
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt catalyzed carbonylation of unactivated C(sp3)–H bonds†
Nagaraju
Barsu
,
Shyam Kumar
Bolli
and
Basker
Sundararaju
*
Fine Chemical Laboratory, Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur, Uttar Pradesh, India. E-mail: basker@iitk.ac.in
First published on 20th December 2016
Abstract
A general efficient regioselective cobalt catalyzed carbonylation of unactivated C(sp3)–H bonds of aliphatic amides was demonstrated using atmospheric (1–2 atm) carbon monoxide as a C1 source. This straightforward approach provides access to α-spiral succinimide regioselectively in a good yield. Cobalt catalyzed sp3 C–H bond carbonylation is reported for the first time including the functionalization of (β)-C–H bonds of α-1°, 2°, 3° carbons and even internal (β)-C–H bonds. Our initial mechanistic investigation reveals that the C–H activation step is irreversible and will possibly be the rate determining step.
Introduction
Catalytic carbonylation is one of the most straightforward processes for the production of carbonyl compounds both in academia and industry.1 Insertion of CO into carbon–hydrogen bonds has become even more interesting provided that the catalyst has the ability to activate both inert C–H bonds and to bind π-acidic carbon monoxide.2 In this regard, Shunsuke Murahashi reported the first effective catalytic C–H carbonylation of benzaldimine in 1955 using a low valent cobalt(0) complex.3 Since then, several reports have been documented for catalytic C(sp2)–H bond carbonylation.4 However the corresponding extension to carbonylation of C(sp3)–H bonds is rare and only a handful of examples have been reported in the literature.5 After the pioneering work of Tanaka et al. for photocatalytic carbonylation of alkanes,6 Chatani reported the directing group assisted C–H carbonylation of α-C(sp3)–H bonds adjacent to nitrogen.7 In 2010–11, Yu and Chatani independently reported the site-selective C(sp3)–H carbonylation of aliphatic amide using Pd and Ru based catalysts, while the former used oxidative conditions, the latter proceeded through a metal-hydride pathway (Scheme 1a).8–10 We assumed that it would be more rational to choose a metal abundantly present in nature compared to precious metals in terms of cost effectiveness and the unexploited inherent properties exhibited by them.11 Utilization of 8-aminoquinoline as a bidentate auxiliary for C–H bond functionalization was first reported by Daugulis,12 and later developed by Chatani13 and others14 for several profitable applications using first row late transition metal precursors.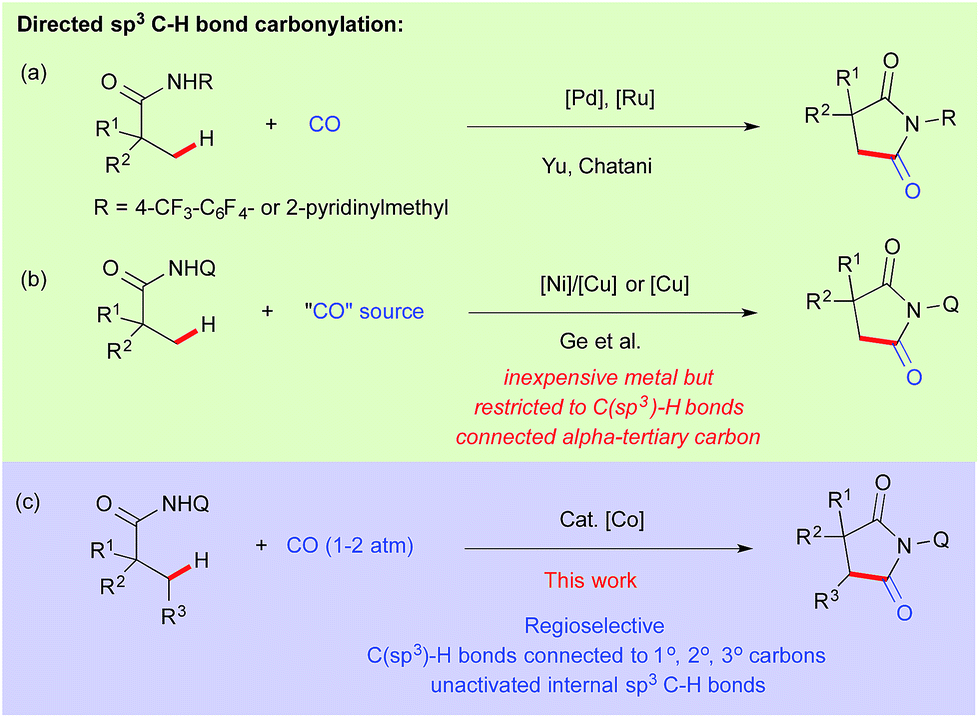 | ||
| Scheme 1 Carbonylation of unactivated C(sp3)–H bonds. | ||
Using this approach, Ge et al. reported recently the carbonylation of unactivated C(sp3)–H bonds with Ni–Cu/Cu as a catalyst using DMF and nitromethane as a carbonyl sources.15 But the presented nickel–copper/copper catalyzed carbonylation was limited to β-C(sp3)–H bonds connected to α-tertiary carbon (Scheme 1b). On the other hand, cobalt seems to be a very promising alternative metal catalyst for various C(sp2)–H bond functionalizations.11a,b,16 Very recently Ge and Zhang independently reported cobalt catalyzed intramolecular cyclization through activation of directed C(sp3)–H bonds.17 Daugulis reported lately, cobalt catalyzed o-C(sp2)–H bond carbonylation of benzamides at room temperature.16n We have previously demonstrated the activation of C(sp3)–H bonds using Cp*Co(III) for C–H alkenation with alkynes and C–H amidation with oxazolone.18 Inspired by these results and based on our continuous efforts on the development of cobalt catalyzed C–H bond functionalization,19 we report herein the first regio- and site-selective cobalt catalyzed carbonylation of unactivated C(sp3)–H bonds including terminal and internal C–H bond connected α-1°, 2° and 3° carbons (Scheme 1c).
