
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tripodal halogen bonding iodo-azolium receptors for anion recognition†
Sheila Ruiz-Botellaa,
Pietro Vidossichb,
Gregori Ujaque*b,
Eduardo Peris *a and
Paul D. Beer*c
*a and
Paul D. Beer*c
aInstitute of Advanced Materials (INAM), Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universitat Jaume I, Avda. Sos Baynat, 12071 Castellón, Spain. E-mail: eperis@uji.es
bDepartament de Química and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Catalonia, Spain. E-mail: gregori.ujaque@uab.cat
cChemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford OX1 3TA, UK. E-mail: paul.beer@chem.ox.ac.uk
First published on 13th February 2017
Abstract
The synthesis, characterization and anion binding properties of 1,3,5-tri-substituted benzene platform-based tripodal receptors containing halogen bonding (XB) iodo-imidazolium and iodo-triazolium motifs, and hydrogen bonding (HB) analogues are described. Proton NMR anion binding investigations reveal the XB receptors are superior halide complexants compared to hydrogen bonding (HB) receptor analogues, with the iodo-imidazolium tripodal receptor exhibiting notably higher association constant values in comparison to the iodo-triazolium host system. In contrast, the HB tripod receptors, display higher affinities for dihydrogen phosphate than the XB receptors. Computational DFT and molecular dynamics were used to corroborate the experimental observations, and to give a clearer insight into the intimate nature of halide binding. The studies reveal a fundamental different binding behavior for the imidazolium-based and the iodo-imidazolium-based receptors.
Introduction
Anion recognition has attracted significant attention during the last 25 years, and has become one of the fundamental pillars of supramolecular chemistry.1 The reason for the increasing interest in developing effective and selective sensors for anions is the ubiquitous role that anions play in biology and industrial processes, together with their important impact in the environment. Among anion receptors, those derived from azolium salts have been widely used due to their strong affinity with anions through (C–H)+⋯X− interactions, which combine the strength of conventional hydrogen bonding with favourable electrostatic attraction.2 The relatively straightforward synthesis of imidazolium and triazolium motifs has facilitated their integration into a variety of acyclic, macrocyclic and interlocked rotaxane host structural frameworks designed to complement the size and shape of target anion guest species.3 Although much less exploited than hydrogen bonding (HB), halogen bonding (XB), the attractive non-covalent interaction between an electron-deficient polarized halogen atom and an electron-rich Lewis base, has been recently demonstrated to often out-perform HB anion recognition in competitive aqueous solvent media, including pure water.1b,4 This has been attributed to a more stringent preference for linearity and experimental evidence for a significant covalent contribution to the overall XB-anion binding process.5A recent report by Ghosh and co-workers compared the anion binding abilities of a tripodal iodo-imidazolium receptor with a protic-imidazolium receptor analogue in acetonitrile, concluding that the XB receptor displayed not only contrasting selectivity for bromide over chloride but also differing stoichiometries of host:guest complexes formed.6 Prompted by these observations and with the objective of directly comparing XB/HB imidazolium and triazolium anion recognition motifs, herein we report the preparation of four tripodal tris-imidazolium and tris-triazolium receptors and demonstrate the XB receptors are superior halide complexants with the iodo-imidazolium tripodal receptor forming the strongest complexes, displaying selectivity for chloride in DMSO solution.
