
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of intracellular algal organic matter and nitrate on disinfection byproduct formation in chlorinated water after UV/H2O2 and UV/Cl2 advanced oxidation processes†
Fateme
Barancheshme
and
Olya S.
Keen
 *
*
University of North Carolina at Charlotte, Charlotte, NC 28223, USA. E-mail: okeen@charlotte.edu; Tel: +1 704 687 5048
First published on 19th December 2024
Abstract
Advanced oxidation processes (AOPs) are one of the highly effective alternatives for treatment of algal toxins in drinking water. Water that contains algal toxins commonly has organic matter of algal origin and elevated nitrate. Organic matter undergoes transformations during advanced oxidation processes and may change in a way that increases disinfection byproduct (DBP) formation when water is chlorinated post-AOP. Nitrate forms reactive nitrogen species under certain UV wavelengths that can also interact with organic matter and change its properties in a way that increases post-AOP DBP formation. Two types of advanced oxidation processes (UV/H2O2 and UV/Cl2) were compared in their ability to change the formation potential of regulated DBPs [four trihalomethanes (THMs) and nine haloacetic acids (HAAs)] and an unregulated nitrogenous DBP (N-DBP) N-nitrosodimethylamine (NDMA) due to the interaction of the process with algal organic matter (AOM) and nitrate in the water. The two AOPs showed no significant differences in post-treatment DBP formation under any of the tested conditions. Higher levels of treatment with both processes led to slightly higher formation potential of some THMs. AOM made a poor precursor for additional THMs and three HAAs (six not consistently detected), but had a higher NDMA yield than background organic matter (0.59 ng mg−1-C vs. 0.18 ng mg−1-C, p = 0.038). Nitrate suppressed chlorinated THMs and favored increased concentrations of brominated THMs and HAAs, resulting in higher percent incorporation of background bromide into DBPs. Moreover, nitrate addition (20 mg-N L−1 of added nitrate compared to the background level of 0.47 mg-N L−1) led to 11 times higher NDMA formation. Formation of N-DBPs during post-AOP chlorination in the presence of AOM and nitrate warrants additional investigation.
Water impactWith increasing frequency of algal blooms, more water treatment plants are looking for solutions to algal toxins that the blooms may produce, and often UV/H2O2 and UV/Cl2 advanced oxidation processes (AOP) are considered. Source water would be expected to have high nitrate and algal organic matter in such scenarios, and it is important to understand how these treatment processes and water quality parameters would work in tandem to affect the formation of disinfection byproducts (DBPs), both regulated and unregulated. Our study reports some fascinating new insights. We found that elevated bromide in the source water used for the experiments combined with nitrate photochemical reactions favor formation of brominated DBPs over chlorinated DBPs. This important effect of nitrate photochemistry has not been reported before for regulated DBPs, although it has been noted for some other compounds, as discussed in accompanying text. However, it is important information for drinking water treatment plants that may consider this treatment train, because brominated DBPs often lead to permit exceedances. Apart from the practical implications, this finding prompts further investigation into the interaction between nitrate photochemistry and brominated DBP formation. Furthermore, we confirmed that the presence of nitrate and algal organic matter dramatically affects formation of NDMA and found that AOP pretreatment does not destroy NDMA precursors. Additionally, this study demonstrated for the first time that there is no difference in the choice of UV/H2O2 or UV/Cl2 with respect to regulated DBP formation in the presence of nitrate and algal organic matter, and that source water quality is a more important consideration than the type of AOP. |
1. Introduction
Toxic algal blooms are becoming more common globally and present a major threat to drinking water safety.1–3 They are typically associated with a high nutrient content in source water. In particular, nitrogen in the form of nitrate has been shown to be the most essential nutrient for toxic algae, with phosphorus being less impactful. In fact, studies show that nitrate is not only a predictor of algal blooms, but also of toxin production during the bloom.4–6 A typical drinking water treatment train consisting of coagulation, flocculation, sedimentation and filtration has been shown to remove algal cells effectively, while leaving behind extracellular toxins7,8 and nitrate. Intracellular algal organic matter (AOM) can be considerable in water that contains algae, especially at the later stages of treatment processes, due to the disruption and lysis of the algal cells in treatment processes such as pre-oxidation and filtration.8–14 AOM released in this process can be a considerable precursor to the formation of both carbonaceous disinfection byproducts (C-DBPs) and nitrogenous disinfection byproducts (N-DBPs) due to high content of protein-like structures.15–17Advanced oxidation processes (AOPs) are among the most effective options for treating drinking water contaminated with algal toxins.18 A typical full-scale AOP for drinking water treatment involves ultraviolet (UV) irradiation of water containing hydrogen peroxide (H2O2). Lately, replacing H2O2 with aqueous chlorine (Cl2) has been gaining popularity due to its ease of implementation and operation. Additionally, some algal toxins are highly reactive with Cl2 giving UV/Cl2 an advantage.19 However, the formation of disinfection byproducts (DBPs) must be considered in subsequent chlorine disinfection of AOP-treated water. Studies also show that dissolved organic matter (DOM) in general and AOM in particular can transform during AOP which can result in a higher formation potential of regulated and unregulated DBPs when water is chlorinated after AOP treatment.20,21 This is of particular concern in the UV/Cl2 process because of the higher doses of chlorine involved and the potential contribution from the reaction between DOM and Cl˙ and other reactive chlorine species (RCS). The concern of additional DBP formation potential with UV/Cl2 compared to UV/H2O2 has emerged with increased popularity of UV/Cl2, in particular for algal toxin treatment,19,22,23 but very few studies have been done to compare the two processes side-by-side.24
When it comes to the application of AOPs for treatment of algal toxins, other relevant water quality considerations, namely nitrate and AOM, need to be considered for their impact on DBP formation potential. Certain sources of UV used in UV-based AOPs, specifically those emitting wavelengths <240 nm, e.g., medium pressure mercury vapor lamps, can photochemically activate nitrate, resulting in formation of HO˙, NO2˙ and NO˙ radicals, as well as other minor nitrogen radical species.25,26 These radicals can potentially participate in reactions with organic matter and form nitrosamines by providing a reactive nitroso group. The importance of photochemical reactions of nitrate for DBP formation was previously demonstrated with chloropicrin formation in water chlorinated post-UV in nitrate-containing water27 and for other N-DBPs, e.g., halonitromethanes and haloacetonitriles.28 Studies indicate that the presence of AOM in water can lead to increased formation of trihalomethanes (THMs) and haloacetic acids (HAAs).29,30 Formation of nitrogen-containing DBPs, such as haloacetonitriles, has been documented as well in the presence of AOM.31 Intracellular AOM was also shown to be a strong precursor to nitrosamine formation during chloramination of drinking water in another study.32
The objective of this study was to determine how UV/H2O2 and UV/Cl2 treatments affect the yield of THMs, HAAs and N-nitrosodimethylamine (NDMA) when the water containing AOM and nitrate is disinfected with chlorine after AOP. A review of six years of data by the USEPA found that NDMA was the most prevalent nitrosamine in drinking water samples, with all other nitrosamines either nondetectable or present at much lower levels.33 Thus, NDMA was selected to represent nitrosamines for this study. Generally, NDMA forms in reactions between chloramines and organic matter, rather than in chlorination reactions. However, the reaction between nitrite and secondary amines is another major formation pathway, as well as a reaction between free chlorine and dimethylamine.34,35 Thus, it was anticipated that nitrate photochemistry (with nitrite, nitrite radicals and nitroso radicals as major products25) and reactions involving background DOM and especially amine-rich AOM would provide potential pathways for NDMA formation.
While previous work studied formation of these DBPs from AOM (e.g., Li and Rao;17 Li and Gao;36 Wert and Rosario-Ortiz37 and others), this study focused on AOM transformed by two of the most commonly used AOPs, UV/H2O2 and UV/Cl2, with UV/Cl2 adding RCS to the process chemistry. Additionally, using source water high in bromide (0.105 mg L−1 due to presence of coal-burning power plants in the watershed) and comparing the results with and without additional nitrate allowed evaluating the combined effect of the water matrix and AOP pre-treatment on changes in bromide incorporation (assimilation of Br− into organic brominated DBPs38), which has toxicity and regulatory compliance implications.