Results and discussion
We began our investigation using aliphatic amide 1a, 2 atm CO gas as a C1 source with a catalytic amount of simple Co(OAc)2, metal carboxylate and silver carbonate as an oxidant at 150 °C for 36 h in chlorobenzene (0.5 mL). The optimization results are summarized in Table 1. Initial screening of different metal carboxylates revealed that a good yield of 2a was obtained with sodium benzoate (50 mol%) as an additive, which is also an optimal reaction parameter for intramolecular β-sp3 C–H bond amidation17a (Table 1, entry 1–4). Lowering the reaction concentration did not provide any further improvement (entry 5). A small amount of sodium benzoate turned out to be crucial for high product formation with cobalt(II) (entry 6 and 7).|
|
||||
|---|---|---|---|---|
| Entry | [Co] | MCO2R (mol%) | Oxidant | Yieldb |
| a All reactions reagents were added under argon atmosphere unless otherwise stated using 1a/[Co]/NaO2CR/Ag2CO3 in 0.2/0.02/0.02–0.05/0.5 mmol using chlorobenzene (0.5 mL) as a solvent at room temperature and then pressurized with CO (2 atm) at 150 °C for 36 h. b Isolated yield. c 1 mL of PhCl was used. d Reaction performed for 24 h. e 20 mol% of [Co] was used. f 3 equiv. of Ag2CO3 was used. g TFT (0.5 mL) was used as a solvent. | ||||
| 1 | Co(OAc)2 | NaOPiv (50) | Ag2CO3 | 27 |
| 2 | Co(OAc)2 | Mn(OAc)3 (50) | Ag2CO3 | n.r. |
| 3 | Co(OAc)2 | NaO2CPh (50) | Ag2CO3 | 67 |
| 4 | Co(OAc)2 | NaO2CMes (50) | Ag2CO3 | 49 |
| 5 | Co(OAc)2 | NaO2CPh (50) | Ag2CO3 | 53c |
| 6 | Co(OAc)2 | NaO2CPh (100) | Ag2CO3 | 43 |
| 7 | Co(OAc)2 | NaO2CPh (20) | Ag2CO3 | 76 |
| 8 | Co(OAc)2 | NaO2CPh (20) | Ag2CO3 | 70d |
| 9 | Co(OAc)2 | NaO2CPh (20) | Ag2CO3 | 79d,e |
| 10 | Co(OAc)2 | NaO2CPh (20) | Ag2CO3 | 87d,f |
| 11 | Co(acac)2 | NaO2CPh (20) | Ag2CO3 | 86d,f |
| 12 | Co(acac)3 | NaO2CPh (20) | Ag2CO3 | 87d,f |
| 13 | Co2(CO)8 | NaO2CPh (20) | Ag2CO3 | 68d,f |
| 14 | CoBr2 | NaO2CPh (20) | Ag2CO3 | 61d |
| 15 | Co(acac)2 | NaO2CPh (20) | Ag2CO3 | 95d,g |
| 16 | [Co(Piv)2]2 | — | Ag2CO3 | 55d,g |
| 17 | Co(acac)3 | — | Ag2CO3 | 92d,g |
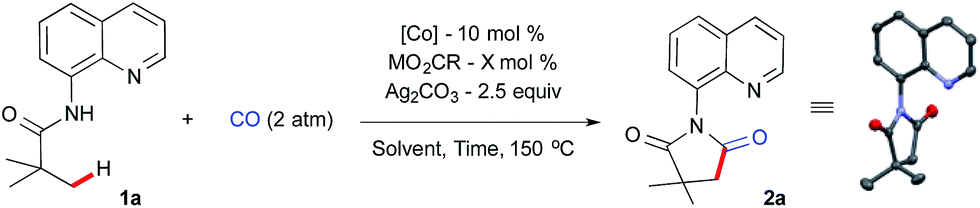
There was no significant change in yield when the reaction time was reduced to 24 h or by increasing the catalyst loading to 20 mol% (entries 8 and 9). To our delight, a small increment in the silver carbonate gave excellent product formation after 24 h (entry 10). Next, we examined various cobalt sources from different oxidations states. It was revealed that Co(II) and Co(III) gave excellent yields whereas Co(0) and Co(II)Br2 gave moderate yields (entries 