
Maneuvering the mineralization of self-assembled peptide nanofibers for designing mechanically-stiffened self-healable composites toward bone-mimetic ECM†
Nimisha A.
Mavlankar
a,
Debasish
Nath
a,
Yadu
Chandran
b,
Nidhi
Gupta‡
 a,
Ashmeet
Singh
a,
Viswanath
Balakrishnan
b and
Asish
Pal
*a
a,
Ashmeet
Singh
a,
Viswanath
Balakrishnan
b and
Asish
Pal
*a
aChemical Biology Unit, Institute of Nano Science and Technology, Knowledge City, Sector – 81, Mohali, Punjab, India. E-mail: apal@inst.ac.in
bSchool of Mechanical and Materials Engineering, Indian Institute of Technology-Mandi, Kamand, Himachal Pradesh, India
First published on 29th July 2024
Abstract
Extracellular matrix (ECM) elasticity remains a crucial parameter to determine cell–material interactions (viz. adhesion, growth, and differentiation), cellular communication, and migration that are essential to tissue repair and regeneration. Supramolecular peptide hydrogels with their 3-dimensional porous network and tuneable mechanical properties have emerged as an excellent class of ECM-mimetic biomaterials with relevant dynamic attributes and bioactivity. Here, we demonstrate the design of minimalist amyloid-inspired peptide amphiphiles, CnPA (n = 6, 8, 10, 12) with tuneable peptide nanostructures that are efficiently biomineralized and cross-linked using bioactive silicates. Such hydrogel composites, CnBG exhibit excellent mechanical attributes and possess excellent self-healing abilities and collagen-like strain-stiffening ability as desired for bone ECM mimetic scaffold. The composites exhibited the formation of a hydroxyapatite mineral phase upon incubation in a simulated body fluid that rendered mechanical stiffness akin to the hydroxyapatite-bridged collagen fibers to match the bone tissue elasticity eventually. In a nutshell, peptide nanostructure-guided temporal effects and mechanical attributes demonstrate C8BG to be an optimal composite. Finally, such constructs feature the potential for adhesion, proliferation of U2OS cells, high alkaline phosphatase activity, and osteoconductivity.
1. Introduction
Bone defects caused by fractures, diseases, or inherited anomalies involve several clinical complexities in both tissue repair to replacement and regeneration.1,2 Although regeneration approaches avoid the risks associated with grafting, such as immunogenicity, graft rejection, and slow and imperfect reconstruction of critical bone, the defects associated with them necessitate the development of reliable alternative biomaterials.3–5 In that regard, designing smart biomaterial to perform guided bone regeneration has led to the fabrication of promising bone substitutes, such as bioceramics, nanohydroxyapatite, sintered TCP, β-wollastonite etc.6–8 The ideal bone cementing materials must be injectable to fit into the irregular-sized defects and should possess load-bearing and adhesion abilities to bind to the bones.9 Recently, bioactive glass has emerged as a viable third-generation scaffold for bone regeneration with the basic requirements of being bioinert, biocompatible, and most importantly providing active and responsive synthetic hydroxyapatite for osteogenesis, osteoinduction, and osteoconduction.10–12 Moreover, compared to synthetic hydroxyapatite, bioglass possessing a bioactivity index of >8 exhibits superior properties, such as binding to both soft and hard surfaces, bone regeneration, and unique genetic activation,13 which leads to greater translation with over 25 clinically approved products in bone, teeth, and wound healing domains.14,15 However, approaches of templating bioglass with ceramic or biopolymeric nano/micro-architectures encounter several synthetic and degradability issues, including loss of bone dynamics upon covalent binding. This necessitates an alternative dynamic template that can overcome the aforementioned challenges and offer bone-matching mechanical strength with good in vitro and in vivo activity.Biomineralized collagen constitutes the majority of extracellular matrix (ECM) proteins that surround and support bone tissue and provide it tensile strength,16 while structural density, crosslinking, and porosity of collagen fibers help maintain the dynamic nature of ECM and its homeostasis.17 In that regard, due to their supramolecular polymerization, peptides can potentially encode specific cues that are unique to natural biopolymeric systems that mimic ECM for cell adhesion, migration, spreading, proliferation, and direct cell lineage, and can participate in cell signalling pathways as well.18–20 Moreover, mechanotransduction becomes a vital cue for cell cycle activation of the osteoprogenitor cells for bone repair.21–24 Guler et al. demonstrated how the chirality and morphology of ECM-mimicking peptide nanofibers can regulate the mechanosensory response of stem cells towards varying osteogenic differentiation.25 Park, Stupp, Adler-Abramovich et al. used peptides as templates for calcium phosphate biomineralization and reported interesting findings related to the role of the nano/micro hierarchical architecture related to osteogenic potential.26–29 Mineralization of metals/inorganic components on peptides thus represents an attractive functional material approach.30–33 Recently, our group demonstrated an elegant design of stimuli-responsive peptide nanostructures with precision strategy34 that was exploited for achieving ECM-mimicking hydrogels,35,36 antibacterial activity,37 and catalytic amyloids.38,39 Thermal cue-mediated control over peptide nanostructures exhibits a tuneable control over bioactive glass formation.40 In another work, we showed an interpenetrating network from a peptide–polymer dynamic composite to show mechanical stiffness and load-bearing ability for chondrocyte proliferation.41 Often, the non-linear mechanical properties of such interpenetrating networks, such as strain-stiffening characteristics, play a vital role in guiding cell adhesion and migration by matching matrix elasticity and stiffness.42–44
However, mimicking such cues in a synthetic model capable of matching optimal mechanics, surface chemistry and bioactivity is quite a challenging task. To control the bioactivity of hybrids derived from self-assembling peptides, it is essential to have precise control over the peptide templates. The selection of an ideal self-assembled peptide template capable of generating a biocompatible, biodegradable nano–micro hierarchical hybrid network lies in determining its optimum parameters, such as hydrophobicity, flexibility, and network-forming capability.45,46 In this work, we attempt to design a peptide-templated bioglass hybrid possessing strain-stiffening ability as a quintessential cue in mechanotransduction to not only match bone mechanics but also show active responsive surface chemistry and bioactivity. What sets these systems apart from other bone biomaterials is that they demonstrate superior mechanical attributes, in addition to their capability to circumvent the limitations that are typically associated with dynamic self-healing and brittleness in calcium phosphate cement and conventional polymer-templated bioglasses. Moreover, the exceptional ability of surface chemistry mediation by hydrophobicity to influence material properties extends beyond mere self-assembly and significantly impacts bioactivity, thereby offering promising insights into the function of bone-mimicking biomaterials.
Herein, we rationally designed a series of self-assembling peptides CnPA (n = 6, 8, 10, 12) from the nucleating core of Aβ (1–42) amyloid differing in the hydrophobicity at their N-termini to furnish tuneable nanofiber templates for in situ mineralization of bioactive glass CnBG (Scheme 1). Such organic–inorganic hybrids constitute a strong calcium silicate network with varied pore size, morphology, and mechanical strength as modulated by the spatio-temporal assembly of the peptide templates. Interestingly, the composites exhibit excellent self-healing behaviour and nonlinear mechanical stiffening, as well as compression load bearing akin to collagen biopolymers. These surface-responsive hydrogels generated hydroxyapatite on dissolution in simulated biological fluids that in turn facilitate matrix mineralization and cell proliferation of the osteoblast U2OS cells. Taken together, all such structural variations and functional implications among the CnBG series revealed C8BG to be an ideal composite with respect to the highest osteogenic response. Such artificially designed ECM mimetic scaffolds capable of matching bone microenvironment and mechanics are a step forward for developing implant coatings with broader relevance in bone tissue engineering.