Results and discussion
The four tris-azolium receptors were synthesized according to the procedures depicted in Scheme 1. The tris-imidazolium salts [1-X](PF6) (X = H, I) were prepared from 1,3,5-tris(bromomethyl)benzene, which upon reaction with three equivalents of imidazole (or iodoimidazole) in the presence of potassium carbonate afforded the neutral tris(imidazolmethyl)benzene compounds. Reaction with excess trimethyloxonium tetrafluoroborate afforded the isolation of the tris-azoliums [2-X](BF4) (X = H, I). Anion exchange with NH4PF6 gave the corresponding trimethylated hexafluorophosphate salts. The tris-triazolium salts [2-X](PF6) (X = H, I) were prepared by Cu(I)-mediated cycloaddition (CuAAC) of 1,3,5-tris(azidemethyl)benzene with three equivalents of 4-tert-butylphenylacetylene (or 4-tert-butylphenyl-iodo-acetylene iodide) to afford the neutral tris(triazole)benzene (or tris(iodotriazole)benzene) derivatives. Methylation and anion exchange produced the XB and HB triazolium receptors (Scheme 1).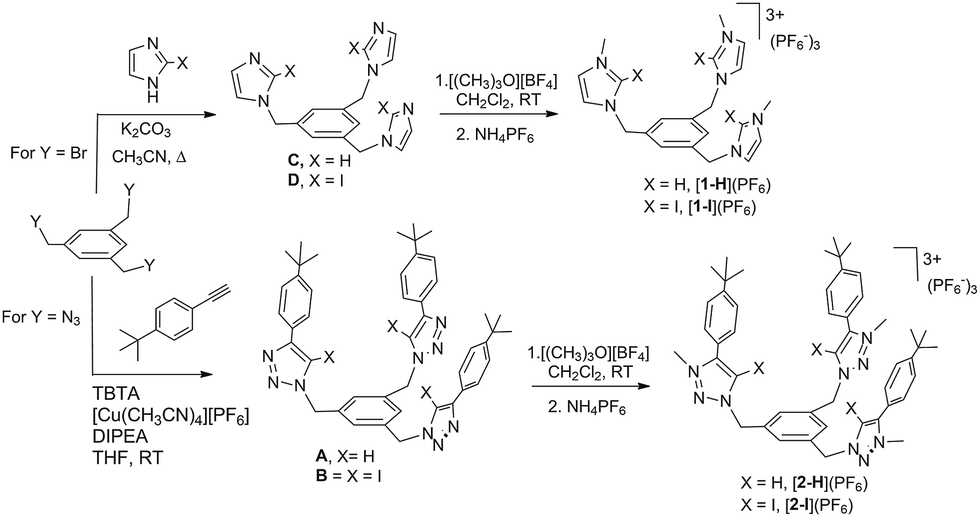 | ||
| Scheme 1 Synthesis of XB and HB tripodal receptors. | ||
The anion recognition properties of the tripod receptors [1-X](PF6) and [2-X](PF6) were studied by 1H NMR titration experiments, by monitoring the shift of the respective receptor's proton signals upon addition of tetrabutylammonium salts (TBAX, X = Cl−, Br−, I−, H2PO4−) in DMSO-d6. Undertaking titration experiments in deuterated acetonitrile, chloroform, methanol or water solutions led to precipitation problems. In general, the addition of anions induced remarkable perturbations in the 1H NMR spectra. In the case of the protic azolium receptors, [1-H](PF6) and [2-H](PF6), the most affected signals were the ones due to the acidic hydrogens of the azolium rings, which experienced maximum downfield shifts of Δδ ≈ 0.43 (for [1-H](PF6)) and Δδ ≈ 0.42 (for [2-H](PF6)) for the titrations with chloride. In all cases, the titrations produced the perturbation of several signals of the hosts, including the one due to the three equivalent protons of the central phenyl ring, therefore suggesting that the interaction of the anion is mainly located in the interior of the tripod-shaped hosts. As an illustrative example, Fig. 1 displays the spectra of the titration of [1-I](PF6) with tetrabutylammonium chloride. The series of spectra show that the signals due to the protons at the phenyl ring and one of the protons at the imidazolium are shifted downfield upon guest addition, while the signal due to the remaining proton of the imidazolium is shifted upfield. We also wanted to know if our iodo-azolium receptors could be suitable for the recognition of fluoride, and for this purpose we also performed a titration of [2-I](PF6) with tetrabutylammonium fluoride (TBAF). The addition of increasing amounts of TBAF did not produce any noticeable change on the spectra for amounts of [F−]/[2-I] < 1, but then new signals appeared, indicating that the receptor slowly decomposes. Unfortunately, we were not able to assign these signals to any new species formed. This preliminary experiment made us conclude that our iodo-azolium receptors are not suitable for fluoride recognition under these conditions.
