
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemically driven dimensionality modulation of hybrid tin(II) halide perovskite microcrystals†
Raúl I.
Sánchez-Alarcón
 a,
Omar E.
Solís
a,
María Cristina
Momblona-Rincon
a,
Teresa S.
Ripolles
*a,
Juan P.
Martínez-Pastor
a,
Rafael
Abargues
*a and
Pablo P.
Boix
*ab
a,
Omar E.
Solís
a,
María Cristina
Momblona-Rincon
a,
Teresa S.
Ripolles
*a,
Juan P.
Martínez-Pastor
a,
Rafael
Abargues
*a and
Pablo P.
Boix
*ab
aInstituto de Ciencia de los Materiales-Universidad de Valencia, Catedrático José Beltrán, 2, 46071, Valencia, Spain. E-mail: teresa.ripolles@uv.es; rafael.abargues@uv.es; Pablo.P.Boix@itq.upv.es
bInstituto de Tecnología Química, Instituto de Tecnología Química, Universitat Politècnica València-Consejo Superior de Investigaciones Científicas, Av. dels Tarongers, 46022 València, Spain
First published on 6th May 2024
Abstract
The lattice arrangement of lead-free perovskites can be altered by synthesis parameters such as concentration, temperature, choice of solvents, and the length of monovalent cations. This effect, often described as dimensionality modulation, can be exploited to develop new materials with better optical properties than their three-dimensional counterpart. This work explores the versatility of the hot-injection synthesis for obtaining 2-thiopheneethylammonium (TEA+) based tin halide perovskite microcrystals. The dimensionality of TEA-based tin bromide perovskite microcrystals can be modulated through changes in the TEA concentration, giving rise to the formation of highly luminescent 0D-TEA4SnBr6 and low-emissive 2D-TEA2SnBr4 microplates. In contrast, 0D-[TEASnCl3][TEACl] and 2D-TEA2SnI4 are thermodynamically preferred as main products when using the chloride and iodide analogues, respectively, limiting the dimensional versatility. It is noteworthy that 0D-TEA4SnBr6 shows the highest PLQY from the low dimensional TEA tin halide family (PLQY = 75% in thin-film) owing to a more confined configuration based on isolated [SnBr6]4− octahedra moieties separated by TEA+ cations, being attractive for optoelectronic applications.
Introduction
Lead halide perovskites show promising properties for semiconductor-based applications, such as efficient energy conversion in solar cells.1 However, one of their major drawbacks is the toxicity of lead, a key component usually present in most efficient compositions. Researchers are actively exploring alternative materials to replace lead in perovskites, aiming to mitigate environmental and health concerns. This has become a key area of research with the aim of developing environmentally friendly and sustainable technologies. The most common approach to form lead-free perovskites is to replace Pb(II) with Sn(II). Since both of them present the same valence electron configuration (ns2sp2), the resulting perovskites can offer similar properties, maintaining the perovskite structure and optoelectronic behaviour, such as high charge carrier mobility, high light absorption properties, and small exciton binding energy.2 However, tin-based perovskites exhibit lower stability than their lead-containing counterparts.3 Even though Sn and Pb present multiple oxidation states, tin forms more commonly Sn(II) and Sn(IV) ions, while lead is often found as Pb(II). The reason for this is attributed to the lanthanide contraction effect in Pb by the presence of 4f14 electrons in its electronic configuration. Conversely, Sn lacks lanthanide shrinkage, leading to easy loss of all valence electrons, and consequently, forming Sn4+ ions.4Reducing the dimensionality of perovskite materials by disaggregation of inorganic moieties with bulky organic cations is a promising approach to mitigate the Sn-based perovskite stability issues.5 For example, in two dimensional perovskites, 2D, layered perovskite moieties are hydrogen bonded with voluminous organic spacers, which are self-assembled by van der Waals forces and π-stacking. If layers of 3D perovskites are stuck within the voluminous organic spacers, the physical properties depend on the thickness of inorganic perovskite moieties, being possible to tune the exciton binding energy and band gap of the resulting materials.6 On the other hand, if the density of bulky organic cations is high enough to separate the structure on individual polyhedron moieties, these conduce to the formation of zero-dimensional perovskite structures, 0D.7
This bulky organic layer can act as a protection from external agents, increasing environmental stability. It also leads to a higher confinement of bounded excitons than their three-dimensional material counterpart,8 which can be advantageous for light emitting applications.9 From the fundamental point of view, manipulating the dimensionality of tin-based perovskites can also impact their bandgap, carrier mobility, and other electronic properties due to the spatial confinement of charge carriers. For example, 2D tin halide perovskites present radiative recombination of free excitons by increasing the exciton 2D binding energy.10,11 On the other hand, 0D tin halide perovskites exhibit radiative recombination of self-trapped excitons (STEs) at energy levels originating from the optical distortion of Sn–X bonds.12,13 In particular, STEs refer to the strong electron (exciton) phonon coupling when a soft lattice is distorted upon photoexcitation.14 Interestingly, 0D-tin halide perovskites show higher PLQYs, better environmental stability, higher Stokes shift, and a major exciton binding energy compared to their 2D and 3D counterparts.13,15,16
However, inorganic and hybrid tin halide compounds can adopt multiple dimensionalities depending strongly on the polyhedron type (owing to the high coordination number of Sn(II) ions), inorganic or organic cations, oxidation number, and preparation conditions (pH, temperature, atmosphere, etc).17,18 For example, zero-dimensional tetrabutylammonium (TBAC) tin(II) chloride could be obtained under acidic conditions with stable pyramidal [SnCl3]− anions.19 If the monovalent cation is changed to Cs+, 3D-CsSnCl3 with [SnCl6]4− octahedrons is stable at high temperature because 5s2 orbitals hybridize with 5pz, resulting in hybrid orbitals with the same energy as that of np halogen orbitals.18,20 It should be mentioned that the octahedral tin chloride configuration is also stable for Sn(IV) ions.21 Different inorganic and organic tin(II/IV) halide compounds and dimensionalities are summarized in Table 1.
| Cation | Compound | Polyhedral unit | Dimensionality | Ref. |
|---|---|---|---|---|
| List of acronyms. TBCA+ = tetrabutylammonium; R/S-α-PEA+ = R/S-α-phenethylammonium; MA+ = methylammonium; FA+ = formamidinium; H4BAPP = 1,4-bis(3ammoniumpropyl)piperazinium; C7H8N3+ = 2-amino benzimidazolium; Cs+ = cesium; DAO2+ = 1,8-octyldiammonium; IPA+: isopropylammonium; (CH3)3S+ = trimethyl sulfonium. | ||||
| TBAC+ | (TBAC)SnCl3 | [SnCl3]− | 0D | 19 |
| R/S-α-PEA+ | R/S-α-PEA SnX3, (X = Cl, Br) | Distorted [SnX6] | 1D | 22 |
| MA+, FA+ | ASnI3 (A = MA+, FA+) | [SnI6]4− | 3D | 23 |
| PEA+ | PEA2SnI4 | [SnI6]4− | 2D | 24 |
| H4BAPP3+ | H4BAPPSnX5 (X = Cl, Br) | [SnX5]3− | 0D | 25 |
| C7H8N3+ | (C7H8N3)4SnBr6 | [SnBr6]4− | 0D | 26 |
| Cs+ | CsSn2Br5 | [Sn2Br5]− | 1D | 27 |
| DAO2+ | DAOSn2I6 | [Sn2I6]2− | 1D | 28 |
| IPA+ | μ-[IPA]3Sn2I7 | [Sn2I7]3− | 2D | 29 |
| (CH3)3S+ | [(CH3)3S]2SnCl6·H2O | [SnCl6]2− | 0D | 21,27 |
| Cs+, MA+, FA+ | A2SnI6 | [SnI6]2− | Undefined | 30 |
Thus, lead-free low-dimensional tin(II) halide perovskites are attracting scientific attention owing to their unique combination of interesting physical properties and lower toxicity.31 They show great potential as visible light emitters on optoelectronic devices32 as well as for enhancing the performance of photovoltaic devices.33 Among multiple organic cations tested on low-dimensional hybrid metal halide perovskites, 2-thiopheneethylammonium (TEA+) has drawn attention as a heteroatomic organic spacer and a passivating agent for lead iodide perovskites, with a stable power conversion efficiency (PCE) of 18.75% for 1000 h34,35 when used in solar cells. Concerning tin-based perovskites, the synthesis of 2D-TEA2SnI4 has been reported at different scales, from nanocrystals36 and thin films37 to single crystals,38 showing a PLQY around 19% for nanodisks in solution prepared by the LARP method36 and 23% for thin films prepared using the antisolvent approach.37 However, to the best of our knowledge, the synthesis and characterization of TEA-based Sn(II) chloride and bromide remain unexplored.