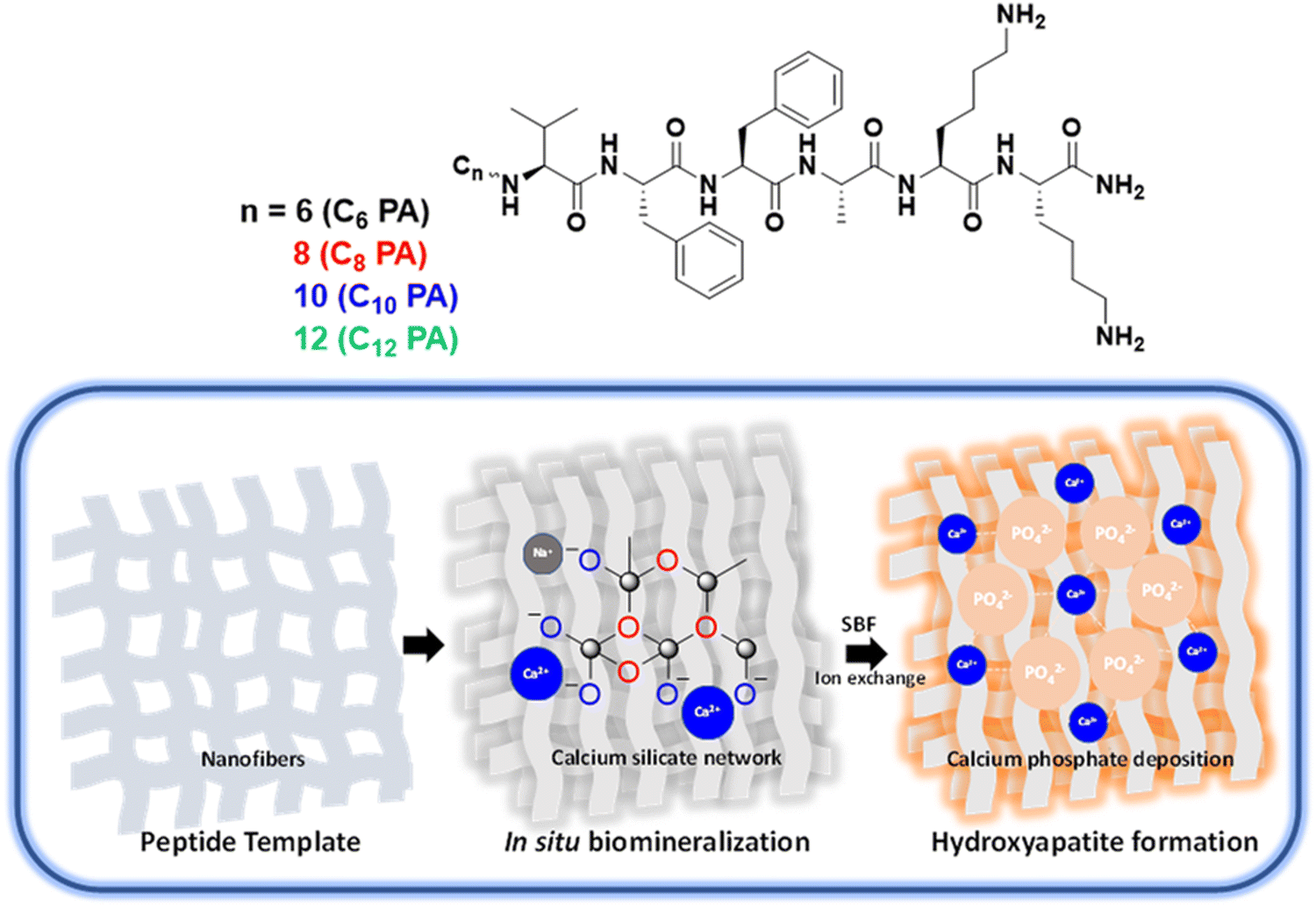 | ||
| Scheme 1 (A) Molecular structure of the amyloid Aβ42-inspired peptide amphiphiles, CnPA composed of hydrophobic chain (n = 6, 8, 10, 12), β-sheet forming VFFA peptide segment and polar lysine segment. (B) Schematic representation of peptide-templated biomineralization of bioactive glass and eventual hydroxyapatite formation upon immersion in simulated body fluid (SBF). | ||
2. Materials and methods
2.1. Design of amphiphilic peptides and self-assembly
Inspired by the nucleating core of Aβ42 (16–22) amyloid, we synthesized ‘NVFFAC’ series of peptides with two lysine motifs flanked at the C-terminal and aliphatic fatty acid spacers from C6–12 at the N-terminal (cf. ESI†). Self-assembly of the amphiphilic peptides was studied by incubating 1 mM peptide at 30 °C unagitated over two days in water and analysed for its fiber formation capability. The effect of ionic strength on self-assembly was assessed using sodium phosphate buffer (SPB) of pH = 8 in sub-stoichiometric and buffering concentrations.2.2. Mineralization of nanofibers to form bioactive glass
To the aqueous solution of peptide nanostructures, different bioglass precursors, namely tetraethyl orthosilicate (TEOS) (223 mM), triethyl phosphate (TEP) (27 mM), sodium sulphate (112 mM), and calcium sulphate (115 mM), were added sequentially at an interval of 30 min at room temperature. The hydrogel composite was formed upon crosslinking the peptide-bioglass fibers with 15 mM SPB (pH 8). The whole system was then incubated for 48 h to complete the in situ mineralization process as mediated by the cross-network formation.2.3. Fourier transform infrared spectroscopy (FTIR)
Bruker Vertex 70 FTIR spectrophotometer was used to record FTIR spectra in the range of 4000 to 400 cm−1 as an attenuated total reflectance (ATR) mode and analysed through Opus Software. Lyophilized peptide/peptide–BG composites were used for structural elucidation. FTIR measurements were carried out with 128 scans while each sample was measured thrice to ensure reproducibility.2.4. Circular dichroism spectroscopy (CD)
CD spectra were recorded using JASCO J-1500 Circular Dichroism Spectrometer, Easton, MD, USA. The wavelength ranged from 190 nm to 350 nm with a scan speed of 200 nm min−1. Spectra were collected using a 2-mm path-length quartz cuvette to record an average of three acquisitions. Secondary structure estimation was computed using Reed's reference on the protein secondary structure estimation using Spectra Manager software.2.5. Fluorescence spectroscopy
Fluorescence emission spectra were recorded using an FS5 spectrofluorometer from Edinburgh instruments. Spectra were recorded with a scanning slit and offset slit of 2 nm each with a dwell time of 0.1 s at 25 °C. Thioflavin-T binding assays were performed to monitor the kinetics of self-assembly using 0.5 mM peptide and 0.025 mM ThT solution (λEx = 440 nm and λEm = 485 nm). Three replicates were included in the experiment.2.6. Raman scattering spectroscopy
Lyophilized powder samples were used to acquire Raman spectra with a WiTEC alpha300R microscope in Raman mode using a 532-nm (Elaser = 2.33 eV) Nd:YAG laser (power ∼75 mW) and 20× (numerical aperture 0.9). Raman data were collected by the WiTEC control software and analysed in the WiTEC project software. Spectra were collected using an integration time of 1 s. The spectra were replicated three times.2.7. X-ray photoelectron spectroscopy (XPS)
XPS spectroscopy was performed on a KAlpha plus XPS system by Thermo Fischer Scientific instruments in an ultrahigh vacuum chamber (7 × 10−9 Torr) using Al-Kα radiation (1.486.6 eV) to know the chemical states at the sample surface and analysed through Advantage software. Samples were prepared by drop casting 20 μL of lyophilized peptide–BG composites (0.1 w/v%). The data is an average of three replicates.2.8. Atomic force microscopy (AFM)
AFM imaging was performed on a Bruker Multimode 8 scanning probe microscope with a silicon cantilever (force constant of 40 N m−1). First, 10 μL of 0.05 mM peptide solutions were drop-cast on a silicon wafer and AFM height images were recorded using tapping mode at scan rates of 0.5–0.7 Hz and were analysed using Nanoscope Analysis 1.4 software. Imaging was conducted on each sample in three different areas to confirm the fiber morphology, while height profiles of 25 different fibers were averaged to confirm the height of fibers.2.9. Transmission electron microscopy (TEM)
An aliquot (6 μL) of diluted peptide bioglass composite gel (0.005 w/v%) was drop-cast on a 200-mesh carbon-coated copper grid. After ∼5 min, excess solution was blotted using a Whatman filter paper from all edges of the grid carefully and dried under vacuum. TEM images and selected area electron diffraction (SAED) patterns were recorded using JEOL JEM 2100 with a tungsten filament at an accelerating voltage of 120 kV. Imaging and measurements were conducted on each sample in three different areas to confirm the uniformity of the sample.2.10. Scanning electron microscopy (SEM)
SEM images were recorded on a JEOL JSM-IT300 attached to an energy-dispersive X-ray spectroscope for elemental determination and quantification. Ten microliters of 0.05 w/v% peptide bioglass composite gel were drop-cast on double-sided carbon tape, followed by platinum/gold coating in the sputtering machine. Images were collected under vacuum using 5 kV accelerating voltage and EDX under 15 kV accelerating voltage with an optimum working distance adjusted for optimal image quality. All EDX measurements were recorded in triplicates and averaged to provide the % Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P ratio.
:P ratio.
