
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe dual capture of AsV and AsIII by UiO-66 and analogues†
Cornelius O.
Audu
a,
Huong Giang T.
Nguyen
a,
Chih-Yi
Chang
a,
Michael J.
Katz
a,
Lily
Mao
a,
Omar K.
Farha
*ab,
Joseph T.
Hupp
*a and
SonBinh T.
Nguyen
*a
aDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208-3113, USA. E-mail: o-farha@northwestern.edu; j-hupp@northwestern.edu; stn@northwestern.edu
bDepartment of Chemistry, Faculty of Science, King Abdulaziz University, Jedah, Saudi Arabia
First published on 16th June 2016
Abstract
UiO-66 and analogues were successfully tailored to chemoselectively capture AsV oxyanions at the hydroxylated node and neutral AsIII species with the thiolated organic linkers. More efficient and faster uptake can be achieved with increasing defect densities, increasing pore aperture sizes, and decreasing particle sizes.
Introduction
Metal organic frameworks (MOFs) are highly porous and crystalline coordination polymers that can be synthesized from a wide variety of metal-cluster nodes and a diverse selection of multitopic organic linkers. As a result, they are highly tailorable and different combinations of functionalities can be incorporated into the nodes and the linkers in an orthogonal manner.1–5 Not surprisingly, these materials have recently garnered increasing interest in capture-and-release studies, where the metal cluster nodes were tailored to capture/release phosphonate-based substrates6,7 or the organic linkers were used to sequester toxic heavy metals.8–10 These precedents led us to propose that MOFs can be used as a model platform to demonstrate the capture of toxic anionic arsenates (AsV) and neutral arsenites (AsIII), both of which exist in ground water (pH 6–8.5),11 in a complementary fashion (Fig. 1); the node can be used for binding anionic AsV and the linkers can be functionalized to capture neutral AsIII. Such a design can serve as a versatile strategy for developing materials that efficiently capture multiple pollutants or toxic agents that exist as diverse species in certain environments. | ||
| Fig. 1 A schematic representation that suggests how MOFs can be tailored to coordinate anionic AsV moieties at the node while binding neutral AsIII with the linkers. | ||
Given their high chemical stability and hydrophilicity,12–17 we deemed MOFs with hexazirconium oxo hydroxo (Zr6O4(OH)4) cluster nodes, such as UiO-66,16–22 to be suitable model targets for modifications to capture both AsV and AsIII from aqueous media. We predicted strong interactions between the nodes of UiO-66 and [AsVO4H3−n]n− oxyanions (Fig. 2 and ESI,† Fig. S1), based on the observed strong coordination of the Zr6O4(OH)4 cluster nodes6,7,24–27 to phosphonates and phosphates, which are isostructural and have similar Brønsted basicity as arsenates.6,7,23–27 In addition, the incorporation of thiol-containing BDC ligands (i.e., 2,5-dimercaptoterephthalic acid) into UiO-66 (ref. 9) should facilitate binding to neutral [AsIII(OH)3]n species,11 akin to the known arsenophilicity of sulfur-containing enzymes and thiol-rich chelators.28–31 Herein, we report the successful use of HCl-UiO-66-(SH)2, a UiO derivative with thiolated linkers and nodes that are “capped” with weakly binding ligands, to efficiently capture both AsIII and AsV from aqueous media (Fig. 1). The missing-linker sites on the Zr6(O)4(OH)4 nodes can serve as excellent binders for AsV oxyanions while the thiolated linkers can selectively coordinate AsIII for dual-capture purposes. The efficiency and capacity of this dual-binding feature is best realized when the binding sites are made easily accessible, either by enlarging the pore aperture size of the MOF or by reducing the particle size of the MOF nanocrystals.
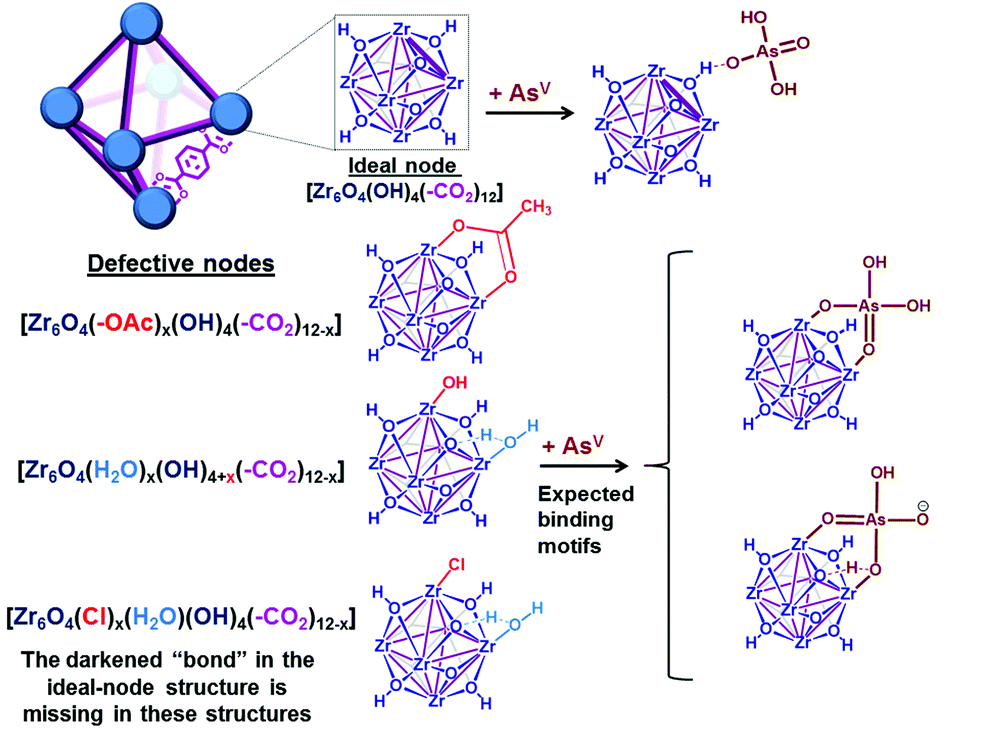 | ||
| Fig. 2 Representative views (see ESI,† Fig. S19 for other possible binding motifs) of a unit cell of UiO-66, with either 12-coordinated ideal nodes [Zr6O4(OH)4(–CO2)12] or imperfect nodes [Zr6O4(OH)x(–CO2)y], which result from missing linkers. Each purple “bond” indicates a coordinating carboxylate from the terephthalate linker. | ||
Results and discussion