11–14). After a brief solvent screening,20 we found out that α,α,α-trifluorotoluene (TFT) was the best solvent, which provided 95% of the succinimide 2a under our optimized reaction parameters (entry 15). A well-defined, isolated cobalt-pivalate dimer21 was used as a catalyst without any additive and this resulted in a 55% yield (entry 16). We further confirmed that the direct use of the Co(III) precursor in the absence of the sodium benzoate provides 2a in 92% yield and the structure of 2a was confirmed by X-ray crystallography (entry 17).22
Prior to the scope of the reaction, we examined the scalability of the reaction. A gram scale reaction was performed with 1a using atmospheric CO gas and provides succinimide 2a in 71% yield after 36 h (Scheme 2) and silver carbonate was regenerated from the residue in 69% (2.5 g) yield using the known literature procedure.16e,23 The recycled silver carbonate was used for C–H carbonylation and resulted in 83% of 2a under standard conditions (Scheme 2).
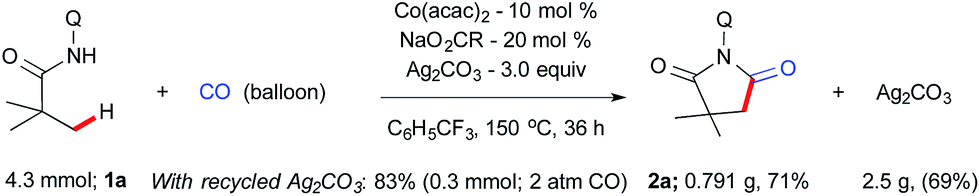 | ||
| Scheme 2 Gram scale synthesis of succinimide. | ||
With the optimized conditions in hand, we next investigated the scope of the aliphatic amides and the results are depicted in Scheme 3. Architecturally interesting spiral succinimides possessing a 4-, 5-, 6-membered ring on the backbone were achieved in good yields (2b–d). Replacing one of the methyl groups from 1a with a benzyl, 4-OMe-benzyl, cyclopentyl, isopropyl or alkyl chain at the α-tertiary center gave the desired succinimides in high yields (2e–j). Phenyl, ester, and –Br substituents on the backbone were tolerated (2k–m). Next, the reactivity difference between C(sp2)–H and C(sp3)–H bonds was investigated for cyclocarbonylation. It was found out that carbonylation of unactivated C(sp3)–H bonds was favoured over C(sp2)–H bonds (2n–o) and only 2o′ was obtained in 15% yield in addition to 2o (50%). This may be due to the fact that the former proceeds via facile 5-membered cyclocobaltation while the latter proceeds through a 6-membered metallocycle. Furthermore, we explore the possibility of activating β-C(sp3)–H bonds next to an α-secondary and primary carbon center, which is much more challenging (this has not been explored before for carbonylation using first row transition metals) than a C–H bond next to an α-tertiary carbon. Gratifyingly, propanamide (1p) and α-methylpropanamide (1q) underwent cyclocarbonylation albeit in a moderate yield (2p, 2q) but the mass of the product can be improved if we prolong the reaction to 36 h with a 20 mol% cobalt catalyst.
