
Immunologically effective biomaterials enhance immunotherapy of prostate cancer
Siqi
Liu
a,
Hui
Guo
a,
Di
Li
 *b and
Chunxi
Wang
*a
*b and
Chunxi
Wang
*a
aDepartment of Urology, General Surgery Center, The First Hospital of Jilin University, 1 Xinmin Street, Changchun 130061, P. R. China
bDepartment of Hepatobiliary and Pancreatic Surgery, General Surgery Center, The First Hospital of Jilin University, 1 Xinmin Street, Changchun 130061, P. R. China
First published on 14th August 2024
Abstract
Prostate cancer (PCa) is one of the most common malignant neoplasms affecting the male population. The onset of the disease is insidious and often associated with severe consequences, such as bone metastases at the time of initial diagnosis. Once it advances to metastatic castration-resistant PCa (mCRPC), conventional treatment methods become ineffective. As research on the mechanism of tumor therapy advances, immunotherapy has been evolving rapidly. However, PCa is a solid tumor type that primarily faces the challenges of poor immunogenicity and inhibitory tumor microenvironment (TME). Fortunately, the extensive use of biomaterials has led to continuous advancement in PCa immunotherapy. These innovative materials aim to address intractable issues, such as immune escape and immune desert, to inhibit tumor progression and metastasis. This detailed review focuses on the regulation of different aspects of tumor immunity by immunologically effective biomaterials, including modulating adaptive immunity, innate immunity, and the immune microenvironment, to enhance the efficacy of PCa immunotherapy. In addition, this review provides a perspective on the future prospects of immunotherapeutic nanoplatforms based on biomaterials in the treatment of PCa.
 Di Li | Di Li is a lecturer in the Department of Hepatobiliary and Pancreatic Surgery, The First Hospital of Jilin University. She obtained her PhD degree at Northeast Normal University in 2016. Following her PhD, she worked at Peking University as a postdoctoral research fellow. From 2018 to 2019, she worked with Prof. Jinjun Shi from Brigham and Women's Hospital, Harvard Medical School, as a visiting scholar. Her research focuses on developing tumor microenvironment-responsive polymer nanoformulations for cancer therapy. |
1. Introduction
Prostate cancer (PCa) is a prevalent form of malignant neoplasm. As societal living standards improve, the incidence and mortality of PCa are increasing.1 The high mortality rate of PCa is mainly attributed to diagnostic delay, because patients with early PCa often lack typical symptoms and are often only diagnosed via serum prostate-specific antigen (PSA) screening.2 However, the present level of public awareness regarding PCa screening is insufficient, and it is difficult to mitigate the incidence of PCa through early diagnosis.3The conventional treatment methods for PCa include surgical intervention, radiotherapy, endocrine therapy, and chemotherapy. Radical prostatectomy is the preferred treatment for early PCa. However, some patients have bone metastasis at the time of initial diagnosis and miss the opportunity for surgery.4–6 Endocrine therapy is a common strategy for patients with postoperative biochemical recurrence and metastatic PCa.7 Unfortunately, tumor-generated androgen demonstrates resistance to endocrine therapy in the case of disease progression to mCRPC.
With the advent of the autologous dendritic cell (DC) vaccine sipuleucel-T, immunotherapy opens a new chapter in the treatment of PCa.8 Immune checkpoint inhibitors have shown considerable efficacy in different cancer types, while the effect on PCa remains questionable.9 The challenge with immunotherapy for PCa lies in the poor tumor immunogenicity, the immune desert within the TME, and the off-target phenomenon of drugs.10 The ideal drug would have the ability to address this dilemma. Biomaterials are ideal carriers for drug delivery due to their remarkable biocompatibility, stable physical and chemical properties, and appropriate surface modification ability.11 Moreover, certain materials exhibit immune-activating properties that make them highly promising in the field of tumor immunotherapy.12 Nanotechnology has emerged as a tool for drug delivery systems. Traditional PCa drugs such as bicalutamide, docetaxel or other drugs loaded in liposomes, and magnetic and hybrid nanoparticles reduce systemic cytotoxicity and delay or overcome the development of drug resistance. Some researchers have used poly(amino acid) nanoparticles as drug delivery systems to treat PCa, and have achieved clear therapeutic effects, such as prolonging the blood circulation time of drugs, the ability of drugs to passively target tumors, and good blood compatibility.13 Therefore, the application prospect of nanoparticles in the immunotherapy of PCa is considerable.
In this review, we discuss the impact of biomaterials on PCa immunotherapy through different immune mechanisms (Fig. 1). In particular, we focus on the precise regulation of immune cycle steps to promote adaptive immunity development, enhance immunity efficacy and duration, and modulate a suppressive TME (Table 1). In addition, we discuss the opportunities and challenges for immunotherapeutic nanoplatforms based on biomaterials to improve the efficacy of immunotherapy.
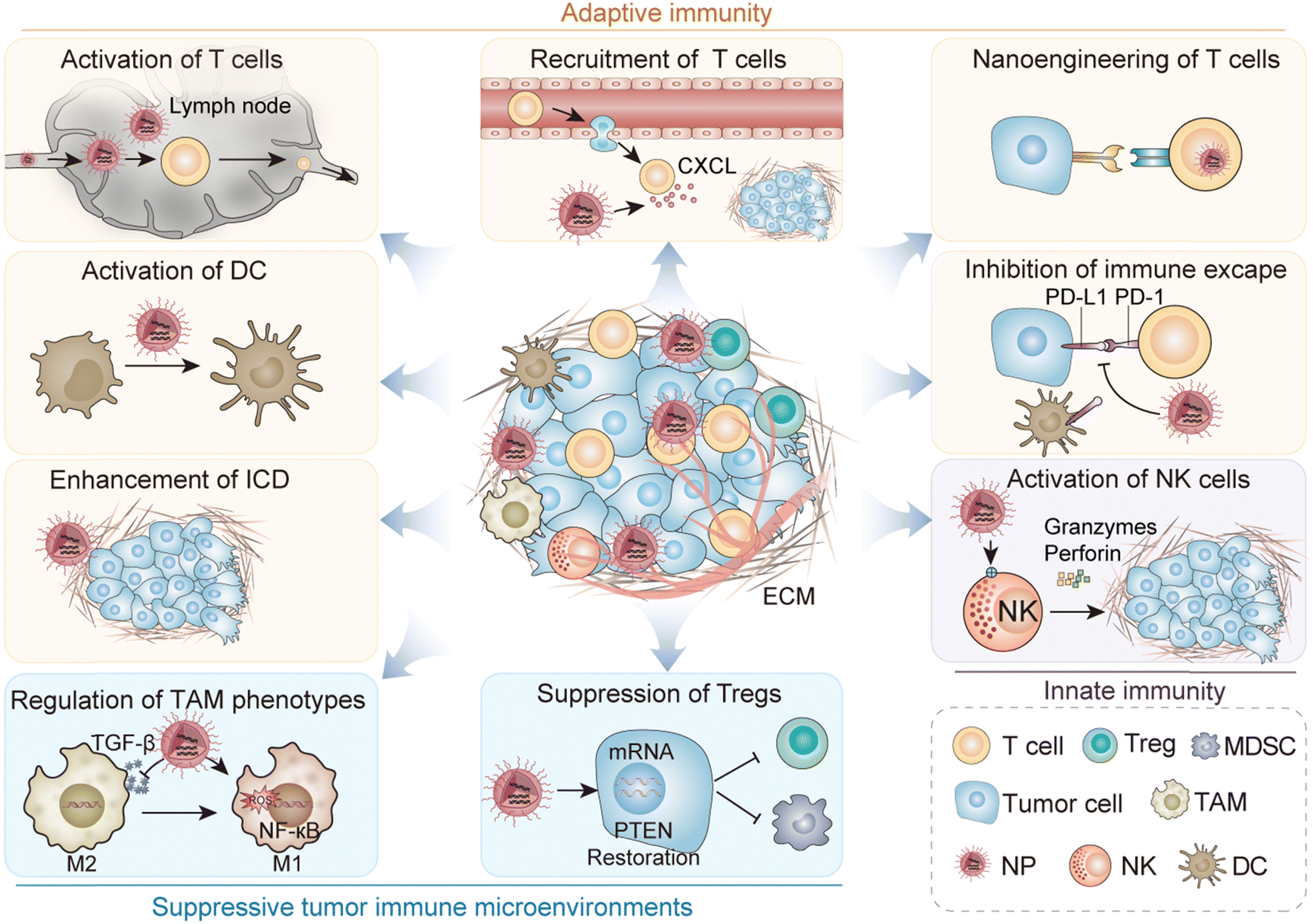 | ||
| Fig. 1 Schematic illustration of immune regulation for PCa therapy. | ||
| Regulatory site | Mechanism | Function | Agent | Ref. |
|---|---|---|---|---|
| Adaptive immunity | Enhance tumor antigen release | Magnetic thermal therapy-induced ICD | Magnetic iron oxide NPs | 14 |
| Chemotherapy-induced ICD | Target delivery of DOX and I3A by PLA-PEG-ACUPA/TPP NPs | 15 | ||
| Codelivery of shikonin and anthracyclines by liposomes | 16 | |||
| Delivery of chemotherapeutic drugs by liposome-microvesicle complexes | 17 | |||
| Photodynamic therapy-induced ICD | YP mesoporous silica-coated NPs | 18 | ||
| Radiotherapy and IBM co-induce ICD | Delivery of anti-CD40 agonist by polylactic-coglycolic acid and alginate complexes | 19 | ||
| Enhance tumor antigen presentation | Direct activation of DCs with TLR agonists | Codelivery of ovalbumin-coded mRNA and TLR7/8 agonist R848 derivative by lipid-PEG NPs | 20 | |
| Codelivery of ICG and R848 by PLGA NPs | 21 | |||
| CpG as a TLR agonist to activate DCs | Delivery of CpG and antigenic peptides by liposomes | 22 | ||
| Delivery of CpG by sodium alginate hydrogel | 23 | |||
| Delivery of CpG by multi-walled CNT to prolong the drug residence time | 24 | |||
| Delivery of CpG by hybrid silk MS2KN spheres | 25 | |||