The dimensionality of perovskite nanocrystals synthesized by the hot injection method can be modulated by means of changes in the precursor concentrations.39–41 Dimensional modulation improves the optical properties of lead-free perovskites and their PLQY owing to the separation and distortion of metal halide octahedrons with lone electron pair ns2.42 For instance, Cs-oleate solution precursor concentration is a cornerstone parameter that determines whether 0D-Cs4SnBr6 or 3D-CsSnBr3 nanocrystals are obtained.43,44 Appropriate dimensionality manipulation of lead-free perovskites expands the range of these less toxic materials with a full gamut of wavelength emission depending on the type of inorganic moiety and organic cation. Therefore, the hot injection approach could be re-imagined as a controlled method to achieve high luminescent perovskite nanostructures.39,43
Here, we report the synthesis of 2D and 0D TEA-based Sn(II) halide perovskite microplates by the hot injection method. The control of the [TEA]/[Sn2+] ratio for the bromide-based perovskite adjusts the resulting perovskite dimensionality from 2D to 0D. The PLQY was strongly affected by this factor, ranging from 0.1% for 2D-TEA2SnBr4 to 75% for orange emitting 0D-TEA4SnBr6. Interestingly, TEA/Sn(II)-driven dimensionality control only takes place with TEA-based tin(II) bromide derivatives. Conversely, the reaction between SnCl2 and SnI2 with TEA provides main products pure 0D-[TEASnCl3][TEACl] and 2D-TEA2SnI4 microplates, respectively.
Materials and methods
Chemicals
Mesitylene (97% Pure) and iodotrimethylsilane (TMSI, 95–97% stabilized) were purchased from Thermo Scientific. Bromotrimethylsilane (TMSBr, 98%) and n-octane were acquired from Acros Organics. Chlorotrimethylsilane (TMSCl, 98%), trioctylphosphine (TOP, 97%), tin(II) ethylhexanoate (95–100%), n-hexane and oleic acid (OLAC, technical grade of 90%) were obtained from Sigma Aldrich. 2-Thiopheneethylamine (TEA, > 98%) was purchased from TCI Chemicals. All the chemicals were used without further purification.Experimental procedure
Characterisation
X-ray diffraction (XRD) diffractograms were measured with an Empyrean diffractometer from PANalytical and the measurements were conducted in the range of 2θ = 4°–60° with a 2θ step of 0.026° and using a Cu target as an X-ray source (Kα Cu radiation, λ = 0.15406 nm). For XRD measurements, 10 μm films were prepared by drop-casting lead-free microcrystal dispersions on n-octane over 0.5 cm × 0.5 cm glass substrates and drying at room-temperature under an inert atmosphere.1H and 13C nuclear magnetic resonance (NMR) spectra in the liquid phase for 0D-TEA4SnBr6 and 2D-TEA2SnX4 (X = Br, I) were recorded with a Bruker AV400 spectrometer, with an Oxford Magnet of 400 MHz (9.4 T). 1H and 13C NMR spectra are reported in ppm (δ) relative to the chemical shift of solvent residual signals for DMSO-d6 at 2.51 ppm and 39.99 ppm, respectively.
13C and 119Sn magic angle solid state nuclear magnetic resonance (MAS-NMR) spectra of 0D-[TEASnCl3][TEACl], 0D-TEA4SnBr6, and 2D-TEA2SnX4 (X = Br, I) were measured with a Bruker Advance III 400 WB spectrometer, with an ultra-shielded Widemouth magnet (89 mm), 400 MHz (9.4T), two RF channels and high-power amplifiers for solid-state experiments. For 119Sn MAS-NMR spectra measurements, the SnO powder sample was used as a reference. On the other hand, for 13C and 119Sn MAS-NMR and liquid state NMR, perovskite microcrystals were isolated from n-octane suspension by centrifugation and dried under an inert atmosphere overnight. 119Sn MAS-NMR spectra of 0D-[TEASnCl3][TEACl] were measured at different spinning rates (8 kHz and 10 kHz).
Transmission electron microscopy (TEM) images were acquired from a Hitachi HT7800 microscope with a high-resolution filament of LaB6 with an acceleration voltage of 100 kV. TEA-based tin(II) halide microcrystal suspensions in n-octane were dropped on Cu/C TEM grids.
Energy-dispersive X-ray spectroscopy (EDS) microanalysis was performed using an Oxford Ultim Max 170 detector monitored with Aztec software coupled to a SCIOS2 FESEM microscope from Thermo Scientific. For these measurements, TEA-based tin(II) halide microcrystals were deposited on Si substrates, and sample borders were coated with Ag paste.
Photoluminescence (PL) and photoluminescence excitation (PLE) measurements were conducted in an FLS1000 spectrophotometer from Edinburgh Instruments. A xenon lamp (Xe2) was used as an excitation source. PL spectra were recorded with a PMT-980 detector from Edinburgh Instruments. PL and PLE spectra were measured with a dwell time of 1 s and a wavelength step of 1 nm. The PL quantum yield (PLQY) was measured with an integrating sphere coupled to an FLS1000 spectrophotometer from Edinburgh Instruments. TEA based tin(II) halide microcrystal films were prepared over 0.25 cm2 quartz substrates by drop cast deposition to measure PLE and PL spectra and PLQY.