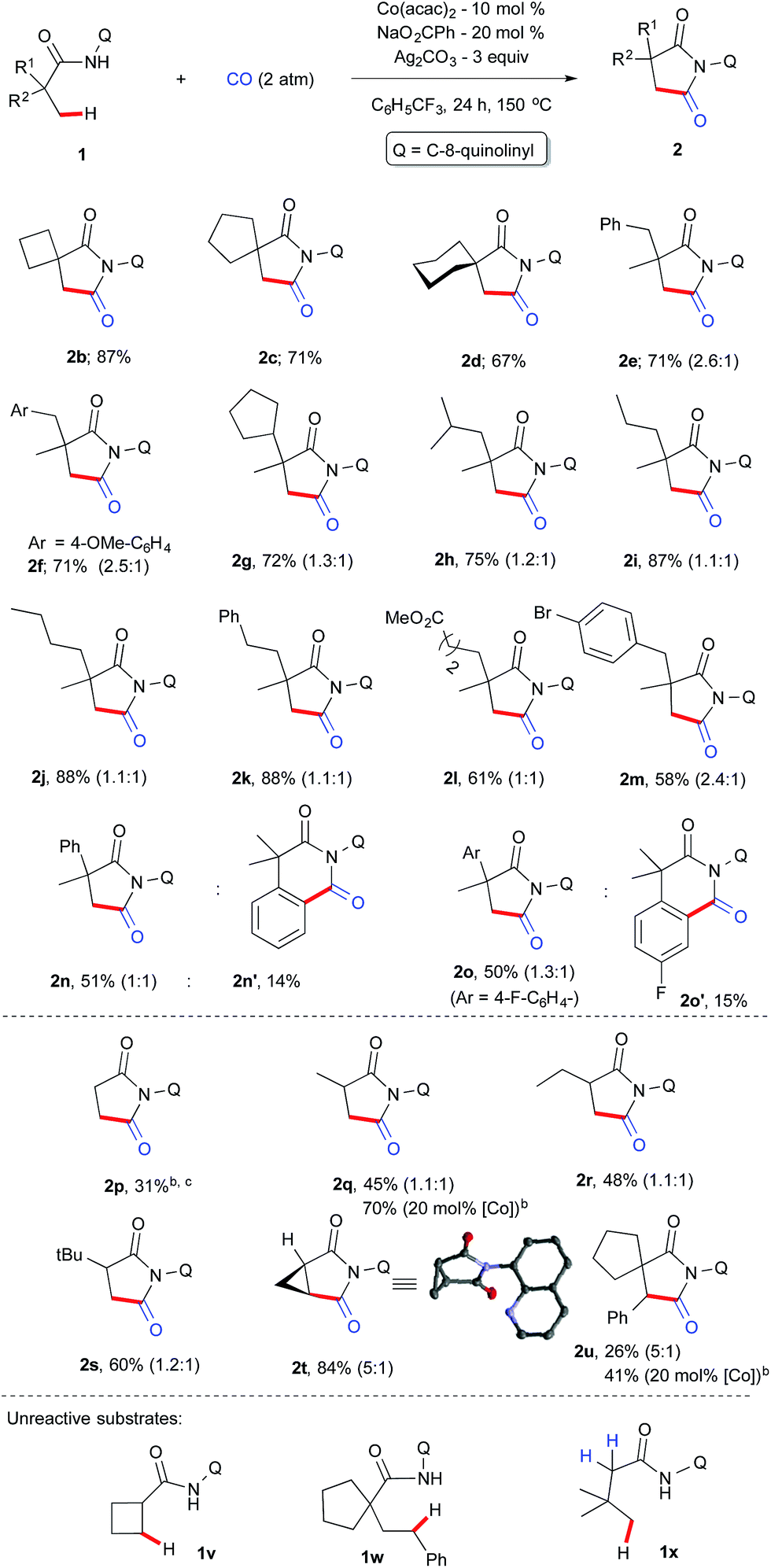 | ||
| Scheme 3 Scope and limitation. aThe number in parenthesis is the ratio of diastereomer. b36 h instead of 24 h. cPhCl as a solvent. | ||
Systematic increase of bulkiness by incorporating a –Et and –tBu group did not significantly alter the isomeric ratio (2r–s). Furthermore, to our delight, an internal C(sp3)–H bond was carbonylated in the case of 1t and 1u, and it resulted in the formation of substituted and structurally strained succinimides (2t & 2u) and X-ray crystallography further confirmed the structure of 2t.22 The reactivity of 1u was documented for the first time via internal β-C(sp3)–H carbonylation. To understand the reaction mechanism, several experiments were carried out as displayed in Scheme 4.