| Activation of DCs with an antigenic peptide vaccine | STEAP1 peptides encapsulated with PLGA microspheres | 26 | ||
| PLGA NPs coated with cancer cell membranes as a source of multiple antigens | 27 | |||
| Preparation of specific antibodies using DCs | LHRH peptides and vaccine adjuvants encapsulated with liposomes | 28 | ||
| Regulation of T cell priming | Utilizing cytokines as stimulators of T cell activation | Delivery of IL15 by PEG-hydrogel microspheres | 29 | |
| Regulation of T cells infiltrating into tumor sites | Inhibit the expression of VEGF | CRGDyK–polyamidoamine dendrimer–rapamycin conjugate | 30 | |
| Reprograming of circulating T cells | Tackling tumor escape caused by MHC molecules using adoptive T cell technology | mRNA encapsulated by poly(β-aminoester) polymer NPs with a polyglutamic acid shell | 31 | |
| Regulation of T cell-killing tumor cells | Prolonging the action time of αPD-1/PD-L1 inhibits the immune escape | Delivery of Ferumoxytol (Fer) capped PD-1 by silica MS | 32 | |
| Co-encapsulating αPD-1 peptide and hollow gold nanoshells by PLGA NPs | 33 | |||
| Innate immunity | Enhance the immune activity of innate immune cells | Utilizing cytokines as stimulators of NK cell activation | Delivery of IL2 by isoDGR-tagged gold NPs | 34 |
| Inhibitory TME | Regulation of TAM phenotypic transformation | Delivering drugs capable of modulating TAM phenotypes | Co-encapsulating TGF-β kinase inhibitor SD-208 and R848 by serum-derived exosomes | 35 |
| Modulating the NF-κB pathway | Mangiferin-functionalized gold NPs | 36 | ||
| Modulating the ROS in the TME | Manganese-doped eumelanin NPs | 37 | ||
| Suppression of Tregs | Modulating the PTEN pathway | Delivery of PTEN mRNA by lipid-polymer hybrid NPs | 38 |
2. Regulation of adaptive immunity
As the concept of the cancer-immunity cycle was introduced,10 it became increasingly evident that T cell-mediated cancer cell destruction represents merely one aspect of this comprehensive process. In the process of adaptive immunity exerting its anticancer effect, the simultaneous regulation of one or more links in the cycle can enhance tumoricidal efficacy by promoting a greater adaptive immune response. The process of the adaptive immune response mediated by a tumor is extremely complex. Succinctly summarized, tumor cells release antigen signals presented to T cells by antigen-presenting cells (APCs), leading to T cell initiation. Subsequently, activated T cells are recruited to the tumor site via chemokine-mediated circulation.39 These cells extravasate through the blood vessels and infiltrate the tumor bed. Upon recognition of the tumor signal, they initiate apoptosis in the tumor cells, releasing new tumor antigen molecules and initiating a fresh immune cycle.10 The application of biomaterials enhances the precision and efficacy of immune regulation in the aforementioned interconnections.402.1. Enhancement of tumor immunogenicity
The challenging issue of enhancing the immunogenicity of PCa serves as a pivotal starting point for modulating the cancer-immunity cycle and is manifested in various strategies employed in immunotherapy. The stimulation of immunogenic cell death (ICD) in tumor cells is a current research focus among scholars because of its dual tumor-killing effects: direct cytotoxic effects on the tumor and consequential antitumor immune responses. Damage-associated molecular patterns (DAMPs), which are danger signals released in ICD-induced tumor cell death, manifest as the increased expression of three signals: calreticulin (CRT) on the membrane of dying tumor cells, the release of adenosine triphosphate (ATP), and excretion of high mobility group box 1 protein (HMGB1).41 DCs, upon encountering these DAMPs, undergo maturation and mediate cellular immune responses by presenting antigens for recognition by cytotoxic T lymphocytes.42 Therefore, enhancing tumor immunogenicity by modulating ICD may be a promising strategy in the treatment of PCa.Magnetic hyperthermia (MHT) achieves rapid tumor ablation by exposing magnetic nanoparticles (NPs) to an alternating magnetic field. It has the advantages of strong tissue penetration, excellent therapeutic effect and non-invasive, and is the focus of scholars. The application of MHT may activate antitumor immunity through ICD-mediated enhancement of antigen release and enhance the therapeutic effect on tumors.14 Activation of antitumor immunity by MHT has been validated in other tumor types. Some scholars have also evaluated the efficacy of this approach in clinical trials for PCa. The trial utilized laser-excited gold–silica nanoshells in combination with magnetic resonance ultrasound fusion imaging for the local ablation of PCa.43 MHT based on inorganic NPs may be an effective method for the treatment of PCa. It can not only kill prostate tumor cells efficiently but also cause ICD of tumor cells, triggering adaptive immunity, and amplify antitumor efficacy. In addition to magnetothermal therapy, chemoimmunotherapy is a more commonly employed treatment. Despite doxorubicin (DOX) is a widely used chemotherapeutic drug in clinics and has been proved to cause ICD, therefore, its limited efficacy hinders its effectiveness in chemoimmunotherapy. Ingenol-3-angelate (I3A) is an emerging antitumor agent with dual chemotherapeutic and immune response-inducing effects, which synergistically enhances the efficacy of DOX when used in combination. Wang's team developed a dual-targeting delivery system, polylactide-poly(ethylene)glycol-2-(3-((S)-5-amino-1-carboxymethyl)-ureido)pentanedioate/triphenylphosphonium (PLA-PEG-ACUPA/TPP), which targeted prostate-specific membrane antigen (PSMA) and mitochondria. The administration of these co-loading nanomedicines resulted in favorable antitumor immune responses, providing a valuable strategy for the application of biomaterials in chemoimmunotherapy.15 It is worth mentioning that refractory PCa is prone to chemotherapy resistance in the face of immunogenic chemotherapy, which is related to B cells. It has been confirmed that the chemokine CXCL13 recruits B cells in PCa tumors to produce lymphotoxin, activates the IkB kinase a (IKKa)-BMI1 module in PCa stem cells, and promotes the progression of castration-resistant PCa. The role of B cells in the resistance toward immunogenic chemotherapy in PCa has been investigated, and the results show that three different mouse PCa models do not respond to oxaliplatin unless B cells are genetically or pharmacologically depleted.44 Immunosuppressive B cells express IgA, interleukin-10, and programmed death ligand 1 (PD-L1). By removing these cells, the tumor can be killed using oxaliplatin mediated immunogenic chemotherapy.