2.11. Brunauer–Emmett–Teller (BET) analysis
Autosorb IQ Quantachrome was used to assess the textural parameters of the lyophilized bioglass composite by first degassing the samples under vacuum for 6 h at 423.15 K. Analysis was performed at 77 K by the adsorption–desorption of N2 (cross-sectional area 0.162 nm2) using the instrument. Sample surface area and pore size distribution were calculated using Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) equation. Data were collected and averaged from three replicates.2.12. X-ray diffraction spectroscopy (XRD)
Powder X-ray diffraction (PXRD) studies were performed for the phase determination on lyophilized powder samples using Bruker D8 Advance X-ray diffractometer having Cu-Kα radiation (Ni-filtered) with a wavelength of 1.54 Å at a scan rate of 0.017 s−1 and a step time of 0.30 s. The sample size was n = 3.2.13. Rheology
Rheological investigations were performed on Anton Paar Rheometer MCR 302 equipped with Rheocompass software using parallel plate (PP-25) geometry at a measuring distance of 0.2 mm at 37 °C.47,48 For all measurements, the measuring plate area was covered with a solvent trap to avoid evaporation of the solvent during a longer experiment time. Each experiment was conducted thrice for reproducible results. Frequency sweep oscillatory rheology was performed from 0.01 to 100 rad s−1 at a constant 1% strain in the linear viscoelastic region. Amplitude sweep studies were performed from 0.01% to 200% strain at 10 rad s−1 to find the cross-over point, critical strain, and yield stress. The self-healing ability of the hydrogels was evaluated by dynamic thixotropic studies upon subjecting the gel network to three consecutive cycles of high (100%) and low strain (1%) at a constant angular frequency of 10 rad s−1. The load-bearing ability of the composites was then quantitatively evaluated through stress vs. strain analysis. For nonlinear rheological studies, a parallel superposition of steady pre-stress (σ) along with 10% of oscillatory stress at a constant angular frequency of 6.28 rad s−1 gave the corresponding oscillatory strain (Δγ). The average linear G′ readings obtained from the corresponding flow curve impart a differential moduli, K′ (δσ/δγ), which was plotted against stress, σ. values to estimate the strain-stiffening behaviour.2.14. Nanoindentation
Nanoindentation measurements were performed in duplicates on a calibrated TI 950 Triboindenter by Bruker using a flat punch nanoindenter (diameter = 490 μm). Samples were prepared by drop casting 50 μL of 1 w/v% sample (native peptide/biomineralized sample/SBF treated sample) on fresh silicon wafer and air drying to minimize any crack formation (dried film thickness ∼3 μm). To alleviate surface roughness, the samples were subjected to increasing load from 25–175 μN at intervals of 25 μN. Load displacement curves were measured at a maximum load of 175 μN (maximum displacement <10% of sample thickness to avoid any substrate effects) and a load profile of 5 s–2 s–5 s with the sequence of load–hold–unload. The hardness (H) and reduced modulus (Er) were then computed using Triboscan software from the unloading contact stiffness and the indenter contact area (Ac) based on the Oliver–Pharr theory.492.15. Bioactivity test
As a bone cementing material, the primary bioactivity of the bioglass scaffold was evaluated by its ability to generate bone-mimicking surfaces in contact with body fluids. Hydroxyapatite formation was applied at the surface of bioglass in the presence of simulated body fluid (SBF) as a marker to evaluate in vitro bone-forming activity. To check the bioactivity of the CnBG, hydrogels were immersed in Hanks’ Balanced Salt solution that matches the ionic composition of human blood plasma for 7, 15, and 25 days on a shaker at 37 °C under sterile conditions. The formation of hydroxyapatite was monitored on the samples of the surface through TEM, SEM-EDX, XRD, and FTIR, to evaluate the bone-forming activity of the samples. Data represented were the mean of triplicate experiments with less than 0.005% standard deviation.2.16. Cell culture studies
MTT assay was carried out to evaluate the biocompatibility of the bare peptides and bioglass composites. Cell viability and cell proliferation were examined by taking the gels in 96-well plates under sterile conditions at 37 °C, followed by incubation in DMEM with 10% FBS for 24 h in a 5% CO2 incubator to optimize the conditions for growth. The following day, media were replaced with fresh FBS medium containing pre-counted 5000 U2OS cells. The images of the cells were recorded using an Invitrogen bright-field microscope. A quantitative assessment of cellular proliferation within the hydrogel matrix was conducted using the Alamar Blue assay. Live/dead staining was performed using fluorescein diacetate (FDA) and propidium iodide (PI), and the images were captured using confocal laser scanning microscopy (CLSM).Alizarin red-S (ARS) staining was used to study the osteogenic differentiation of U2OS cells by growing U2OS cells with up to 80% confluency on the bioglass matrix for 14 days. Media were changed every 3 days, and the cell monolayer was stained with ARS solution in ddH2O, washed with cold phosphate buffer saline, and ethanolic destaining was performed to finally visualize the calcium deposits.
For the alkaline phosphatase (ALP) assay, U2OS cells were seeded on each scaffold for 7 days followed by staining according to the manufacturer's instruction (alkaline phosphatase assay kit, Immunotag). The scaffolds were washed with phosphate-buffered saline thrice and lysed in 1 mL of assay buffer, sonicated, and centrifuged at 8000 rpm at 4 °C for 10 min. The supernatant was kept on ice, with the sequential addition of 40 μL of the reaction buffer and substrate followed by 90 μL of reagent 1 and 20 μL of reagent 2 as instructed for the kit. The solution was incubated for 15 min at 37 °C, and the absorbance was recorded at 510 nm. Statistical analyses were conducted on datasets derived from a sample size of three, and results are presented as mean ± standard deviation (SD). Data analysis was performed using GraphPad Prism 6. Statistical significance was determined by applying one-way ANOVA followed by Tukey's tests, considering a 95% confidence interval. Results were deemed statistically significant when P ≤ 0.05.
3. Results and discussion
3.1. Self-assembly of peptide nanofibers
We designed peptide amphiphiles, C6PA, C8PA, C10PA, and C12PA, with an Aβ (1–42) amyloid-inspired VFFA nucleating core tethered to fatty acid chains of varying lengths from hexanoic acid (C6) to dodecanoic acid (C12) at the N termini. The lysines at the C termini provided the optimal hydrophobic/hydrophilic balance to render them amphiphilic, which influenced the self-assembly with respect to fiber formation propensity via mediation by π–π stacking of phenylalanine, hydrogen bonding along the amide motifs, and van der Waals interactions among the hydrophobic chains.We studied the self-assembly of the CnPA peptides (1 mM) in aqueous media by varying ionic strengths (zero, substoichiometric, and excess) with sodium phosphate buffer adjusted to pH 8. The secondary structure of CnPA was evaluated using time-dependent CD spectra under three different conditions: (a) water, (b) with 0.5 mM sodium phosphate buffer (pH = 8), and (c) with 5 mM sodium phosphate buffer (pH = 8) (Fig. 1A–C and Fig. S2, ESI†). C6PA exhibited a random-coil signature in water that remained unaltered upon increasing ionic strength even after 48 h. Similarly, C8PA showed a random-coil signature in water and lower ionic strength of buffer; however, at higher ionic strength C8PA exhibited intense bisignate β-sheet signature with negative and positive Cotton bands at 218 nm and 198 nm, respectively, suggesting that hydrophobicity and ionic strength concomitantly drive the self-assembly pattern. Further, C10PA and C12PA with hydrophobic fatty acid chains demonstrated majorly random coil signatures in water; nevertheless, a lower ionic strength was sufficient for them to assemble into β-sheets with characteristic bisignate cotton bands at 218 nm and 201 nm. Thus, the assemblies exhibited the absence of a peak at 218 nm (n → π* electronic transition of peptide backbone) owing to suboptimal experimental conditions for the peptides to self-assemble that were facilitated via increased ionic strength (Fig. 1A–C). Next, we investigated the kinetics of self-assembly by monitoring the ellipticity at 201 nm and 218 nm (Fig. 1D–F and Fig. S2, ESI†). Thus, with time, θ201 exhibited sigmoidal growth for only C12PA in water indicating nucleation-elongation characteristics. However, upon increasing the ionic strength, both C12PA and C10PA showed faster self-assembly kinetics as seen from the time-dependent θ218 plots. Interestingly, at higher ionic strength of SPB, C8PA exhibited optimal hydrophobicity and a much higher increase in ellipticity at 218 nm. As the peptide C8PA assembly matured with time, we also noticed a sudden increase in the chiral signal at 201 nm that might account for late-stage error correction mechanisms via dynamic monomer exchange to render a highly ordered β-sheet arrangement (Fig. S2, ESI†).50 This indicated the role of VFFA fragments and hydrophobic chains in promoting self-assembly. To understand the contribution of ionic strength, we analysed the CD data using Reed's method to estimate the percentage of secondary structures present in these peptides after 48 h (Fig. S3, ESI†).51 Thus, at higher ionic strengths, while 100% of the C6PA appeared in random coil orientation, a significant increase in the β-sheet content was observed in C8PA (10.8% in water to 68.1% at high ionic strength). However, we did not notice such an increase for C10PA or C12PA, indicating that this synergistic behaviour is specific to C8PA. We propose that on increasing the ionic strength, C8PA achieved a perfect balance between VFFA core stacking and packing among the hydrophobic tails. However, in C10/C12 chains, interactions among the hydrophobic tails began to dominate, thereby deviating further from the optimal balance required for an ordered assembly. Such a trend in β-sheet content and kinetics of self-assembly for CnPA was further corroborated using a Thioflavin T (ThT) binding assay that showed an increase in the ThT emission at 485 nm depending on the ionic strength and hydrophobic tails of CnPAs akin to the CD spectral analysis (Fig. S4, ESI†). Both the ThT and CD results offered a detailed understanding of the kinetics of the hierarchical assembly process as mediated by the hydrophobic chains and peptide segments to render nanoscale fibrillar organization. Next, we monitored the morphological differences in the fibrillation of CnPA using atomic force microscopy. Thus, C6PA formed short polydisperse nanofibers (height 10 ± 2 nm); C8PA assembled into twisted bundles (height 3 ± 1 nm) while C10PA and C12PA assembled into fibers of 8 ± 1 and 5 ± 1 nm height, respectively (Fig. 1G–J). The twisted bundle morphology of C8PA observed in the height profile (Fig. S5, ESI†) could be corroborated with its intense CD spectra validating highly ordered assembly. Thus, we exploited such nanofibers to form an interpenetrating 3D porous hydrogel network. For hydrogel formation with native peptides, we utilized 1 w/v% of CnPA that was self-assembled for 48 hours and crosslinked using salt–bridge interactions with 15 mM SPB to form hydrogels.
