 2855 CrossRef CAS PubMed.
- S. Kato, J. Jung, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2013, 6, 3756 CAS.
-
(a) Y. Isaka, S. Kato, D. Hong, T. Suenobu, Y. Yamada and S. Fukuzumi, J. Mater. Chem. A, 2015, 3, 12404 RSC;
(b) Y. Isaka, K. Oyama, Y. Yamada, T. Suenobu and S. Fukuzumi, Catal. Sci. Technol., 2016, 6, 681 RSC.
-
(a) A. Das, V. Joshi, D. Kotkar, V. S. Pathak, V. Swayambunathan, P. V. Kamat and P. K. Ghosh, J. Phys. Chem. A, 2001, 105, 6945 CrossRef CAS;
(b) D. Hong, J. Jung, J. Park, Y. Yamada, T. Suenobu, Y.-M. Lee, W. Nam and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7606 RSC.
- It should be noted, however, that the contribution from the catalytic hydroxylation of benzene with [RuIII(bpy)3]3+ in competition with H2O oxidation cannot be ruled out.
-
(a) S. Fukuzumi and K. Ohkubo, Chem.–Eur. J., 2000, 6, 4532 CrossRef CAS PubMed;
(b) S. Fukuzumi, M. Patz, T. Suenobu, Y. Kuwahara and S. Itoh, J. Am. Chem. Soc., 1999, 121, 1605 CrossRef CAS;
(c) S. Fukuzumi, H. Ohtsu, K. Ohkubo, S. Itoh and H. Imahori, Coord. Chem. Rev., 2002, 226, 71 CrossRef CAS.
-
W. L. F. Armarego and C. L. L. Chai, in Purification of Laboratory Chemicals, Pergamon Press, Oxford, U. K., 6th edn, 2009 Search PubMed.
-
(a) S. Hong, F. F. Pfaff, E. Kwon, Y. Wang, M.-S. Seo, E. Bill, K. Ray and W. Nam, Angew. Chem., Int. Ed, 2014, 53, 10403 CrossRef CAS PubMed;
(b) B. Wang, Y.-M. Lee, W.-Y. Tcho, S. Tussupbayev, S.-T. Kim, Y. Kim, M. S. Seo, K.-B. Cho, Y. Dede, B. C. Keegan, T. Ogura, S. H. Kim, T. Ohta, M.-H. Baik, K. Ray, J. Shearer and W. Nam, Nat. Commun., 2016, 8, 14839 CrossRef PubMed.
- C. Turro, J. M. Zaleski, Y. M. Karabatsos and D. G. Nocera, J. Am. Chem. Soc., 1996, 118, 6060 CrossRef CAS.
- C. Matsubara, N. Kawamoto and K. Takamura, Analyst, 1992, 117, 1781 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Table S1 and Fig. S1–S8. See DOI: 10.1039/c7sc02495a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Wonwoo
Nam
a,
Wonwoo
Nam

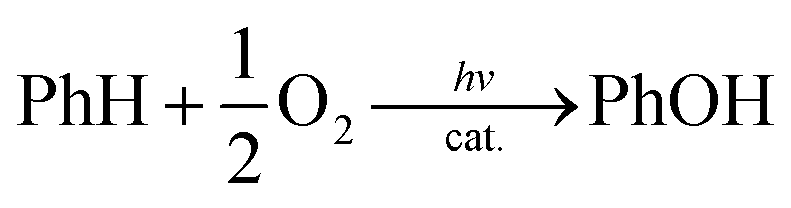
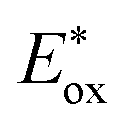 = −1.01 V vs. NHE) is more negative than that of [RuII(bpy)3]2+* (
= −1.01 V vs. NHE) is more negative than that of [RuII(bpy)3]2+* (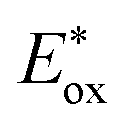 = −0.84 V vs. NHE).23 The UV-vis spectrum of the photocatalyst, [RuII(Me2phen)3]2+, during the photocatalytic oxidation remained almost identical to that of the initial stage at 445 nm, indicating that the overall reactivity decreased with time due to oxidative degradation of the catalyst 1 (see Experimental section; Fig. S1, ESI†).
= −0.84 V vs. NHE).23 The UV-vis spectrum of the photocatalyst, [RuII(Me2phen)3]2+, during the photocatalytic oxidation remained almost identical to that of the initial stage at 445 nm, indicating that the overall reactivity decreased with time due to oxidative degradation of the catalyst 1 (see Experimental section; Fig. S1, ESI†).