2. Materials and methods
The following methods were adapted in part from Barancheshme.392.1. Sample collection and water matrix
The water for use as a background matrix was collected from a local drinking water treatment plant treating surface water from the Catawba River. The treatment train at the plant consisted of coagulation, sedimentation, filtration, and chlorine disinfection, and the water sample was collected prior to disinfection. The plant uses no powdered activated carbon or pre-oxidation. Water volume sufficient for the entire experiment was collected in December when no background algal blooms were present. Water samples were filtered through a 0.45 μm mixed cellulose ester filter within hours of collection. The filter was selected because it had very low extractables, which was confirmed experimentally by running ultrapure water (18.2 MΩ cm resistivity) through the filter and measuring total organic carbon (TOC) and total nitrogen (TN) content before and after filtration (Table S1 in ESI†). Despite having low extractables, the filters were prewashed with ultrapure water. The filtered water sample was stored at 4 °C for future use. Relevant water quality parameters for the sample can be found in Table 1. Nitrate (NO3-N), nitrite (NO2-N), ammonia (NH3-N) and TN were measured using Hach Test-in-Tube kits (TNT835, 839, 830 and 10071, respectively) and Hach DR6000 spectrophotometer. TOC was measured with a Shimadzu TOC-LCPN instrument using a high temperature combustion method (Standard Method 5310-B). Total organic nitrogen (TON) was calculated as TN minus nitrate, nitrite and ammonia. Total Kjeldahl nitrogen (TKN) was calculated as TON plus ammonia. Bromide (Br−) was measured using a Dionex ICS-3000 ion chromatography system with a Dionex IonPAc AS22 4 × 250 mm capillary column and a Dionex IonPAc AG22 4 × 50 mm guard column (Thermo Scientific, Waltham, MA). The eluent consisted of 1.7 mM sodium bicarbonate and 1.8 mM carbonate in ultrapure water. Ultrapure water was also used as a method blank. Standards were prepared from a bromide standard solution (Sigma-Aldrich).| Parameter (mg L−1) | NO3−-N | NO2-N | NH3-N | TN | TON | TKN | TOC | Br− |
|---|---|---|---|---|---|---|---|---|
| Background | 0.473 | 0.013 | ND | 0.600 | 0.114 | 0.114 | 0.633 | 0.105 |
| Algal organic matter | 0.328 | 0.024 | 0.13 | 14.00 | 13.52 | 13.65 | 33.63 | NA |
Nitrate (as NaNO3, Sigma Aldrich) or AOM (extraction described in the next section) was added to this matrix as needed.
2.2. AOM extraction
AOM was generated in the laboratory from non-toxin-producing Microcystis sp. algae (Carolina Biological Supply, Item No. 151840, Burlington, NC) using protocols available in the literature.31 Briefly, Microcystis sp. was cultivated at 22 °C under a fluorescent lamp with light/dark cycle of 12 h/12 h until reaching the stationary growth phase in 125 mL flasks containing 100 mL of Gibco BG11 media optimized for cyanobacteria (Thermo Fisher Scientific, Waltham, MA). New cultures were set up by transferring 5 mL of a stock culture into 100 mL of fresh medium under fluorescent light for ten days to allow the alga to grow. Algal mass was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min and the cells separated during the centrifugation were washed and re-suspended with 100 mL of ultrapure water three times. Next, the cells were subjected to three freeze/thaw cycles at −80 °C and 37 °C to lyse the cells. Finally, the solution was filtered through prewashed 0.45 μm cellulose acetate membranes. The organic matter in the filtrate was intracellular AOM which was stored in the dark at −20 °C for long term storage and 4 °C for short term storage.
000 rpm for 10 min and the cells separated during the centrifugation were washed and re-suspended with 100 mL of ultrapure water three times. Next, the cells were subjected to three freeze/thaw cycles at −80 °C and 37 °C to lyse the cells. Finally, the solution was filtered through prewashed 0.45 μm cellulose acetate membranes. The organic matter in the filtrate was intracellular AOM which was stored in the dark at −20 °C for long term storage and 4 °C for short term storage.
The intracellular organic matter was characterized by measuring TOC, NO3-N, NO2-N, NH3-N and TN, and calculating TON and TKN using the same methods as described for background water characterization. Table 1 lists the characteristics of AOM.
2.3. AOP experiments
H2O2 (30% w/w) and Cl2 as a 10–15% solution of sodium hypochlorite were purchased from Sigma-Aldrich (St. Louis, MO). The concentration of H2O2 was measured using the triiodide method40 for which ammonium molybdate, potassium iodide, and potassium hydrogen phthalate were obtained from Sigma-Aldrich (St. Louis, MO). Cl2 was measured using N,N-diethyl-p-phenylenediamine (DPD) Hach powder pillows. Both Cl2 and H2O2 methods are spectrophotometric, and absorbance was measured using a Hach DR6000 UV/Vis spectrophotometer.AOP experiments were conducted in a bench-scale quasi-collimated beam apparatus equipped with a 1 kW medium pressure mercury vapor lamp (Ace Glass, Vineland, NJ) emitting a polychromatic UV spectrum in the 200–400 nm range. The UV apparatus setup is similar to the schematic shown in Bolton and Linden41 and was additionally equipped with a fan for cooling and ventilation of ozone. AOP experiments were performed at the ambient pH of the water matrix (6.8) using UV doses of 500, 1000 and 2000 mJ cm−2, which captures the typical range of full-scale UV doses in AOPs used for drinking water treatment. The time to achieve the highest UV dose was <17 min and is comparable to the typical residence time in a full-scale AOP reactor which is on the order of a few minutes. Because the timescale of chlorine reaction with organic matter during the DBP formation experiment was 24 hours, any additional contact time between chlorine and organic matter during the AOP experiment would be negligible. Changes to the water matrix and parameters such as pH, TOC, alkalinity, etc. are generally negligible within the range of the UV doses applied, and the experimental conditions can be regarded as pseudo-steady-state. The irradiance was measured by a NIST-calibrated International Light IL-1400 radiometer (Peabody, MA), and the UV dose was calculated using the method by Bolton and Linden.41 The spectrum of the medium pressure lamp was measured by an Ocean Optics USB2000+ spectral radiometer (Dunedin, FL). It must be noted that the dose was calculated based on the background water matrix. Samples with additional AOM or nitrate would require slightly longer exposure time to achieve the same UV dose (approximately 19 min vs. 17 min). Thus, the exposure to the radicals generated by H2O2 or Cl2 in samples with additional AOM or nitrate was slightly lower (by <14%), but it was expected to have minimal impact on the outcome of the experiments. This was taken into consideration while interpreting the results where appropriate.
Three water matrices were used in the experiments: background, background + 20 mg-N L−1 of NO3−, and background + 3 mg-C/L of AOM. Based on the previous studies,42–44 AOM in natural waters experiencing an algal bloom is between 2 and 9.5 mg L−1 TOC with intracellular organic matter dominating the mix. Nitrate in surface water affected by human activity is typically in the range of 1–10 mg-N L−1.45 The oxidants were added at either 5 or 10 mg L−1 for H2O2 and 2 or 4 mg L−1 for Cl2 which is also within the range of concentration for Cl2 and H2O2 used in full-scale processes (typically, 1–5 mg L−1 and 3–20 mg L−1, respectively46). Each sample was exposed to three UV doses (500, 1000 and 2000 mJ cm−2) to evaluate the effects of increased AOP treatment on DBP yield in subsequent chlorination in each of the three matrices. UV doses selected are also within the range used for full scale AOP which are typically, 400–2000 mJ cm−2.47 These experiments were performed in triplicate and analyzed for THMs, HAAs, and NDMA.
2.4. Post-AOP chlorination
All bottles and glassware were precleaned using standard procedures for DBP analysis. Samples were prepared under uniform formation conditions (UFC)48 to analyze DBPs. Based on the UFC standard operating procedure, each sample was dosed with borate buffer and hypochlorite buffer, and the pH was adjusted to 8.0 ± 0.2. Residual chlorine was 1.0 ± 0.4 mg L−1, and the incubation time was 24 h in the dark at 20.0 ± 1.0 °C.48 A preliminary test was conducted to determine the 24 h chlorine demand of each water matrix and chlorine dose necessary to achieve 1.0 ± 0.4 mg L−1 of residual chlorine after 24 h. Cl2-to-TOC dosage of 2.5:1 (based on the TOC of the water matrix) worked best with the residual chlorine after 24 h of 0.93 and 1.16 mg L−1 for water samples with additional AOM and water samples without added AOM, respectively.For post-AOP samples that include chlorinated DBP analysis, chlorine is the recommended option to quench H2O2.49 UV/H2O2 samples needed an additional stoichiometric chlorine dose of 2.09 mg Cl2/mg H2O2. The reaction between chlorine and H2O2 is almost instantaneous (<30 s required to quench H2O2) compared to the reaction between chlorine and DOM.20,49 Consequently, additional chlorine for quenching H2O2 was consumed within seconds, and no increase in DBP yield was expected in UV/H2O2 samples because of the increased initial chlorine dose required to quench H2O2.49 There is a possibility that this approach can result in slightly underdosing or overdosing the stoichiometric concentration required to quench H2O2. Because the stoichiometric requirement is considerable, an error in H2O2 value of 0.1 mg L−1 would change the initial chlorine concentration by ±0.2 mg L−1. These challenges in precise H2O2 quenching may result in an increased degree of variability in data which may obscure the ability to capture more subtle differences between data sets, in particular when comparing UV/H2O2 and UV/Cl2. It is advisable that future work with DBP formation in post-AOP samples has a larger granularity of Cl2 concentrations during UFC experiments to ascertain that the integrated chlorine exposure is maintained in the narrow range across the experiments, and if not, the results can be normalized to the integrated chlorine exposure, which may allow capturing more subtle differences in DBP formation between the sample sets, if there are any.