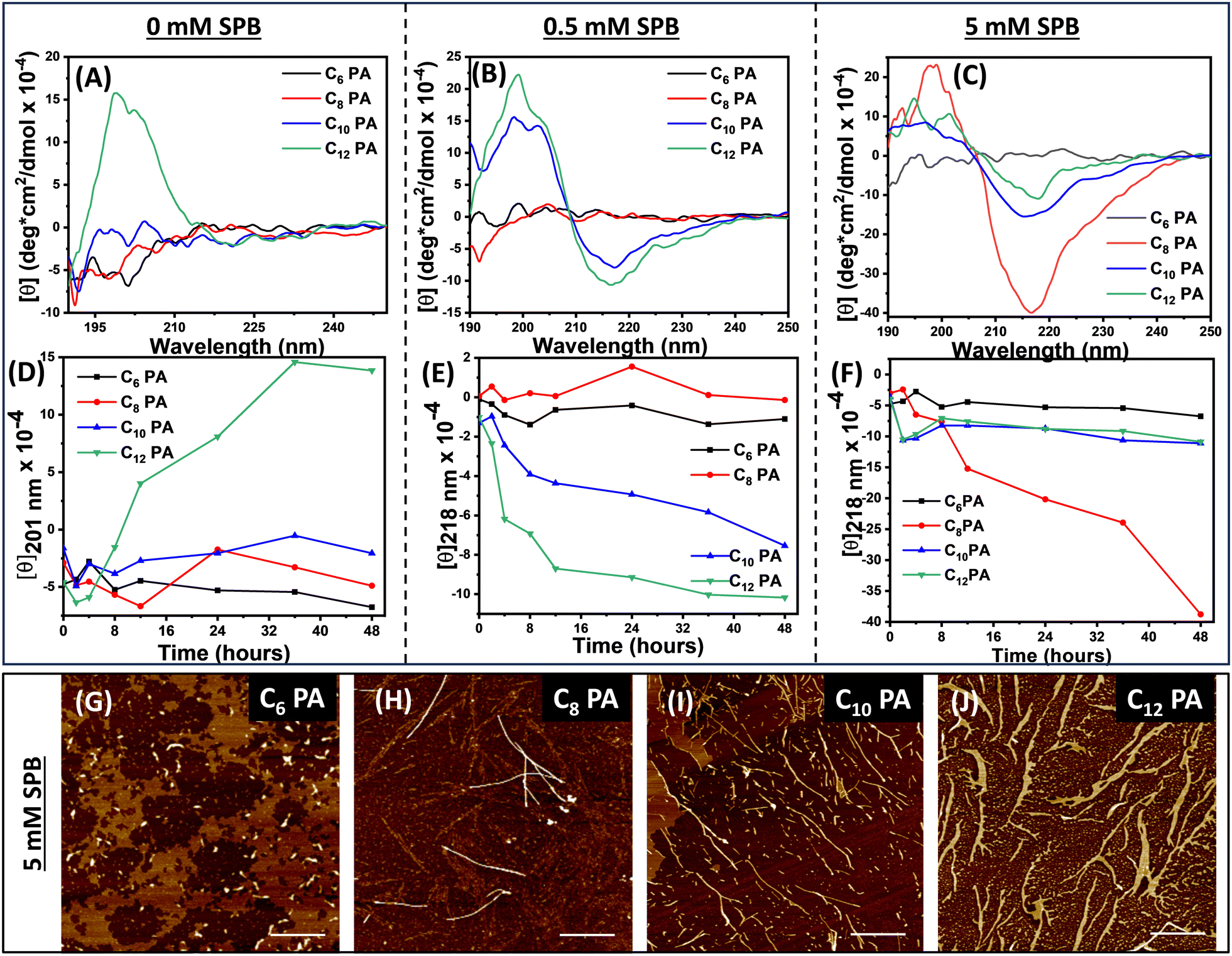 | ||
| Fig. 1 Self-assembly and kinetics for CnPA peptides (n = 6, 8, 10, 12) in SPB solutions of varying ionic strength after 48 h. (A)–(C) CD spectra and (D)–(F) ellipticity plots at 201 nm and 218 nm depicting the evolution of CD signal with growing assemblies monitored for 48 h, (G)–(J) AFM images showing nanostructures of CnPA after incubation in the high ionic strength of the SPB solution for 48 h (scale bar = 1 μm). (c = 0.05 mM). | ||
3.2. Mineralization and self-assembled peptide nanofibers as templates for bioactive glass
Next, the peptide nanofibers, CnPA, in water (8.6 mM) were utilized as a template for bioactive glass synthesis to eventually produce CnBG composites. Using the peptide nanostructures as a template, a silicate network was constructed with tetraethylorthosilicate to furnish glassy composites having continuous Si–O–Si bonding. Triethyl phosphate (TEP) facilitated the glass network formation by incorporating phosphorous into the siloxane network, which aided the desirable degradation behaviour, release kinetics, and bioactivity. Subsequently, we introduced sodium and calcium ions to enhance the interaction with the open structure of the Si–O− bonds (oxygens not bridged to two silicon atoms henceforth termed as non-bridging oxygen, NBO). The presence of phosphate and sulphate counter ions ensured the formation of Ca–(Na)–P clusters and better cross-linking within the peptide nanofibers and between the peptide template and silicate network (Fig. S6, ESI†). Finally, we added sodium phosphate buffer (15 mM; pH 8), followed by incubation at 25 °C for 48 h to complete the in situ mineralization process and furnish CnBG hydrogels (Fig. 2A). The templating process involved an array of non-covalent and covalent interactions, viz. electrostatic interactions, between protonated lysine amines and silicates/sulphates, hydrogen bonding, van der Waals interactions through peptide backbone with silica, ionic interactions via alkali/alkaline earth metals, and covalent siloxane linkages. We investigated the mineralized hydrogels for their structural, morphological, and textural characterization to get a clear insight into the mineralization process. The twisted bundle morphology of C8PA resembled a highly ordered assembly with tight packing confers as its unique features that were also true for its corresponding bioactive glass, C8BG, in contrast to other peptide templated bioglass hybrids (CnBG). Comparative FTIR spectra of CnPA and CnBG showed decreased intensity and peak shifting for the characteristic amide I peak at 1629 cm−1 and amide II peak at 1541 cm−1 of the CnPA upon CnBG composite formation (Fig. 2B). Emergence of new peaks corresponding to Si–O–Si (stretch at 1107 cm−1 and bend at 459 cm−1), ring structure of the silicate network (798 cm−1), and P–O (619 cm−1) bond along with Si–O (non-bridging oxygen NBO) at 995 cm−1 indicated the formation of a silicate network in the bioglass. However, the weaker intensity of peaks at 798 cm−1 and 619 cm−1 accounted for the less symmetric bending vibrations of Si–O–Si and lower concentration of TEP in the bioglass, respectively. Comparative structural analysis with PXRD suggested a transformation of the amorphous peptides, CnPA (Fig. S7, ESI†), into crystalline biomineralized composites of CnBG (Fig. 2C). Thus, the peaks at 11.68, 32.07, 29.33, and 21.07 indicated a presence of Ca2SiO4 and CaSiO3 phases and confirmed the incorporation of Ca2+ ions into the silica network of the bioglass. Raman spectra of all the CnPA showed sharp peaks at 1618 cm−1 and 1033 cm−1 for anti-symmetric bending of NH3+ and C–C band, respectively, in lysine. In addition, characteristic peaks of phenylalanine (1005 cm−1 and 1160 cm−1), valine (CH3 bending 1452 cm−1), and alanine (C–C stretch 1350 cm−1) were observed, along with carbonyl stretching (1677 cm−1) (Fig. S8, ESI†). However, upon biomineralization to CnBG, the decrease in intensity of the Raman peaks at 1033 and 1618 cm−1 indicated lysine to be a high-propensity site for the mineralization process (Fig. 2D).40 The surface chemistry in bioactive glasses prompted us to conduct detailed surface analysis with X-ray photoelectron spectroscopy that confirmed the presence of Si, O, P, S, Ca, Na, N, and C in all the CnBG samples (Fig. 2E and Fig. S9A–D, ESI†). Notably, the Si 2p photoelectron spectra exhibited a marginal increasing trend of binding energy from 103.34 eV for C6BG to 104.42 eV for C12BG. This indicated relatively higher network modifier content in C6BG that could result in a higher proportion of non-bridging oxygen. The deconvoluted XPS spectra demonstrated characteristic peaks for bridging and non-bridging oxygens and their ratio (Fig. S9E–H (ESI†) and Table 1). The presence of such non-bridging oxygen (NBO) facilitated an open network structure allowing for Si–O–Na and Si–O–Ca bonding sites that could eventually result in faster release of ions or a larger mineral phase deposition. | ||
| Fig. 2 (A) Schematic representation of the bioglass fabrication process and the structural network formed. (B) FTIR spectra of the peptides, CnPA, and corresponding bioglass composites, CnBG. (C) XRD spectra of the crystalline bioglass composites and (D) Raman spectra showing the structural features of the bioglass composites compared to the native peptides CnPA owing to biomineralization. (E) Surface elemental analysis by XPS Si 2p photoelectron spectra shows an increase in the binding energy from C6BG to C12BG. | ||
| CnBGn | Binding energy for Si 2pa | NBO:BO from O 1s photoelectron spectraa | Ca:P before immersion in SBFb | Ca:P after immersion in SBFb | Surface areac (m2 g−1) |
|---|---|---|---|---|---|
|
a Binding energy obtained from deconvoluted XPS spectra for Si 2p and O 1s and the ratio of non-bridging to bridging oxygens in the silicate network.