 | ||
| Fig. 1 Representative region of the 1H NMR (500 MHz) spectra of the titration of [1-I](PF6) (2 mM) with tetrabutylammonium chloride (100 mM) in DMSO-d6. The plot represents the binding isotherms. | ||
For the titrations with all other anions, global non-linear regression analysis of the 1H NMR titration data determined 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric receptor: anion association constants (Ka)7 shown in Table 1 and as a graphical representation of the quantitative data in Fig. 2.8
:1 stoichiometric receptor: anion association constants (Ka)7 shown in Table 1 and as a graphical representation of the quantitative data in Fig. 2.8
| Anion | [1-H](PF6) | [2-H](PF6) | [1-I](PF6) | [2-I](PF6) |
|---|---|---|---|---|
| a K11 values calculated by global nonlinear regression analysis.7a,9 Titrations were carried out at 298 K, using starting concentrations of host 2 mM and of guest of 100 mM, in DMSO-d6 at 298 K. Titrations in DMSO-d6:D2O (9:1).b Data taken from ref. 10. |
||||
| Cl− | 1341 ± 4% | 589 ± 3% | 2480 ± 9% | 829 ± 8% |
| Br− | 884 ± 4% | 377 ± 6% | 1078 ± 6% | 595 ± 2.7% |
| I− | 300 ± 5% | 329 ± 5% | 551 ± 9% | 522 ± 6.3% |
| H2PO4− | 1950b | 2543 ± 7% | 311 ± 2%a | 514 ± 5%/378 ± 4a |
 | ||
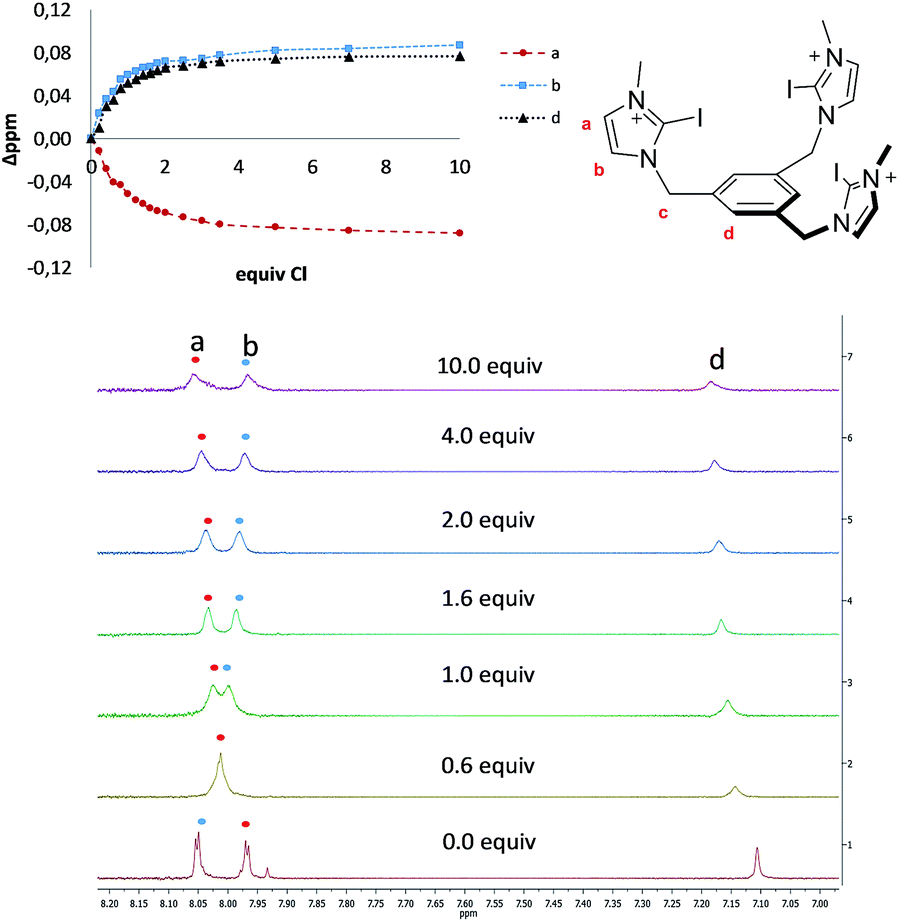
| Fig. 2 Molecular structure of [1-I](BF4). Solvent (CHCl3) and hydrogen atoms have been omitted for clarity. Ellipsoids are at 50% of probability. Selected bond distances (Å) and angles (°): I(1)–Cl(1) 2.996(2), I(1)–Cl(1) 3.127(1); C(2)–I(1)–Cl(1) 176.88 and C(2)–I(1)–Cl(1)177.44°. | ||
Amongst the halides, all receptors form the strongest complexes with chloride followed by bromide with iodide forming the weakest association, in agreement with the basicity trend of the halide anions.