Results and discussion
The hot injection method was employed to synthesize 0D-TEA4SnBr6 and 2D-TEA2SnBr4 microcrystals by adjusting the precursor concentration, as detailed in the Experimental section, with [TEA]/[Sn2+] ratios of 1 and 0.3, respectively. Thin films fabricated from 2D-TEA2SnBr4 microcrystals exhibited flakes with an average particle size of 7.66 μm ± 5.2 μm, and a mean thickness of 280 nm (Fig. 1a–c). On the other hand, quasi-rectangular plates with a length of ∼32± 8 μm and a thickness of ∼3.55 μm were predominantly observed on 0D-TEA4SnBr6 thin films (Fig. 1d–f). | ||
| Fig. 1 Top-view SEM (a) and (d), cross-sectional SEM (b) and (e) and TEM images (c) and (f) of 2D-TEA2SnBr4 (a)–(c) and 0D-TEA4SnBr6 (d)–(f) microcrystals. | ||
Fig. 2a shows the XRD diffractograms of TEA-based Sn(II) bromide perovskite films for different [TEA]/[Sn2+] ratios. From [TEA]/[Sn2+] < 0.5, the XRD diffractogram shows the multiple reflections peaks at 2θ of 5.62°, 11.32°, 17.02°, 22.78°, 28.64° and 34.56°. These reflections are spaced apart by a 2θ interval of 5.7°, as expected from a Ruddlesden–Popper phase for pure 2D TEA2SnBr4.36 Additionally, EDS analysis (Table S1, ESI†) reveals that Br/Sn and S/Sn atomic ratios are in good agreement with the expected stoichiometry. As the [TEA]/[Sn2+] ratio increases, isolated metal halide octahedra [SnBr6]4− or clusters are generated. From [TEA]/[Sn2+] > 0.5, new non-periodically XRD peaks are observed at 8.52°, 17.13°, 18.23° and 25.84°, which are attributed to 0D-TEA4SnBr6. Atomic percentages of Br are higher than those measured for 2D-TEA2SnBr4 microplates (Table S1, ESI†), which supports the proposed chemical formula.
 | ||
| Fig. 2 (a) XRD patterns of 2D-TEA2SnBr4 and 0D-TEA4SnBr6 samples synthesized at different molar ratios [TEA]/[Sn2+]; PL and PLE spectra of: (b) 2D-TEA2SnBr4 and (c) 0D-TEA4SnBr6; (d) 2D/0D ratio (red line), PLQY % (blue line) and Stokes shift (green line) as a function of [TEA]/[Sn2+] ratio. PLQYs of 2D-TEA2SnBr4 and 0D-TEA4SnBr6 thin film samples were measured at a fixed wavelength of 280 nm. | ||
Concurrently, the increase of TEA molarities results in a decrease in the formation of the 2D-TEA2SnBr4 phase. From [TEA]/[Sn2+] > 2, the diffractograms reveal only reflection peaks of 0D-TEA4SnBr6 along with those of pure TEABr at 2θ values of 4.5°, 9°, 14°, 19°, 24°, 29° and 34°. As expected, 0D-TEA4SnBr6 reflection peaks do not exhibit the same periodic ordering observed in the 2D-TEA2SnBr4.
The differences between 2D-TEA2SnBr4 and 0D-TEA4SnBr6 crystal phases and band structures also generate remarkable differences in their optical properties. PL emission and PL excitation (PLE) spectra of TEA–Sn(II) bromide films synthesized at different molar ratios are shown in Fig. S1 (ESI†). In particular, the [TEA]/[Sn2+] ratio of 0.5, for 2D-TEA2SnBr4 films, is represented in Fig. 2b. Its PL spectra can be deconvoluted into two bands; one sharp band at 470 nm, and another broader band centered at 730 nm. The shape of the PL emission bands is similar to that reported for 2D-PEA2PbI4,45 thus mainly determined by the radiative recombination of free excitons. Additionally, the PLE spectrum exhibits a sharp band at 470 nm. From the spectral difference in the position of the band maximum of the PL and PLE spectra, we measured the Stokes shift energy, which is around 113 meV. The sharp PL and PLE bands shown in Fig. 2b, along with the high order observed in XRD, may be associated with the synthesis of lamellar-like microcrystals. These structures consist of intercalated layers comprising [SnBr6]4− octahedra and organic layers of TEA+ cations, as reported for 2D-TEA2SnI4 nanocrystals and thin films.46 In contrast, the PL emission and PLE spectra of 0D-TEA4SnBr6 films synthesized with a [TEA]/[Sn2+] ratio of 3 are displayed in Fig. 2c. When the perovskite was exposed to UV light (wavelength of 282 nm), 0D-TEA4SnBr6 exhibited a PL spectrum characterized by a band centered at 610 nm and an FWHM of 130 nm. The PLE spectrum shows two sharp bands with a maximum at 282 nm and 310 nm. From PL and PLE spectra, we measured a strong Stokes shift of 2.38 eV, which is attributed to the radiative recombination of self-trapped excitons.47,48
The larger bandgaps and Stokes shifts when transitioning from the 2D to 0D TEA-based Sn bromide perovskite are attributed to a reduction of the orbital overlap between [SnX6]4− octahedra as the 0D phase becomes more predominant with increasing [TEA]/[Sn2+] ratio. This occurs because TEA+ cations separate the [SnBr6]4− octahedra of 2D layers to form isolated [SnBr6]4− octahedra. This results in a higher exciton binding energy, lower carrier mobility, and increased stability. The isolated structure of 0D perovskites also imparts a strong quantum confinement effect, contributing to a higher PL emission compared to that of higher dimensionality.
Fig. 2d shows the 0D/2D phase ratio, PLQY, and Stokes shift of TEA-based Sn(II) bromide perovskite films depending on the [TEA]/[Sn2+] molar ratio. To estimate the phase ratio between 2D and 0D materials, we used eqn (1):
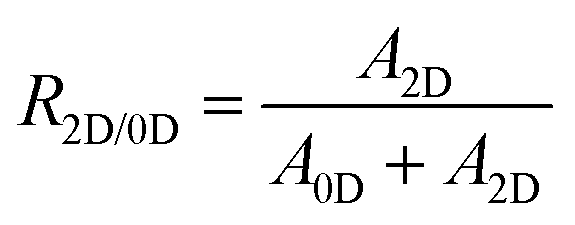 | (1) |
Based on our experimental results, the growth of 0D-TEA4SnBr6 is strongly influenced by the isolation of [SnBr6]4− octahedra layers from the 2D-TEA2SnBr4 material onto individual clusters resulting from the increase of TEA+ concentration. Also, the growth of 0D-TEA4SnBr6 is not influenced by coupling with mesitylene or oleic acid molecules as it is the case of the phenethylammonium tin(II) bromide perovskite, and thus the separation of inorganic moieties is influenced mostly by the increment in the TEA+ concentration.49 As a consequence, highly luminescent 0D-TEA4SnBr6 microcrystals can be synthesized by controlling the concentration of TEA.50
A qualitative magic angle solid state (MAS) NMR analysis was carried out to investigate the local environment and chemical bonding of carbon (13C) and tin (119Sn) nuclei in solid-state materials (Fig. 3). 2D-TEA2SnBr4 and 0D-TEA4SnBr6 are compared with TEABr to determine the internal structure of low dimensional TEA-based Sn(II) bromide perovskites.