As a model platform, UiO-66 is highly attractive given its excellent synthetic tunability: functionalized derivatives of p-benzene dicarboxylate (BDC) can be easily incorporated into the framework either through de novo synthesis17,32 or post-synthetically.33,34 In addition, the degree of coordinative unsaturation of the nodes can be tuned with the use of organic18,19,35 or inorganic17 acid-modulators. In the current study, we select nearly defect-free AcOH-UiO-66|11/12 (i.e., AcOH-“capped” UiO-66; see Fig. 2, top structure in the lower left corner) as a control sample. This material was prepared from ZrCl4 and H2BDC in dimethylformamide (DMF) and in the presence of acetic acid as a modulator. It comprises well-defined octahedral particles with a Brunauer–Emmett–Teller (BET) surface area of ∼1150 m2 g−1, and a powder X-ray diffraction (PXRD) pattern identical to that of crystalline UiO-66 (ESI,† Fig. S2–S4). Thermogravimetric analysis (TGA) data for this material suggested a formula unit of Zr6O4(OH)4(BDC)5.5, alternatively referred to as Zr6O4(OH)4(–CO2)11/12, which suggests that only a small amount of defects (i.e., missing-linker sites) are present (ESI,† Fig. S7). (The “(–CO2)11/12” notation is used to indicate 1 missing carboxylate per node or 0.5 missing BDC linker per formula unit). As mentioned above, these missing-linker sites are known to bind well to phosphonates6 and vanadates,36 and should also be susceptible toward arsenate binding (Fig. 2).To elucidate the ability of the Zr6O4(OH)4 node to bind AsV, we additionally synthesized HCl-UiO-66|(12−x)/12, a series of UiO-66 materials where the amount of missing linker (x) was systematically varied using the HCl-modulator strategy.17 These materials were also prepared from ZrCl4 and H2BDC in DMF, but with different molar ratios and with HCl as the modulator (see details in ESI,† Section S2 and Table S1). Notably, we successfully obtained HCl-UiO-66|9/12, with ∼3 available missing-linker sites per node, presumably being “weakly capped” by either HO−, Cl−, or H2O (see Fig. 2, last two structures in the lower left corner).
AsV uptake by UiO-66 derivatives
AsV-adsorption experiments were conducted at pH ∼7,37 to simulate the middle range of ground water pH, by exposing samples of the MOFs (10 mg each) to separate 50 ppm solutions (30 mL portions38) of Na2HAsO4·7H2O as the AsV source. The amount of AsV in the supernatant is monitored using inductively coupled plasma optical emission spectroscopy (ICP-OES) and the per-node uptake of AsV by the MOF at time t can then be calculated. As expected, AcOH-UiO-66|11/12 showed good uptake of AsV (Fig. 3, green profile) from the Na2HAsO4·7H2O test solution. The near-stoichiometric AsV![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zr6 uptake ratio (1.1:1 AsV:Zr6) at 6 h strongly suggests a preferential binding of AsV to the missing-linker sites on the Zr6 nodes. Assuming that missing-linker sites are the most easily accessible, half of these sites (0.5:1 AsV:Zr6), presumably those that are close to the surface of the MOF nanocrystals, would be saturated within the first 30 min (Fig. 3, green profile). The rate of adsorption slows down as the remaining, internal sites (i.e., deeper inside the MOF nanocrystals) are saturated over the next few hours, consistent with a diffusion-limited behavior. That the 24 h uptake ratio (1.2:1 AsV:Zr6) slightly exceeds the estimated available binding site suggests the possible involvement of secondary binding pathways, such as formation of As oligomers11 on the node (see ESI,† Fig. S21a for an illustration), linker displacement by the incoming AsV species (see below and ESI,† Section S3 for additional discussions),39 and/or anion exchange with the bridging hydroxyl sites of the nodes,40,41 although delineating these pathways is beyond the scope of this manuscript.
:Zr6 uptake ratio (1.1:1 AsV:Zr6) at 6 h strongly suggests a preferential binding of AsV to the missing-linker sites on the Zr6 nodes. Assuming that missing-linker sites are the most easily accessible, half of these sites (0.5:1 AsV:Zr6), presumably those that are close to the surface of the MOF nanocrystals, would be saturated within the first 30 min (Fig. 3, green profile). The rate of adsorption slows down as the remaining, internal sites (i.e., deeper inside the MOF nanocrystals) are saturated over the next few hours, consistent with a diffusion-limited behavior. That the 24 h uptake ratio (1.2:1 AsV:Zr6) slightly exceeds the estimated available binding site suggests the possible involvement of secondary binding pathways, such as formation of As oligomers11 on the node (see ESI,† Fig. S21a for an illustration), linker displacement by the incoming AsV species (see below and ESI,† Section S3 for additional discussions),39 and/or anion exchange with the bridging hydroxyl sites of the nodes,40,41 although delineating these pathways is beyond the scope of this manuscript.