2.5. THM and HAA extraction and analysis
Four regulated THMs and nine regulated HAAs were analyzed using the EPA methods 551.1 and 552.3, respectively, both optimized by Liu et al.50 The extraction method can be found in ESI.†Gas chromatography with electron capture detector (GC-ECD, 63Ni) was used for THM and HAA analysis (Shimadzu-QP2010 GC, Shimadzu, Japan) in splitless mode with the injector temperature at 230 °C. Helium and nitrogen were used as the carrier and makeup gases at a flow rate of 1.4 mL min−1 and 30 mL min−1, respectively. THMs were separated on a fused silica DB-1301 capillary column (30 m length, 0.25 mm inner diameter, and 1 μm film thickness with a temperature range between −20 °C and 280–300 °C) (Agilent Technologies, Palo Alto, CA, USA). The oven temperature was held at 35 °C for 15 min, and then increased at 25 °C min−1 to 145 °C where it stayed for an additional 3 min, followed by a 35 °C min−1 ramp to 240 °C, where it was held for another 5 min. The temperature of the detector was held at 260 °C. GC-ECD analysis of HAAs was the same as for THMs except a SH-Rtx-1701 capillary column (30 m length, 0.25 mm inner diameter, and 1 μm film thickness with a temperature range of −20 to 270/280 °C) (Shimadzu, Japan) was used. The oven temperature was held at 40 °C for 10 min, followed by a ramp at 10 °C min−1 to 85 °C, and another ramp at 30 °C min−1 to 205 °C where it was held for another 5 min.
The concentration of THM4 standard solution (Restek, Center County, PA) was 200 μg mL−1 for each THM. The linearity range of the GC-ECD analysis was 0.02–200 μg L−1 for chloroform (CF), 0.002–200 μg L−1 for bromodichloromethane (BDCM) and dibromochloromethane (DBCM), and 0.2–200 μg L−1 for bromoform (BF). Linearity R2 range for the calibration curves was 0.94–0.97. The retention times were 8.4 min for CF, 11.4 min for BDCM, 22.3 min for DBCM and 28.6 min for BF. The lowest linear calibration standard was considered to be the lowest limits of quantification (0.02 μg L−1 for CF, 0.2 μg L−1 for BF and 0.002 μg L−1 for BDCM and DBCM). Additional information on the standard and the calibration curves is available in ESI.†
The HAA9 standard solution was purchased from Restek (Center County, PA). The concentrations of each HAA varied from 200 to 2000 μg L−1 (Table S3 of ESI†). The retention times and lower limits of quantification (lowest linear calibration standard concentration) were 5.1 min and 0.012 μg L−1 for monochloroacetic acid (MCAA), 9.6 min and 0.08 μg L−1 for monobromoacetic acid (MBAA), 16.7 min and 1.2 μg L−1 for dichloroacetic acid (DCAA), 18.1 min and 0.04 μg L−1 for trichloroacetic acid (TCAA), 20.6 min and 8 μg L−1 for bromochloroacetic acid (BCAA), 23.5 min and 4 μg L−1 for dibromoacetic acid (DBAA), 24.9 min and 0.08 μg L−1 for bromodichloroacetic acid (BDCAA), 25.4 min and 0.2 μg L−1 for chlorodibromoacetic acid (CDBAA), and 26.9 min and 0.04 μg L−1 for tribromoacetic acid (TBAA). The retention time for the 1,2-dichloropropane internal standard was 12.6 min. R2 values ranged from 0.88 to 0.998. ESI† shows the details of the standard and the calibration curves of each HAA compound.
2.6. NDMA extraction and analysis
NDMA was measured based on modified EPA method 521 using solid-phase extraction (SPE), followed by liquid chromatography separation and tandem triple quadrupole mass spectrometry (LC-MS/MS) method developed by Zhao et al.51Samples were extracted using Supelclean™ Coconut Charcoal SPE cartridges (bed weight of 2 g and a volume of 6 mL) and a vacuum system (−30 kPa). The cartridges were rinsed with 15 mL each of hexane and dichloromethane, and the residual organic solvents were removed under vacuum. Next, the cartridges were conditioned with 15 mL of methanol and 15 mL of water. Sodium bicarbonate (0.5 g) was added to 250 mL of the water sample to bring the pH to 8. The sample was then spiked with 40 ng L−1 of NDMA-d6 and passed through the SPE cartridge at a flow rate of 3–5 mL min−1. The analyte adsorbed on the SPE cartridge was eluted with 15 mL of dichloromethane, concentrated to 1 mL under vacuum, transferred to an autosampler vial and stored at −20 °C until analysis.51,52 The internal standard of 40 ng L−1 NDPA-d14 was added to the samples before the LC-MS/MS analysis. Ultrapure water was used as a blank and was extracted to ensure all reagents were NDMA-free. NDMA standard was purchased from Sigma-Aldrich (Missouri, US) while NDMA-d6 and NDPA-d14 were purchased from Restek (Centre County, PA). It must be noted that in 2024, the US EPA banned the use of dichloromethane (also known as methylene chloride) in most applications. Other methods may be available in literature that call for different solvents for NDMA extraction.
The Agilent 6400 series triple quadrupole LC–MS/MS system was used. The LC separation was performed using a ZORBAX Eclipse XDB-C8 column (4.6 × 150 mm, 3.5 μm, Agilent Technologies, Palo Alto, CA) with 10 mM ammonium acetate and 0.01% acetic acid in water as mobile phase Solvent A and 100% methanol as mobile phase Solvent B.51 All solvents were HPLC-grade or higher. Injection volume was 100 μL and eluent flow rate was 0.3 mL min−1. The solvent gradient was held at 60% of solvent B for 1 min, which was then ramped to 90% solvent B over 5 min followed by decreasing solvent B back to 60% and a 3 min re-equilibration before injecting the next sample.
The MS/MS method was run in the positive electrospray ionization and multiple-reaction monitoring mode. MassHunter Quantitation software was used for quantification. The peaks of standards and extracted samples were monitored automatically and results were reproducible. MS/MS parameters including collision energy and cell accelerator voltage are shown in Table S4 of ESI.† Gas flow rate was 10 L min−1 at 350 °C. LC-MS/MS analysis was linear in the range of 0.001–1000 μg L−1 for NDMA (Fig. S3 in ESI†).
2.7. T-test analysis of DBP results
Two-sample t-tests were performed in JMP Pro 16 to evaluate the significance of any differences in samples means of DBP yields between the samples. A two-tailed distribution and unequal variances were assumed. A p-value of less than 0.05 (at 95% confidence level) was statistically significant. First, the effects of the UV dose were analyzed. The data for each matrix and each oxidant type and dose were considered separately. At this most granular level, each set consisted of three data points. Because slightly different levels of DBPs formed at all conditions, to capture the effects of the higher UV dose, the data sets were evaluated as yields (μg-DBP/mg-C) as well as the concentrations values at 1000 and 2000 mJ cm−2 normalized to the value at 500 mJ cm−2. Trends of increase or decrease in DBP yields with UV dose were easier to identify in normalized data. If no effect of the UV dose was observed, then the data sets were compared based on the oxidant type and oxidant concentration for all UV doses combined. In the rare instances when the UV dose showed a significant effect, the data sets were evaluated at each UV dose separately. Finally, to test the effects of the matrix change, all samples regardless of UV dose and oxidant dose were combined (UV/H2O2 and UV/Cl2 datasets evaluated separately if the oxidant type showed significant impact in the previous set of tests) and compared to the same set of data in a spiked matrix. For rare instances when the UV dose showed an effect, the data sets were analyzed separately at each UV dose. Pooling together the data from sets that did not show a significant difference in the previous rounds of t-tests allowed for a larger number of sets and increased the strength of statistics and the ability to find significant influences from each of the variables considered.3. Results
Experimental data was analyzed to answer the following questions regarding DBP yields when water is chlorinated after AOP:1. Is there a difference in DBP yields after water is treated with UV/H2O2vs. UV/Cl2 AOP?
2. Does increasing the level of treatment with AOP (500 mJ cm−2vs. 1000 mJ cm−2vs. 2000 mJ cm−2) change the yields of DBPs?