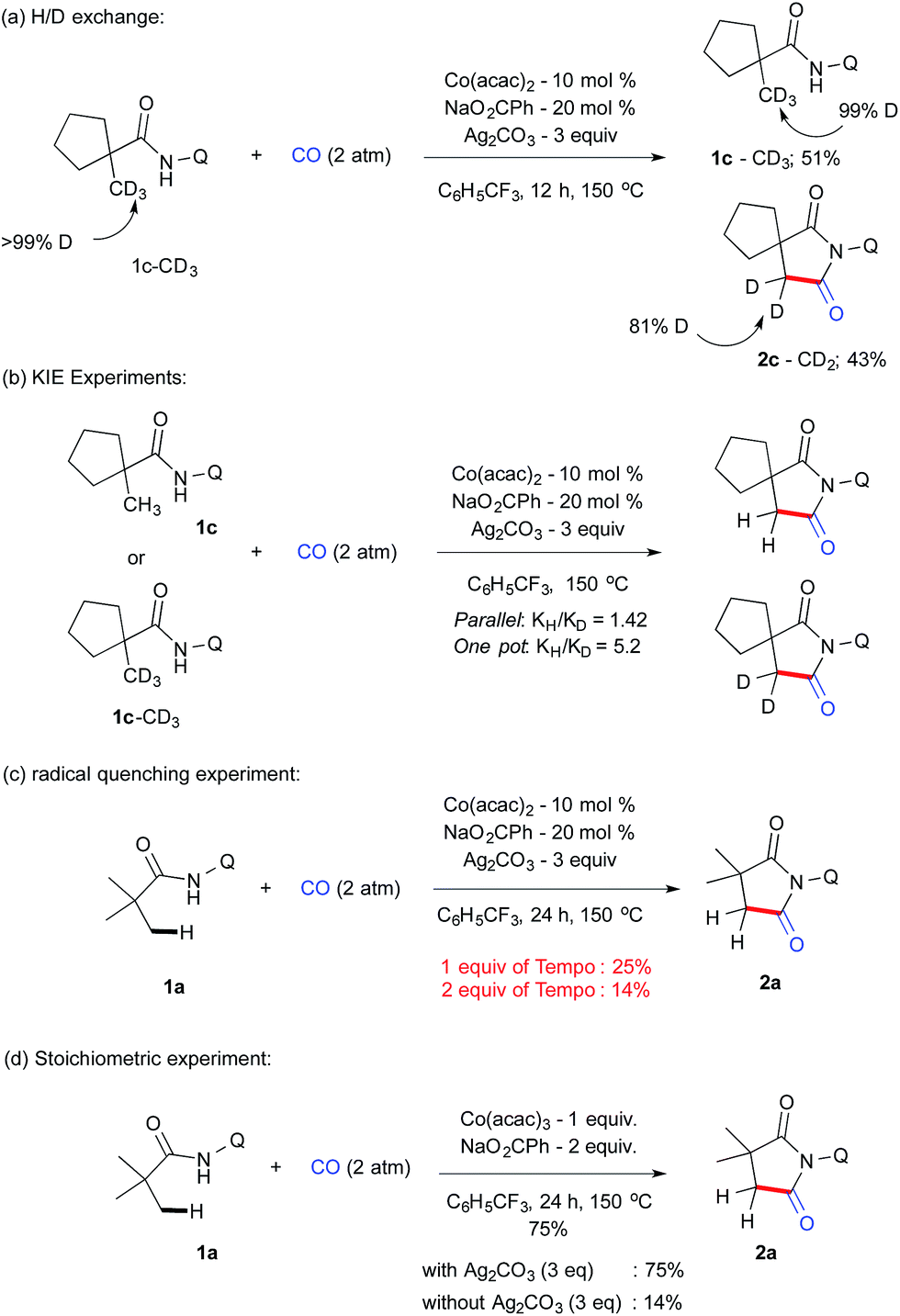 | ||
| Scheme 4 H/D exchange, KIE and control experiments. | ||
1c–CD3 employed under our reaction conditions gave the corresponding carbonylation product 2c–CD2 (43%) along with the recovered starting material 1c–CD3 (51%) after 12 h (Scheme 4a). Analysis of the isolated compound revealed that 1c–CD3 did not have any H/D exchange, which indicates that the C–H activation step is irreversible. KIE values of 1.42 and 5.2 were determined from parallel and one-pot experiments using 1c–CH3 and 1c–CD3 (Scheme 4b). This value possibly suggests that the cleavage of C–H bonds is the rate-determining step. Next, we performed a series of control experiments e.g. addition of a stoichiometric amount of a radical scavenger such as TEMPO (1 eq.), which drastically reduces the yield of 2a, which further reduced to 14% when 2 equiv. of TEMPO was employed (Scheme 4c). Although there is no adduct possible to isolate from the mixture, the present result suggests that the possibility of the SET pathway may also to be considered. In addition, we have also carried out the reaction with a stoichiometric amount of Co(III) to gain insight into the possibility of Co(III) or Co(IV) intermediates involved in the catalytic cycle with and without Ag2CO3. In line with radical experiments, 1 eq. of CoIII(acac)3 without silver carbonate gave 14% of 2a compared to a 75% mass of 2a which was isolated with silver carbonate. These results indicate that Co(IV) was involved in reductive elimination instead of Co(III) (scheme 4d).
Based on the control experiments and previous reports on β-C(sp3)–H bond activation,17 we propose the plausible mechanism as shown in Scheme 5. Initial oxidation of Co(II) to Co(III) instigated by Ag+ (confirmed by UV-Vis spectra),20 which subsequently coordinated with 1avia N,N-coordination (initial deprotonation possibly by silver carbonate) led to intermediate A. Then, carboxylate/carbonate assisted deprotonation provides cobaltacycle B. Co(III) intermediate B coordinates with CO followed by insertion into the C–Co(III), which gave the intermediate C, which may either undergo reductive elimination to Co(I) and reoxidized by silver to active Co(III) species as depicted in path a.16a–g Alternatively, intermediate C will undergo one electron oxidation to Co(IV), which will undergo reductive elimination to 2a and regenerate Co(II) (path b). At the same time, one may also consider that intermediate B will undergo one electron oxidation to C′ which then subsequently insert CO between Co–C and lead to D (path c), which further undergoes reductive elimination. Although the final solution cannot be given for the mechanism at this stage, control experiments suggest that path b or c may be favoured over path a.