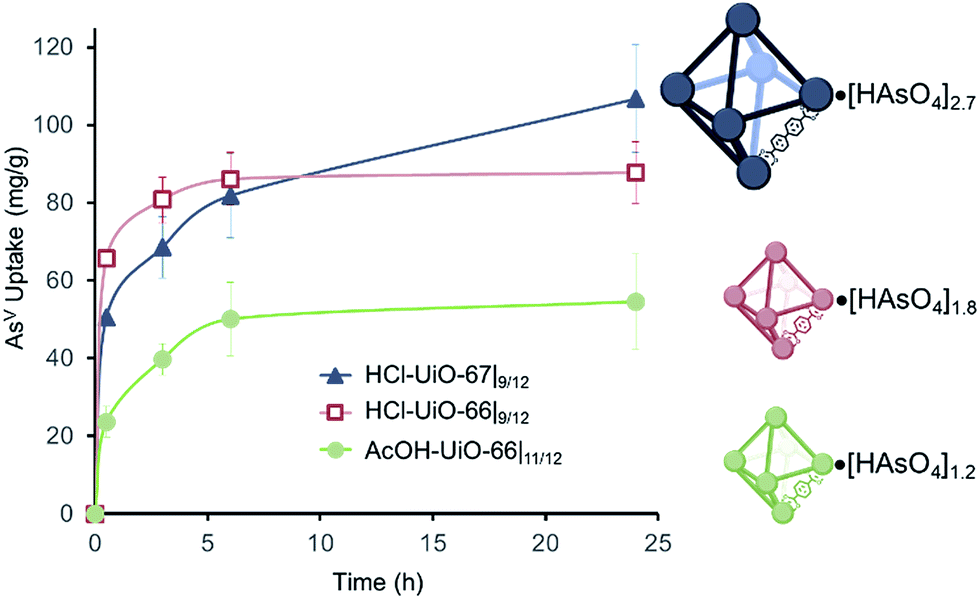 | ||
| Fig. 3 AsV-uptake profiles for MOF samples (10 mg) that have been exposed to AsV-containing solutions (30 mL; 50 ppm initial concentration). The total amounts of bound AsV per node is indicated on the right. Each data point is an average of 3 to 4 different experiments. | ||
Consistent with our hypothesis that missing-linker sites can also bind arsenates well, HCl-UiO-66|9/12, which has 3 missing-linker sites per node, captured AsV substantially faster and more (1.4:1 AsV:Zr6 or ∼46% of the missing-linker sites after 30 minutes) than the nearly defect-free AcOH-UiO-66|11/12 (Fig. 3, cf. red and green profiles). Interestingly, the uptake ratio for HCl-UiO-66|9/12 in our standard 50 ppm initial As-exposure experiment levels out relatively quickly and does not change after 6 h: the AsV uptake ratio at this time point, as well as after 24 h of exposure, only amounts to ∼60% of the available missing-linker sites (1.8:1 AsV:Zr6) even though there is still excess AsV in solution.38 These data suggests that the AsV uptake in HCl-UiO-66|9/12 is probably dominated by an equilibrium-driven process, as demonstrated for organophosphorus6 and selenates42 uptakes in UiO-type MOFs. This is indeed the case: as the As-exposure concentration is increased to 100 ppm, all of the available missing-linker sites (3:1 AsV:Zr6) for HCl-UiO-66|9/12 can be filled after 24 h (Fig. 4).
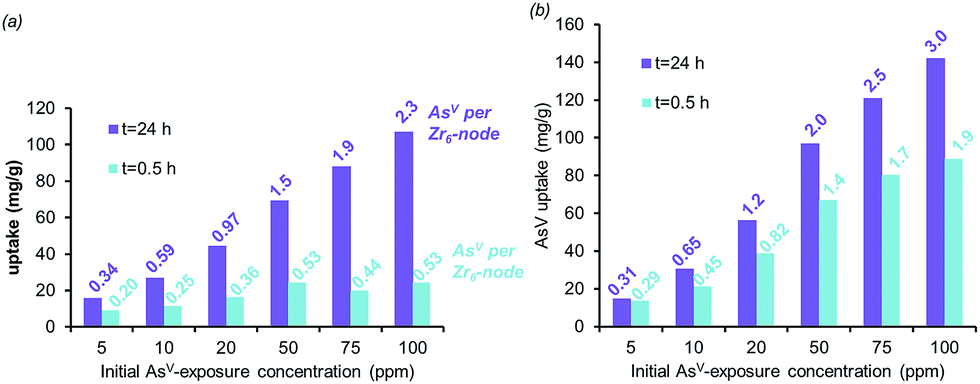 | ||
| Fig. 4 The AsV-adsorption isotherms for AcOH-UiO-66|11/12 (a) and HCl-UiO-66|9/12 (b) at short (0.5 h) and long (24 h) exposure time, plotted as bar graphs with the AsV:Zr6 ratios indicated at the top of each bar. Experimental conditions: batch exposure of a sample of MOF (10 mg) to the appropriate AsV-containing solution (30 mL). | ||
The combination of equilibrium-driven and secondary uptake mechanisms lead to quite different behaviors for AcOH-UiO-66|11/12 and HCl-UiO-66|9/12 as the initial As-exposure concentration was varied from 5 to 100 ppm (Fig. 4). While the AsV adsorption profiles for HCl-UiO-66|9/12 at different time points all increased in the same manner (see also ESI,† Fig. S10d), the AsV:Zr6 uptake ratios never exceeded the per-node number of missing-linker sites, presumably due to sterics.43 In contrast, the AsV adsorption profiles for AcOH-UiO-66|11/12 varied significantly depending on the timing of the measurements: at 0.5 h the AsV:Zr6 uptake ratios did not vary much beyond 53% of the available missing-linker sites (0.20–0.53:1 AsV:Zr6) while that at 24 h varied over a very large 34–235% range (0.34–2.35:1 AsV:Zr6) (see also ESI,† Fig. S10c as well as the accompanying discussion that follows Fig. S10).