3. Does the addition of nitrate or AOM and their photochemical reactions during AOP change the yields of DBPs?
3.1. Trihalomethanes
Mean yield of CF in the background matrix combining the data for both oxidants and all oxidant doses used was 82% higher when chlorinated after exposure to 2000 mJ cm−2 UV dose than after 500 mJ cm−2 and the difference was statistically significant (p < 0.001). Mean yield in samples chlorinated after 1000 mJ cm−2 was 16% higher than after 500 mJ cm−2 and the difference was not statistically significant (p = 0.47). The results were particularly pronounced after water was pretreated with higher oxidant doses (4 mg L−1 of Cl2 and 10 mg L−1 of H2O2) where CF yield at 1000 mJ cm−2 was 56% higher than at 500 mJ cm−2, and at 2000 mJ cm−2 it was 245% higher, with p-values of 0.059 and <0.0001, respectively. The same trends were observed for BDCM with approximately the same formation in the background matrix after exposure to 500 and 1000 mJ cm−2 but 33% higher after 2000 mJ cm−2 (p < 0.001). Similarly, when looking at higher doses of both oxidants, the increase was more pronounced at 57% (p = 0.011). These results are shown in Fig. 1 and suggest that higher exposure of organic matter to hydroxyl radicals leads to higher yield of some THMs when samples are subsequently chlorinated, which is consistent with what was found in previous studies.20 Similar trends for increase in concentration with increasing oxidation of organic matter by hydroxyl radicals were not observed for CDBM or BF, suggesting that brominated THMs are less affected than chlorinated THMs by these oxidative changes in organic matter. These trends were lost in the matrix spiked with AOM with the exception of CF in the UV/H2O2 treated samples where a statistically significant increase of 14% was observed after 2000 mJ cm−2 compared to 500 mJ cm−2 (p = 0.022). AOM was added at 3 mg-C L−1 compared to 0.6 mg-C L−1 of the background DOM and was a predominant source of carbon in spiked samples, so the loss of the trend for increased chlorinated THM formation with increased oxidation in AOM-spiked samples indicates that oxidation does not affect THM formation from AOM. It is most likely due to the more protein-like nitrogen-rich structure of AOM compared to the more humic-like structure of typical surface water DOM,16,36 which affects the potential of AOM to serve as a precursor for each DBP.53 The primary mechanism of THM formation is believed to be the result of electrophilic substitution of aromatic rings within the bulk organic matter, which then further break down into smaller molecules, some of which form THMs.54 Phenols and aromatic amines were shown to be particularly effective precursors.55 While AOM is rich in amino groups, it is deficient in aromatic rings compared to typical DOM of terrestrial origin present in surface water. Additionally, hydrophobic fraction of DOM is primarily responsible for THM formation,56 but it only accounts for a very small fraction of intracellular AOM.36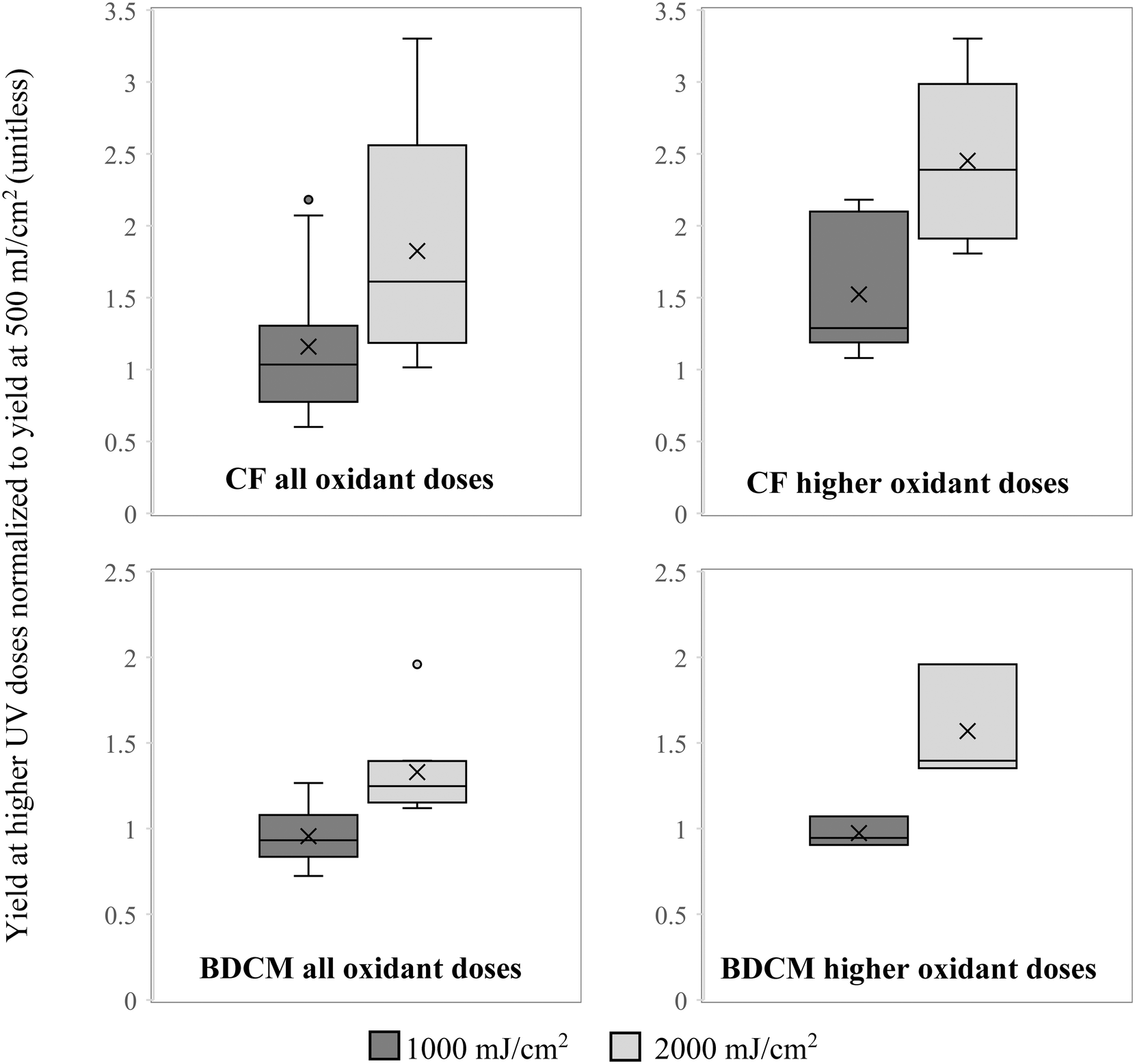 | ||
| Fig. 1 Yield of chloroform (CF) and bromodichloromethane (BDCM) in background matrix when chlorinated after 1000 mJ cm−2 normalized to 500 mJ cm−2 (dark gray) and at 2000 mJ cm−2 normalized to 500 mJ cm−2 (light gray). Figures on the left combine the results of both UV/H2O2 and UV/Cl2 at all oxidant concentrations, and figures on the right show results with higher oxidant doses. | ||
Because differences were observed in CF and BDCM yield after chlorination post-exposures to higher doses of UV, formation of these two THMs in different matrices was analyzed separately at 2000 mJ cm−2 and together for the samples exposed to 500 and 1000 mJ cm−2. For BF and CDBM the results from all UV doses were combined for analysis. When results were analyzed separated by the type of oxidant used to generate hydroxyl radicals (Cl2 or H2O2), no difference was observed in formation of any of the 4 THMs (p > 0.05, data not shown).
AOM was a poor precursor for THM formation. The yield of CF with just the background organic matter was 1.29 μg mg−1-C when data from samples pretreated by all oxidants and UV doses were combined. In contrast, CF yield from AOM was 0.31 μg mg−1-C (calculated based on the difference in concentrations in the samples with and without AOM divided by the additional mg-C from AOM). For the other THMs their actual concentration remained the same in the samples with and without additional AOM indicating that THMs other than CF do not form readily from AOM. AOM is very nitrogen-rich and likely does not easily fragment into precursor molecules for THMs. The fact that pretreatment with UV/Cl2 and UV/H2O2 showed no difference in THM formation indicates that reactions with RCS do not lead to chlorine incorporation into the structure of AOM or background DOM in a way that leads to formation of THMs, and reaction with Cl2 rather than RCS is the main mechanism of THM formation.
The yield of CF and BDCM was suppressed by nitrate, and the effect was more pronounced at higher UV doses (Fig. 2). The average yield in the background matrix for CF was 1.09 μg mg−1-C in chlorination after exposure to AOP at lower UV doses (500 and 1000 mJ cm−2) but 0.49 μg mg−1-C when nitrate was added (p = 0.0037). At 2000 mJ cm−2 the respective average values were 1.69 μg mg−1-C and 0.40 μg mg−1-C (p < 0.0001). For BDCM the difference was not significant in samples exposed to AOP at lower UV doses, but significant after exposure to 2000 mJ cm−2 (51.2 μg mg−1-C and 20.6 μg mg−1-C without and with additional nitrate, respectively, p < 0.001). It must be noted that nitrate addition resulted in inner filter of UV. However, the effect was minor, reducing overall UV dose reaching H2O2 or Cl2 by < 14%, while the decrease in yields of CF and BDCM were 55–76%. The role of nitrate in the suppression of CF and BDCM formation may result from the reactions associated with nitrogen-containing radicals when nitrate is irradiated by UV. This reaction may be scavenging THM precursors or modifying them chemically into substances that form something other than THMs upon subsequent chlorination. For example, it may add nitrogen to the structure of organic matter and result in higher formation of N-DBPs instead. Interestingly, the effect of nitrate was more pronounced with more chlorine vs. bromine in the THM structure (most pronounced for CF, less so for BDCM, and not significant for CDBM and BF).