Effective ICD requires high ICD inducer stimulant doses. However, these stimulant doses may produce undesirable systemic toxicity. To tackle this issue, Li et al. developed a co-delivered liposome that combined shikonin with anthracyclines to induce a synergistic effect of ICD and cytotoxicity against tumor cells. In contrast with liposomes loaded with a single therapeutic agent, dual-loaded liposomes exhibited the capacity to enhance antitumor efficacy by inducing a strong immune response by lower drug doses, thereby enhancing safety profiles. Liposomes loaded with ICD inducers demonstrate release profiles sensitive to pondus hydrogenii (pH) and glutathione (GSH) or exhibit dual-response release profiles (Fig. 2). The immunogenic effect of copper ion-mediated shikonin (LipSHK) loaded liposomes in vivo was evaluated. Compared with the normal saline group, CRT and HMGB1 in the high-concentration drug-loaded NP group increased 3.0 and 3.2 times, respectively. Although LipSHK can effectively induce ICD, the high dose results in liver toxicity. The efficacy of the dual-loaded liposomal treatment was then evaluated in a PCa model. Compared with single-loaded liposomes, dual-loaded liposomes have a better antitumor effect. The results showed that dual-loaded liposomes had a better therapeutic effect than single-loaded liposomes.16
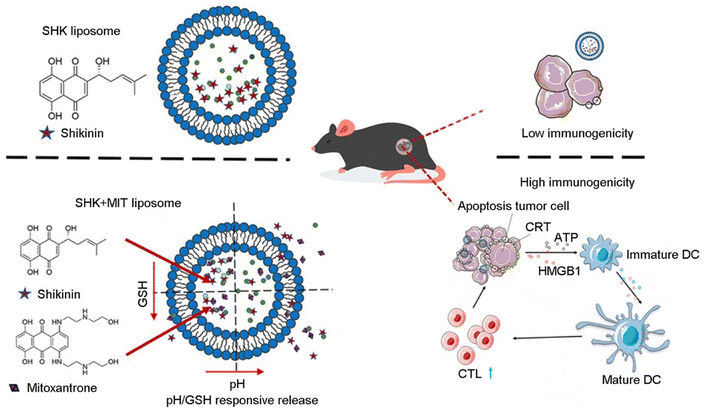 | ||
| Fig. 2 Enhancing the immunogenicity in cold tumor cells. Illustration of liposome NP-mediated enhancement of immunotherapy via ICD. Reproduced with permission from ref. 15, © Elsevier 2021. | ||
Photodynamic therapy, another advanced technology, is a photochemical-based approach that utilizes a nontoxic photosensitizer accumulated in tissues. Upon exposure to light of a specific wavelength, the photosensitizer produces cytotoxicity by generating reactive molecules. The emergence of nanomedicine has further augmented the potential of photodynamic therapy by integrating targeted delivery of photosensitizers to tumor sites and inducing ICD to trigger immune responses.45 Sengar et al. developed mesoporous silica-coated yttrium–aluminum garnet NPs doped with praseodymium and modified with porphyrin and folic acid. This strategy employs photodynamic therapy to induce tumor cell death, with folic acid serving as a targeting molecule by increasing the affinity to the folate receptor Folr1 in cancer cells, thereby increasing the tissue penetration depth and improving the efficiency of photodynamic therapy.18 To induce ICD through physical methods, some researchers employed ultrasound technology to control the release of ICD inducers in liposome-microbubble complexes.17 Using the principle that hyperthermia can induce tumor cells to release antigens, some scholars have linked the potential mechanism of photothermal therapy and immune activation, and developed a platform of black phosphate nanobinding photothermal therapy (PTT) containing DOX. The results confirmed that this drug combination stimulated the ICD process and DC maturation in PC-3 cells. In addition, the modification of zinc ions improved the killing ability of the nanosystem on PCa cells.46
Radiotherapy stands as the primary treatment for PCa, and compelling evidence has emerged demonstrating that radiotherapy induces ICD in tumor cells.47 Yasmin-Karim's team developed a novel approach integrating radiotherapy with immunogenic biomaterials (IBMs), such as poly(lactide-co-glycolide) (PLGA) and alginate. The combination strategy effectively overcame the immunosuppressive TME, resulting in a more pronounced infiltration of various immune cell populations.19
Collectively, these results demonstrate the potential of biomaterials in modulating the release of tumor antigens. Currently, ICD-induced biomaterials are receiving increasing attention due to their biosafety and multifunctional modifications. ICD induced by some biomaterials that do not contain anticancer drugs has gradually attracted scholars' interest.41
2.2. Activation of APCs
Following the release of tumor-associated antigens (TAAs), APCs process TAA-derived polypeptides through the major histocompatibility complex (MHC) and present immune signals recognizable by tumor-specific T lymphocytes. This is the second step in the cancer-immunity cycle and serves as a crucial link between tumor cells and T lymphocytes.10 DCs are the most important and extensively investigated APCs. Due to their distinctive properties, DCs have been harnessed to develop cell-based antitumor vaccines.48 Furthermore, the incorporation of T cell-activating ligands on biocompatible materials has been employed to construct artificial APCs, which exhibit significant potential in tumor immunotherapy.49Messenger RNA (mRNA) tumor vaccines are nucleic acid vaccines designed to target TAAs for effective cancer immunotherapy. The principle of mRNA vaccines is that after one or more target proteins have been identified, the corresponding transcript carrying the antigenic information of the tumor is transfected into the cytoplasm of the host cell, usually the APC. The encoded protein activates the immune system by binding to MHC molecules and being displayed on the surface of the APC. However, the inefficiency of in vivo mRNA delivery and the requirement for immune co-stimulation are major obstacles to achieving antitumor efficacy. NP-based vaccines are gaining increasing attention for their ability to elicit an effective anticancer immune response. Islam et al. demonstrated an adjuvant-pulsed mRNA vaccine NP composed of ovalbumin-encoded mRNA and palmitic acid-modified TLR7/8 agonist R848 derivative, coated in a lipid-PEG shell. Immature APCs phagocytose NPs, migrate through the lymphatic system, and reach the nearest lymph node for maturation. Upon full maturation, APCs present antigens on their membranes, activate CD4+ and CD8+ T cells, and generate specific immunity. The NP-based mRNA vaccine significantly increases the transfection efficiency of mRNA and subsequently augments the presentation of MHC-I antigens derived from mRNA in APCs.20
In addition, using adjuvants to improve the efficiency of APC presentation is also an effective strategy to improve immunotherapy. DCs express Toll-like receptors (TLR), which enables them to recognize biomolecules of pathogen-associated molecular patterns or DAMPs. Therefore, enhancing tumor antigen presentation by modulating DCs is a crucial strategy.