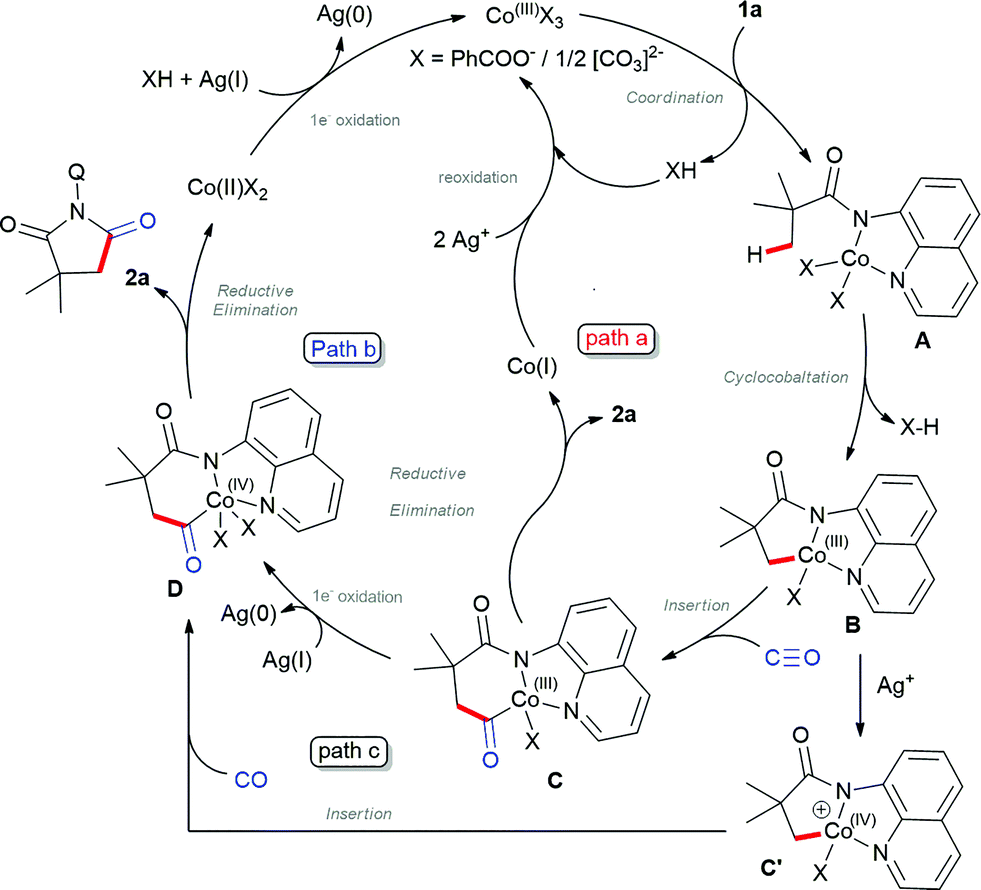 | ||
| Scheme 5 Proposed mechanism. | ||
To validate the synthetic utility of this method, we have performed a two ring opening reaction with 1a to obtain 1,4-dicarbonyl compound 3 and 1,4-dicarboxylicacid 4 in good yields under the standard reaction conditions (Scheme 6).
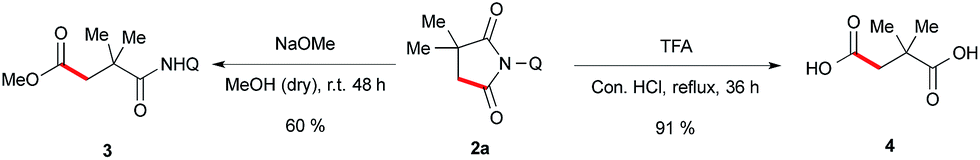 | ||
| Scheme 6 Synthetic applications. | ||
Conclusions
We have developed a new protocol for cobalt catalyzed site-selective carbonylation of unactivated C(sp3)–H bonds under atmospheric CO pressure to access various succinimide derivatives that are active against seizures (anticonvulsant ethosuximide). Our initial mechanistic investigation reveals that the involvement of a one electron process operated in the catalytic cycle and the reaction may proceed through Co(IV) to Co(II) (Scheme 5, path b or c). Studies to further extend this new reactivity to develop new medicinal compounds and materials are currently underway. Further understanding of the mechanism including the SET process, isolation of stoichiometric intermediates is presently on going in our laboratory.Acknowledgements
Financial support provided by SERB (EMR2016/000136) to support this research work is gratefully acknowledged. NB thanks to CSIR for his fellowship.Notes and references
- M. Beller, Catalytic carbonylation reactions, topic in organometallic Chemistry, Springer, 2006, pp. 1–283 Search PubMed.