 | ||
| Fig. 2 Yield of chloroform (CF) and bromodichloromethane (BDCM) in background matrix (dark gray) and with added nitrate (light gray). Figures on the left combine the results of 500 mJ cm−2 and 1000 mJ cm−2 irradiations for all oxidants, and figures on the right show results after irradiation with a higher UV dose (2000 mJ cm−2). | ||
The maximum contaminant level (MCL) for total THMs (TTHMs) of 80 μg L−1 was not exceeded in any of the samples, with the highest TTHM concentration of 46.3 μg L−1 (in the background matrix that was treated with Cl2 = 4 mg L−1 and UV = 2000 mJ cm−2) and the average of 23.9 μg L−1. TTHM yield in this study was 0.2–90.4 μg mg−1-C (27.9 average and 21.0 median) and is comparable to the range reported across a number of other studies involving chlorination of AOM (0–176.8 μg mg−1-C) as summarized by Leite, Daniel and Bond.57
3.2. Haloacetic acids
Nine HAAs were analyzed in this study, among which only three HAAs were detected relatively consistently in the treated samples: MCAA, CDBAA and TBAA. The detection frequency was as follows: MCAA 68% of samples, MBAA 23%, CDBAA 64%, DBAA 11%, DCAA 1%, MBAA 0%, BCAA 18%, TCAA 11% and TBAA 68%. Only three HAAs with detection frequency above 50% were used in the analysis. Generally, TCAA and DCAA are the dominant HAAs,58 but due to high detection limits and other analytical challenges this study was not able to assess the treatment and matrix effects on these important HAAs. At high bromide concentrations of ≥20 μM, HAAs tend to speciate more towards the brominated HAAs observed in this study.59 The bromide concentration in the source water used in this study, while elevated, was only 1.3 μM. Brominated HAAs are expected to represent approximately 10% of the total HAAs (THAAs) by molar concentration at this level of bromide.59 The prevalence of CDBAA and TBAA is likely due to the analytical challenges with the other HAAs that would have been more predominant if consistently detected. Thus, HAA results can only be analyzed in a limited way in this study by observing the effect of the treatment processes and the background matrix on the three individual HAAs. The relative concentrations of MCAA, CDBAA and TBAA with respect to each other, when excluding the six HAA that were not consistently detected, are consistent with what is reposted in literature for source waters with elevated bromide.59–61Higher level of AOP pretreatment did not show any increase in the yield of any of the three HAAs in subsequent chlorination. Just as with chlorinated THMs, MCAA formation was also suppressed by nitrate (27% decrease on average from 0.29 μg mg−1-C to 0.21 μg mg−1-C, p = 0.0039), Fig. 3. However, the effect was minor and could have been impacted by the 14% decrease in the effective UV dose that resulted from the addition of nitrate. On the other hand, CDBAA yield increased by 45% on average from 1.02 μg mg−1-C to 1.47 μg mg−1-C in the presence of nitrate (p = 0.046) and TBAA increased by 213% from 1.52 μg mg−1-C to 3.24 μg mg−1-C (p = 0.042), Fig. 3. Again, similar to what was observed with THMs, nitrate favored formation of brominated DBPs.
 | ||
| Fig. 3 Effect of nitrate on yield of individual HAAs. | ||
AOM did not make a good precursor for additional formation of the three HAAs, similar to what was observed with THMs. While visual inspection of the data appeared to show a few higher yields of the three HAAs in chlorination of the UV/H2O2 treated samples, compared to UV/Cl2 treated samples, the overall differences were not statistically significant with the exception of MCAA yield in the background matrix (average and standard deviation of 0.21 ± 0.08 μg mg−1-C after treatment with UV/H2O2 compared to 0.10 ± 0.02 μg mg−1-C after UV/Cl2, p < 0.001). Work by other researchers showed that HAAs form readily from AOM, with some studies showing variability by species from which AOM was obtained62 and some showing no effect of the species.63 It is possible, that AOP treatment changes AOM in a way that transforms HAA precursors as has been shown in several studies with UV/persulfate treatment of AOM.64 Additionally, with only three of nine HAAs consistently detected in this study, the ability to draw broader conclusions is limited.
3.3. Brominated DBPs
The background water matrix had a relatively high bromide concentration (0.105 mg L−1) due to the presence of coal burning power plants upstream in the watershed of the source water of the drinking water treatment facility from which the samples were obtained. High bromide leads to higher formation of brominated DBPs that contribute to higher TTHMs and THAAs because of the larger molecular weight than chlorinated DBPs. Overall, bromide incorporation in this study was 38% on average. No trends were present for bromide incorporation with increasing levels of AOP treatment. There was also no statistical difference in bromide incorporation for samples treated with UV/H2O2vs. UV/Cl2. Addition of nitrate increased bromide incorporation when compared to the background matrix, and the difference was statistically significant (p = 0.02). The average and the standard deviation for bromide ion incorporation into DBPs in all of the background matrix samples, nitrate-spiked and AOM-spiked samples were 29 ± 11%, 50 ± 29%, and 36 ± 17%, respectively. As observed with THMs and HAAs yields, the presence of nitrate and the resulting photochemical reactions decreased formation of chlorinated THMs which could possibly lead to bromination reactions happening preferentially. Overall, percent bromide incorporation is consistent with previous studies on this subject.38,65 A shift towards higher level of bromine substitution of THMs and HAAs formed from AOM has been previously reported for water pre-oxidized with ozone prior to chlorination.66 Potentially, additional radicals formed in the photolysis of nitrate increase the state of oxidation of organic matter compared to samples with no nitrate. Nitrate radicals (NO3˙) can also react with halides (e.g. Cl− and Br−) to form respective chlorine and bromine radicals (Cl˙ and Br˙), and the bimolecular second-order reaction rate with bromide is two orders of magnitude faster than with chloride (4 × 109 M−1 s−1vs. 7.1 × 107 M−1 s−1).67 While the overall radical chemistry of combined nitrate and halides under UV is rather complex, the general shift towards a higher concentration of reactive bromine species vs. reactive chlorine species would support a higher degree of bromination in DBPs.68 Higher bromine incorporation into DBPs after exposure of natural samples to UV in the presence of nitrate with subsequent chlorination was also shown in another study where addition of nitrate to water containing bromide increased formation of bromopicrin by a factor of 4 or more.693.4. N-Nitrosodimethylamine (NDMA)
The levels of NDMA in the background matrix were very low (0.11 ng L−1 on average), and likely were influenced by the photochemistry of the background nitrate, which was present at approximately 0.5 mg-N L−1. Such a concentration of nitrate in combination with the UV source used in this study can result in considerable formation of nitrite and nitrogen radicals.26 NDMA concentrations as high as 6 ng L−1 were reported in chlorination of drinking water (2 mg L−1 DOC, 8 mg L−1 Cl2, pH 7, over 72 hours) by Kristiana and Tan.70NDMA yield was not affected by the UV dose or the type of process (UV/Cl2 or UV/H2O2). Fig. 4 shows the NDMA yield comparison for the two AOPs separated by matrix (background, with additional nitrate or with additional AOM). Visually, it appears that UV/Cl2 tends towards higher NDMA yield than UV/H2O2, except when elevated nitrate is present. However, statistical analysis was not able to confirm the significance of this difference (p > 0.05). When data at 5 mg L−1 of H2O2 are compared to data from UV/Cl2 exposure at either Cl2 concentration, the p-value is 0.02, which suggests statistical significance, however, this significance is not present when data at 10 mg L−1 of H2O2 is compared to the UV/Cl2 data sets. Therefore, despite the appearance of the difference and the statistical significance in some subsets of the data, it cannot be stated conclusively that NDMA yield after UV/Cl2 is higher than after UV/H2O2. Further investigation into this topic is warranted.
 | ||
| Fig. 4 NDMA yield in chlorination after UV/H2O2 (dark gray) vs. after UV/Cl2 (light gray) in different matrices. | ||
Yield of NDMA from AOM was higher on average than from background organic matter. It was calculated by taking the difference in the NDMA average concentration in the matrix spiked with AOM and the background matrix and dividing that value by the additional carbon from the AOM spike. Algal DOM is very nitrogen-rich with a C:N ratio (by mass) of 2.5 in this study compared to a 5.6 ratio of the background matrix or, for example, a 7.9 ratio reported in another study that reported composition of surface water NOM.71 Therefore, yield of NDMA per mg of organic nitrogen was also evaluated. The average NDMA yield from the background organic matter was 0.18 ng mg−1-C and 1.0 ng mg−1-N compared to 0.68 ng mg−1-C and 1.7 ng mg−1-N from the AOM. The difference in ng mg−1-C was statistically significant (p = 0.038), while the difference in ng mg−1-N was not. Nitrogen in AOM has been previously demonstrated to be an important precursor for N-DBP formation.72 Additionally, studies have shown that preoxidation with ozone breaks AOM molecules into smaller molecular size fractions increasing the yields of N-DBPs.73 A similar effect is likely taking place when AOM is oxidized by radicals forming in UV/H2O2 and UV/Cl2 exposures.
The effect of nitrate was very dramatic, increasing NDMA yield by a factor of 11 from the average of 0.18 ng mg−1-C in background matrix to 1.9 ng mg−1-C with nitrate (Fig. 5).
 | ||
| Fig. 5 NDMA yield per mg of organic carbon in the background matrix and in matrices spiked with nitrate and AOM. | ||
In all cases, the NDMA concentration was lower than the USEPA health-related advisory level of 10 ng L−1 ranging from 0.007 to 5.3 ng L−1. Nevertheless, the impact of nitrate on the formation of N-DBPs, especially in combination with UV where it can generate reactive nitrogen species, is a topic that warrants further investigation. Nitrate forms nitrite radicals among other reactive nitrogen species under UV irradiation, and it is possible that they can add to the structure of background organic matter74 providing nitroso groups that become part of nitrosamines when they react with organic amines in the water. Organic amines are an important precursor of N-DBP formation.75 It is also possible that nitrate alone may have an effect without the presence of UV. This study did not evaluate samples with no UV exposure, so the direct contribution from nitrate or dark reaction with nitrite76 that would have formed after UV irradiation cannot be separated from the photochemical reactions with reactive nitrogen species77 and should be investigated further. Increased UV dose did not have an obvious effect, like it did with THMs and HAAs, confirming that dark reactions involving nitrate and nitrite (a stable byproduct of nitrate photolysis) may be important. Chlorine–nitrite combination leads to the formation of nitrosating species such as N2O4.78 While a sufficient level of nitrite would typically not be present in the source water, nitrate photochemistry can generate the nitrite necessary for the increased NDMA formation.26 NDMA yield ranged from 0.01–6.3 ng mg−1-C (0.9 average and 0.5 median). A review by Leite, Daniel and Bond57 shows a wide range or reported values from 0.8 to 3700 ng mg−1-C, but the median for the reported studies is 10 ng mg−1-C, which is comparable to what was observed in this study.