TLR agonists such as Requimod (R848) are commonly used as immune adjuvants to enhance tumor antigen presentation by promoting the maturation of DCs. However, it is important to note that inflammatory cytokines produced by free R848 induction result in potential adverse events and safety risks. Thus, Huang et al. designed PLGA NPs co-loaded with indocyanine green (ICG) and R848 (PLGA-ICG-R848) to synergistically treat PCa with PTT and immunotherapy (Fig. 3A).21 PLGA-ICG-R848 showed no significant difference in cell viability under different concentrations of ICG. However, after combined photothermal treatment, cell survival decreased significantly in a concentration-dependent manner (Fig. 3B). In the experiment to test drug promotion of DC maturation, the results showed that the R848 group was significantly higher than other groups, and the effects of the NP group and free drug group were comparable (Fig. 3C). To evaluate the in vivo photothermal intensity of PLGA-ICG-R848 NPs, a RM9-Luc subcutaneous tumor xenograft model was established. During laser irradiation, the subcutaneous temperature of the NPs+ laser group was consistently higher than that of the PBS+ laser group (Fig. 3D). On the basis of photothermal treatment, bifunctional PLGA-ICG-R848 NPs showed immune activity both in vitro and in vivo. These findings may therefore have important implications for developing therapeutic strategies against PCa by combining PTT with immunotherapy. Unmethylated cytosine-phosphate-guanine (CpG) oligonucleotides, renowned for their immunostimulatory functions, are commonly utilized as Toll-like receptor 9 (TLR9) agonists.22 As an immune adjuvant, CpG increases the expression of co-stimulatory molecules on DCs, facilitating antigen presentation and augmenting the efficacy of immunotherapy.50 To prolong the retention time of CpG within tumor sites, Chao et al. developed a multifunctional sodium alginate (ALG)-catalase (Cat)-oligonucleotide (CpG) fluid hydrogel (131I-Cat/CpG/ALG).23 The in situ injection of the hydrogel effectively directs the drug toward the tumor lesion and concurrently prolongs the residence time of the immune adjuvant in the tumor site. The low dose of 131I labeled Cat alleviates the hypoxic environment and improves radiotherapy efficiency in tumor lesions by decomposing hydrogen peroxide to produce oxygen. Notably, 131I-Cat/CpG/ALG triggered stronger systemic antitumor immune responses, regulated DC activation results in immune system stimulation, substantial amplification of tumor-specific killer T lymphocytes, and elevated levels of antitumor cytokines. The significant antitumor effects of this strategy in a PCa model were verified.
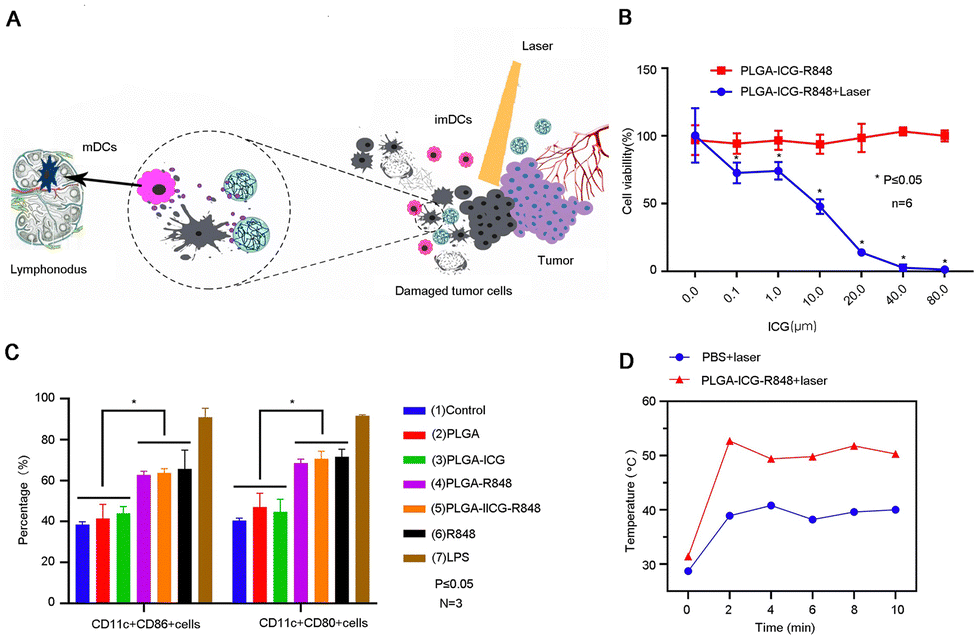 | ||
| Fig. 3 Photothermal therapy combined with immunotherapy for PCa. (A) Illustration of photothermal therapy combined with immunotherapy for PCa. (B) A comparison of the tumor cell inhibitory effect of photothermal therapy combined with immunotherapy and single therapy. (C) Flow cytometry detected NPs promoting BMDCs maturation. (D) The photothermal intensity of the NP group was measured using an FLIR thermal imager. Reproduced with permission from ref. 20, © International J. Nanomed., 2021, 16, 2775–2787 originally published by and used with permission from Dove Medical Press Ltd. | ||
Inorganic materials, such as carbon nanotubes (CNTs), have been reported to promote the aggregation of major MHC-I and the co-stimulatory ligand anti-CD28 (CD28). Xia et al. developed multi-walled CNT-coupled cell-penetrating peptide nanocarriers for CpG delivery.24 The application of nanomaterials and TLR agonists improves drug stability and enhances the cellular uptake of CpG. The application of nanocomposites showed a strong immune-stimulating ability. Kozlowska et al. employed functionalized bioengineered spider silk balls to deliver oligonucleotides containing unmethylated CpG motifs to prevent degradation of the oligonucleotides in serum, improved nuclease resistance, and promoted DCs maturation.25
In addition to the above-mentioned mRNA vaccines that solve the problem of antigen-specific presentation and the application of TLR agonists to enhance the function of APCs, tumor heterogeneity and alterations in the antigenicity of TAAs are other problems that tumor vaccines need to address. PCa exhibits favorable characteristics for vaccine-based immunotherapy, partly due to the presence of TAAs. These TAAs are typically expressed in normal prostate tissue and overexpressed in PCa. PSA is predominantly expressed in prostate epithelial cells and is substantially elevated in cancer tissue. It is an important serological indicator for the current clinical diagnosis of PCa. Another marker of normal prostate tissue is PSMA, which plays an important role in the clinical imaging diagnosis of prostate bone metastases. Prostatic acid phosphatase is a glycoprotein mainly secreted by prostate epithelial cells and plays an important role in the production of sipuleucel-T vaccines. In addition, prostate stem cell antigen (PSCA), TRPM8, and prostate epithelial antigen 1 (STEAP1), etc., are all highly desired antigens in immunotherapy.51 Herrmann et al. encapsulated STEAP1 as an antigenic peptide into PLGA microspheres and elicited a strong immune response in mice. In this study, the researchers evaluated different antigenic peptides encapsulated in NPs and found only PLGA microspheres containing STEAP1 peptides demonstrated effective induction of cytotoxic T lymphocyte (CTL) in vivo. It is also proved that vaccination with PLGA microspheres is more effective than incomplete Freund's adjuvant.26 However, monovalent antigen vaccines usually fail to elicit robust antitumor responses. Chen and his co-workers loaded imiquimod R837 as a vaccine adjuvant with PLGA NPs to address this limitation. Furthermore, they incorporated multiple antigens by coating the particles with cancer cell membranes, serving as a source of diverse antigens.27 This strategy applies the cell membrane to trigger a strong multi-antigenic immune response, concurrently evading immune rejection while enhancing targeting. In addition to regulating the use of specifically toxic T lymphocytes to kill tumor cells using DCs, some scholars also use vaccine technology for endocrine therapy. Androgen-specific antibodies were produced by encapsulating luteinizing hormone-releasing hormone (LHRH) peptides and TLR adjuvants using nanoliposomes, which possess the ability to selectively target FcRs (receptors for IgG Fc fragments) expressed on DCs.28 The novelty of this study lies in the use of nanotechnology to successfully induce endocrine therapy using immunotherapy, thereby presenting novel avenues for combined therapy in different fields.