For the recognition of halides, the association constants determined for the XB iodo-substituted hosts ([1-I](PF6) and [2-I](PF6)) were found to be significantly larger in magnitude than those for the related HB analogues. Also the tripodal imidazolium receptors formed notably stronger complexes with chloride and bromide than the tripodal triazolium analogues. Hence the iodo-imidazolium tripod receptor [1-I](PF6) displays the highest affinity toward Cl− versus Br− and I−, among all the tripodal receptors under study. This contrasts the quantitative binding data for the recognition of the H2PO4− (Table 1), where receptor [2-H](PF6) exhibits the highest affinity. This may be explained as a consequence of the innate preference of XB donor groups for softer halide over harder oxoanion receptors, probably as a consequence of the charge transfer covalent and/or dispersion contributions of the C–I⋯A− XB interaction.4e
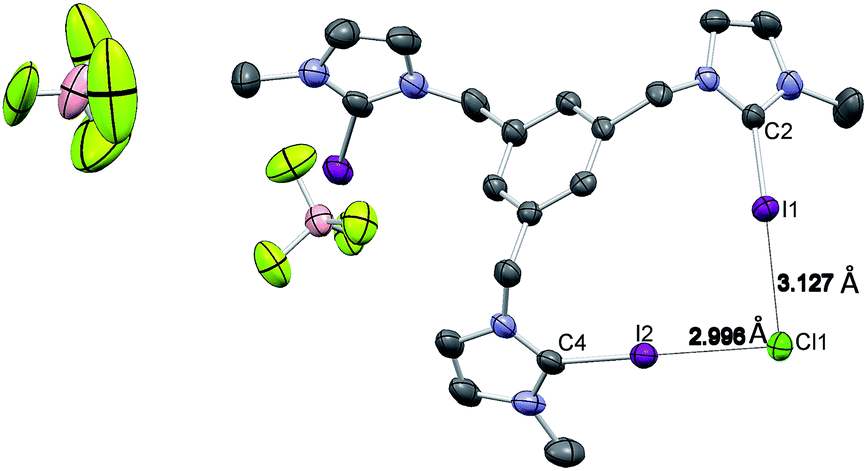
Single crystals of [1-I](BF4) were grown by slow diffusion of methanol into a concentrated chloroform solution of [1-I](BF4) to which one equivalent of tetrabutylammonium chloride was added. Fig. 2 shows the structure of the compound, where a clear interaction between two iodo-imidazoliums and the chloride anion is observed. The C–I⋯Cl distances are 2.996 (I1) and 3.127 (I2) Å, which reflect a 24.3 and 21.0% shortening with respect to the sum of the van der Waals radii, respectively. The C–I–Cl angles are 176.88 (I1) and 177.44° (I2), therefore very close to 180° expected for a halogen bonding interaction. By expanding the structure, it is possible to observe that the chloride anion is also interacting with one of the imidazolium arms of a second receptor molecule (Fig. 3), therefore all three iodines from the imidazoliums are halogen-bonded to chloride, thus yielding an overall host:guest stoichiometry of 1:1.
 | ||
| Fig. 3 One section of the crystal packing of [1-I](BF4). Solvent (CHCl3), BF4− anions and hydrogen atoms have been omitted for clarity. The crystal packing is showing the halogen bonding interaction between the iodo atoms from two molecules with the chloride. | ||
A molecular modeling study was undertaken to characterize the conformational dynamics of receptors [1-H] and [1-I] and their interactions with counterions. Each arm of the receptor has two rotatable bonds, potentially giving rise to a high number of conformers. Furthermore, several different interaction modes with the counterions may be envisioned. Because of this, we used classical molecular dynamics (MD) simulations to efficiently explore the conformational space. Simulations were performed in explicit solvent and starting from different conformations of the receptor. Two counterions were considered, PF6− and Cl−. About 100 ns simulations were collected for each system (see ESI† for a description of the computational methods and an expanded account of the results obtained).
Simulation revealed that the nature of the counterion has a remarkable impact on the receptor's conformational properties due to the strength of the interactions they establish. For both receptors, PF6− displayed unspecific and short lived interactions with the receptor's arms, which experience rapid interchange of orientation. On the contrary, chloride anions show longer lived binding to the imidazolium and iodo-imidazolium rings. In the case of [1-H], simulation revealed the formation of a tri-coordinated species (Fig. 4a), in which Cl− is trapped within the interior of the tripod. This species appears to have a lifetime of the order of tens of ns (Fig. 4b, central part of the graph).