- S. T. Gadge, P. Gautam and B. M. Bhanage, Chem. Rec., 2016, 16, 835–856 CrossRef CAS PubMed and references therein.
- S. Murahashi, J. Am. Chem. Soc., 1955, 77, 6403–6404 CrossRef CAS.
- Selected recent reviews see: (a) R.-Y. Zhu, M. E. Farmer, Y.-Q. Chen and J. Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578–10599 CrossRef CAS PubMed; (b) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef CAS PubMed; (c) X.-F. Wu, H. Neumann and M. Beller, ChemSusChem, 2013, 6, 229–241 CrossRef CAS PubMed; (d) X.-F. Wu, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1–35 CrossRef CAS PubMed; (e) D. A. Colby, R. G. Bergmann and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed; (f) A. Brennuführer, H. Neumann and M. Beller, ChemCatChem, 2009, 1, 28–41 CrossRef.
- (a) H. Li, B.-J. Li and Z.-J. Shi, Catal. Sci. Technol., 2011, 1, 191–206 RSC; (b) R. H. Crabtree, J. Organomet. Chem., 2004, 689, 4083–4091 CrossRef CAS.
- T. Sakakura, T. Sodeyama, K. Sasaki, K. Wada and M. Tanaka, J. Am. Chem. Soc., 1990, 112, 7221–7229 CrossRef CAS.
- N. Chatani, T. Asaumi, T. Ikeda, S. Yorimitsu, Y. Ishii, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2000, 122, 12882–12883 CrossRef CAS.
- E. J. Yoo, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 17378–17380 CrossRef CAS PubMed.
- (a) N. Hasegawa, V. Charra, S. Inoue, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 133, 8070–8073 CrossRef CAS PubMed; (b) N. Hasegawa, K. Shibata, V. Charra, S. Inoue, Y. Fukumoto and N. Chatani, Tetrahedron, 2013, 69, 4466–4472 CrossRef CAS.
- Selected other recent reports on Pd Catalyzed sp3 C–H bond carbonylation, see: (a) P.-L. Wang, Y. Li, Y. Wu, C. Li, Q. Lan and X.-S. Wang, Org. Lett., 2015, 17, 3698–3701 CrossRef CAS PubMed; (b) C. Wang, L. Zhang, C. Chen, J. Han, Y. Yao and Y. Zhao, Chem. Sci., 2015, 6, 4610–4614 RSC; (c) A. McNally, B. Haffemayer, B. S. L. Collings and M. J. Gaunt, Nature, 2014, 510, 129–133 CrossRef CAS PubMed; (d) P. Xie, Y. Xie, B. Qian, H. Zhou, C. Xia and H. Huang, J. Am. Chem. Soc., 2012, 134, 9902–9905 CrossRef CAS PubMed; (e) S. S. Stahl, J. A. Labinger and J. E. Bercaw, Angew. Chem., Int. Ed., 1998, 37, 2180–2192 CrossRef; (f) M. Asadullah, T. Kitamura and Y. Fujiwara, Chem. Lett., 1999, 28, 449–450 CrossRef.
- Selected recent reviews on first row transition metal catalysed C–H bond functionalization see: (a) D. Wei, X. Zhu, J.-L. Niu and M.-P. Song, ChemCatChem, 2016, 8, 1242–1263 CrossRef CAS; (b) N. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef; (c) N. Yoshikai, ChemCatChem, 2015, 7, 732–734 CrossRef CAS; (d) T. Hyster, Catal. Lett., 2015, 145, 458–467 CrossRef CAS; (e) I. Bauer and H. J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed; (f) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed; (g) A. A. Kulkarni and O. Daugulis, Synthesis, 2009, 4087–4109 CAS.
- V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS PubMed.
- (a) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2014, 136, 898–901 CrossRef CAS PubMed; (b) Y. Aihara, M. Tobisu, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2014, 136, 15509–15512 CrossRef CAS PubMed; (c) T. Kubo, Y. Aihara and N. Chatani, Chem. Lett., 2015, 44, 1365–1367 CrossRef CAS.
- Selected recent reviews on the use of 8-aminoquinoline as a directing group, see: (a) L. C. Misal Castro and N. Chatani, Chem. Lett., 2015, 44, 410–421 CrossRef; (b) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed; (c) M. Corbet and F. Decampo, Angew. Chem., Int. Ed., 2013, 52, 9896–9898 CrossRef CAS PubMed; see also (d) T. Matsubara, S. Asako, E. IIies and E. Nakamura, J. Am. Chem. Soc., 2014, 136, 646–649 CrossRef CAS PubMed.