DCs play a pivotal role in tumor antigen presentation. The use of biomaterials enables tumor vaccines to have more modalities to modulate DCs.52,53 The co-delivery of adjuvants also increases the strength of vaccine-elicited tumor-specific immunity.
2.3. Regulation of T cell-mediated cellular immunity
Interleukin-15 (IL-15) is an important cytokine indispensable in the proliferation and maintenance of CD8+ T cells (Fig. 4A). However, due to its short half-life, a single injection makes it hard to provide the sustained exposure necessary for optimal stimulation of the target immune cells. Excitingly, biomaterials provide a secure platform for its continuous stimulation of immune cells. Hangasky and colleagues prepared covalently linked IL-15 PEG-hydrogel microspheres (MS-IL-15) for long-acting IL-15, which significantly increases CD8+ T cells and exhibited a remarkable tumor inhibitory effect in a mouse model of PCa.29 MS-IL-15 significantly extended the elimination time of IL-15, which was t1/2 of 168 hours within 120 hours, and the drug release time was more durable and stable (Fig. 4B). The immunotherapeutic effect of MS-IL-15 in a mouse model of PCa was evaluated. When combined with anti-CD40, it can stimulate the proliferation and activation of T cells (Fig. 4C). The combination administration shows strong anti-tumor activity, significantly prolongs survival, and the advantage of long-acting drug agonists compared with multiple single drug injections (Fig. 4D). These data suggest that hydrogels are promising cytokine carriers and can potentially be loaded with various agents for antitumor therapy.55
 | ||
| Fig. 4 Application of biomaterials to activated T cells. (A) Illustration of the effect of IL-15 on T cells in immunotherapy. (B) Comparison of drug release between hydrogel-supported and free drugs. (C) Activation effect of drug-loaded hydrogel microspheres and anti-CD40 on CD8+ T cells. (D) The antitumor effect and survival curve of a single dose of IL-15 hydrogel microsphere subcutaneous injection (below) instead of multiple IL-15 injections (above). The data are the tumor relative median volume ± median SE. (A) Reproduced with permission from ref. 10, © Elsevier 2013. (B)–(D) Reproduced with permission from ref. 29, © BMJ Publishing Group Ltd. 2022. | ||
 | ||
| Fig. 5 Application of biomaterials in the destruction of tumor blood vessels and the extracellular matrix. (A) Illustration of T cell trafficking. (B) Illustration of VEGF as a hindrance factor for T-cell trafficking. (C) Biomaterials inhibit mTOR signaling in fibroblasts and cancer cells. (D) Biomaterials decreased VEGF expression in PCa. (E) Biomaterials inhibit fibroblast-mediated PCa progression. (F) Biomaterials inhibit angiogenesis. (A) and (B) Reproduced with permission from ref. 61, © Elsevier 2013 (C)–(F) Reproduced with permission from ref. 31, © John Wiley and Sons 2018. | ||
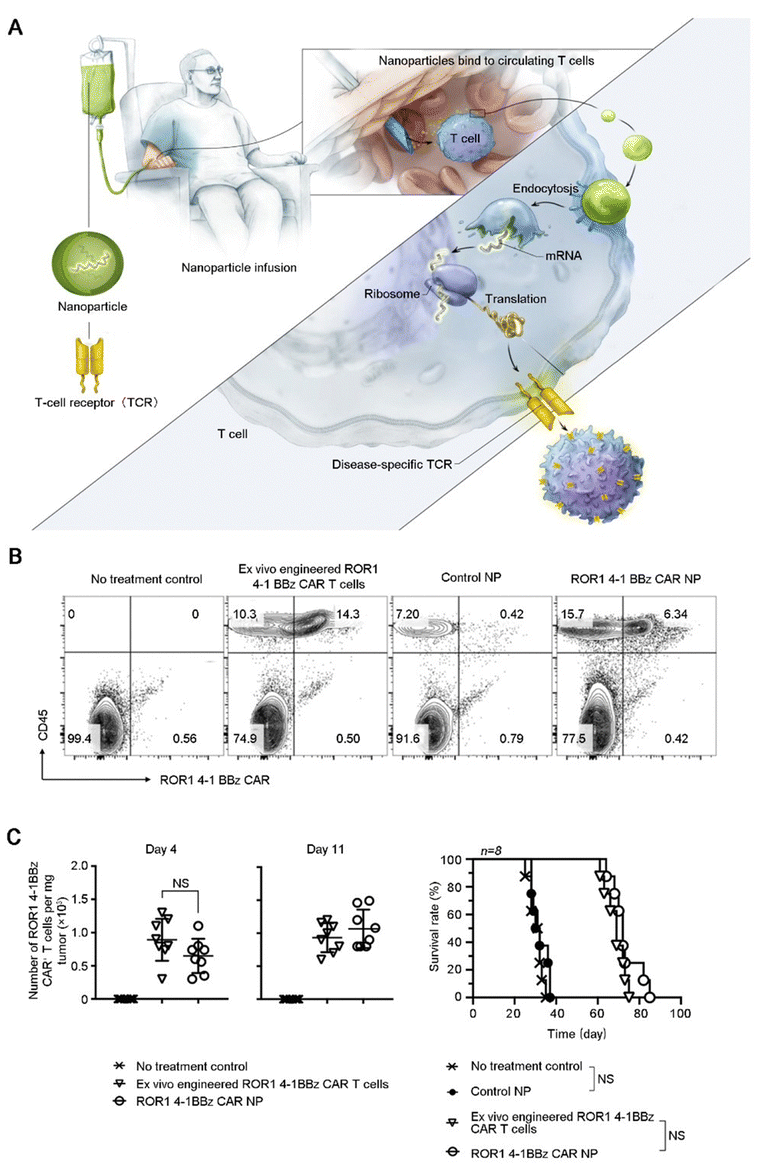 | ||
| Fig. 6 Applications of biomaterials in the regulation of T cells. (A) Illustrating how to reprogram T cells in situ to express disease-specific CARs or TCRs using IVT mRNA carried by polymeric NP. (B) Multicolor flow cytometry was used to detect bioengineered T cells in prostate tumors after treatment. (C) Quantification of ROR1-CAR+T cells in tumors isolated at days 4 and 7 after initiation of treatment and survival results. Shown are mean ± SD; two-tailed unpaired t-test, n = 8. Reproduced with permission from ref. 32, © Springer Nature 2020. | ||
After recognizing tumor cells, T cells execute cytotoxic functions. However, in order to avoid damage and destruction of normal tissues, immune checkpoints serve as a class of immunosuppressive molecules that counteract the killing activity of T cells. Tumors exploit this mechanism for immune evasion; induced T cells fail to recognize and kill tumor cells, thereby establishing a state of immune tolerance. Specifically, programmed cell death protein-1 (PD-1) and programmed cell death ligand-1 (PD-L1) molecules are located on the surface of activated T lymphocytes and tumor cells, respectively. Upon binding of PD-1 with PD-L1, mature T lymphocytes perceive the tumor cell as a non-threatening entity and fail to initiate an attack. Immune checkpoint blockade (ICB) through the use of anti-PD-1/anti-PD-L1 antibodies has produced effective therapeutic responses in a large number of patients with various cancer types.64 However, the strategy encountered two major obstacles: low delivery efficiency and systemic exposure. In order to solve these problems, Choi et al. utilized mesoporous silica nanocarriers as a vehicle for drug delivery, and the surface was coated with nano-iron oxide (Fig. 7A). The nanocarriers prolonged the in vivo sustained release time of anti-PD-L1, thereby augmenting the number of CTLs and improving tumor suppression efficacy (Fig. 7B).32
 | ||
| Fig. 7 Suppression of immune escape in prostate PCa cells. (A) Illustration of αPD-L1 ICB loading and ferumoxytol capping in NP. (B) Analysis of the number of tumor-infiltrating T lymphocytes and tumor volume after NP treatment. Reproduced with permission from ref. 33, © John Wiley and Sons 2019. | ||
Immunologically effective biomaterials have been applied throughout the cancer-immunity cycle, demonstrating their potential to modulate adaptive immunity for enhanced tumor cell elimination with antigen specificity. By precisely regulating the intricate interplay between the immune system and cancer cells, these interventions based on biomaterials hold great promise in advancing therapeutic strategies.