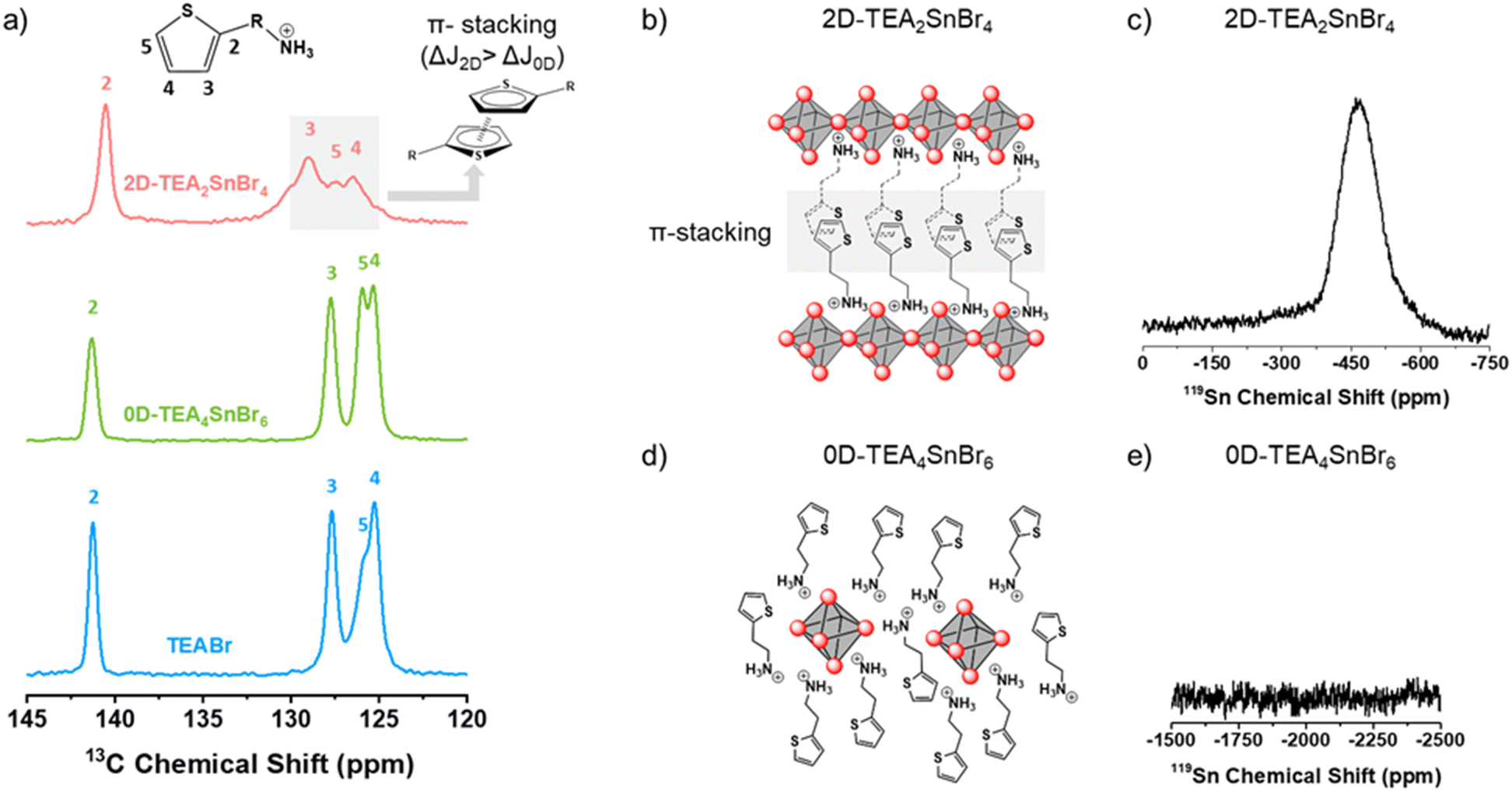 | ||
| Fig. 3 (a) 13C MAS-NMR spectra of TEABr, 0D-TEA4SnBr6, and 2D-TEA2SnBr4 powders. Crystalline structures and 119Sn NMR of (b) and (c) 2D-TEA2SnBr4 and (d) and (e) 0D-TEA4SnBr6. | ||
The 13C NMR spectra of 2D-TEA2SnBr4, 0D-TEA4SnBr6, and TEABr are shown in Fig. 3a. A set of four signals, ranging from 145 to 120 ppm, has been ascribed to the aromatic carbons of TEA+. J1, J2, and J3 signals observed in 2D-TEA2SnBr4 are shifted to a higher field (ΔJ = 1.21 ppm) than those of 0D-TEA4SnBr6 (ΔJ = 0.41 ppm) in comparison with TEABr. A characteristic signature of π-stacking interactions often manifests in changes to signal intensity and peak broadening in NMR spectra. The π-stacking interactions among aromatic rings lead to dynamic processes, causing an exchange between stacked and unstacked conformations. This equilibrium results in broader peaks in the NMR spectra due to variations in the chemical environments experienced by the interacting nuclei.
At a low [TEA]/[Sn2+] ratio, TEA+ can be distributed between layers of [SnBr6]4− octahedra, leading to the formation of a 2D tin bromide perovskite. In this structure, [SnBr6]4− octahedra are interconnected through four corner-sharing, creating 2D perovskite monolayers. Under these conditions, we can assume that thiophene rings self-assemble by π–π stacking interaction between thiophenes of adjacent 2D perovskite monolayers in 2D-TEA2SnBr4.51 This combination of structural features contributes to the unique properties and characteristics of the resulting material.52 Conversely, when the [TEA]/[Sn2+] ratio is high, the TEA+ cations form a surrounding layer around individual [SnBr6]4− octahedra, resulting in a lack of interconnection between the octahedra. The high [TEA]/[Sn2+] ratio influences the structural organization, and the isolation between [SnBr6]4− octahedra enables their strong quantum confinement effect, which has implications for the properties of the material, such as radiative recombination by self-trapped excitons.44 The TEA+ cations randomly distributed around the [SnBr6]4− octahedra reduce the π stacking interaction. As a result, the chemical shifts of aromatic carbons observed in 0D-TEA4SnBr6 are very similar to those of TEABr.53
Regarding the 119Sn MAS-NMR spectra, 2D-TEA2SnBr4 (Fig. 3d and Fig. S2, ESI†) shows two different signals at −465 ppm and −1983 ppm assigned to Sn(II) and Sn(IV) respectively,20 with a lower intensity for the latter. In the case of 0D-TEA4SnBr6, no significant signal was observed between −1000 and −2000 ppm. This is fully aligned with the better ambient stability displayed by the 0D-TEA4SnBr6 compared to its bidimensional counterpart.