The aforementioned large discrepancy in behaviors accentuates the differences in As-uptake mechanisms between HCl-UiO-66|9/12 and AcOH-UiO-66|11/12. While the As-uptake by the former is presumably based on filling up missing-linker sites, that for the latter changes between short and long exposure times. We speculate that the As-uptake mechanism for AcOH-UiO-66|11/12 during the short exposure is also based on filling up missing-linker sites but that for the long exposure is dominated by the secondary binding pathways mentioned above. We note in passing that the large number of defect sites, and presumably larger pores (ESI,† Fig. S5), in HCl-UiO-66|9/12 are highly advantageous for capturing purposes: the adsorption profiles measured at 3, 6, and 24 h are quite similar (ESI,† Fig. S10d), suggesting that most of the removal occurs during the first 3 h. Under our experimental conditions (Fig. 4), this means that >90% of the AsV oxyanions from a 5 ppm solution can be removed after 0.5 h (0.29:1 AsV:Zr6; see also ESI,† Fig. S10f), and complete removal (0.31:1 AsV:Zr6) occurred after 3 h.
TEM-based EDS analyses of AcOH-UiO-66|11/12 and HCl-UiO-66|9/12 samples that have been exposed to 100 ppm AsV solutions for 24 h are also consistent with the aforementioned uptake contrast. While no visible morphological changes can be observed, the latter sample clearly showed a much higher AsV uptake based on the relative As/Zr signal ratios (ESI,† Fig. S18). The PXRD data for AsV-exposed materials are identical to the data for the corresponding as-synthesized materials (ESI,† Fig. S1), indicating that the crystallinities of the MOF samples are mostly retained even after significant AsV uptake and prolonged (24 h) shaking. Interestingly, while ICP-OES analysis of the supernatants from the batch-adsorption experiments shows no evidence of ZrIV ions, concurrent analyses of these samples by ESI-MS and high-resolution water-suppression 1H NMR spectroscopy reveals the presence of some H2BDC linker. Although these data support the occurrence of the aforementioned linker-displacement secondary binding mechanism, and thus possible partial degradation of the initial MOF structure, a quantitative assessment is not possible at the present time (see ESI,† Section S3 for further discussion). Additionally, it is worthwhile to note that the As concentrations that we explored for the uptake experiments herein are much higher than those that exists in natural water sources (1 ppb to 3 ppm),44 which could accelerate secondary linker-displacement mechanisms such as those mentioned above.
The importance of site accessibility is also reflected in the faster initial-uptake behavior by HCl-UiO-66|9/12, which have larger pores (ESI,† Fig. S5) and whose particles are about four times smaller than those of AcOH-UiO-66|11/12 (ESI,† Fig. S6). The larger pores of HCl-UiO-66|9/12, in comparison to AcOH-UiO-66|11/12, can be attributed a combination of higher number of missing linkers and smaller “capping” ligands (HO−, Cl−, or H2O). In the aqueous uptake experiments, these weak-binding ligands can also be more easily displaced by the incoming AsV moieties in contrast to the chelating acetate “capping” ligand for AcOH-UiO-66|11/12. In addition to the increase in accessibility that comes with larger pores, samples with smaller particles should have higher external surface area (ESI,† Table S2) that also enables faster AsV chemisorption. Partially supporting this conjecture is the similar initial uptake rates for all three HCl-UiO-66|x/12 samples (ESI,† Fig S13a and b and Table S3), which have nearly identical average particle sizes (ESI,† Fig S6).
AsV uptake by UiO-67 derivative
The aforementioned data prompted us to hypothesize that enlarging the pore aperture of UiO-66, through the use of a longer linker, would enhance diffusion and facilitate accessibility to binding sites that are deeper inside a MOF crystal, as demonstrated by Li and coworkers45 for the capture of AsV by mesoporous ZIF-8. Thus, we synthesized HCl-UiO-67|9/12, a UiO-66 analogue with the longer biphenyl-4,4′-dicarboxylate linker and a similar number of missing linkers as our HCl-UiO-66|9/12 sample (based on TGA estimation of missing-linker sites46), again using HCl as a modulator. Fortuitously, this sample has similar particle sizes as HCl-UiO-66|9/12 (∼280 nm vs. ∼250 nm, see ESI,† Fig. S6), allowing us to compare their AsV uptake behaviors without the need to account for the effects of particle size differences.Similar to HCl-UiO-66|9/12, HCl-UiO-67|9/12 displayed a high initial AsV uptake ∼56% (1.7:1 AsV:Zr6) within 30 min of exposure to the AsV testing solution (Fig. 3, dark blue curve). However, after 6 h, the AsV uptake for HCl-UiO-67|9/12 has risen above that of HCl-UiO-66|9/12 (70% binding sites vs. 60%). The uptake continues to rise, albeit at a slow rate, over the next 18 h, presumably due to the gradual diffusion of AsV into the internal binding sites of the HCl-UiO-67|9/12 particles, filling 90% of binding sites (2.7:1 AsV:Zr6). While the uptake clearly has not reached equilibrium at 24 h, this capacity is very close to that of the expected 3 AsV oxyanions per Zr6 node. As in the cases for AcOH-UiO-66|11/12 and HCl-UiO-66|9/12, the PXRD pattern of the AsV-exposed HCl-UiO-67|9/12 does not differ from that the corresponding as-synthesized materials (ESI,† Fig. S3a), which is consistent with a retention of some sample crystallinity. Together, these data clearly indicate that the larger pores in HCl-UiO-67|9/12 (ESI,† Table S2) can definitely facilitate uptake by sites that are deeper inside a MOF nanocrystal (see further discussion below).