3. Regulation of innate immunity
Another type of immune response is innate immunity, distinguished by its lack of antigen specificity, but rapid response capabilities. Once innate immune cells are activated by the environment, they can directly kill tumor cells. Natural killer (NK) cells represent a prominent subset of innate immune cells known for their ability to eliminate tumor cells. While certain other innate immune cells may not possess direct cytotoxicity against tumor cells, they play a critical role in presenting proteins expressed by tumor cells, thereby facilitating the immune response toward the tumor.3.1. Regulation of the activity of innate immune cells
As innate immune cells in the human body, NK cells produce cytokines with anti-tumor effects and do not need to be “trained” with specific antigens to achieve activation. In contrast with neutrophils and macrophages, which belong to the innate immune cell family, NK cells have a wider range of bacterial clearance and defense against viruses. Therefore, NK cells hold significant promise in the realm of tumor immunotherapy. NK cells naturally kill tumor cells by releasing perforin and granzyme, respectively, which permeate the cytoplasmic membrane and initiate apoptosis.65 The activation of NK cells depends on the “third signal,” encompassing pro-inflammatory cytokines (multiple interleukin molecules) produced by myeloid cells. Among these, IL-12 plays a central role in facilitating NK cell effector function, which is originally named NK cell stimulating factor. IL-12 facilitates the expansion and maturation of NK cells, participates in the development of memory NK cells, and enhances cell performance.66 However, high-dose application of IL-12 is associated with severe systemic toxicity, in rare cases, fatal events, which hinder its direct application in immunotherapy.67 To address the toxicity issue, Gasparri's team prepared cell-targeting peptide Iso1-labeled gold nanospheres (Iso1/Au/IL-12) containing IL-12 for improving tumor therapeutic effects. The findings suggest that this nanoformulation can deliver extremely low IL-12 doses to tumors, increase tumor infiltration by innate immune cells, such as NK cells, monocytes and neutrophils, resulting in tumor growth inhibition (Fig. 8A–C).34 Cytokines regulate a variety of cells involved in the innate immune response, but the direct systemic injection of cytokines will bring great toxic side effects to the body. The use of nanotechnology to deliver drugs can be the most effective immune activation on the basis of acceptable safe drug doses. | ||
| Fig. 8 Activation of innate immune cells. (A) High-dimensional bhSNE plot of tumor-infiltrating immune cells in mice treated with NPs. (B) The number of NK cells in untreated and NP-treated mice was determined by flow cytometry. (C) Effect of treatments on tumor growth. Reproduced with permission from ref. 35, © John Wiley and Sons 2019. | ||
3.2. Innate immunity initiated through the stimulator of IFN genes pathway (STING)
STING, also known as a transmembrane protein, IFN regulator, etc., is a class of transmembrane proteins located in the endoplasmic reticulum. STING is important in the innate immune response, the body's tumor immune process, and cell autophagy. It regulates protein synthesis and IFN expression through chemical modifications such as its own phosphorylation, thus playing a key role in multiple immune links in the body effect.68 The STING pathway differs from other innate immune sensing pathways proposed to enhance tumor immunogenicity. The reason is that it not only produces IFN and other pro-inflammatory cytokines through downstream signaling cascades but also indirectly stimulates tumor APCs.69 This subsequently facilitates the mobilization of tumor-specific CD8+ T cells; thereby, the STING pathway serves as a bridge to initiate tumor adaptive immune responses.70 The combination of the STING agonist and cytokine IL-15 was confirmed to enhance NK and T cell activity in PCa.71 The synergistic effect benefited from IFN production through the innate immune pathway. Furthermore, the augmented potency of NK cells was also attributable to the increasing expression of perforin and CD69. Techniques such as intratumoral injection of STING agonists in conjunction with ICB, or adoptive cell therapy have achieved considerable therapeutic outcomes in the treatment of PCa.72 The powerful immune-activating capacity of STING agonists appears to be an excellent strategy for immunotherapy. However, the inherent shortcomings of small-molecule STING agonists limit their in vivo application and ultimate therapeutic efficacy due to low bioavailability in targeted tissues. Drug delivery systems play an important role in enhancing the efficiency of STING activation. At present, STING agonists use lipids, polymers, and inorganic materials as carriers, and their advantages are reflected in the use of stabilizing groups, biomimetic coatings, targeting ligands, and response parts for functionalization.73 The application of biomaterials optimizes the single treatment of STING agonists by intratumoral injection, providing more possibilities for tumor immunotherapy. It was reported that PLGA NPs successfully ameliorate the problem of an extensive first-pass effect and intricate drug delivery, while concurrently exhibiting synergistic therapeutic benefits. This strategy offers the capability to activate the innate immunity of tumor cells and significantly enhance the tumor inhibition when combined with STING agonists.74 Researchers have synthesized PLGA NPs loaded with cell metabolism inhibitors to further activate the cGAS/STING signaling pathway in tumor cells by triggering mitochondrial DNA release caused by mitochondrial metabolism disorders. This strategy combined with STING agonists has been confirmed to effectively promote the activation of CD8 T cells and synergistically amplify the effect of immunotherapy.754. Regulation of the inhibitory TME
Despite the enhanced cytotoxic activity of tumor-specific T cells in the cancer-immunity cycle, tumors still develop, which is closely related to the tumor-suppressive microenvironment. Immunosuppressive cells, such as TAMs, Tregs, and myeloid-derived suppressor cells (MDSCs), infiltrate around tumors and exert immunosuppressive effects by inhibiting cytotoxic T lymphocytes, thereby facilitating tumor expansion. In light of this challenge, the use of biomaterials for targeted intervention against these immunosuppressive cells has emerged as a promising avenue.4.1. Transformation of TAMs phenotypes
TAMs are one of the most abundant immune cell populations in the TME and play an important role in the immune regulation of PCa. Depending on the polarization state, TAMs typically differentiate into an antitumor M1 phenotype and a tumor-promoting M2 phenotype.76 Generally, TAMs predominantly exhibit an M2 phenotype, which is extremely important for the inhibitory effect of immunotherapy. Currently, there are two ways to target TAMs for cancer immunotherapy: depleting TAMs and regulating phenotypic transformation of TAMs. However, arbitrary depletion of TAMs stifles the potential immune-stimulatory effects of phagocytosis and presentation by M1-type macrophages. Therefore, it is of great interest to develop new methods to inhibit the function of M2 macrophages and promote their conversion to the M1 phenotype. Since TLR agonists have demonstrated the ability to induce the conversion of macrophages toward an M1 phenotype, some scholars have used exosomes isolated from serum as a small-molecule drug platform to co-deliver the TGF-β kinase inhibitor SD-208 and the TLR7/8 agonist R848 (Fig. 9A). The experimental results show that this strategy inhibits the invasion of PCa cells (Fig. 9B), triggers the polarization of macrophages toward an antitumor phenotype (Fig. 9C), releases a large number of pro-inflammatory cytokines, and manifests an obvious antitumor efficacy.35 Furthermore, compelling evidence has emerged to highlight that exosomes play a crucial role in long-distance communication between cells, facilitated by their ability to reach other cells and tissues through the circulatory system, resulting in