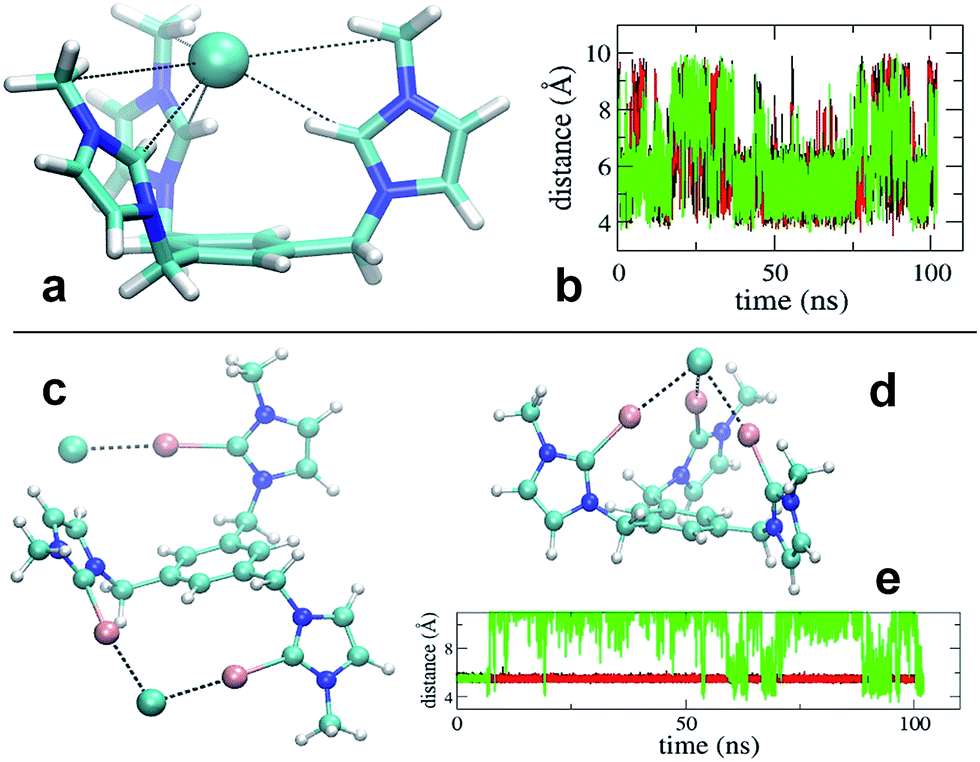 | ||
| Fig. 4 Molecular modeling studies of the recognition properties of the [1-H] (a) and [1-I] (c and d) receptors. In (b), the time series of the distances between the carbenium carbons of the imidazolium rings of [1-H] are shown. In (e), the distances between one chloride anion and the carbenium carbon of the iodoimidazolium rings of [1-I] are shown. | ||
For [1-I] an equivalent structure of the receptor/Cl− complex is not actually possibly due to the steric bulk of the iodine atoms. A tri-coordinated species may actually form (Fig. 4d), although this is not the preferred binding mode, which consists of the formation of halogen bonding interactions with two arms of the receptor. The simulations show that halogen bonding are highly stable interactions, so once formed, they are maintained for the rest of the simulation (Fig. 4e). Binding of Cl− to two arms was observed in simulations of models [1-I](Cl)3 and [1-I](Cl)(PF6)3, thus suggesting that it is independent of the number of equivalents of chloride. A second anion binds the third arm of the receptor as shown in Fig. 4c, therefore affording a situation which is fully consistent with the solid state structure.
Quantum chemical calculations DFT(B3LYP) (see ESI†) support the picture arising from the MD simulations of [1-I]. Representative structures from the MD simulations were optimized and used to compare the energetics of Cl binding to one, two and three arms of the receptor. Successive binding of the arm's receptor is favourable (Table S17 from ESI†). However, when increasing from two to three arm's coordination the gain in stability is small, due to the cost of bringing together the positively charged arms. In fact, the energy difference between the open and closed conformations of [1-I] is 9.9 kcal mol−1 in favor of the open structure (Fig. S62 from ESI†), which is similar to the binding energy of Cl− to iodoimidazolium (−10.6 kcal mol−1, Fig. S63, ESI†). We also compared the energies of the receptor interacting with two Cl− in different conformations (Fig. S61, ESI†). In line with the above results and those from the MD study, binding of one Cl− to two arms and another Cl− to one arm (Fig. S61b and c, ESI†) is favoured compared to the binding of one Cl− to three arms and one Cl− interacting unspecifically with the receptor (Fig. S61a, ESI†), in a situation that clearly resembles our experimental solid state structure.