It is worth noting that the initial uptake profiles (over the first 30 minutes) of all five of our MOF samples discussed thus far fit well to the pseudo-first-order Lagergren kinetic model while the total uptake profiles (over a 24 h period) fit best to the pseudo-second-order Lagergren kinetic model (ESI,† Fig. S12 and S13). These results are consistent with the adsorption process being governed initially by the chemisorption of AsV to the readily accessible binding sites near the surfaces of the nanocrystals and becoming diffusion-limited over time as AsV anions migrate into the MOF nanoparticles. Among the HCl-UiO-66|x/12 samples, this diffusion-limited behavior becomes most apparent after 3 h, with the sample having the most missing linkers displaying the highest equilibrium capacity. Presumably, the samples with more missing linkers will also have larger pores that facilitate diffusion (see ESI,† Fig. S13 and its caption for further discussion).
As mentioned earlier, phosphonates,6,7 and vanadates,36 which have similar structures to arsenates, have been reported to bind strongly to the missing-linker sites on the Zr6(O)4(OH)4 node of UiO-type MOFs through Zr–O–M motifs (M = V, P). As such, we also expect arsenates to displace any weakly bound monotopic “capping” ligand (acetates, chlorides, and/or water) at the missing-linker sites (Fig. 2, bottom section) in our HCl-UiO-type MOF crystals and form strong Zr–O–M bonds through the so-called anion-exchange mechanism.39,42 Analyses of the diffuse-reflectance infrared Fourier-transformed spectroscopy (DRIFTS) data revealed a broad peak (800–900 cm−1) indicative of the adsorbed AsV. The blue-shifted shoulder peak at 898 cm−1 could be due to formation of As–OZr stretch as observed for zirconium arsenate crystals.47 The As3d XPS spectrum of AsV-treated HCl-UiO-67|9/12 reflected a ∼0.5 eV blue shift in binding energy in comparison to that for powder Na2HAsO4·7H2O (Fig. 5a), in agreement with formation of As–O–Zr species41,47,48 (see ESI,† Fig. S13–S16 and their captions for further discussion). Interestingly, there is a new peak (3654 cm−1) in the bridging hydroxide region of the DRIFTS spectra for the AsV-treated MOF samples (Fig. 5a and ESI,† Fig. S14b), which we attribute to a combination of As(OH) and AsO⋯H⋯OZr species, similar to the PO⋯H⋯OZr species reported by Deria et al.26 Together, these data leads us to believe that the arsenate oxyanions are coordinated to the Zr6(O)4(OH)4 node (possible binding motifs shown in Fig. 2 and ESI,† Fig. S14).
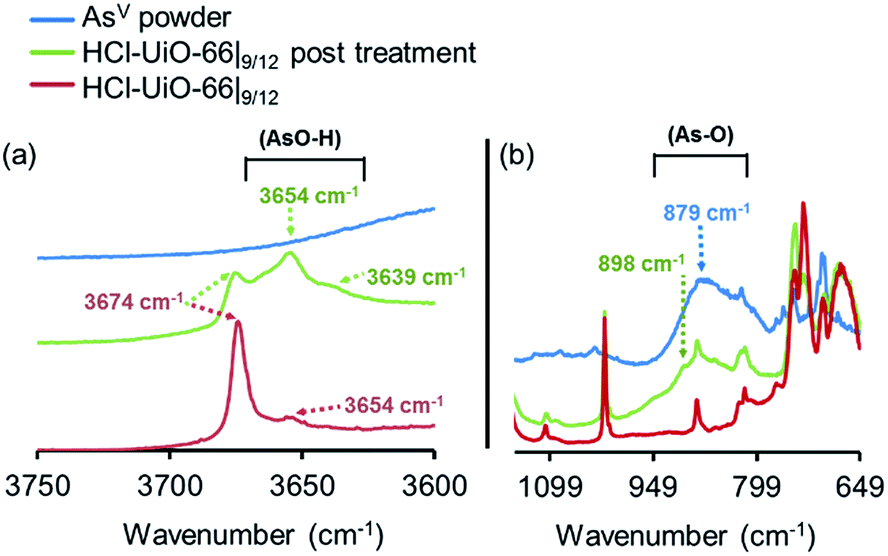 | ||
| Fig. 5 DRIFTS spectra of HCl-UiO-66|9/12 sample before and after AsV treatment, showing the presence of –As–O–H bonds after exposure. Spectra for powder Na2HAsO4·7H2O are included as reference. Data for AcOH-UiO-66|11/12 can be found in the ESI,† Fig. S14. | ||
AsIII and AsV uptake by thiolated UiO-66 derivatives
To demonstrate that neutral AsIII species can also be captured by the UiO-66 platform using thiolated ligand sites, we prepared UiO-66(SH)2 using both AcOH and HCl modulators (see ESI,† Section S2 for experimental details), which afforded thiolated MOFs with the same amount of defects, Zr6O4(OH)4(–CO2)10.5/12 (see ESI,† Section S5). When exposed to aqueous solutions of AsIII,48 both AcOH-UiO-66(SH)2 and HCl-UiO-66(SH)2 showed much higher uptake compared to the non-thiolated controls (Fig. 6a), confirming our hypothesis that thiol can be used to chemoselectively capture AsIII. Further supporting the importance of the thiol functionalities is the observation that HCl-UiO-66(OH)2, the hydroxylated analogue of HCl-UiO-66(SH)2, does not uptake any significant amount of AsIII under the same condition (Fig. 6a). While HCl-UiO-66(SH)2, with higher total pore volume and higher micropore surface area (see ESI,† Table S2), is expected to have a higher AsIII capacity, the uptake amount (40 mg g−1, 1:1 AsIII:Zr6) is twice that obtained for AcOH-UiO-66(SH)2. We attribute this to the non-negligible size of the acetate “capping” ligand in the latter, which can significantly narrow the surface apertures of the MOF crystals and reduce the amount of uptake by subsurface sites. As mentioned above, the chelating nature of the acetate ligand made it less likely to be displaced by HO− or H2O ligand during the uptake experiments, making this mechanism of pore-aperture-narrowing more significant for the “acetate-capped” materials. Notably, sequential exposure of both HCl-UiO-66(SH)2 and HCl-UiO-66|9/12 to AsV (Fig. 6b) and then to AsIII (Fig. 6c), showed a stark contrast of the two materials: the latter only bind AsV while the former can still bind a notable amount of AsIII (10 mg g−1 at 6 h, 0.26:1 AsIII:Zr6) after significant AsV uptake (40 mg g−1 at 6 h, 1:1 AsV:Zr6). This 1:1 AsV:Zr6 stoichiometry corresponds to 67% of the defect sites on HCl-UiO-66(SH)2 and is comparable to that of AcOH-UiO-66|11/12 (cf.Fig. 6b and 3). Consistent with its low level of defects, exposing HCl-UiO-66(SH)2 to AsV solution (30 mL of 50 ppm AsV) for 24 h does not appear to degrade it (<1% BDC-SH linker seen in solution via ICP-OES sulfur analysis; see ESI,† Section S3).