Optical and structural properties of 0D-[TEASnCl3][TEACl] and 2D-TEA2SnI4 microcrystals
It is key to understand if a similar approach can be carried out to selectively control the dimensionality of the expanded TEA-based Sn(II) halide family. Microcrystals based on chloride can be synthetized with different [TEA]/[Sn2+] ratios. The XRD patterns of TEA-based Sn(II) chloride films (Fig. 4a) displayed reflection peaks at 8.63°, 17.26°, 18.09°, and 26.03° for ratios ranging from 0.125 to 1. These peaks show slight shifts compared to those of 0D-TEA4SnBr6 (2θ values of 8.52°, 17.13°, 18.23°, and 25.84°) (Fig. 2a), which is attributed to a decrease in the lattice parameters resulting from the use of chloride.54 For [TEA]/[Sn2+] ratios between 2 and 4, XRD is dominated by the reflections of the TEACl salt, which is in excess. Noticeably, PL spectra show a broad emission band between 450 and 850 nm, with a high Stokes shift energy of 2.38 eV for all the [TEA]/[Sn2+] ratios tested (Fig. S3, ESI†). This is a characteristic fingerprint of 0D TEA-based Sn(II) chloride.47,48 However, in contrast to the high values obtained for the 0D Br-based materials, the PLQY increases slightly from 14.77% (microcrystals thin films) for samples synthesized under Sn(II) rich conditions ([TEA]/[Sn2+] = 0.125) to a maximum of 25% at high TEA concentrations ([TEA]/[Sn2+] = 3). These XRD patterns and emission properties indicate that only 0D-TEA-based Sn(II) chloride can be synthesized regardless of the [TEA]/[Sn2+] ratio.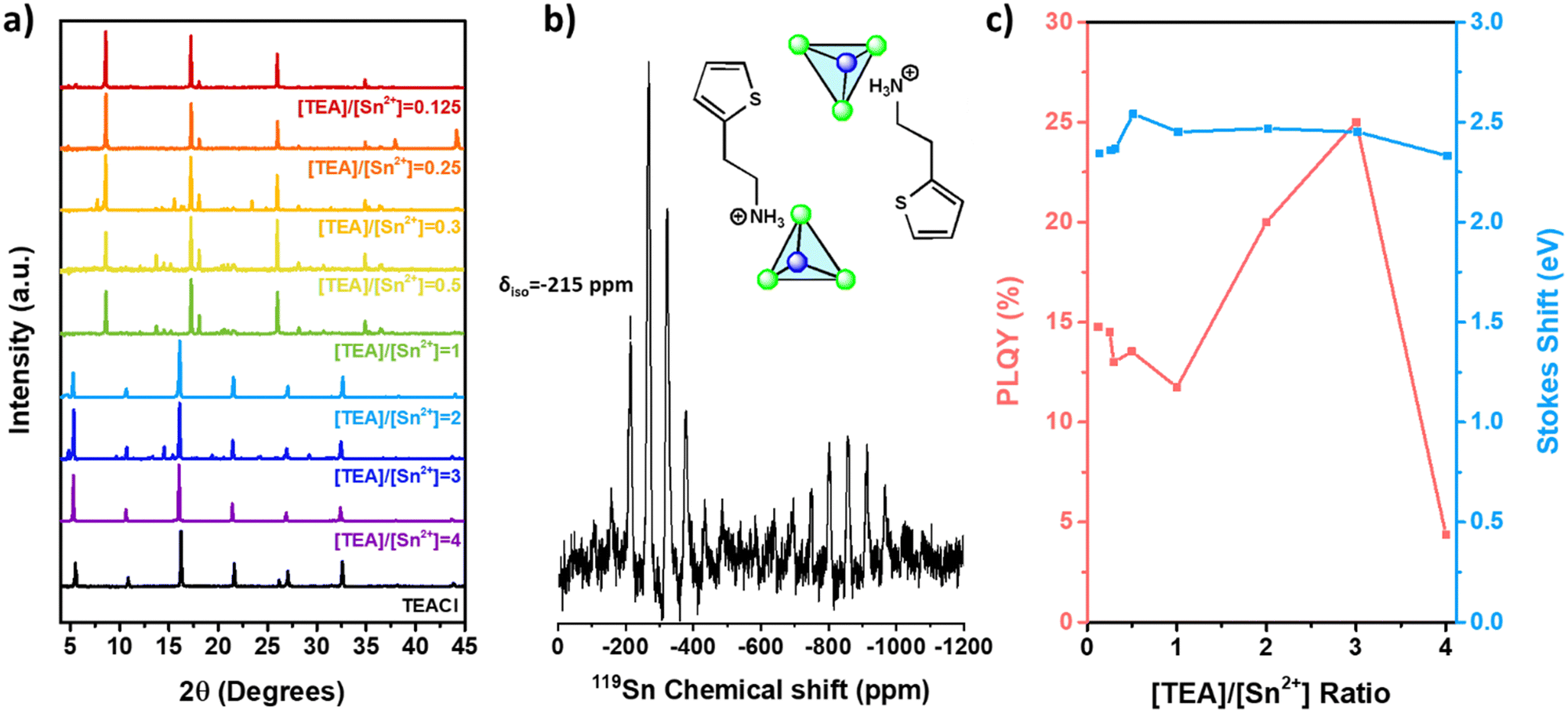 | ||
| Fig. 4 (a) XRD patterns, (b) 119Sn MAS-NMR spectra measured at room temperature (Inset: probable crystalline structure) and (c) variation of the PLQY and Stokes shift of 0D-[TEASnCl3][TEACl] microcrystals as a function of the molar ratio [TEA]/[Sn2+]. | ||
Interestingly, the 119Sn MAS-NMR spectrum of TEA-based Sn(II) chloride microcrystals (Fig. 4b) revealed a series of peaks between −100 ppm and −800 ppm related to the chemical shift anisotropy (CSA) effect (δiso = −215 ppm) of Sn(II). This can be attributed to the formation of [SnCl3]− trigonal pyramids instead of [SnCl6]4− octahedra.55 To complement 119Sn MAS-NMR measurements, EDS analysis shows that Br/Sn and S/Sn atomic ratios are 4.4 and 2.5, respectively. This may initially suggest a stoichiometry similar to the 2D counterpart. However, the CSA effect depicted in Fig. 4b indicates the presence of a low symmetry site for Sn(II). Therefore, we propose a chemical formula of 0D-[TEASnCl3] [TEACl]. A similar stoichiometry has been reported for different 0D Sn(II) chloride perovskites (A = Cs+, MA+, FA+, (CH3)3S+, C6H22N4Cl3+, and TBA+).19,20,56–58 Some authors have reported the formation of 0D-Cs2SnCl6 with high symmetry [SnCl6]2− octahedra, which showed a single signal on 119Sn NMR around −708 ppm.20,56,59–61 Additionally, other reports suggest a possible phase transition between [SnCl3]− trigonal pyramids to [SnCl6]4− octahedra for Cs-based Sn(II) chloride, with a characteristic NMR signal at −561 ppm observed after annealing the sample above 500 °C under vacuum.56,62 In our case, distinguishing between [SnCl6]4− (from phase transition) and [SnCl6]2− (produced by oxidation of Sn(II) ions) is not directly accessible due to the CSA effect observed in Fig. 4b. Moreover, the signals observed in Fig. S4 (ESI†) change in intensity at different spinning rates in NMR experiments, yet the chemical shifts are the same. As a consequence, we can conclude that TEA-based Sn(II) chloride microcrystals are synthesized as a 0D material with well-defined [SnCl3]− pyramidal anions. EDS analysis (Table S1, ESI†) is in good agreement with the proposed stoichiometry, which confirms that Sn is mainly present as Sn(II).