Conclusions
Four tripodal acyclic tris-azolium receptors containing XB iodoimidazolium or iodo-triazolium and protic HB imidazolium and triazolium appendages were prepared for a comparative study of their recognition abilities towards a series of anions. 1H NMR titration experiments undertaken in DMSO-d6 revealed all four receptors to form 1:1 stoichiometric anion complexes with the XB receptors exhibiting the highest binding affinities towards halides which is a clear indication that halides are best recognized by halogen bonding interactions, compared to hydrogen bonding (HB). By contrast, a reversal of selectivity is observed with H2PO4−, where the HB receptors form the strongest associations. The nature of the halogen bonding and hydrogen bonding interactions of the tripodal iodoimidazolium and imidazolium receptors was also investigated by means of computational DFT and molecular dynamics studies. These studies corroborate the experimental results, and give important information about the preferred relative conformation of the three arms of the receptors. While the imidazolium receptor uses all three arms of the tripod to bind the halide, the iodoimidazolium receptor prefers to bind the halide by only two of the three arms, with the remaining arm bound to a second halide ion. This different behaviour is possibly due to the steric bulk of the iodine atoms, which prevent the formation of the tricoordinated structure.
Experimental section
General comments
All manipulations were carried out under nitrogen using standard Schlenk techniques. Anhydrous solvents were either distilled from appropriate drying agents (SPS) or purchased from Aldrich and degassed. Triazidemethylbenzene,11 4-tert-butylphenyl-iodoacetylene,12 1,3,5-tris((1H-imidazol-1-yl)methyl)benzene13 (C) were prepared according to the literature procedures. NMR spectra were recorded on Varian Mercury 300, Varian Unity Plus 500 and Bruker AVII 500 (with 13C Cryoprobe) spectrometers. Mass spectra were carried out on Micromass LCT and Bruker micro-TOF spectrometers.Synthesis of A
A mixture of triazidemethylbenzene (68.9 mg, 0.28 mmol), 4-tert-butylphenylacetylene (174.9 mg, 1.10 mmol), [Cu(CH3CN)4](PF6) (50.0 mg, 0.13 mmol), TBTA (23.9 mg, 0.045 mmol) and DIPEA (252 μL, 1.5 mmol) in 3 mL CH2Cl2 was stirred during 48 h at room temperature. The resulting suspension was washed with NH4OH and brine. The product was purified by column chromatography by eluting with a mixture of CH2Cl2:AcOEt (7:3). The compound was obtained as a white solid in 65% yield (130 mg). 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 8.3 Hz, 8H, CHbenz), 7.68 (s, 3H, CHbenz), 7.42 (d, J = 8.4 Hz, 8H, CHbenz), 7.19 (s, 3H, CHtriaz), 5.50 (s, 6H, CH2), 1.33 (s, 81H, CH3). 13C NMR (101 MHz, CDCl3) δ 151.66, 148.63, 137.28, 127.51, 127.42, 125.92, 125.61, 119.63, 53.48, 34.82, 31.42. Electrospray accurate mass analysis (positive mode): 718.43296 [M + H]+.
Synthesis of B
A mixture of triazidemethylbenzene (54.4 mg, 0.22 mmol), 4-tert-butylphenyl-iodoacetylene (222.9 mg, 0.73 mmol), [Cu(CH3CN)4](PF6) (37.51 mg, 0.1 mmol), TBTA (17.8 mg, 0.0355 mmol) in 3 mL CH2Cl2 was stirred during 48 h at room temperature. The resulting suspension was washed with NH4OH and brine. The product was purified by column chromatography by eluting with CH2Cl2:AcOEt (7:3). The compound was obtained as a white solid in 70% yield (170 mg). 1H NMR (400 MHz, CDCl3) δ 7.87 (d, J = 8.2 Hz, 6H, CHbenz), 7.48 (d, J = 8.2 Hz, 6H, CHbenz), 7.23 (s, 3H, CHbenz), 5.65 (s, 6H, CH2), 1.35 (s, 27H, CH3). 13C NMR (101 MHz, CDCl3) δ 151.93, 150.43, 137.53, 136.10, 127.65, 127.37, 127.22, 127.14, 125.65, 76.23, 54.14, 53.96, 34.88. Electrospray accurate mass analysis (positive mode): 1096.12354 [M + H]+.