b EDX values to estimate the Ca:P ratio of the biomineralized hybrids before and after immersion in SBF depicting hydroxyapatite phase formation.
c The surface area is calculated from BET. The standard deviation of the triplicate dataset for the above studies was very close to the mean with a difference of only 0.005%.
|
|||||
| 6 | 103.34 | 0.202 | 0.92 | 1.41 | 19.55 |
| 8 | 103.39 | 0.195 | 0.81 | 1.56 | 53.94 |
| 10 | 103.50 | 0.171 | 0.99 | 1.59 | 10.06 |
| 12 | 104.42 | 0.21 | 1.16 | 1.45 | 7.59 |
Further, we performed SEM to visualize the topology of the peptide bioglass composites that exhibited porous three-dimensional fibrous networks (Fig. 3A–D) owing to the formation of a highly crosslinked silicate network. Ca2+ ions might also have a role in enhancing the aggregation propensity and network formation during the biomineralization process. To predict whether such morphological features accounted for the distinct potential to create an apatite mineral phase, we calculated the atomic percentage of Ca:P before and after immersing the scaffold in simulated biological fluids (Fig. S10, ESI†). EDX showed an increase in the percent of the Ca:P ratio for the CnBG samples after immersing in SBF (Fig. 5). Interestingly, C8BG showed the highest percent change in the Ca:P ratio, which might account for the high network modifier content. The dissolution of critical ions, such as Ca, Na, PO4, and Si, largely depends on the porous network of the matrix making it indispensable to analyse the pore size, pore volume, and surface area. We performed BET analysis through nitrogen adsorption–desorption pattern of the CnBG composites that exhibited type IV isotherm pattern with H1 hysteresis that is distinctive of the mesoporous structures (Fig. 3E–H). On fitting in the BJH equation, C8BG exhibited the highest surface area which was also evident from the isotherms having the maximum volume of gas adsorption (Table 1). However, C10BG and C12BG showed reduced surface area presumably due to the tight packing and interaction among the long aliphatic chains.
 | ||
| Fig. 3 (A)–(D) SEM Images of biomineralized peptide composites for C6BG, C8BG, C10BG, and C12BG. (E)–(H) Nitrogen adsorption/desorption isotherms showing type IV BET isotherm pattern. | ||
3.3. Biomechanical investigations from rheology and nanoindentation
Since natural biomaterials have a quintessential damage tolerance mechanism with dual-mode intrinsic and extrinsic toughening, we performed detailed mechanical investigations for peptide-templated bioactive glass composites using rheology.52CnPA and CnBG hydrogels demonstrated a frequency-independent behavior albeit with the CnBG composites exhibiting two-fold higher elastic moduli in comparison to the native CnPA hydrogels (Fig. 4A and B). The presence of strong interactive silicate networks acted as reinforcement fillers to impart such enhancement in the elastic moduli. Further, the CnPA and CnBG hydrogels with longer aliphatic chains (C12 chain) exhibited the highest storage moduli (∼3.8 × 105 Pa) that might account for the role of the percent content of β-sheets in determining gel strength. Dynamic oscillatory amplitude sweep experiments exhibited yield stress values for the hydrogels, indicating the breakage of the network upon increasing deformation (Fig. 4C and Fig. S11, ESI†). High yield stress values for C6BG and C8BG provided hints toward their resistance to flow on applied stress despite the presence of a significant amount of network modifier as seen from the higher NBO:BO ratios. All CnBG bioactive glasses were found to be thixotropic in nature and showed a progressive decrease in storage and loss moduli with time under the application of constant shear stress, followed by a gradual recovery upon removal of stress for three repeated cycles (Fig. 4D and Fig. S12, ESI†). For an irregular-sized bone defect, the bone cementing ability of the bioglass would depend on its injectability, the capacity to adapt to the shape of the defect without losing its integrity, and to self-heal in case of any damages occurring during the process. To evaluate the self-healing ability of the hydrogels, we moulded the dye (safranin O) doped and undoped gels and kept them in the vicinity to observe dynamic supramolecular interaction and self-healing of the two blocks into one whole block over 2 hours (Fig. 4E and Fig. S13A, ESI†). The comparative frequency sweep of self-healed vs. intact gels with similar moduli also supported these findings (Fig. S13B, ESI†). Moreover, the hybrid gels exhibited injectability, enabling their passage through a syringe, thereby enhancing their applicability, particularly in the realm of bone tissue engineering (Fig. S13C, ESI†). Further, we explored ubiquitous strain-stiffening for the CnBG hydrogel composites as found in biopolymers consisting of extracellular matrix filaments, and microtubules. The strain-stiffening properties were determined by an increased elastic modulus under small to moderate strains, which is crucial for ECM mimicking as it confers compliance to small forces and resistance to large deformations, thereby maintaining shape dynamics and functioning of the bone. It also allows long-range cell–cell communication owing to the interconnected fiber alignment that is essential between the sparsely present osteoblast progenitor cells. To evaluate the non-linear elastic response, we examined K′, which symbolized the elastic component as a function of stress by strain (δσ/δγ) in two distinct regimes. At the early pre-stress stage (σ), K′ = Go, representing the equilibrium state; beyond the critical stress (σc), an exponential increase occurs in K′ exhibiting a non-linear elastic response. σc serves as a distinguishing parameter for the strain stiffening response. CnBG with n = 8, 10, 12 exhibited early onset (∼0.05 Pa) of stiffening response relative to C6BG with higher σc value (∼0.4 Pa) for the onset of exponential response (Fig. 4F). As evident from higher σc, a relatively less dense fiber network in C6BG was responsible for imparting higher network flexibility, underlining the alteration over the strain-stiffening behaviour. Such stiffening response at a lower stress regime is a characteristic of mineralized collagen biopolymers with hydroxyapatite crystallization in the interstitial space and is thus absent in the native peptides (Fig. S14, ESI†).42,53 Next, we investigated the load-bearing capacity of the hydrogels using stress–strain compressibility curves that would validate their suitability to be used as artificial matrices (Fig. 4G and Table S1, ESI†). While the native peptide hydrogels deformed upon compression at ∼7 kPa, the bioglass composites could bear a load as high as ∼55 kPa. This was presumably due to the presence of additional Si–O–Si crosslinking to render an intricate network of peptide nanostructures with silicates. Such compression softening behaviour is stabilized by secondary bio-silicate network and results in the augmentation of the breaking strength, highlighting one of its crucial attributes as an adaptable biomaterial. It was thus evident from the extensive mechanical studies that CnBG deposition on CnPA templates renders mechanically enhanced features to it which are desirable for matching bone mechanics.
 | ||
| Fig. 4 (A) Comparative oscillatory frequency sweep rheology, (B) the corresponding bar diagram shows a comparison between the storage moduli, G′ and (C) comparative amplitude sweep oscillatory rheology for CnPA hydrogels (1 w/v%, pH 8) and CnBG composites. Data presented are the mean ± standard deviation (SD) of triplicate experiments. (D) Thixotropic studies on C8BG upon application of alternative cycles of high (100%) and low strains (0.1%) at a constant angular frequency of 10 rad s−1. (E) A pictorial depiction of the self-healing ability of the bioglass composite. (F) Differential moduli, K′, against stress σ for CnBG showing strain-induced stiffening behavior. (G) Compressive test comparing the load-bearing ability of C8PA and C8BG. (H) Load displacement curves for C8 hydrogel composites from nanoindentation and (I) corresponding reduced moduli (Er), hardness (H). | ||
Next, we used a nanoindentor to investigate the changes in surface hardness and mechanical properties of the materials upon mineralization which is reminiscent of dental enamel dissolution and remineralization.54 As a classical bone cementing material, the bioactivity of CnBG composites was evaluated in vitro with respect to the hydroxyapatite (HAp) forming ability that in turn reflected in vivo bone-bonding ability. Thus, C8BG was immersed in simulated body fluid for 25 days to promote calcium phosphate hydroxyapatite deposition to form C8HAp (Fig. 5). We compared the mechanical performance of C8PA, C8BG, and C8HAp by depositing films that were indented on the surface using the force required to penetrate <10% of the film thickness to avoid any substrate effects. A comparative load–displacement curve at 175 μN force demonstrated very high displacement of the native peptide C8PA (∼250 nm), indicating the presence of soft elastic peptide nanofibers, as compared to the biomineralized C8BG (∼140 nm) and C8HAp (∼30 nm) (Fig. 4H). Surface deposition of calcium phosphate in C8HAp made the surface stiff enough with minimal displacement at the same force. This was also evident from the increase in reduced moduli (Er) upon biomineralization to C8BG (∼151.67 MPa) from C8PA (∼99.07 MPa) (Fig. 4I). The hardness (H) values for these samples were proportional to the Er depicting average bond strength and limit of plasticity. Furthermore, we exploited Oliver Pharr's Theory to get the ductility index (Er/H) reflecting the relative amount of plastic indentation work that was found to be the highest for C8HAp (Fig. S15, ESI†). These observations were corroborated well with the fracture toughness of the indented biomaterial for bone integrity.