long-distance regulation. Therefore, exosomes present great potential as a kind of “natural nanoparticle” to deliver cargo such as drugs and genes for small-molecule therapy benefiting from their high stability in blood, natural hydrophilicity, natural targeting, low immunogenicity, etc.77 The upregulation of nuclear factor kappa-B (NF-κB) signaling within the TME emerges as another crucial determinant driving the polarization of macrophages toward the M2 phenotype.78 NF-κB, a key transcription factor implicated in cancer initiation and progression, also exhibits associations with drug resistance mechanisms. Therefore, immunomodulating the NF-κB pathway is a viable strategy for the regulation of macrophages. However, the main challenge lies in achieving optimal bioavailability while ensuring safe dosage levels. To solve this problem, Khoobchandani et al. employed nanotechnology by utilizing mangiferin-functionalized gold NPs (MGF-AuNPs) to treat PCa. The MGF-AuNPs increased the bioavailability of loaded phytochemicals. Furthermore, this innovative strategy focuses on the transformation of tumor-promoting M2 macrophages into the antitumor M1 phenotype, with the potential to significantly improve the treatment outcomes of patients with CRPC.36 It was reported that the presence of reactive oxygen species (ROS) within the TME exerts a notable influence on the polarization and immunosuppressive function of M2 macrophages.79 Liu et al. reported a synergistic thermoimmunotherapy approach for PCa utilizing cyclic RGD peptides and manganese-doped eumelanin nanocomposites (RMnMels). The use of RGD peptides facilitated the targeting accumulation of RMnMels within tumors. The incorporation of metal enabled T1-weighted magnetic resonance/photoacoustic imaging, allowing for visualization of tumors in vivo. The experimental results demonstrated that RMnMels promoted the repolarization of M2-type macrophages to M1-type macrophages by scavenging ROS in PCa mice.37 Biomimetic nanomaterials mimic natural cells by adjusting the size, surface charge, shape, and material consistency.80 In particular, the inherent presence of abundant functional groups within biomimetic nanomaterials confers specific biological activities, which make them an ideal nanoplatform. The development of novel NP engineering approaches that enable targeted delivery or modulating TAMs has great promise for cancer treatment.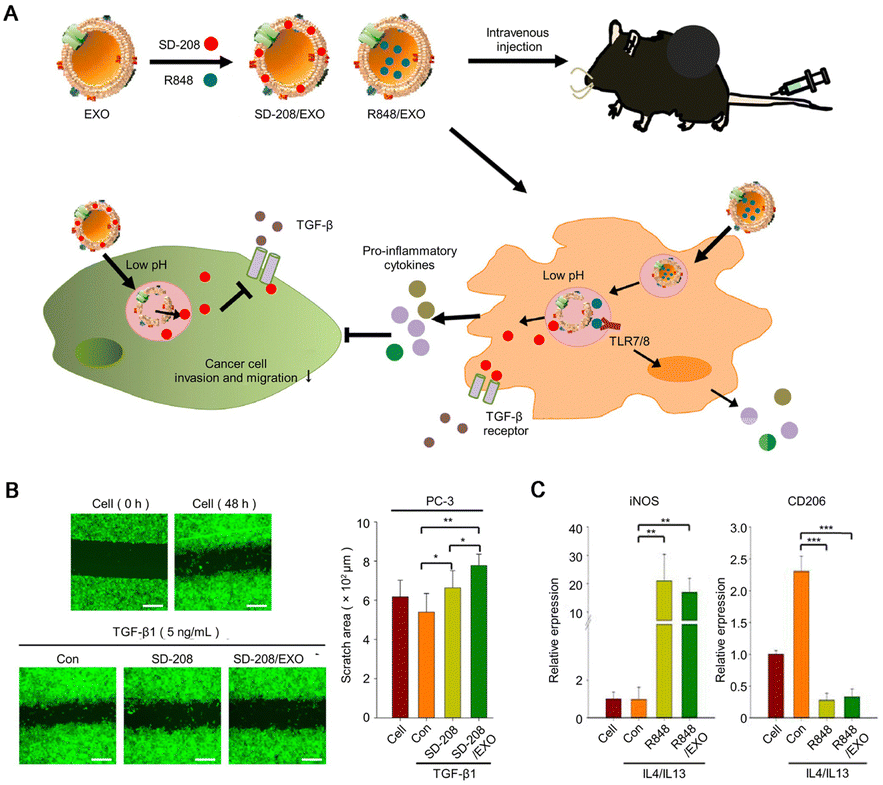 | ||
| Fig. 9 Regulation of the tumor-suppressive immune microenvironment. (A) Schematic of the therapeutic mechanism of drug-loaded NPs inhibiting tumor immune escape. (B) Cell migration assay was performed by adding transforming growth factor TGF-β (5 ng mL−1) (scale bar: 500 μm) to free drug and drug-loaded NP, respectively. The figure shows the scratch area of PCa cells after drug treatment. (C) Relative mRNA expression of the inflammatory macrophage polarization marker iNOS and anti-inflammatory macrophage polarization marker CD206 in cells. Reproduced with permission from ref. 36, © Elsevier 2022. | ||
4.2. Suppression of Tregs
Tregs are a subtype of CD4+ T cells. In order to maintain the dynamic balance of the body's immune system and prevent the immune system from excessive activation, its role is to inhibit the adaptive immune response. Obviously, this property is extremely disadvantageous in the immunotherapy of tumors. In the course of tumor treatment, the antagonism of Tregs to CTL leads to the weakening of immunotherapy.81 Although the use of anti-CTLA-4 monoclonal antibodies represents a current strategy to inhibit Tregs and enhance immune effectiveness, the clinical benefits remain limited.82 Studies have found that the deletion of the tumor suppressor gene phosphatase and tensin homolog deleted on chromosome ten (PTEN) promotes a suppressive immune microenvironment, leading to the accumulation of suppressive immune cells such as Tregs.83 Therefore, modulating the activation of Tregs via the PTEN pathway is a promising concept for tumor immunotherapy. Transfection of host genes using synthetic mRNA technology is a common technique in gene therapy.84 Lin et al. developed a lipid-polymer hybrid NP platform for the targeted delivery of PTEN mRNA to tumor sites. This innovative platform was formulated based on the self-assembly of mPEG-PLGA copolymer, with mRNA encapsulated within the complex comprising cationic lipid-like material 1,2-epoxytetradecane-modified polyamidoamine (G0-C14). The experimental results showed that, through PTEN gene repair, the expression of Tregs and other immunosuppressive cells was reduced, and the immunosuppressive microenvironment was reversed.384.3. Targeted regulation of MDSCs
MDSCs represent a subset of immunosuppressive cells in the TME and are associated with poor prognosis in PCa patients.85 MDSCs are derived from myeloid progenitor cells, which are transformed from immature myeloid cells under pathological conditions.86 In cancer patients, a variety of cytokines mediate the transformation and recruitment of MDSCs.87 MDSCs exhibit a range of biological behaviors, including the generation of ROS and the consumption of amino acids, contributing to their immunosuppressive effects. More specifically, the increased production of ROS by MDSCs leads to DNA damage within CTLs, ultimately impairing their capacity to effectively eliminate tumor cells. Furthermore, MDSCs weaken the number and tumor-suppressing function of T cells by removing metabolites such as arginine, tryptophan, and cysteine from the microenvironment. These metabolites are necessary for sustaining T cell biological activity, and MDSCs employ multiple pathways, such as arginase and NOS2, to achieve this detrimental effect.88 In a mouse model of PCa, Lu and collaborators elucidated the potent immunotherapeutic effects of kinase inhibitors that specifically target MDSCs. The effects of kinase inhibitors were particularly pronounced when combined with immune checkpoint inhibitors.89 It is believed that MDSCs have potential as a target for PCa immunotherapy. The utilization of nanotechnology in this field remains largely unexplored. There is a growing belief that nanotechnology exhibits considerable promise for precise targeting of MDSCs.5. Future prospectives
PCa, as a recognized “cold tumor,” still faces many challenges in the process of immunotherapy. Despite the remarkable efficacy demonstrated by immune checkpoint inhibitors and monoclonal antibodies in treating various tumors, the objective response rate of PCa remains suboptimal. With the development of science and technology and the integration of multiple disciplines, biomaterials with immunological effects have been developed in the past few years. Biomaterial-based immunotherapy strategies or utilizing biomaterials to improve immunotherapy efficiency have attracted extensive attention.The conception of the cancer-immunity cycle has provided valuable insights into the intricate regulation of adaptive immunity. Increased exposure to pathogen molecular pattern signals such as antigens and increased processing of antigens by APCs are the initial steps of adaptive immunity. Therefore, strategies to load therapeutic drugs with inherently IBMs have been extensively explored. Interestingly, the use of biomaterials to increase cellular ICD has also shown encouraging results.90 However, for PCa immunotherapy, only activating the immune effect through biological materials is limited, and it needs to be combined with radiotherapy and other technologies to achieve the desired effect. After ICD-activated immune mediation, improving the efficiency of APC immunotherapy is another important issue. The application of immune adjuvants such as TLR agonists to enhance the ability of DCs to process antigens has promoted the development of cancer vaccines. Further research should focus on developing biomaterials loaded with various therapeutic drugs, such as siRNA, mRNA, and oligonucleotides, to modulate the antigen-presenting ability of APCs.91 Furthermore, the synergistic approach of combining multiple immune adjuvants and therapeutic drugs has demonstrated superior antitumor efficacy compared with monotherapy utilizing single agents.