Synthesis of D
Iodoimidazole (200.0 mg, 1.031 mmol) was dissolved in dry CH3CN and potassium hydroxide (160.0 mg, 2.85 mmol) was added and stirred at room temperature during 2 h. Then, 1,3,5-tris(bromomethyl)benzene (121.3 mg, 0.34 mmol) was added and stir at room temperature during 6 h. After this time the mixture was diluted with acetonitrile and the compound was filtered. The solvent was removed in under vacuum and the product was obtained as a white solid in 27% yield (60 mg).1H NMR (400 MHz, DMSO-d6) δ 7.37 (s, 3H, CHimidazole), 6.99 (d, 3H, CHimidazole), 6.90 (d, 3H, CHbenzene), 5.12 (s, 6H, CH2). 13C NMR (101 MHz, DMSO-d6) δ 138.47, 132.50, 125.68, 124.56, 93.37, 51.88, 40.65, 40.44, 40.23, 40.02, 39.81, 39.60, 39.40.Synthesis of [2-H](PF6)
Compound A (100 mg, 0.139 mmol) was dissolved in dry CH2Cl2 (5 mL) and trimethyloxonium tetrafluoroborate (67.8 mg, 0.459 mmol) was added. The mixture was stirred at room temperature during 48 h. Diethyl ether was added to the mixture to produce the precipitation of the compound, which was filtered and redissolved in CHCl3 and washed several times with a saturated solution of NH4PF6. After removal of the solvent the compound was obtained as an off-white solid in 90% yield (149 mg). 1H NMR (400 MHz, CDCl3): δ 8.11 (s, 3H, CHtriz), 7.83 (s, 3H, CHbenz), 7.48 (broad signal, 12H, CHbenz), 5.79 (s, 6H, CH2), 4.28 (s, 9H, CH3), 1.28 (s, 27H, CH3). 13C NMR (101 MHz, CD3CN) δ 156.39, 144.56, 134.93, 131.83, 129.91, 129.26, 127.45, 120.22, 56.97, 39.57, 35.64, 31.12. Electrospray accurate mass analysis (positive mode): 1052.42292 [M − 2PF6−]+.Synthesis of [2-I](PF6)
Compound B (39.1 mg, 0.036 mmol) was dissolved in dry CH2Cl2 (5 mL) and trimethyloxonium tetrafluoroborate (19.0 mg, 0.13 mmol) was added. The mixture was stirred at room temperature during 48 h. Diethyl ether was added to the mixture to produce the precipitation of the compound, which was filtered and redissolved in CHCl3 and washed several times with a saturated solution of NH4PF6. After removal of the solvent the compound was obtained as an off-white solid in 82% yield (41 mg). 1H NMR (400 MHz, CDCl3) δ 7.79 (s, 3H, CHtriaz), 7.57 (d, J = 8.2 Hz, 6H, CHbenz), 7.47 (d, J = 8.1 Hz, 6H, CHbenz), 5.81 (s, 6H, CH2), 4.11 (s, 9H, CH3), 1.34 (s, 27H, CH3). 13C NMR (101 MHz, CD3CN) δ 156.31, 148.03, 134.08, 130.55, 130.20, 127.13, 119.92, 57.42, 39.71, 35.31, 30.73. Electrospray accurate mass analysis (positive mode): 642.57419 [M − 2PF6]2+.Synthesis of [1-I](PF6)
Compound C (36.5 mg, 0.052 mmol) was dissolved in dry CH2Cl2 (5 mL) and trimethyloxonium tetrafluoroborate (25.6 mg, 0.17 mmol) was added. The mixture was stirred at room temperature during 24 h. Diethyl ether was added to the mixture to produce the precipitation of the compound, which was filtered and redissolved in CHCl3 and washed several times with a saturated solution of NH4PF6. After removal of the solvent the compound was obtained as an off white solid in 96% yield (36.6 mg), 1H NMR (400 MHz, DMSO) δ 8.26 (d, 3H, CH), 8.20 (d, 3H, CH), 7.77 (s, 3H, CHaromatic), 5.91 (s, 6H, CH2), 4.42 (s, 9H, CH3). 13C NMR (101 MHz, CD3CN) δ 146.11, 138.68, 137.77, 136.49, 109.44, 65.33, 50.24. Electrospray accurate mass analysis (positive mode): 246.97277 [M − 3PF6−]+.Synthesis of [1-H](PF6)
Compound D (50 mg, 0.157 mmol) was dissolved in dry CH2Cl2 (3 mL) and trimethyloxonium tetrafluoroborate (76.7 mg, 0.52 mmol) was added. The mixture was stirred at room temperature during 24 h. Diethyl ether was added to the mixture to produce the precipitation of the compound, which was filtered and redissolved in CHCl3 and washed several times with a saturated solution of NH4PF6. After removal of the solvent the compound was obtained as an off-white solid in 53% yield (52 mg). 1H NMR (400 MHz, DMSO): δ 8.38 (s, 3H, NCHN), 6.93 (s, 3H, CHimidazole) 6.90 (s, 3H, CHimidazole), 6.63 (s, 3H, CHbenzene), 4.61 (s, 6H, CH2), 3.03 (s, 9H, CH3). 13C NMR (101 MHz, DMSO) δ 137.26, 136.73, 128.90, 124.59, 122.83, 51.82, 36.40. Electrospray accurate mass analysis (positive mode): 653.40780 [M − PF6−]+.Acknowledgements
We gratefully acknowledge financial support from MEC of Spain (CTQ2014-51999-P and CTQ2014-54071-P) and the Universitat Jaume I (P11B2014-02). We are grateful to the Serveis Centrals d'Instrumentació Científica (SCIC) of the Universitat Jaume I for providing with spectroscopic facilities. We would also like to thank the Generalitat Valenciana for a fellowship (S-R.-B.).Notes and references
- (a) A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed; (b) T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC; (c) N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed; (d) P. A. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205–241 RSC; (e) P. A. Gale and C. Caltagirone, Chem. Soc. Rev., 2015, 44, 4212–4227 RSC; (f) M. J. Langton, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 1974–1987 CrossRef CAS PubMed.