While NDMA was selected as a single N-DBP in this study because of its regulatory status, it is important to consider that nitrate (and its photochemical reactions) in combination with AOM transformed by AOPs can be a major source of other N-DBPs. N-DBPs such as haloacetonitriles, haloacetamides, halonitromethanes, halonitroalkanes and others may form at higher levels than NDMA or other nitrosamines, and present higher toxicity risks.79 While some work has been done on other N-DBPs in scenarios that involve AOM or UV irradiation,75 further research into N-DBP formation in water containing AOM and nitrate and chlorinated post-AOP is warranted, as well as into unregulated non-N-DBPs (e.g., haloacetaldehydes) that are known to form at high levels when AOM is present.63
4. Conclusions
Overall, the choice of treatment with UV/Cl2 or with UV/H2O2 did not affect the outcome of DBP yield upon subsequent chlorination for the tested DBP categories (THMs, HAAs and NDMA). The effect of UV/Cl2 on formation of chlorinated DBPs as compared to UV/H2O2 is an often-speculated topic because of the RCS chemistry involved in the former process. While many studies evaluated each process separately, this study shows through a large number of samples with various matrices that UV/Cl2 and UV/H2O2 deliver the same result with respect to subsequent yield of regulated DBPs. Nevertheless, regardless of the AOP type, increased oxidation of organic matter resulted in higher formation of THMs, in particular of those more chlorinated than brominated (CF and BDCM). The same effect on HAAs was not observed, however, only three of nine HAAs were consistently detected in this study. AOM proved to be a poor precursor for THMs or HAAs with much lower yield per mg-C compared to the background organic matter for CF and essentially no measurable contribution to the other THMs and HAAs. In terms of NDMA yield, AOM was slightly more efficient in NDMA generation (both per mg-C and per mg-N). The difference was not statistically significant on the per mg-N basis but significant on the per mg-C basis, compared to the background organic matter. Nitrate presence significantly inhibited formation of chlorinated THMs and HAAs giving rise to the brominated THMs and HAAs instead. Because of the higher mass of bromine atom compared to chlorine atom, formation of brominated DBPs at the same molar concentration as chlorinated DBPs will have a higher mass per volume (μg L−1) concentration. MCLs for TTHMs and THAAs are set in μg L−1, and thus presence of nitrate in combination with elevated background bromide can lead to permit exceedances for the regulated DBPs. Additionally, nitrate had a dramatic effect on NDMA yield increasing it by a factor of 11. Nitrate was spiked at 20 mg-N L−1, which would be on the high end of what is seen in a typical surface water, but it is within the realm of possibility for water sources significantly impacted by agricultural runoff or wastewater effluent. Concentrations within a more typical range of contaminated water sources (1–10 mg L−1) can still have a considerable impact on formation of N-DBPs. In water sources that also have a high level of organic matter, especially of algal origin, it can lead to NDMA levels exceeding local action limits. The role of UV and reactive nitrogen species in this process should be further investigated as well as formation of other N-DBPs apart from NDMA. Nitrate was a more consequential water constituent than AOM for DBP formation in this study. Utilities considering using UV-based AOPs for treatment of algal toxins, especially in water high in Br−, should use UV sources that minimally activate nitrate photochemistry, such as traditional low-pressure mercury vapor lamps. Nitrate removal may be needed to minimize the risk of additional NDMA formation. Additionally, TON rather than TOC would be a better predictor of NDMA formation in waters affected by algal bloom.Abbreviations and acronyms
| ACS | American Chemical Society |
| AOM | Algal organic matter |
| AOP | Advanced oxidation process |
| BCAA | Bromochloroacetic acid |
| BDCAA | Bromodichloroacetic acid |
| BDCM | Bromodichloromethane |
| BF | Bromoform |
| CDBAA | Chlorodibromoacetic acid |
| C-DBP | Carbonaceous disinfection by product |
| CF | Chloroform |
| DBAA | Dibromoacetic acid |
| DBCM | Dibromochloromethane |
| DBP | Disinfection byproduct |
| DCAA | Dichloroacetic acid |
| DOM | Dissolved organic matter |
| DPD | N,N-Diethyl-p-phenylenediamine |
| EPA | Environmental Protection Agency |
| GC-ECD | Gas chromatography with electron capture detector |
| HAA | Haloacetic acid |
| LC-MS/MS | Liquid chromatography tandem mass spectrometry |
| MBAA | Monobromoacetic acid |
| MCAA | Monochloroacetic acid |
| MCL | Maximum contaminant level |
| MTBE | Methyl tert-butyl ether |
| N-DBP | Nitrogenous disinfection byproduct |
| NDMA | N-Nitrosodimethylamine |
| NIST | National Institute of Standards and Technology |
| PTFE | Polytetrafluoroethylene |
| RCS | Reactive chlorine species |
| SPE | Solid phase extraction |
| TBAA | Tribromoacetic acid |
| TCAA | Trichloroacetic acid |
| THAAs | Total haloacetic acids |
| THM | Trihalomethane |
| TKN | Total Kjeldahl nitrogen |
| TN | Total nitrogen |
| TOC | Total organic carbon |
| TON | Total organic nitrogen |
| TTHMs | Total trihalomethanes |
| UFC | Uniform formation conditions |
| UV | Ultraviolet |
Data availability
Data for this article, including yields of THMs, HAAs and NDMA, are available at Dataverse at https://doi.org/10.7910/DVN/TW5HCN.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this project was provided by the North Carolina Water Resources Research Institute (NC WRRI) Urban Water Consortium grant 2009-1343-13. The authors gratefully acknowledge Heather Oakes for her assistance with the disinfection byproduct extraction, and the staff at the water utility that provided the sample.References
- K. A. Loftin, J. L. Graham, E. D. Hilborn, S. C. Lehmann, M. T. Meyer, J. E. Dietze and C. B. Griffith, Cyanotoxins in inland lakes of the United States: Occurrence and potential recreational health risks in the EPA National Lakes Assessment 2007, Harmful Algae, 2016, 56, 77–90 CrossRef CAS.
- K. A. Loftin, J. M. Clark, C. A. Journey, D. W. Kolpin, P. C. Van Metre, D. Carlisle and P. M. Bradley, Spatial and temporal variation in microcystin occurrence in wadeable streams in the southeastern United States, Environ. Toxicol. Chem., 2016, 35(9), 2281–2287 CrossRef CAS.
- J. R. Beaver, E. E. Manis, K. A. Loftin, J. L. Graham, A. I. Pollard and R. M. Mitchell, Land use patterns, ecoregion, and microcystin relationships in U.S. lakes and reservoirs: A preliminary evaluation, Harmful Algae, 2014, 36, 57–62 CrossRef CAS.
- M. Beaulieu, F. Pick and I. Gregory-Eaves, Nutrients and water temperature are significant predictors of cyanobacterial biomass in a 1147 lakes data set, Limnol. Oceanogr., 2013, 58(5), 1736–1746 CrossRef CAS.
- C. J. Gobler, J. M. Burkholder, T. W. Davis, M. J. Harke, T. Johengen, C. A. Stow and D. B. Van de Waal, The dual role of nitrogen supply in controlling the growth and toxicity of cyanobacterial blooms, Harmful Algae, 2016, 54, 87–97 CrossRef CAS.
- D. Z. Wang and D. Hsieh, Effects of nitrate and phosphate on growth and C2 toxin productivity of Alexandrium tamarense CI01 in culture, Mar. Pollut. Bull., 2002, 45(1–12), 286–289 CrossRef CAS.
- M. Drikas, C. W. K. Chow, J. House and M. D. Burch, Using coagulation, flocculation and settling remove toxic cyanobacteria, J. - Am. Water Works Assoc., 2001, 93(2), 100–111 CrossRef.
- D. A. Karner, J. H. Standridge, G. W. Harrington and R. P. Barnum, Microcystin algal toxins IN SOURCE AND FINISHED DRINKING WATER, J. - Am. Water Works Assoc., 2001, 93(8), 72–81 CrossRef CAS.
- T. Hall, J. Hart, B. Croll and R. Gregory, Laboratory-Scale Investigations of Algal Toxin Removal by Water Treatment, Water Environ. J., 2000, 14(2), 143–149 CrossRef CAS.
- J. Rositano, G. Newcombe, B. Nicholson and P. Sztajnbok, Ozonation of nom and algal toxins in four treated waters, Water Res., 2001, 35(1), 23–32 CrossRef CAS PubMed.
- P. T. Orr, G. J. Jones and G. R. Hamilton, Removal of saxitoxins from drinking water by granular activated carbon, ozone and hydrogen peroxide—implications for compliance with the Australian drinking water guidelines, Water Res., 2004, 38(20), 4455–4461 CrossRef CAS PubMed.