 | ||
| Fig. 5 In vitro bioactivity ascertained from microscopy and XRD. TEM images for C8BG (A) before and (B) and (C) after immersion in SBF for 7 and 25 days, respectively (inset showing the SAED pattern for deposited multiphasic HCA). The yellow line depicts biomineralized fibers of C8BG without any hydroxyapatite deposition, (D) SEM images for C8BG after immersion in SBF for 25 days showing the precipitated mineral phase. (E) The XRD data for C8BG after SBF immersion shows octacalcium phosphate and hydroxycarbonate apatite diffraction peaks. (F) FTIR spectra show an increase in CO32− and PO43− peaks of C8BG upon SBF immersion. | ||
3.4. Bioactivity of the peptide-templated bioglass scaffolds
After validating the mechanical properties, we substantiated the bone-forming ability of the bioglass scaffolds by estimating the bioactivity in simulated body fluid. The surface chemistry of the bioglass composites promotes rapid bone formation via the dissolution of critical concentrations of Si (17–20 ppm), P, Ca (88–100 ppm), and Na ions from the surface at a rate similar to the kinetics of collagen formation and bone growth in vivo. Interestingly, bioactive glass dictated in situ deposition of hydroxyapatite to exert direct control over genes that regulate cell cycle induction and progression and possess a high level of bioactivity and bone regenerative capability. We immersed the composites in simulated body fluid to promote HAp formation in five consecutive stages i.e., quick ionic exchange of Na+ with H+/H3O+, followed by glass network dissolution, polycondensation of silanols to form a silica gel layer with high surface area, and nucleation-crystallization of hydroxyl carbonate apatite (HCA) layer.55 TEM images exhibited structurally and compositionally altered surfaces of C8BG before and after immersion in SBF for 7 and 25 days (Fig. 5A–C). Selected area electron diffraction (SAED) analysis (inset) furnished the crystallographic details of the layer with d-spacing to indicate the deposition of phases resembling hkl values of hydroxyapatite (JCPDS 26-1056 and JCPDS 19-0272). Further, elemental mapping showed the distribution of bioglass ions, namely Si, O, Na, Ca, P, and N, on the surface of C8BG with the presence of Si, O, and Na at the periphery as potential dissolution sites owing to the open NBO network (Fig. S16A, ESI†). Upon immersing C8BG in SBF for 25 days, we observed colocalization of Ca and P on the surface, indicating CaP deposition (Fig. S16B, ESI†). SEM depicted the morphological changes from porous network features to multiphasic deposition in CnBG after immersion in SBF (Fig. 5D and S17, ESI†). Moreover, XRD corroborated the biphasic calcium phosphate deposition of octacalcium phosphate (hkl 2![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 1, 331,
1, 331, ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 2) and carbonate hydroxyapatite (hkl 210, 112, 203) upon immersion in SBF (Fig. 5E). The diffraction peaks for bioglass composites at 11.68°, 21.07°, and 29.33° disappeared and new peaks appeared at 32° and 45°, suggesting fast surface reactive ionic exchange for the growing phases of deposited calcium phosphate (Fig. S18, ESI†). FTIR spectra confirmed the increase in characteristic vibration modes of carbonate (800 cm−1) and phosphate (570 cm−1), with a steady increase up to 1.59, indicating prominence of carbonate apatite to signify hydroxyapatite with a parallel decrease in Si–O (NBO) peak of bioglass (Fig. 5F and Fig. S19, ESI†). Comparative Ca:P ratios present on the surface of CnBG, as evaluated from SEM-EDX, indicated that the composition of bone mimicked the release kinetics of natural hydroxyapatite (Ca:P = 1.67) (Fig. S20, ESI†). This indicated the presence of higher NBO in C8BG to exhibit the highest % change in Ca:P to eventually render a faster silica dissolution process, thereby enabling the deposition of different forms of hydroxyapatites. The octacalcium phosphate phase found in these composites after being soaked in SBF could act as a precursor to HAp and was indicative of its bone regenerative capability for mineralizing the collagen matrix.
2) and carbonate hydroxyapatite (hkl 210, 112, 203) upon immersion in SBF (Fig. 5E). The diffraction peaks for bioglass composites at 11.68°, 21.07°, and 29.33° disappeared and new peaks appeared at 32° and 45°, suggesting fast surface reactive ionic exchange for the growing phases of deposited calcium phosphate (Fig. S18, ESI†). FTIR spectra confirmed the increase in characteristic vibration modes of carbonate (800 cm−1) and phosphate (570 cm−1), with a steady increase up to 1.59, indicating prominence of carbonate apatite to signify hydroxyapatite with a parallel decrease in Si–O (NBO) peak of bioglass (Fig. 5F and Fig. S19, ESI†). Comparative Ca:P ratios present on the surface of CnBG, as evaluated from SEM-EDX, indicated that the composition of bone mimicked the release kinetics of natural hydroxyapatite (Ca:P = 1.67) (Fig. S20, ESI†). This indicated the presence of higher NBO in C8BG to exhibit the highest % change in Ca:P to eventually render a faster silica dissolution process, thereby enabling the deposition of different forms of hydroxyapatites. The octacalcium phosphate phase found in these composites after being soaked in SBF could act as a precursor to HAp and was indicative of its bone regenerative capability for mineralizing the collagen matrix.