 | ||
| Fig. 6 (a) AsIII uptake profiles49 by thiolated UiO-66 samples and non-functionalized analogues confirming the important role of the soft thiol ligands in capturing AsIII. While the AcOH-UiO-66|11/12 and HCl-UiO-66|9/12 controls show some initial uptake of AsIII, this appears to be semi-reversible, not unexpectedly if we consider the weaker binding nature of the soft AsIII ion to the hard missing-linker sites of the nodes, especially under slightly acidic conditions. (b and c) In sequential exposures to AsV (b) and AsIII (c), HCl-UiO-66(SH)2 shows good uptakes for both AsV and AsIII compared to HCl-UiO-66|9/12, which only binds AsV. See ESI† for data concerning the reverse exposure order. Experimental conditions: batch exposure of a sample of MOF (10 mg) to the appropriate As-containing solution (30 mL; 50 ppm initial concentration). | ||
Interestingly, sequential exposure of HCl-UiO-66(SH)2 to AsIII and then to AsV solutions (ESI,† Fig. S9) shows a reverse ordering of uptake capacities (40 mg g−1 at 6 h of the first exposure plus 10 mg g−1 of AsV at 6 h of the second exposure). These data suggest that while HCl-UiO-66(SH)2 has capability for binding both AsIII and AsV, the total capacity may again be limited by sterics. Full uptake of the first species, regardless of the oxidation state, invariably results in a narrowed pore that lowers the accessibility of the binding sites deeper inside the nanocrystals. Such a case would result in lower uptakes of the second species than available binding sites.
The reversibility of AsIII and AsV binding
To identify some possible post-adsorption regeneration strategies for these promising MOF sorbents, we examined the reversibility of As binding in these materials (see additional discussion in ESI,† Section S9). Given that [AsIII(OH)3]n species are known to bind to sulfur-containing enzymes and thiol-rich chelators through the exchange of the hydroxyl group with RSH moieties, we hypothesized that the (BDC-S)xAs(OH)3−x species present in AsIII-treated HCl-UiO-66(SH)2 is best decomposed into soluble AsIII moieties and intact MOF via competitive ligand exchange with excess soluble thiols (eqn (1)). Indeed, about 30% of AsIII can be removed from AsIII-treated UiO-66(SH)2 within 3 h when treated with an ∼0.5 M solution of thiophenols (∼90 equiv in excess) at 50 °C under stirring. No As was removed without stirring or heating, consistent with a thermodynamically driven equilibrium that is limited by slow diffusion through the crystal. Also consistent with this hypothesis are the observations that treatments with a smaller excess of thiophenols and the use of less acidic alkylthiols did not work as well (ESI,† Table S5). Notably, ZrIV ions and BDC-(SH)2 linkers were not observed in the treatment solution, suggesting that HCl-UiO-66(SH)2 is completely stable under these conditions. The stability of this MOF to regeneration is highly beneficial as it can be reused in applications that aim to remove AsIII, which is the more toxic and prevalent form of arsenic in anaerobic ground water streams.11| RSH (xs) + BDC-S-AsIII ⇌ RS-AsIII + BDC-SH | (1) |
The desorption of AsV from AsV-treated AcOH-UiO-66|11/12 and HCl-UiO-66|9/12 is slightly more problematic given their strong chelation to the nodes (Fig. 1), which can only be disrupted by treatment with either a strong acid or base (eqn (2a) and (b)); however such treatments may also facilitate some decomposition of the MOF.50 Indeed, while subjecting AsV-treated HCl-UiO-66|9/12 to either a 3.3 M HCl (∼16000 equiv per node) or a 3.3 M NaOH (>1000 equiv per node) solution led to the desorption of a significant amount of AsV, a small amount of ZrIV ions was also released (ESI,† Table S4). From the limited set of data that we obtained to date, we are optimistic that this loss of ZrIV ions can be minimized with proper optimization of exposure time and acid (or base) concentrations.