- E. Rodríguez, G. D. Onstad, T. P. J. Kull, J. S. Metcalf, J. L. Acero and U. von Gunten, Oxidative elimination of cyanotoxins: Comparison of ozone, chlorine, chlorine dioxide and permanganate, Water Res., 2007, 41(15), 3381–3393 CrossRef.
- S. Merel, M. Clément and O. Thomas, State of the art on cyanotoxins in water and their behaviour towards chlorine, Toxicon, 2010, 55(4), 677–691 CrossRef CAS.
- A. A. de la Cruz, A. Hiskia, T. Kaloudis, N. Chernoff, D. Hill and M. G. Antoniou, et al., A review on cylindrospermopsin: the global occurrence, detection, toxicity and degradation of a potent cyanotoxin, Environ. Sci.: Processes Impacts, 2013, 15(11), 1979–2003 RSC.
- J.-L. Lin and A. R. Ika, Pre-oxidation of Microcystis aeruginosa-laden water by intensified chlorination: Impact of growth phase on cell degradation and in situ formation of carbonaceous disinfection by-products, Sci. Total Environ., 2022, 805, 150285 CrossRef CAS PubMed.
- R. K. Henderson, A. Baker, S. A. Parsons and B. Jefferson, Characterisation of algogenic organic matter extracted from cyanobacteria, green algae and diatoms, Water Res., 2008, 42(13), 3435–3445 CrossRef CAS PubMed.
- X. Li, N. R. H. Rao, K. L. Linge, C. A. Joll, S. Khan and R. K. Henderson, Formation of algal-derived nitrogenous disinfection by-products during chlorination and chloramination, Water Res., 2020, 183, 116047 CrossRef CAS PubMed.
- E. J. Rosenfeldt, UV advanced oxidation for treatment of taste and odor and algal toxins. Ohio AWWA Annual Conference Research Workshop, 2011.
- F. Barancheshme, K. Sikon and O. Keen, Loss of toxicity of microcystins in UV/H2O2 and UV/Cl2 treatment, J. Water Process Eng., 2024, 57, 104707 CrossRef.
- A. D. Dotson, V. S. Keen, D. Metz and K. G. Linden, UV/H2O2 treatment of drinking water increases post-chlorination DBP formation, Water Res., 2010, 44(12), 3703–3713 CrossRef CAS PubMed.
- Y. Wu, D. Sheng, Y. Wu, J. Sun, L. Bu, S. Zhu and S. Zhou, Molecular insights into formation of nitrogenous disinfection byproducts from algal organic matter in UV-LEDs/chlorine process based on FT-ICR analysis, Sci. Total Environ., 2022, 812, 152457 CrossRef CAS PubMed.
- X. Duan, T. Sanan, A. de la Cruz, X. He, M. Kong and D. D. Dionysiou, Susceptibility of the Algal Toxin Microcystin-LR to UV/Chlorine Process: Comparison with Chlorination, Environ. Sci. Technol., 2018, 52(15), 8252–8262 CrossRef CAS PubMed.
- C. K. Remucal and D. Manley, Emerging investigators series: the efficacy of chlorine photolysis as an advanced oxidation process for drinking water treatment, Environ. Sci.:Water Res. Technol., 2016, 2, 565–579 RSC.
- C. Wang, N. Moore, K. Bircher, S. Andrews and R. Hofmann, Full-scale comparison of UV/H2O2 and UV/Cl2 advanced oxidation: The degradation of micropollutant surrogates and the formation of disinfection byproducts, Water Res., 2019, 161, 448–458 CrossRef CAS PubMed.
- J. Mack and J. R. Bolton, Photochemistry of nitrite and nitrate in aqueous solution: a review, J. Photochem. Photobiol., A, 1999, 128(1–3), 1–13 CrossRef CAS.
- O. S. Keen, N. G. Love and K. G. Linden, The role of effluent nitrate in trace organic chemical oxidation during UV disinfection, Water Res., 2012, 46(16), 5224–5234 CrossRef CAS PubMed.
- D. A. Reckhow, K. G. Linden, J. Kim, H. Shemer and G. Makdissy, Effect of UV treatment on DBP formation, J. AWWA, 2010, 102(6), 100–113 CrossRef CAS.
- A. D. Shah, A. D. Dotson, K. G. Linden and W. A. Mitch, Impact of UV Disinfection Combined with Chlorination/Chloramination on the Formation of Halonitromethanes and Haloacetonitriles in Drinking Water, Environ. Sci. Technol., 2011, 45(8), 3657–3664 CrossRef CAS PubMed.
- S. Zhou, Y. Shao, N. Gao, S. Zhu, L. Li, J. Deng and M. Zhu, Removal of Microcystis aeruginosa by potassium ferrate (VI): Impacts on cells integrity, intracellular organic matter release and disinfection by-products formation, Chem. Eng. J., 2014, 251, 304–309 CrossRef CAS.
- A. A. M. Gad and S. El-Tawel, Effect of pre-oxidation by chlorine/permanganate on surface water characteristics and algal toxins, Desalin. Water Treat., 2016, 57(38), 17922–17934 CrossRef CAS.
- J. Fang, J. Ma, X. Yang and C. Shang, Formation of carbonaceous and nitrogenous disinfection by-products from the chlorination of Microcystis aeruginosa, Water Res., 2010, 44(6), 1934–1940 CrossRef CAS.
- Y. Wang, F. Li, J. Du, X. Shi, A. Tang and M.-L. Fu, et al., Formation of nitrosamines during chloramination of two algae species in source water—Microcystis aeruginosa and Cyclotella meneghiniana, Sci. Total Environ., 2021, 798, 149210 CrossRef CAS PubMed.
- Environmental Protection Agency. Six-year review 3 technical support document for nitrosamines. 2016, EPA 810-R-16-009.
- J. Choi and R. L. Valentine, Formation of N-nitrosodimethylamine (NDMA) from reaction of monochloramine: a new disinfection by-product, Water Res., 2002, 36(4), 817–824 CrossRef CAS.
- W. A. Mitch and D. L. Sedlak, Formation of N-Nitrosodimethylamine (NDMA) from Dimethylamine during Chlorination, Environ. Sci. Technol., 2002, 36(4), 588–595 CrossRef CAS.
- L. Li, N. Gao, Y. Deng, J. Yao and K. Zhang, Characterization of intracellular & extracellular algae organic matters (AOM) of Microcystic aeruginosa and formation of AOM-associated disinfection byproducts and odor & taste compounds, Water Res., 2012, 46(4), 1233–1240 CrossRef CAS PubMed.
- E. C. Wert and F. L. Rosario-Ortiz, Intracellular Organic Matter from Cyanobacteria as a Precursor for Carbonaceous and Nitrogenous Disinfection Byproducts, Environ. Sci. Technol., 2013, 47(12), 6332–6340 CrossRef CAS PubMed.
- J. Sohn, G. Amy and Y. Yoon, Bromide Ion Incorporation Into Brominated Disinfection By-Products, Water, Air, Soil Pollut., 2006, 174(1), 265–277 CrossRef CAS.
- F. Barancheshme, Treatment of Algal Toxins in Drinking Water with UV-Based Advanced Oxidation Processes, Ph.D. thesis, The University of North Carolina at Charlotte, United States, North Carolina, 2020 Search PubMed.
- N. V. Klassen, D. Marchington and H. C. E. McGowan, H2O2 Determination by the I3- Method and by KMnO4 Titration, Anal. Chem., 1994, 66(18), 2921–2925 CrossRef CAS.
- J. Bolton and K. Linden, Standardization of Methods for Fluence (UV Dose) Determination in Bench-Scale UV Experiments, J. Environ. Eng., 2003, 129(3), 209–215 CrossRef CAS.
- Y. Deng, M. Wu, H. Zhang, L. Zheng, Y. Acosta and T. D. Hsu, Addressing harmful algal blooms (HABs) impacts with ferrate(VI): Simultaneous removal of algal cells and toxins for drinking water treatment, Chemosphere, 2017, 186, 757–761 CrossRef CAS PubMed.
- J. L. Adams, E. Tipping, H. Feuchtmayr, H. T. Carter and P. Keenan, The contribution of algae to freshwater dissolved organic matter: implications for UV spectroscopic analysis, Inland Waters, 2018, 8(1), 10–21 CrossRef CAS.
- D. Lee, M. Kwon, Y. Ahn, Y. Jung, S.-N. Nam, I.-h. Choi and J.-W. Kang, Characteristics of intracellular algogenic organic matter and its reactivity with hydroxyl radicals, Water Res., 2018, 144, 13–25 CrossRef CAS.
- R. M. Kreiling, W. B. Richardson, L. A. Bartsch, M. C. Thoms and V. G. Christensen, Denitrification in the river network of a mixed land use watershed: unpacking the complexities, Biogeochemistry, 2019, 143(3), 327–346 CrossRef CAS.
- E. Mackey, R. Hofmann, A. Festger and C. Vanyo, UV-Chlorine AOP in potable reuse: A guidance manual to assessment and implementation, 2022.
- N. McLellan, K. Bell and M. Holmer, UV-AOP 101 for potable reuse: Design considerations and practical applications. IUVA Americas Conference, Redondo Beach, CA, 2018.