13C MAS-NMR spectra for 0D-[TEASnCl3][TEACl] (Fig. S5, ESI†) do not show any diminution in the signal's intensity, such as 0D-TEA4SnBr6 compounds, so π-stacking is negligible.51 In Fig. S3 and Table S3 (ESI†), multiple displacements are observed in the PL wavelength of the 0D-[TEASnCl3][TEACl] microcrystal, between 630 nm for [TEA]/[Sn2+] = 1 and 595 nm for [TEA]/[Sn2+] = 2. This displacement may be due to tilting of the [SnCl3]− tetrahedra induced by the increase in TEA+ concentration.56 The increase in the PLQY of 0D-TEA-based tin(II) chloride synthesized at a [TEA]/[Sn2+] ratio between 2 and 4, and the reflections related to TEACl could suggest the formation of [TEASnCl3][TEACl]X (where X is the number of TEACl molecules associated with the principal structure TEASnCl3).55
In the synthesis of TEA-based Sn(II) iodide, only pure 2D-TEA2SnI4 microcrystals were predominantly obtained at all the explored molar ratios (Fig. S7, ESI†), without any evidence of dimensionality change. The XRD diffraction pattern shows multiple peaks at 5.53°, 11.23°, 16.98°, 22.73°, and 28.53° and separated by a period of ∼5.7° (Fig. S7, ESI†), which indicates the formation of the Ruddlesden–Popper phase for 2D-TEA2SnI4.46 These could be confirmed with EDS analysis shown in Table S1 (ESI†). The absence of peaks was observed in the XRD patterns of 0D TEA-based tin(II) iodide. PL and PLE spectra (Fig. S7 and Table S3, ESI†) of TEA-based Sn(II) iodide display similar results for all the [TEA]/[Sn2+] ratios tested. PLE and PL spectra are composed of a narrow band at 620 nm and 660 nm, respectively, and a small Stokes shift of around 80 meV, which is characteristic of radiative recombination of free excitons. The PLQY for 2D-TEA2SnI4 microcrystals (Table S3, ESI†) obtained in this work is relatively low (PLQY = 3%) compared with the record value reported by other authors (18.85% for nanodisk solution in n-hexane46 and 23% in thin films63). This suggested the formation of multiple native superficial defects such as Sn(II) vacancies that affect the emissive properties of 2D-TEA2SnI4 microcrystals, as observed on different metal–organic and inorganic tin(II) halide nanocrystals synthesized by the hot injection method.63,64
Conclusions
In summary, the hot injection approach has proven to be an optimal platform to explore the dimensionality modulation in lead-free perovskites. Different low dimensionality 0D and 2D microplates based on 2-thiopheneethylammonium tin(II) bromide were successfully synthesized at 160 °C in low-polarity solvents. Experiments conducted at different molar ratios of precursors showed the possibility of modulating the dimensionality of 2D-TEA2SnBr4 microplates into 0D-TEA4SnBr6 with an increase of TEA molarity in solution, giving access to an orange light lead-free material with a PL emission at 610 nm, a strong Stokes shift of 2.3 eV and a high PLQY of 75% in thin films.A similar dimensional transition was not observable for 0D-[TEASnCl3][TEACl] and 2D-TEA2SnI4. It is worth noting that 0D-[TEASnCl3][TEACl] showed good optical properties with a maximum PLQY of 25%. The PLQY of 0D-TEA4SnBr6 is higher than those of 0D-[TEASnCl3][TEACl] and 2D-TEA2SnI4 because of the confinement effects and separation between inorganic moieties. Although the superior PLQYs for 0D-TEA4SnBr6 and 0D-[TEASnCl3][TEACl] are related to the recombination of self-trapped excitons, the first of them exhibited a higher stable PL emission compared to its chloride analogue. This dimensional modulation can be a useful approach to obtain and design high luminescent lead-free materials with interesting properties for practical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No. 862656 (project DROP-IT) by the Spanish MICINN through project no. PID2020-120484RB. We acknowledge the support from the Spanish MINECO through the project Nirvana (no. PID2020-119628RB-C31) by MCIN/AEI/10.13039/501100011033. Also, we acknowledge the financial support from Generalitat Valenciana through the CIDEGENT contract (ref: CIDEGENT/2021/044) and CIAPOS 2022/018 grant. The work was partially funded by MCIN/AEI through project TED2021-131600B-C32.References
- E. Aydin, T. G. Allen, M. De Bastiani, A. Razzaq, L. Xu, E. Ugur, J. Liu and S. De Wolf, Pathways toward commercial perovskite/silicon tandem photovoltaics, Science, 2024, 383, eadh3849 CrossRef CAS PubMed.
- M. Pitaro, E. K. Tekelenburg, S. Shao and M. A. Loi, Tin Halide Perovskites: From Fundamental Properties to Solar Cells, Adv. Mater., 2022, 34, 2105844 CrossRef CAS PubMed.
- M. Malekshahi Byranvand, W. Zuo, R. Imani, M. Pazoki and M. Saliba, Tin-based halide perovskite materials: properties and applications, Chem. Sci., 2022, 13, 6766 RSC.
- M. Awais, R. L. Kirsch, V. Yeddu and M. I. Saidaminov, Tin Halide Perovskites Going Forward: Frost Diagrams Offer Hints, ACS Mater. Lett., 2021, 3, 299–307 CrossRef CAS.
- Y. Lin, Y. Bai, Y. Fang, Q. Wang, Y. Deng and J. Huang, Suppressed Ion Migration in Low-Dimensional Perovskites, ACS Energy Lett., 2017, 2, 1571–1572 CrossRef CAS.
- S. Ghimire and C. Klinke, Two-dimensional halide perovskites: synthesis, optoelectronic properties, stability, and applications, Nanoscale, 2021, 13, 12394 RSC.
- B. Su, G. Song, M. S. Molokeev, Z. Lin and Z. Xia, Synthesis, Crystal Structure and Green Luminescence in Zero-Dimensional Tin Halide (C8H14N2)2SnBr6, Inorg. Chem., 2020, 59, 9962–9968 CrossRef CAS PubMed.
- L. Mao, C. C. Stoumpos and M. G. Kanatzidis, Two-Dimensional Hybrid Halide Perovskites: Principles and Promises, ACS Energy Lett., 2018, 3, 54–62 CrossRef.
- Y. Chen, Y. Sun, J. Peng, J. Tang, K. Zheng and Z. Liang, 2D Ruddlesden–Popper Perovskites for Optoelectronics, Adv. Mater., 2018, 30, 1703487 CrossRef.
- J. Hu, L. Yan and W. You, Two-Dimensional Organic–Inorganic Hybrid Perovskites: A New Platform for Optoelectronic Applications, Adv. Mater., 2018, 30, 1802041 CrossRef PubMed.
- D. B. Straus and C. R. Kagan, Electrons, Excitons, and Phonons in Two-Dimensional Hybrid Perovskites: Connecting Structural, Optical, and Electronic Properties, J. Phys. Chem. Lett., 2018, 9, 1434–1447 CrossRef CAS PubMed.
- M. G. Ju, J. Dai, L. Ma, Y. Zhou and X. C. Zeng, Zero-Dimensional Organic−Inorganic Perovskite Variant: Transition between Molecular and Solid Crystal, J. Am. Chem. Soc., 2018, 140, 10456–10463 CrossRef CAS PubMed.
- B. M. Benin, D. N. Dirin, V. Morad, M. Wçrle, S. Yakunin, G. Rainm, O. Nazarenko, M. Fischer, I. Infante and M. V. Kovalenko, Highly Emissive Self-Trapped Excitons in Fully Inorganic Zero- Dimensional Tin Halides, Angew. Chem., Int. Ed., 2018, 57, 11329–11333 CrossRef CAS PubMed.
- H. Shi, D. Han, B. Saparov, Y. Z. Ma, S. Chen, W. Ming, C. Zhou, B. Ma and M. H. Du, Unraveling luminescence mechanisms in zero-dimensional halide perovskites, J. Mater. Chem. C, 2018, 6, 6398 RSC.
- J. Yin, P. Maity, M. De Bastiani, I. Dursun, O. M. Bakr, J. L. Brédas and O. F. Mohammed, Molecular behavior of zero-dimensional perovskites, Sci. Adv., 2017, 3, e1701793 CrossRef PubMed.
- S. Seth and A. Samanta Photoluminescence, of Zero-Dimensional Perovskites and Perovskite-Related Materials, J. Phys. Chem. Lett., 2018, 9, 176–183 CrossRef CAS PubMed.
- J. L. Wardell, Tin: Inorganic Chemistry, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, 2011 Search PubMed.
- J. D. Donaldson and S. M. Grimes, in The inorganic chemistry of tin, Chemistry of Tin, ed. P. J. Smith, Springer, Dordrecht, 1998 Search PubMed.
- H. Peng, X. Wang, Z. Zhang, Y. Tian, Y. Xiao, J. Hu, J. Wang and B. Zou, Bulk assembly of a 0D organic tin(II) chloride hybrid with high anti-water stability, Chem. Commun., 2021, 57, 8162 RSC.