3.5. Cellular response to the surface reactive peptide templated bioglass matrix
Next, we evaluated the biological response exhibited by CnPA and CnBG against the human osteoblast U2OS cell line. The MTT assay showed excellent biocompatibility of CnPA and CnBG hydrogel matrices even for high concentrations (1000 μg mL−1) depicting normal metabolic activity of the cells (Fig. 6A). In fact, more than 80% of cells were viable indicating good cellular proliferation toward peptide hydrogels and bioglass composites. Further, we investigated the cell adhesion propensity of the C8BG matrix cultured with U2OS cells under an optical microscope that showed a spread-out morphology with developing cellular projections from rounded cells (Fig. 6B and Fig. S21, ESI†). Such biocompatibility and proliferation in the matrices might stem from the interesting strain-stiffening contractile behaviour of the biomaterials that provides essential mechanical cues to promote cell–matrix interactions.36,56,57 Alamar blue assay provided a quantitative estimation of the cell proliferation over time as monitored by increasing fluorescence intensity at 590 nm to exhibit better adaptation of the cells to microenvironment in the bioglass hybrids (Fig. 6C). Significant increase in cellular proliferation towards C8BG, in comparison to C8PA, was noted, as obtained from statistical analysis using one-way ANOVA followed by Tukey's test. Further, live/dead assay using FDA/PI staining provided information about the cell viability for a longer culture period of 5 days (Fig. S22, ESI†). Thus, the majority of the U2OS cells on the C8BG scaffold exhibited green FDA staining with almost no traces of red stains of PI even after 5 days (Fig. 6D–F) of incubation, suggesting healthy cells with negligible cell death. However, cells seeded on the bare peptide C8PA exhibited significantly less cell proliferation (Fig. S22, ESI†). Next, we exploited the Alizarin red-S assay to monitor calcium deposition in U2OS cell-laden C8PA and C8BG matrices incubated over 14 days to validate the bioactivity data (Fig. 6G and H). The appearance of brick-red precipitates suggested the presence of calcium-containing osteocytes in differentiated osteoblasts (Fig. S23, ESI†).58,59 Quantitative estimation of ossification was evaluated from the absorbance at 550 nm and depicted the highest matrix mineralization with regard to calcium phosphate deposition for C8BG (Fig. 6I). One-way ANOVA and Tukey's test established significant differences among the native peptides vs. the peptide bioglass hybrids. Finally, we investigated the alkaline phosphatase (ALP) activity as an early marker of osteogenic differentiation and observed enhanced activity in the presence of CnBG scaffolds after 7 days of cell seeding. Interestingly, the bioglass scaffolds exhibited significantly greater ALP activity (∼5–10 times) as compared to CnPA hydrogels (Fig. 6J). Such high ALP enzyme expression reconciles the role of bioactive glasses in providing favourable microenvironment cues for osteogenesis, which is aided by the ionic dissolution products and in situ HAp deposition. Further, cells on the C8BG matrices showed maximum enhancement of the ALP activity (252 U mL−1), among all the bioglass matrices. Although C10PA and C12PA and their corresponding bioglass exhibited higher hydrophobicity, mechanical strength, and higher Ca:P before immersion in SBF, the tight packing probably decelerated the release of ions from its matrix. On the contrary, C6BG formed the weakest hydrogel owing to the modulation of hydrophobic/hydrophilic balance in the peptide to result in higher open network structure and exhibited better ARS and ALP activity albeit with weak mechanical attributes. In that regard, synergistic self-assembly in C8BG endows optimal hydrophobicity along with temporal and structural attributes. Thus, C8BG exhibited a high NBO:BO ratio and the highest percent change in Ca:P and high surface area, along with suitable mechanics (tensile strength and collagen-like stiffening), which was reflected in the bioactivity with favourable cellular attachment, spreading, and growth vitals for tissue development. Interestingly, all our bioglass constructs favourably supported multi-layer cellular growth with minimum cell death or detachment from the matrix, encouraging their use as excellent biomaterials. However, a factorial analysis to establish a direct correlation among the hydrogels is challenging although factors, such as cross-over point and strain-stiffening, were seen to influence osteogenic expression, indicating elastic gels to store the deformation work necessary for cellular activity.60 It is interesting to note that, while the majority of the recent reports focus on bioglass utilize natural polymers, such as collagen (poor rigidity and mechanics), silk fibroin (sericin limiting osteogenesis), cellulose (poor hydrophilicity), or hyaluronic acids (expensive purification), our study demonstrates biopolymer-like templating efficacy with self-assembled peptide scaffold with tailorable hydrophobic–hydrophilic balance, improved osteogenesis and reliable synthetic and purification protocols.6,12 Additionally, hydroxyapatite deposition using peptide scaffolds usually incorporates acidic amino acids while our approach adopts a collagen-mimicking lysine-based hydroxyapatite deposition strategy.29,58 Moreover, although amino acid-based bioglass composites have gained recent interest, the effect of hydrophobicity as a crucial factor dictating the bioactive responses has not been investigated elsewhere. Thus, the present work offers a multidimensional advancement in the clinically important bioactive glasses for a more rationalized and tailorable, precision biomaterial. It is important to acknowledge that our study has limitations that should be addressed in future research. One major limitation is the lack of in vivo testing with animal models, which is essential to assess the effectiveness and safety of biomaterials for bone tissue regeneration. Additionally, the absence of mesenchymal stem cells and 3D cell culture in our study may limit the understanding of the system. These aspects are currently beyond the scope of our research, but they are important considerations for future studies to enhance the translational potential of our findings. Our laboratory is actively working towards addressing these limitations and improving the design of biomaterial-based systems for tissue regeneration.
 | ||
| Fig. 6 Osteogenic potential of the CnPA and corresponding CnBG composites on the subsistence of U2OS cells. (A) MTT assay for the CnPA hydrogels and CnBG composites after 48 h (1000 μg mL−1). Untreated wells were considered as a positive control. Data represents mean ± SD from three experiments performed in triplicates. (B) The spread morphology of the cells under a bright field after incubation with C8BG for 48 h. Red arrows depict cells adhered to the matrix initiating spreading marked by the change in morphology. (C) Alamar Blue assay to estimate cellular proliferation over C8PA and C8BG for 24, 28, and 72 hours. (D)–(F) Live/dead staining using FDA/PI assay depicting the cellular interactions with C8BG after 5 days. (G) and (H) Alizarin red-S-stained optical images of C8PA and C8BG incubated with U2OS cells for 14 days to identify calcium-containing osteocytes in differentiated osteoblasts and (I) quantitative estimation of ossification of matrices with respect to Ca deposits for CnPA and CnBG. (J) Alkaline phosphatase activity of the cells seeded on CnBG matrices for 7 days to mark early osteogenic differentiation. All scale bars = 100 μm. Data represented are the mean ± standard deviation (SD) of triplicate experiments. | ||
4. Conclusions
In summary, we demonstrated a rational design with a series of minimalist peptide amphiphiles CnPA (n = 6, 8, 10, 12) with varying hydrophobicity in the N-termini that exhibited modulation in the propensity to form β-sheet and self-assembly behaviour in the presence of ionic strength. Interestingly, the synergy of ionic strength and hydrophobicity drove the self-assembly of C8PA. We exploited the peptide nanostructure as a template to construct a functional 3D network by incorporating precursors for bioactive glass and CnBG formation. All the CnBG constructs demonstrated efficient in situ biomineralization to form a strong silicate network with a calcium silicate phase. Interestingly, template morphology and secondary structures influenced the in situ mineralization of the bioactive glass to eventually control pore size, morphology, and textural features and dictate the viscoelastic nature of the bioglass composites. These composites possessed high load-bearing ability, collagen-like stiffening, and demonstrated thixotropic self-healing behaviour. The bundling of the fibers and consecutive spiky biomineralization in C8BG prompted us to study its mechanical response in vitro using indentation to achieve bone-like strength in the order of MPa and appreciable ductility. It was evident that both structural features that were guided by temporal effect and mechanical strength dictated the bioactivity and cellular response. The hydroxyapatite-forming ability and optimal osteogenic response of C8BG confirmed the utility of the supramolecular hydrogel composite towards ECM mimicking bone tissue engineering scaffold. Ultimately, developing supramolecular inorganic–organic hybrid hydrogel composites with variation in mechanical stiffness of the scaffold in the context of matrix microenvironment cues especially in terms of strain stiffening response is one of its kind. This research presents a fresh perspective on advancing bone biomaterials by emphasizing the significance of hydrophobicity as a fundamental yet crucial factor in directing the outcomes of self-assembly, surface chemistry, and mechanics of peptide-templated bioactive glass. This, in turn, has profound implications for bioactivity. Such mesoporous, dynamic, self-healable biomaterials with topological, structural, and mechanical control can be potential candidates for implanting coating for bone tissue engineering applications. Further, its application in bone regeneration or wound healing requires elaborate in vivo studies as an efficient model for tissue engineering that is underway in our laboratory.Author contributions
A. P. conceived the idea of the work, along with N. M. who performed most of the work with the other coauthors. D. N. performed the cell studies and strain-stiffening studies. Y. C. and B. V. contributed to nanoindentation measurements. N. G. and A. S. contributed to the initial studies of biomineralization and rheology.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to INST, Mohali, for providing infrastructure and characterization facilities. N. M. and N. G. gratefully acknowledge the INST PhD and post-doctoral fellowship. A. S. thanks UGC for the SRF fellowship (SR no. 2121310082). The work was financially supported by the Department of Science & Technology (DST, SERB project CRG/2020/004251).Notes and references
- H. Petite, V. Viateau, W. Bensaïd, A. Meunier, C. De Pollak, M. Bourguignon, K. Oudina, L. Sedel and G. Guillemin, Nat. Biotechnol., 2000, 18, 959–963 CrossRef CAS.
- A. Salhotra, H. N. Shah, B. Levi and M. T. Longaker, Nat. Rev. Mol. Cell Biol., 2020, 21, 696–711 CrossRef CAS PubMed.
- N. Xue, X. Ding, R. Huang, R. Jiang, H. Huang, X. Pan, W. Min, J. Chen, J.-A. Duan, P. Liu and Y. Wang, Pharmaceuticals, 2022, 15, 879 CrossRef CAS.
- O. J. G. M. Goor, S. I. S. Hendrikse, P. Y. W. Dankers and E. W. Meijer, Chem. Soc. Rev., 2017, 46, 6621–6637 RSC.
- B. Bhattacharjee, S. Ghosh, D. Patra and J. Haldar, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2022, 14, e1745 Search PubMed.
- I. Elgali, O. Omar, C. Dahlin and P. Thomsen, Eur. J. Oral Sci., 2017, 125, 315–337 CrossRef PubMed.
- A. Galotta, F. Agostinacchio, A. Motta, S. Dirè and V. M. Sglavo, J. Eur. Ceram. Soc., 2023, 43, 639–647 CrossRef CAS.
- P. S. Yavvari, A. K. Awasthi, A. Sharma, A. Bajaj and A. Srivastava, J. Mater. Chem. B, 2019, 7, 2102–2122 RSC.
- Ö. Demir-Oğuz, A. R. Boccaccini and D. Loca, Bioact. Mater., 2023, 19, 217–236 Search PubMed.
- W. Vogel and W. Höland, Angew. Chem., Int. Ed. Engl., 1987, 26, 527–544 CrossRef.
- L. Souza, J. H. Lopes, D. Encarnação, I. O. Mazali, R. A. Martin, J. A. Camilli and C. A. Bertran, Sci. Rep., 2018, 8, 12808 CrossRef PubMed.