- S. R. Summers, M. S. Hooper and M. H. Shukairy, et al., Assessing DBP yield: uniform formation conditions. American Water Works Association, ETATS-UNIS, Denver, CO, 1996.
- O. Keen, A. Dotson and K. Linden, Evaluation of Hydrogen Peroxide Chemical Quenching Agents following an Advanced Oxidation Process, J. Environ. Eng., 2013, 139(1), 137–140 CrossRef CAS.
- X. Liu, X. Wei, W. Zheng, S. Jiang, M. R. Templeton, G. He and W. Qu, An optimized analytical method for the simultaneous detection of iodoform, iodoacetic acid, and other trihalomethanes and haloacetic acids in drinking water, PLoS One, 2013, 8(4), e60858 CrossRef CAS PubMed.
- Y.-Y. Zhao, J. Boyd, S. E. Hrudey and X.-F. Li, Characterization of New Nitrosamines in Drinking Water Using Liquid Chromatography Tandem Mass Spectrometry, Environ. Sci. Technol., 2006, 40(24), 7636–7641 CrossRef CAS PubMed.
- J. W. A. Charrois, M. W. Arend, K. L. Froese and S. E. Hrudey, Detecting N-Nitrosamines in Drinking Water at Nanogram per Liter Levels Using Ammonia Positive Chemical Ionization, Environ. Sci. Technol., 2004, 38(18), 4835–4841 CrossRef CAS PubMed.
- K. M. H. Beggs, R. S. Summers and D. M. McKnight, Characterizing chlorine oxidation of dissolved organic matter and disinfection by-product formation with fluorescence spectroscopy and parallel factor analysis. Journal of Geophysical Research, Biogeosciences, 2009, 114(G4), 1–10 Search PubMed.
- A. Adin, J. Katzhendler, D. Alkaslassy and C. Rav-Acha, Trihalomethane formation in chlorinated drinking water: A kinetic model, Water Res., 1991, 25(7), 797–805 CrossRef CAS.
- M. D. Arguello, C. D. Chrisweii, J. S. Fritz, L. D. Kissinger, K. W. Lee, J. J. Richard and H. J. Svec, Trihalomethanes in Water: A Report on the Occurrence, Seasonal Variation in Concentrations, and Precursors of Trihalomethanes, J. AWWA, 1979, 71(9), 504–508 CrossRef CAS.
- L. Liang and P. C. Singer, Factors Influencing the Formation and Relative Distribution of Haloacetic Acids and Trihalomethanes in Drinking Water, Environ. Sci. Technol., 2003, 37(13), 2920–2928 CrossRef CAS PubMed.
- L. S. Leite, L. A. Daniel and T. Bond, Algal organic matter as a disinfection by-product precursor during chlor(am)ination: a critical review, Environ. Sci.: Water Res. Technol., 2023, 9(11), 2787–2802 RSC.
- E. Malliarou, C. Collins, N. Graham and M. J. Nieuwenhuijsen, Haloacetic acids in drinking water in the United Kingdom, Water Res., 2005, 39(12), 2722–2730 CrossRef CAS.
- G. A. Cowman and P. C. Singer, Effect of Bromide Ion on Haloacetic Acid Speciation Resulting from Chlorination and Chloramination of Aquatic Humic Substances, Environ. Sci. Technol., 1996, 30(1), 16–24 CrossRef CAS.
- C. M. M. Bougeard, E. H. Goslan, B. Jefferson and S. A. Parsons, Comparison of the disinfection by-product formation potential of treated waters exposed to chlorine and monochloramine, Water Res., 2010, 44(3), 729–740 CrossRef CAS PubMed.
- C. M. Villanueva, M. Kogevinas and J. O. Grimalt, Haloacetic acids and trihalomethanes in finished drinking waters from heterogeneous sources, Water Res., 2003, 37(4), 953–958 CrossRef CAS.
- H. Zhai, S. Cheng, L. Zhang, W. Luo and Y. Zhou, Formation characteristics of disinfection byproducts from four different algal organic matter during chlorination and chloramination, Chemosphere, 2022, 308, 136171 CrossRef CAS.
- C. Liu, M. S. Ersan, M. J. Plewa, G. Amy and T. Karanfil, Formation of regulated and unregulated disinfection byproducts during chlorination of algal organic matter extracted from freshwater and marine algae, Water Res., 2018, 142, 313–324 CrossRef CAS.
- F. Dong, Q. Lin, C. Li, G. He and Y. Deng, Impacts of pre-oxidation on the formation of disinfection byproducts from algal organic matter in subsequent chlor(am)ination: A review, Sci. Total Environ., 2021, 754, 141955 CrossRef CAS PubMed.
- J. Tan, S. Allard, Y. Gruchlik, S. McDonald, C. A. Joll and A. Heitz, Impact of bromide on halogen incorporation into organic moieties in chlorinated drinking water treatment and distribution systems, Sci. Total Environ., 2016, 541, 1572–1580 CrossRef CAS PubMed.
- M. Zhu, N. Gao, W. Chu, S. Zhou, Z. Zhang, Y. Xu and Q. Dai, Impact of pre-ozonation on disinfection by-product formation and speciation from chlor(am)ination of algal organic matter of Microcystis aeruginosa, Ecotoxicol. Environ. Saf., 2015, 120, 256–262 CrossRef CAS PubMed.
- P. Neta and R. E. Huie, Rate constants for reactions of nitrogen oxide (NO3) radicals in aqueous solutions, J. Phys. Chem., 1986, 90(19), 4644–4648 CrossRef CAS.
- T. Huang, L. Deng, T. Wang, X. Liao, J. Hu, C. Tan and R. P. Singh, Effects of bromide ion on the formation and toxicity alteration of halonitromethanes from nitrate containing humic acid water during UV/chlor(am)ine disinfection, Water Res., 2022, 225, 119175 CrossRef CAS PubMed.
- B. A. Lyon, A. D. Dotson, K. G. Linden and H. S. Weinberg, The effect of inorganic precursors on disinfection byproduct formation during UV-chlorine/chloramine drinking water treatment, Water Res., 2012, 46(15), 4653–4664 CrossRef CAS PubMed.
- I. Kristiana, J. Tan, C. A. Joll, A. Heitz, U. von Gunten and J. W. A. Charrois, Formation of N-nitrosamines from chlorination and chloramination of molecular weight fractions of natural organic matter, Water Res., 2013, 47(2), 535–546 CrossRef CAS PubMed.
- P. K. Egeberg, M. Eikenes and E. T. Gjessing, Organic nitrogen distribution in NOM size classes, Environ. Int., 1999, 25(2), 225–236 CrossRef CAS.
- X.-X. Wang, B.-M. Liu, M.-F. Lu, Y.-P. Li, Y.-Y. Jiang and M.-X. Zhao, et al., Characterization of algal organic matter as precursors for carbonaceous and nitrogenous disinfection byproducts formation: Comparison with natural organic matter, J. Environ. Manage., 2021, 282, 111951 CrossRef CAS.
- S. Zhou, S. Zhu, Y. Shao and N. Gao, Characteristics of C-, N-DBPs formation from algal organic matter: Role of molecular weight fractions and impacts of pre-ozonation, Water Res., 2015, 72, 381–390 CrossRef CAS.
- D. Vione, V. Maurino, C. Minero, M. Vincenti and E. Pelizzetti, Formation of nitrophenols upon UV irradiation of phenol and nitrate in aqueous solutions and in TiO2 aqueous suspensions, Chemosphere, 2001, 44(2), 237–248 CrossRef CAS PubMed.
- A. D. Shah and W. A. Mitch, Halonitroalkanes, Halonitriles, Haloamides, and N-Nitrosamines: A Critical Review of Nitrogenous Disinfection Byproduct Formation Pathways, Environ. Sci. Technol., 2012, 46(1), 119–131 CrossRef CAS PubMed.
- C. Liu, Z. Liang, C. Yang, F. Cui and Z. Zhao, Nitrite-enhanced N-nitrosamines formation during the simulated tetracycline polluted groundwater chlorination: Experimental and theoretical investigation, Chem. Eng. J., 2022, 431, 133363 CrossRef CAS.
- M. Masuda, H. F. Mower, B. Pignatelli, I. Celan, M. D. Friesen, H. Nishino and H. Ohshima, Formation of N-Nitrosamines and N-Nitramines by the Reaction of Secondary Amines with Peroxynitrite and Other Reactive Nitrogen Species: Comparison with Nitrotyrosine Formation, Chem. Res. Toxicol., 2000, 13(4), 301–308 Search PubMed.
- S. W. Krasner, W. A. Mitch, D. L. McCurry, D. Hanigan and P. Westerhoff, Formation, precursors, control, and occurrence of nitrosamines in drinking water: A review, Water Res., 2013, 47(13), 4433–4450 CrossRef CAS.
- S. D. Richardson, M. J. Plewa, E. D. Wagner, R. Schoeny and D. M. DeMarini, Occurrence, genotoxicity, and carcinogenicity of regulated and emerging disinfection by-products in drinking water: A review and roadmap for research, Mutat. Res., Rev. Mutat. Res., 2007, 636(1), 178–242 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ew00749b |
| This journal is © The Royal Society of Chemistry 2025 |