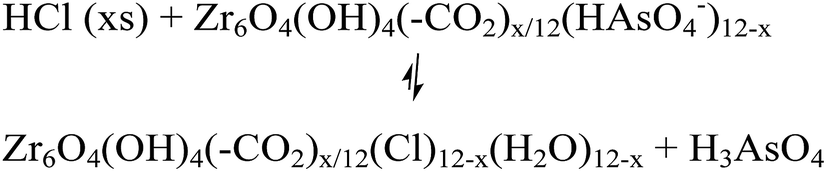 | (2a) |
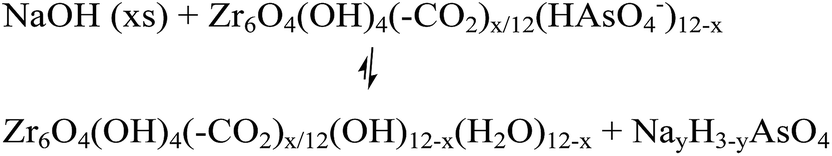 | (2b) |
We note that exposing AsV-treated HCl-UiO-66|9/12 to solutions with pH 2, 7, or 12 did not lead to any noticeable As release until pH 12 (ESI,† Fig. S22). This observation is in agreement with a previous report by Wang et al. where UiO-66 exhibits the lowest AsV uptake at pH 11 in the 2–11 pH range that was studied.39 Interestingly, exposing AsV-treated AcOH-UiO-66|11/12 to the same series of pH solutions led to a more noticeable desorption of AsV that increases proportionally with the basicity of the solution (ESI,† Fig. S22). This process may be attributed to the removal of the weakly bound AsV that originally adsorbed onto AcOH-UiO-66|11/12 through secondary binding mechanisms (see discussion above).
Conclusions
In summary, we have successfully demonstrated the respective use of the nodes and linkers in a series of UiO MOFs to chemoselectively capture anionic AsV and neutral AsIII. Missing-linker sites on the Zr6(O)4(OH)4 nodes are excellent binders for AsV oxyanions, thiolated linkers can selectively coordinate AsIII, and both of these recognition motifs can be incorporated into the same framework for dual-capture purposes. Our results also suggest that the full capacity of this dual-binding feature is best realized when the binding sites internal to the MOF crystals are made easily accessible, either by enlarging the pore aperture size or by reducing the particle sizes. Notably, the binding of both AsV and AsIII appear to be reversible with the proper treatments, suggesting that the dual-capture strategy can be incorporated into the design of regenerable/reusable adsorbents capable of efficiently capturing multiple pollutants or toxic agents that exist as diverse species in aqueous environments.Acknowledgements
S. T. N., J. T. H., and O. K.F. acknowledge financial support from DTRA (HDTRA1-14-1-0014). C.O.A is an NSF Graduate Research Fellow (Grant No. DGE-1324585). Experimental facilities at the Integrated Molecular Structure Education and Research Center (IMSERC) and the Northwestern University Atomic- and Nanoscale Characterization Experimental Center (EPIC, Keck-II) at Northwestern University (NU) were purchased with grants from NSF-NSEC, NSF-MRSEC, the Keck Foundation, the state of Illinois, and NU. ICP-OES analyses were carried out at either IMSERC or the Quantitative Bio-element imaging center (QBIC) at NU. This work also made use of the J.B. Cohen X-Ray Diffraction Facility supported by the MRSEC program of the National Science Foundation (DMR-1121262) at the Materials Research Center of Northwestern University. We thank Dr Sanjiban Chakraborty for his help with the HF digestion and Prof. Justin Notestein for the use of the TGA equipment.Notes and references
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Soc. Rev., 2014, 43, 5933–5951 RSC.
- P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- J. B. DeCoste, T. J. Demasky, M. J. Katz, O. K. Farha and J. T. Hupp, New J. Chem., 2015, 39, 2396–2399 RSC.
- X. Zhu, B. Li, J. Yang, Y. Li, W. Zhao, J. Shi and J. Gu, ACS Appl. Mater. Interfaces, 2015, 7, 223–231 CAS.
- X. Zhu, J. Gu, Y. Wang, B. Li, Y. Li, W. Zhao and J. Shi, Chem. Commun., 2014, 50, 8779–8782 RSC.
- M. Carboni, C. W. Abney, S. Liu and W. Lin, Chem. Sci., 2013, 4, 2396–2402 RSC.
- K. K. Yee, N. Reimer, J. Liu, S. Y. Cheng, S. M. Yiu, J. Weber, N. Stock and Z. Xu, J. Am. Chem. Soc., 2013, 135, 7795–7798 CrossRef CAS PubMed.
- B.-C. Luo, L.-Y. Yuan, Z.-F. Chai, W.-Q. Shi and Q. Tang, J. Radioanal. Nucl. Chem., 2015, 307, 269–276 CrossRef.
- W. R. Cullen and K. J. Reimer, Chem. Rev., 1989, 89, 713–764 CrossRef CAS.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- J. M. Taylor, K. W. Dawson and G. K. H. Shimizu, J. Am. Chem. Soc., 2013, 135, 1193–1196 CrossRef CAS PubMed.
- H. Furukawa, F. Gandara, Y. B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- J. J. Low, A. I. Benin, P. Jakubczak, J. F. Abrahamian, S. A. Faheem and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 15834–15842 CrossRef CAS PubMed.
- J. E. Mondloch, M. J. Katz, N. Planas, D. Semrouni, L. Gagliardi, J. T. Hupp and O. K. Farha, Chem. Commun., 2014, 50, 8944–8946 RSC.
- M. J. Katz, Z. J. Brown, Y. J. Colon, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- J. B. DeCoste, G. W. Peterson, B. J. Schindler, K. L. Killops, M. A. Browe and J. J. Mahle, J. Mater. Chem. A, 2013, 1, 11922–11932 CAS.
- R. Ameloot, M. Aubrey, B. M. Wiers, A. P. Gomora-Figueroa, S. N. Patel, N. P. Balsara and J. R. Long, Chem.–Eur. J., 2013, 19, 5533–5536 CrossRef CAS PubMed.