- P. A. Dash, S. Mohanty and S. K. Nayak, J. Non-Cryst. Solids, 2023, 614, 122404 CrossRef CAS.
- A. C. Profeta and G. M. Prucher, Dent. Mater. J., 2015, 34, 559–571 CrossRef CAS.
- A. Shearer, M. Montazerian, J. J. Sly, R. G. Hill and J. C. Mauro, Acta Biomater., 2023, 160, 14–31 CrossRef CAS PubMed.
- L. L. Hench, J. Mater. Sci.: Mater. Med., 2006, 17, 967–978 CrossRef CAS PubMed.
- X. Zhu, C. Wang, H. Bai, J. Zhang, Z. Wang, Z. Li, X. Zhao, J. Wang and H. Liu, Mater. Today Bio, 2023, 20, 100660 CrossRef CAS PubMed.
- W. Xie, X. Wei, H. Kang, H. Jiang, Z. Chu, Y. Lin, Y. Hou and Q. Wei, Adv. Sci., 2023, 10, 2204594 CrossRef CAS PubMed.
- K. S. Hellmund and B. Koksch, Front. Chem., 2019, 7, 172 CrossRef CAS PubMed.
- H. Shin, S. Jo and A. G. Mikos, Biomaterials, 2003, 24, 4353–4364 CrossRef CAS PubMed.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- X. Wang, L. Ji, J. Wang and C. Liu, Bioact. Mater., 2023, 27, 138–153 CrossRef CAS.
- E. Arslan, I. C. Garip, G. Gulseren, A. B. Tekinay and M. O. Guler, Adv. Healthcare Mater., 2014, 3, 1357–1376 CrossRef CAS.
- K. Kapat, S. Kumbhakarn, R. Sable, P. Gondane, S. Takle and P. Maity, Biomedicines, 2024, 12, 313 CrossRef CAS PubMed.
- J. M. Anderson, M. Kushwaha, A. Tambralli, S. L. Bellis, R. P. Camata and H.-W. Jun, Biomacromolecules, 2009, 10, 2935–2944 CrossRef CAS.
- E. Arslan, M. Hatip Koc, O. Uysal, B. Dikecoglu, A. E. Topal, R. Garifullin, A. D. Ozkan, A. Dana, D. Hermida-Merino, V. Castelletto, C. Edwards-Gayle, S. Baday, I. Hamley, A. B. Tekinay and M. O. Guler, Biomacromolecules, 2017, 18, 3114–3130 CrossRef CAS PubMed.
- J. Ryu, S. H. Ku, M. Lee and C. B. Park, Soft Matter, 2011, 7, 7201 RSC.
- T. D. Sargeant, C. Aparicio, J. E. Goldberger, H. Cui and S. I. Stupp, Acta Biomater., 2012, 8, 2456–2465 CrossRef CAS PubMed.
- M. Ghosh, M. Halperin-Sternfeld, I. Grigoriants, J. Lee, K. T. Nam and L. Adler-Abramovich, Biomacromolecules, 2017, 18, 3541–3550 CrossRef CAS.
- C. J. Newcomb, R. Bitton, Y. S. Velichko, M. L. Snead and S. I. Stupp, Small, 2012, 8, 2195–2202 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- J. P. Joseph, C. Miglani, A. Maulik, S. R. Abraham, A. Dutta, A. Baev, P. N. Prasad and A. Pal, Angew. Chem., Int. Ed., 2023, 62, e202306751 CrossRef CAS PubMed.
- Z. A. C. Schnepp, R. Gonzalez-McQuire and S. Mann, Adv. Mater., 2006, 18, 1869–1872 CrossRef CAS.
- M. C. Mañas-Torres, G. B. Ramírez-Rodríguez, J. I. García-Peiro, B. Parra-Torrejón, J. M. Cuerva, M. T. Lopez-Lopez, L. Álvarez De Cienfuegos and J. M. Delgado-López, Inorg. Chem. Front., 2022, 9, 743–752 RSC.
- D. Gupta, A. Bhatt, V. Gupta, C. Miglani, J. P. Joseph, J. Ralhan, D. Mandal, Md. E. Ali and A. Pal, Chem. Mater., 2022, 34, 4456–4470 CrossRef CAS.
- J. P. Joseph, N. Gupta, C. Miglani, D. Nath, A. Singh, D. Gupta and A. Pal, Chem. Mater., 2022, 34, 4364–4374 CrossRef CAS.
- D. Nath, J. Ralhan, J. P. Joseph, C. Miglani and A. Pal, Biomacromolecules, 2024, 25, 853–863 CrossRef CAS.
- Umesh, J. Ralhan, V. Kumar, H. Bhatt, D. Nath, N. A. Mavlankar, H. N. Ghosh and A. Pal, Langmuir, 2024, 40, 2754–2763 CrossRef CAS PubMed.
- A. Singh, J. P. Joseph, D. Gupta, C. Miglani, N. A. Mavlankar and A. Pal, Nanoscale, 2021, 13, 13401–13409 RSC.
- S. Patra, N. A. Mavlankar, L. Ramesan, A. Singh and A. Pal, Chemistry, 2023, 5, 1190–1202 CrossRef CAS.
- N. Gupta, A. Singh, N. Dey, S. Chattopadhyay, J. P. Joseph, D. Gupta, M. Ganguli and A. Pal, Chem. Mater., 2021, 33, 589–599 CrossRef CAS.
- J. Thomas, N. Gupta, J. P. Joseph, V. Chopra, A. Pal and D. Ghosh, ACS Biomater. Sci. Eng., 2021, 7, 5798–5809 CrossRef CAS PubMed.
- S. Van Helvert and P. Friedl, ACS Appl. Mater. Interfaces, 2016, 8, 21946–21955 CrossRef CAS.
- R. C. Ollier, Y. Xiang, A. M. Yacovelli and M. J. Webber, Chem. Sci., 2023, 14, 4796–4805 RSC.
- C. Storm, J. J. Pastore, F. C. MacKintosh, T. C. Lubensky and P. A. Janmey, Nature, 2005, 435, 191–194 CrossRef CAS PubMed.
- P. Y. W. Dankers, M. C. Harmsen, L. A. Brouwer, M. J. A. Van Luyn and E. W. Meijer, Nat. Mater., 2005, 4, 568–574 CrossRef CAS PubMed.
- U. Blache, E. M. Ford, B. Ha, L. Rijns, O. Chaudhuri, P. Y. W. Dankers, A. M. Kloxin, J. G. Snedeker and E. Gentleman, Nat. Rev. Methods Primers, 2022, 2, 98 CrossRef CAS PubMed.
- N. A. Mavlankar, A. K. Awasthi, J. Ralhan and A. Pal, ChemNanoMat, 2022, 8, e202200368 CrossRef CAS.
- R. J. Mart, R. D. Osborne, M. M. Stevens and R. V. Ulijn, Soft Matter, 2006, 2, 822 RSC.
- W. C. Oliver and G. M. Pharr, J. Mater. Res., 1992, 7, 1564–1583 CrossRef CAS.
- E. Mattia, A. Pal, G. Leonetti and S. Otto, Synlett, 2016, 103–107 Search PubMed.
- J. Reed and T. A. Reed, Anal. Biochem., 1997, 254, 36–40 CrossRef CAS.
- U. G. K. Wegst, H. Bai, E. Saiz, A. P. Tomsia and R. O. Ritchie, Nat. Mater., 2015, 14, 23–36 CrossRef CAS.
- I. K. Piechocka, A. S. G. Van Oosten, R. G. M. Breuls and G. H. Koenderink, Biomacromolecules, 2011, 12, 2797–2805 CrossRef CAS.
- Z. Kovács, M. Fábián, N. Szász, I. Székács and V. K. Kis, J. Non-Cryst. Solids, 2022, 581, 121416 CrossRef.
- L. L. Hench, R. J. Splinter, W. C. Allen and T. K. Greenlee, J. Biomed. Mater. Res., 1971, 5, 117–141 CrossRef.
- Q. Wen and P. A. Janmey, Exp. Cell Res., 2013, 319, 2481–2489 CrossRef CAS PubMed.
- I. K. Piechocka, K. A. Jansen, C. P. Broedersz, N. A. Kurniawan, F. C. MacKintosh and G. H. Koenderink, Soft Matter, 2016, 12, 2145–2156 RSC.
- S. Chowdhuri, M. Ghosh, L. Adler-Abramovich and D. Das, Pharmaceutics, 2021, 13, 1602 CrossRef CAS.
- M. Ghosh, M. Halperin-Sternfeld, I. Grinberg and L. Adler-Abramovich, Nanomaterials, 2019, 9, 497 CrossRef CAS.
- E. Y. Du, F. Ziaee, L. Wang, R. E. Nordon and P. Thordarson, Polym. J., 2020, 52, 947–957 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00810c |
| ‡ Current address: CSIR-Indian Institute of Toxicology Research, Vishvigyan Bhawan, 31, Mahatma Gandhi Marg, Lucknow, Uttar Pradesh, India. |
| This journal is © The Royal Society of Chemistry 2024 |