- (a) X. Wu, J. Miao, Y. Li, G. Li and H. Ge, Chem. Sci., 2016, 7, 5260–5264 RSC; (b) X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2015, 137, 4924–4927 CrossRef CAS PubMed.
- Selected references high valent in cobalt catalyzed C(sp2)–H bond functionalization see: (a) S. Prakash, K. Muralirajan and C.-H. Cheng, Angew. Chem., Int. Ed., 2016, 55, 1844–1848 CrossRef CAS PubMed; (b) C. Du, P.-X. Li, X. Zhu, J.-F. Suo, J.-L. Niu and M.-P. Song, Angew. Chem., Int. Ed., 2016, 55, 13571–13575 CrossRef CAS PubMed; (c) S. Maity, R. Kancherla, U. Dhawa, E. Hoque, S. Pimparkar and D. Maiti, ACS Catal., 2016, 6, 5493–5499 CrossRef; (d) A. Lerchen, S. Vásquez-Céspedes and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 3208–3211 CrossRef CAS PubMed; (e) G. Tan, S. He, X. Huang, X. Liao, Y. Cheng and J. You, Angew. Chem., Int. Ed., 2016, 55, 10414–10418 CrossRef CAS PubMed; (f) R. Manoharan, G. Sivakumar and M. Jeganmohan, Chem. Commun., 2016, 52, 10533–10536 RSC; (g) T. Yamaguchi, Y. Kommagalla, Y. Aihara and N. Chatani, Chem. Commun., 2016, 52, 10129–10132 RSC; (h) L. Kong, S. Yu, X. Zhou and X. Li, Org. Lett., 2016, 18, 588–591 CrossRef CAS PubMed; (i) J. R. Hummel and J. A. Ellman, J. Am. Chem. Soc., 2015, 137, 490–498 CrossRef CAS PubMed; (j) P. Patel and S. Chang, ACS Catal., 2015, 5, 853–858 CrossRef CAS; (k) H. Wang, J. Koeller, W. Liu and L. Ackermann, Chem.–Eur. J., 2015, 21, 15525–15528 CrossRef CAS PubMed; (l) Z.-Z. Zhang, L. Bin, C.-Y. Wang and B.-F. Shi, Org. Lett., 2015, 17, 4094–4097 CrossRef CAS PubMed; (m) L. Grigorjeva and O. Daugulis, Angew. Chem., Int. Ed., 2014, 53, 10209–10212 CrossRef CAS PubMed; (n) L. Grigorjeva and O. Daugulis, Org. Lett., 2014, 16, 4688–4690 CrossRef CAS PubMed; (o) T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 2207–2211 CrossRef CAS PubMed.
- (a) X. Wu, K. Yang, Y. Zhao, H. Sun, G. Li and H. Ge, Nat. Commun., 2015, 6, 6462–6471 CrossRef CAS PubMed; (b) J. Zhang, H. Chen, C. Lin, Z. Liu, C. Wang and Y. Zhang, J. Am. Chem. Soc., 2015, 137, 12990–12996 CrossRef CAS PubMed.
- (a) M. Sen, B. Emayavaramban, N. Barsu, J. R. Premkumar and B. Sundararaju, ACS Catal., 2016, 6, 2792–2796 CrossRef CAS; (b) N. Barsu, Md. A. Rahman, M. Sen and B. Sundararaju, Chem.–Eur. J., 2016, 22, 9135–9138 CrossRef CAS PubMed.
- (a) D. Kalsi, R. A. Laskar, N. Barsu, J. R. Premkumar and B. Sundararaju, Org. Lett., 2016, 18, 4198–4201 CrossRef CAS PubMed; (b) N. Barsu, M. Sen and B. Sundararaju, Chem. Commun., 2016, 52, 1338–1341 RSC; (c) N. Barsu, D. Kalsi and B. Sundararaju, Chem.–Eur. J., 2015, 21, 9364–9368 CrossRef CAS PubMed; (d) M. Sen, D. Kalsi and B. Sundararaju, Chem.–Eur. J., 2015, 21, 15529–15533 CrossRef CAS PubMed; (e) D. Kalsi and B. Sundararaju, Org. Lett., 2015, 17, 6118–6121 CrossRef CAS PubMed.
- See ESI.†.
- G. Aromi, A. S. Batsonov, P. Christian, M. Helliwell, A. Parkin, S. Parsons, A. A. Smith, G. A. Timco and R. E. P. Winpenny, Chem.–Eur. J., 2003, 9, 5142–5161 CrossRef CAS PubMed.
- See notes for more information.†.
- Catalytic carbonylation was performed using recycled silver carbonate, which resulted in 83% isolated yield of succinimide 2a under the standard reaction conditions.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1507203 (2t) & 1507204 (2a). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc05026c |
| This journal is © The Royal Society of Chemistry 2017 |