- D. J. Kubicki, D. Prochowicz, E. Salager, A. Rakhmatullin, C. P. Grey, L. Emsley and S. D. Stranks, Local Structure and Dynamics in Methylammonium, Formamidinium, and Cesium Tin (II) Mixed-Halide Perovskites from 119Sn Solid-State NMR, J. Am. Chem. Soc., 2020, 142, 7813–7826 CrossRef CAS PubMed.
- Y. Lin, Y. Zhong, Y. Lin, J. Lin, L. Pang, Z. Zhang, Y. Zhao, X. Y. Huang and K. Z. Du, White light emission in 0D halide perovskite [(CH3)3S]2SnCl6·H2O crystals through variation of doping ns2 ions, Front. Optoelectron., 2024, 17, 6 CrossRef PubMed.
- K. Tao, Q. Li and Q. Yan, 1D Tin (II)-Based Chiral Hybrid Perovskite Single Crystals with Extremely Distorted Inorganic Chains for Second Harmonic Generation, Adv. Opt. Mater., 2024, 2400018 CrossRef.
- Y. Dang, Y. Zhou, X. Liu, D. Ju, S. Xia, H. Xia and X. Tao, Formation of Hybrid Perovskite Tin Iodide Single Crystals by Top-Seeded Solution Growth, Angew. Chem., Int. Ed., 2016, 55, 3447–3450 CrossRef CAS PubMed.
- Y. Ju, X. Wu, S. Huang, G. Dai, T. Song and H. Zhong, The Evolution of Photoluminescence Properties of PEA2SnI4 Upon Oxygen Exposure: Insight into Concentration Effects, Adv. Funct. Mater., 2022, 32, 2108296 CrossRef CAS.
- J. N. Lv, J. Zhang, Y. M. Liu, S. Y. Zhang, X. Y. Deng, M. Xu, X. W. Lei, Z. W. Chen and C. Y. Yue, Zero-dimensional hybrid tin halides with stable broadband light emissions, Dalton Trans., 2024, 53, 4698 RSC.
- D. S. Zheng, S. B. Xiao, H. J. Yang, Z. Deng, L. J. Xu and Z. N. Chen, J. Lumin., 2024, 270, 120530 CrossRef CAS.
- S. Bonomi, M. Patrini, G. Bongiovanni and L. Malavasi, Versatile vapor phase deposition approach to cesium tin bromide materials CsSnBr3, CsSn2Br5 and Cs2SnBr6, RSC Adv., 2020, 10, 28478 RSC.
- I. Spanopoulos, I. Hadar, W. Ke, P. Guo, S. Sidhik, M. Kepenekian, J. Even, A. D. Mohite, R. D. Schaller and M. G. Kanatzidis, Water- Stable 1D Hybrid Tin(II) Iodide Emits Broad Light with 36% Photoluminescence Quantum Efficiency, J. Am. Chem. Soc., 2020, 142, 9028–9038 CrossRef CAS PubMed.
- C. C. Stoumpos, L. Mao, C. D. Malliakas and M. G. Kanatzidis, Structure−Band Gap Relationships in Hexagonal Polytypes and Low-Dimensional Structures of Hybrid Tin Iodide Perovskites, Inorg. Chem., 2017, 56, 56–73 CrossRef CAS PubMed.
- Y. E. Ajjouri, F. Locardi, M. C. Gélvez-Rueda, M. Prato, M. Sessolo, M. Ferretti, F. C. Grozema, F. Palazon and H. J. Bolink, Mechanochemical Synthesis of Sn(II) and Sn(IV) Iodide Perovskites and Study of Their Structural, Chemical, Thermal, Optical, and Electrical Properties, Energy Technol., 2020, 8, 1900788 CrossRef.
- G. Schileo and G. Grancini, Lead or no lead? Availability, toxicity, sustainability and environmental impact of lead-free perovskite solar cells, J. Mater. Chem. C, 2021, 9, 67 RSC.
- P. Zhu and J. Zhu, Low-dimensional metal halide perovskites and related optoelectronic applications, InfoMat, 2020, 2, 341–378 CrossRef CAS.
- C. Ortiz- Cervantes, P. Carmona- Monroy and D. Solis- Ibarra, Two-Dimensional Halide Perovskites in Solar Cells: 2D or not 2D?, ChemSusChem, 2019, 12, 1560–1575 CrossRef CAS PubMed.
- Y. Du, D. Zhu, Q. Cai, S. Yuan, G. Shen, P. Dong, C. Mu, Y. Wang and X. C. Ai, Spacer Engineering of Thiophene-Based Two-Dimensional/Three-Dimensional Hybrid Perovskites for Stable and Efficient Solar Cells, J. Phys. Chem. C, 2022, 126, 3351–3358 CrossRef CAS.
- K. C. Hsiao, M. H. Jao, B. T. Li, T. H. Lin, S. H. C. Liao, M. C. Wu and W. F. Su, Enhancing Efficiency and Stability of Hot Casting p–i–n Perovskite Solar Cell via Dipolar Ion Passivation, ACS Appl. Energy Mater., 2019, 2, 4821–4832 CrossRef CAS.
- J. T. Lin, D. G. Chen, C. H. Wu, C. S. Hsu, C. Y. Chien, H. M. Chen, P. T. Chou and C. W. Chiu, A Universal Approach for Controllable Synthesis of n-Specific Layered 2D Perovskite Nanoplates, Angew. Chem., Int. Ed., 2021, 60, 7866–7872 CrossRef CAS PubMed.
- H. Jia, H. Shi, R. Yu, H. Ma, Z. Wang, C. Zou and Z. Tan, Biuret Induced Tin-Anchoring and Crystallization Regulating for Efficient Lead-Free Tin Halide Perovskite Light-Emitting Diodes, Small, 2022, 18, 2200036 CrossRef CAS PubMed.
- G. Ding, X. He, H. Zhang and H. Fu, Ethanol-assisted synthesis of two-dimensional tin (II) halide perovskite single crystals for amplified spontaneous emission, J. Mater. Chem. C, 2022, 10, 10902–10907 RSC.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nanocrystals of Cesium Lead Halide Perovskites (CsPbX3, X = Cl, Br, and I): Novel Optoelectronic Materials Showing Bright Emission with Wide Color Gamut, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- Q. A. Akkerman, S. G. Motti, A. R. Srimath Kandada, E. Mosconi, V. D’Innocenzo, G. Bertoni, S. Marras, B. A. Kamino, L. Miranda, F. De Angelis, A. Petrozza, M. Prato and L. Manna, Solution Synthesis Approach to Colloidal Cesium Lead Halide Perovskite Nanoplatelets with Monolayer-Level Thickness Control, J. Am. Chem. Soc., 2016, 138, 1010–1016 CrossRef CAS PubMed.
- A. Pan, B. He, X. Fan, Z. Liu, J. J. Urban, A. P. Alivisatos, L. He and Y. Liu, Insight into the Ligand-Mediated Synthesis of Colloidal CsPbBr3 Perovskite Nanocrystals: The Role of Organic Acid, Base, and Cesium Precursors, ACS Nano, 2016, 10, 7943–7954 CrossRef CAS PubMed.
- K. M. McCall, V. Morad, B. M. Benin and M. V. Kovalenko, Efficient Lone-Pair-Driven Luminescence: Structure–Property Relationships in Emissive 5s2 Metal Halides, ACS Mater. Lett., 2020, 2(9), 1218–1232 CrossRef CAS PubMed.