- (a) F. D'Anna and R. Noto, Eur. J. Org. Chem., 2014, 4201–4223 CrossRef; (b) J. J. Cai and J. L. Sessler, Chem. Soc. Rev., 2014, 43, 6198–6213 RSC; (c) K. Sato, S. Arai and T. Yamagishi, Tetrahedron Lett., 1999, 40, 5219–5222 CrossRef CAS; (d) E. Alcalde, C. Alvarez-Rua, S. Garcia-Granda, E. Garcia-Rodriguez, N. Mesquida and L. Perez-Garcia, Chem. Commun., 1999, 295–296 RSC; (e) H. Ihm, S. Yun, H. G. Kim, J. K. Kim and K. S. Kim, Org. Lett., 2002, 4, 2897–2900 CrossRef CAS PubMed; (f) V. Amendola, M. Boiocchi, B. Colasson, L. Fabbrizzi, M. J. R. Douton and F. Ugozzoli, Angew. Chem., Int. Ed., 2006, 45, 6920–6924 CrossRef CAS PubMed.
- (a) Z. Xu, S. K. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 1457–1466 RSC; (b) M. V. Baker and D. H. Brown, Mini-Rev. Org. Chem., 2006, 3, 333–354 CrossRef CAS.
- (a) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed; (b) B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522–2571 RSC; (c) H. Wang, W. Z. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072–5104 CrossRef CAS PubMed; (d) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed; (e) A. Brown and P. D. Beer, Chem. Commun., 2016, 52, 8645–8658 RSC.
- S. W. Robinson, C. L. Mustoe, N. G. White, A. Brown, A. L. Thompson, P. Kennepohl and P. D. Beer, J. Am. Chem. Soc., 2015, 137, 499–507 CrossRef CAS PubMed.
- S. Chakraborty, R. Dutta and P. Ghosh, Chem. Commun., 2015, 51, 14793–14796 RSC.
- (a) P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC; (b) K. Hirose, J. Inclusion Phenom. Macrocyclic Chem., 2001, 39, 193–209 CrossRef CAS; (c) D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 53, 12792–12805 RSC.
- Analysis of the binding isotherms showed that the curves were best fitted to the formation of 1:1 stoichiometric
complexes. This conclusion was also supported by the fact that the fittings to a 1:1 stoichiometry gave the lowest residuals compared to other potential stoichiometries. The association constants, K11, were calculated by global nonlinear regression analysis by simultaneously including all protons showing chemical shift variations.
- A. J. Lowe, F. M. Pfeffer and P. Thordarson, Supramol. Chem., 2012, 24, 585–594 CrossRef CAS.
- T. Fahlbusch, M. Frank, J. Schatz and H. Schmaderer, Eur. J. Org. Chem., 2006, 1899–1903 CrossRef CAS.
- F. Sander, U. Fluch, J. P. Hermes and M. Mayor, Small, 2014, 10, 349–359 CrossRef CAS PubMed.
- L. C. Gilday, T. Lang, A. Caballero, P. J. Costa, V. Felix and P. D. Beer, Angew. Chem., Int. Ed., 2013, 52, 4356–4360 CrossRef CAS PubMed.
- F. D'Anna, H. Q. N. Gunaratne, G. Lazzara, R. Noto, C. Rizzo and K. R. Seddon, Org. Biomol. Chem., 2013, 11, 5836–5846 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1521906. For ESI and crystallographic data in CIF or other electronic format See DOI: 10.1039/c6ra28082j |
| This journal is © The Royal Society of Chemistry 2017 |