- Dissociation Constants of Inorganic Acids and Bases, in CRC Handbook of Chemistry and Physics [On line], ed. W. M. Haynes, CRC Press/Taylor and Francis, Boca Raton, FL, USA, 96th edition, Internet Version 2016, section 5 Search PubMed.
- G. K. Shimizu, R. Vaidhyanathan and J. M. Taylor, Chem. Soc. Rev., 2009, 38, 1430–1449 RSC.
- R. Vivani, G. Alberti, F. Costantino and M. Nocchetti, Microporous Mesoporous Mater., 2008, 107, 58–70 CrossRef CAS.
- P. Deria, W. Bury, I. Hod, C.-W. Kung, O. Karagiaridi, J. T. Hupp and O. K. Farha, Inorg. Chem., 2015, 54, 2185–2192 CrossRef CAS PubMed.
- K.-Y. A. Lin, S.-Y. Chen and A. P. Jochems, Mater. Chem. Phys., 2015, 160, 168–176 CrossRef CAS.
- T. Watanabe and S. Hirano, Arch. Toxicol., 2013, 87, 969–979 CrossRef CAS PubMed.
- A. M. Spuches, H. G. Kruszyna, A. M. Rich and D. E. Wilcox, Inorg. Chem., 2005, 44, 2964–2972 CrossRef CAS PubMed.
- D. Ramadan, P. C. Rancy, R. P. Nagarkar, J. P. Schneider and C. Thorpe, Biochemistry, 2009, 48, 424–432 CrossRef CAS PubMed.
- H. V. Aposhian and M. M. Aposhian, Chem. Res. Toxicol., 2006, 19, 1–15 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- M. Kim, J. F. Cahill, Y. Su, K. A. Prather and S. M. Cohen, Chem. Sci., 2012, 3, 126–130 RSC.
- O. Karagiaridi, W. Bury, J. E. Mondloch, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2014, 53, 4530–4540 CrossRef CAS PubMed.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem.–Eur. J., 2011, 17, 6643–6651 CrossRef CAS PubMed.
- H. G. T. Nguyen, N. M. Schweitzer, C. Y. Chang, T. L. Drake, M. C. So, P. C. Stair, O. K. Farha, J. T. Hupp and S. T. Nguyen, ACS Catal., 2014, 4, 2496–2500 CrossRef CAS.
- While Wang and coworkers (ref. 39) have found the uptake of aqueous AsV by UiO-66 to be best at pH = 2, neutral pH was selected for our study to simulate ground-water environment.
- This amount of solution was chosen to give an experimental composition of 3.2 AsV per hexazirconium node in HCl-UiO-66ǀ9/12, the most defective materials in our study. This ratio corresponds to a maximum replacement of the three missing carboxylate sites per node; the slight excess of As beyond 3 is intended to facilitate the sampling and analysis for mass balance in the supernatant in case of complete uptake. However, as described later on in the manuscript, this situation is not expect to ever occur in our study. See ESI,† Section S11 for the detailed calcualtions.
- C. Wang, X. Liu, J. P. Chen and K. Li, Sci. Rep., 2015, 5, 16613/1–10 Search PubMed.
- T. M. Suzuki, J. O. Bomani, H. Matsunaga and T. Yokoyama, React. Funct. Polym., 2000, 43, 165–172 CrossRef CAS.
- A. Bortun, M. Bortun, J. Pardini, S. A. Khainakov and J. R. García, Mater. Res. Bull., 2010, 45, 142–148 CrossRef CAS.
- A. J. Howarth, M. J. Katz, T. C. Wang, A. E. Platero-Prats, K. W. Chapman, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 7488–7494 CrossRef CAS PubMed.
- We speculate that the uptake kinetics by the HCl-UiO-66 sample could be strongly influenced by the narrowing of the pore aperture as arsenates are being uptaken (see illustration in the ESI,† Fig. S21b; as well as ref. 6 for a similar discussion).
- A. Gomez-Caminero, P. Howe, M. Hughes, E. Kenyon, D. R. Lewis, M. Moore, J. Ng, A. Aitio and G. Becking, Arsenic and Arsenic compounds [Online], International Programme on Chemical Safety, WHO, Geneva, 2nd edn, 2001, Section 3.1, http://www.inchem.org/documents/ehc/ehc/ehc224.htm#3.1 (accessed May 27, 2016) Search PubMed.
- Y. N. Wu, M. Zhou, B. Zhang, B. Wu, J. Li, J. Qiao, X. Guan and F. Li, Nanoscale, 2014, 6, 1105–1112 RSC.
- See ESI† for data and discussion. In short, TGA data was inconclusive for UiO-67 as it is uncertain where the loss of linkers starts.
- D. M. Poojary, A. I. Bortun, L. N. Bortun, C. Trobajo, J. R. García and A. Clearfield, Microporous Mesoporous Mater., 1998, 20, 77–85 CrossRef CAS.
- A. I. Bortun, A. Clearfield, M. Suárez, R. Llavona and J. Rodríguez, Mater. Chem. Phys., 1998, 55, 152–154 CrossRef CAS.
- AsIII adsorption was carried out at pH 5 to facilitate the conversion of two As-OH moieties on arsenite into stable chelating As-SR bonds as shown in the reaction of thiol-rich proteins with Lewisite, L. A. Stocken and R. H. S. Thompson, Biochem. J., 1946, 40, 529–535 CrossRef CAS PubMed.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018/1–15 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Includes synthesis and preparation of UiO-66 and analogues along with characterization data (PXRD, TGA analysis, N2 isotherms, DRIFTS, XPS analysis), Lagergren kinetic data fittings, leaching studies, recycling studies, and other relevant observations. See DOI: 10.1039/c6sc00490c |
| This journal is © The Royal Society of Chemistry 2016 |