- R. Chiara, Y. O. Ciftci, V. I. E. Queloz, M. K. Nazeeruddin, G. Grancini and L. Malavasi, Green-Emitting Lead-Free Cs4SnBr6 Zero-Dimensional Perovskite Nanocrystals with Improved Air Stability, J. Phys. Chem. Lett., 2020, 11, 618–623 CrossRef CAS PubMed.
- T. C. Jellicoe, J. M. Richter, H. F. J. Glass, M. Tabachnyk, R. Brady, S. E. Dutton, A. Rao, R. H. Friend, D. Credgington, N. C. Greenham and M. L. Böhm, Synthesis and Optical Properties of Lead-Free Cesium Tin Halide Perovskite Nanocrystals, J. Am. Chem. Soc., 2016, 138, 2941–2944 CrossRef CAS PubMed.
- M. Wang, J. Tang, H. Wang, C. Zhang, Y. S. Zhao and J. Yao, Grain Boundary Enhanced Photoluminescence Anisotropy in Two-Dimensional Hybrid Perovskite Films, Adv. Optical Mater., 2020, 8, 1901780 CrossRef CAS.
- J. T. Lin, C. C. Liao, C. S. Hsu, D. G. Chen, H. M. Chen, M. K. Tsai, P. T. Chou and C. W. Chiu, Harnessing Dielectric Confinement on Tin Perovskites to Achieve Emission Quantum Yield up to 21%, J. Am. Chem. Soc., 2019, 141, 10324–10330 CrossRef CAS PubMed.
- J. Almutlaq, J. Yin, O. F. Mohammed and O. M. Bakr, The Benefit and Challenges of Zero-Dimensional Perovskites, J. Phys. Chem. Lett., 2018, 9(14), 4131–4138 CrossRef CAS PubMed.
- S. Seth and A. Samanta Photoluminescence, of Zero-Dimensional Perovskites and Perovskite-Related Materials, J. Phys. Chem. Lett., 2018, 9(1), 176–183 CrossRef CAS PubMed.
- L. J. Xu, H. Lin, S. Lee, C. Zhou, M. Worku, M. Chaaban, Q. He, A. Plaviak, X. Lin, B. Chen, M. H. Du and B. Ma, 0D and 2D: The Cases of Phenylethylammonium Tin Bromide Hybrids, Chem. Mater., 2020, 32, 4692–4698 CrossRef CAS.
- J. C. Dahl, S. Niblett, Y. Cho, X. Wang, Y. Zhang, E. M. Chan and A. Paul Alivisatos, Scientific Machine Learning of 2D Perovskite Nanosheet Formation, J. Am. Chem. Soc., 2023, 145(42), 23076–23087 CrossRef CAS PubMed.
- T. Sheikh, S. Maqbool, P. Mandal and A. Nag, Introducing Intermolecular Cation-p Interactions for Water-Stable Low Dimensional Hybrid Lead Halide Perovskites, Angew. Chem., Int. Ed., 2021, 60, 18265–18271 CrossRef CAS PubMed.
- J. Wang, J. Dong, F. Lu, C. Sun, Q. Zhang. and N. Wang, Two-dimensional lead-free halide perovskite materials and devices, J. Mater. Chem. A, 2019, 7, 23563 RSC.
- L. Zhang, Z. Luo, W. Wang, Y. Liu, X. He and Z. Quan, Organic Cation-Directed Modulation of Emissions in Zero-Dimensional Hybrid Tin Bromides, Inorg. Chem., 2022, 61(37), 14857–14863 CrossRef CAS PubMed.
- M. Roknuzzaman, K. Ostrikov, H. Wang, A. Du and T. Tesfamichael, Towards lead-free perovskite photovoltaics and optoelectronics by ab-initio simulations, Sci. Rep., 2017, 7, 14025 CrossRef PubMed.
- X. Liu, Y. Li, T. Liang and J. Fan, Role of Polyhedron Unit in Distinct Photophysics of Zero Dimensional Organic− Inorganic Hybrid Tin Halide Compounds, J. Phys. Chem. Lett., 2021, 12, 5765–5773 CrossRef CAS.
- M. Ha, A. Karmakar, G. M. Bernard, E. Basilio, A. Krishnamurthy, A. M. Askar, K. Shankar, S. Kroeker and V. K. Michaelis, Phase Evolution in Methylammonium Tin Halide Perovskites with Variable Temperature Solid-State 119Sn NMR Spectroscopy, J. Phys. Chem. C, 2020, 124, 15015–15027 CrossRef CAS.
- M. M. Elsenety, A. Kaltzoglou, I. Koutselas, V. Psycharis, C. P. Raptopoulou, A. G. Kontos, K. G. Papadokostaki, N. K. Nasikas and P. Falaras, Synthesis, Crystal Structure, and Broadband Emission of (CH3)3SSnCl3, Inorg. Chem., 2022, 61, 4769–4777 CrossRef CAS.
- X. Liu, W. Wu, Y. Zhang, Y. Li, H. Wu and J. Fan, Critical Roles of High- and Low-Frequency Optical Phonons in Photodynamics of Zero-Dimensional Perovskite-like (C6H22N4Cl3) SnCl3 Crystals, J. Phys. Chem. Lett., 2019, 10, 7586–7593 CrossRef CAS PubMed.
- A. Karmakar, S. Mukhopadhyay, P. G. B. Gachod, V. A. Mora-Gomez, G. M. Bernard, A. Brown and V. K. Michaelis, Uncovering Halogen Mixing and Octahedral Dynamics in Cs2SnX6 by Multinuclear Magnetic Resonance Spectroscopy, Chem. Mater., 2021, 33, 6078–6090 CrossRef CAS.
- A. Kaiba, M. H. Geesi, Y. Riadi, E. O. Ibnouf, T. A. Aljohani and P. Guionneau, A new Organic–Inorganic hybrid compound (NH3(CH2)2C6H5)2[SnCl6]: Crystal structure, characterization, Hirshfeld surface analysis, DFT calculation, vibrational properties, and biological evaluation, J. Solid-State Chem., 2021, 304, 122587 CrossRef CAS.
- J. Li, Z. Tan, M. Hu, C. Chen, J. Luo, S. Li, L. Gao, Z. Xiao, G. Niu and J. Tang, Antimony doped Cs2SnCl6 with bright and stable emission, Front. Optoelectron., 2019, 12(4), 352–364 CrossRef.
- A. Karmakar, A. Bhattacharya, D. Sarkar, G. M. Bernard, A. Mar and V. K. Michaelis, Influence of hidden halogen mobility on local structure of CsSn(Cl1-xBrx)3 mixed-halide perovskites by solid-state NMR, Chem. Sci., 2021, 12, 3253 RSC.
- J. T. Lin, Y. K. Hu, C. H. Hou, C. C. Liao, W. T. Chuang, C. W. Chiu, M. K. Tsai, J. J. Shyue and P. T. Chou, Superior Stability and Emission Quantum Yield (23% ± 3%) of Single-Layer 2D Tin Perovskite TEA2SnI4 via Thiocyanate Passivation, Small, 2020, 16, 2000903 CrossRef CAS.
- Q. Liu, J. Yin, B. B. Zhang, J. K. Chen, Y. Zhou, L. M. Zhang, L. M. Wang, Q. Zhao, J. Hou, J. Shu, B. Song, N. Shirahata, O. M. Bakr, O. F. Mohammed and H. T. Sun, Theory-Guided Synthesis of Highly Luminescent Colloidal Cesium Tin Halide Perovskite Nanocrystals, J. Am. Chem. Soc., 2021, 143, 5470–5480 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc00623b |
| This journal is © The Royal Society of Chemistry 2024 |