Another approach to enhancing tumor-specific immunity is to modulate the biological behavior of T cells. The use of cytokines to prime T cells has yielded satisfactory results.92 The chemotactic regulation of T cells by chemokines seems to be a novel idea for immunotherapy.93 However, related research on PCa remains relatively scarce, indicating the need for further studies. Notably, the cancer-immunity cycle is a cyclical process, and promoting the development of any link will promote the advancement of the entire cycle. Therefore, expanding our perspectives beyond the conventional focus on research such as APCs and T cells is imperative. The strategy of coordinated regulation of multiple immune processes has promise as a future direction for the regulation of immunotherapy.
Innate immunity is the body's natural immune barrier and has a strong immune function. In recent years, extensive research on the STING pathway has become a new idea for tumor therapy. The STING pathway not only initiates innate immunity to kill tumors through the regulation of IFN but also activates DCs to specifically kill tumors through adaptive immunity. Using the STING pathway to regulate tumor immunity from the perspective of tumor metabolism has great potential in the treatment of PCa. Regarding the regulation of innate immunity by cytokines, they have shown promising efficacy in tumor treatment. However, it is crucial to thoroughly evaluate the safety concerns arising from their dose-dependent effects before considering their actual in vivo application.
In general, immune modulation strategies, including modulation of both adaptive and innate immunity, provide a platform for the delivery of modulatory drugs. Modulator drugs are more targeted and delivered more efficiently than traditional strategies. In light of the rapid advancements in smart biomaterials, there has been a notable emergence of biomaterials capable of modulating the TME, thereby paving the way for their utilization in the field of immunotherapy.
The tumor-suppressive TME greatly limits the above-mentioned immunomodulatory strategies, so the regulation of immunosuppressive cells is a key strategy for PCa immunotherapy. Using biomaterials to modulate the transformation of TAM phenotypes is a viable option. A variety of NPs have achieved the successful repolarization of TAMs to the M1 phenotype that is more conducive to tumor immunotherapy while also effectively inhibiting the occurrence of Tregs. The combination therapy of an MDSC inhibitor and immune checkpoint inhibitor has produced an excellent curative effect on PCa. MDSCs represent a potential target of immunotherapy, and their combination with other strategies appears to produce a better tumor inhibition effect.
In general, different biomaterials, such as polymers, liposomes, NPs, etc., have shown their characteristics in PCa immunotherapy. NPs serve not only as carriers for therapeutic agents but also as immune cell regulators. The cargo-loaded polymers and liposomes showed good biocompatibility and a notable antitumor effect. With appropriate modifications, these immune-responsive biomaterials will show potential for antitumor therapy.
6. Conclusions
In conclusion, as the exploration of adaptive immunity, innate immunity, and the TME continues to deepen, immunologically effective biomaterials have become increasingly utilized in PCa immunotherapy, enabling targeted therapy and precise treatment. In recent years, reports on the immune effects of biomaterials have emerged one after another. Immunomodulatory drug delivery systems based on biomaterial carriers are constantly being developed to address immune homeostasis imbalance in PCa. The use of biomaterials to enhance immunotherapy by their immunomodulatory effects has been increasingly recognized. For example, Ding et al. developed activation-adapted antitumor immune nanoadjuvants using chiral biomaterials poly(lactic acid) (PLLA and PDLA) with the same atomic composition but different spatial structures. PDLA-OVA triggered a more adaptive anti-cancer immune response, leading to more effective suppression of cancer genesis and progression, suggesting that immunologically effective PDLA nanoadjuvants have great potential in cancer immunotherapy.52 Chiral polypeptides were also researched as activation-adapted antitumor immune nanoadjuvants by the same group, and showed the positive role of chiral biomaterials in anti-tumor immunomodulation.53 In addition, emerging biomimetic materials such as cell membranes in recent years are expected to balance biocompatibility and improve the immune effect of synthetic materials in the body. How to guarantee the quality control of biomaterials is a problem worthy of discussion. Although the current first-line treatments for advanced PCa are still novel androgen receptor inhibitors and a few biomaterials have been translated into clinical applications, immunologically effective biomaterials are indeed expected to provide better opportunities for PCa immunotherapy in the foreseeable future.Author contributions
Siqi Liu: resources, writing – original draft, writing – review & editing; Hui Guo: writing – review & editing; Di Li: conceptualization, funding acquisition, supervision, writing – review & editing. Chunxi Wang: conceptualization, funding acquisition, supervision, writing – review & editing.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the National Key Research and Development Program (No. 2021YFC2400603), the National Natural Science Foundation of China (No. 52273159, 52203166), the Science and Technology Development Program of Jilin Province (No. 20220204018YY), the Fundamental Research Funds for the Central Universities, and the Bethune Project of Jilin University (No. 2023B01).References
- S. Sandhu, C. M. Moore, E. Chiong, H. Beltran, R. G. Bristow and S. G. Williams, Lancet, 2021, 398, 1075–1090 CrossRef CAS PubMed.
- X. Gao, T. Niu, Q. Xia, B. Hu, Z. Zhao, X. Feng, J. Yang, B. Tang and K. Xu, Anal. Chim. Acta, 2022, 1210, 339852 CrossRef CAS.
- H. An, L. Li, Y. Wang, Z. Wang, D. Hou, Y. Lin, S. Qiao, M. Wang, C. Yang, Y. Cong, Y. Ma, X. Zhao, Q. Cai, W. Chen, C. Lu, W. Xu, H. Wang and Y. Zhao, Nat. Commun., 2019, 10, 4861 CrossRef.
- S. Yamashita, S. Muraoka, T. Wakamiya, K. Kikkawa, Y. Kohjimoto and I. Hara, Cancers, 2022, 14, 3466 CrossRef PubMed.
- J. Wen, Y. Zhu, L. Li, J. Liu, Y. Chen and R. Chen, Eur. J. Nucl. Med. Mol. Imaging, 2022, 49, 2086–2095 CrossRef CAS PubMed.
- K. Virgo, R. Rumble, R. de Wit, D. Mendelson, T. Smith, M. Taplin, J. Wade, C. Bennett, H. Scher, P. Nguyen, M. Gleave, S. Morgan, A. Loblaw, S. Sachdev, D. Graham, N. Vapiwala, A. Sion, V. Simons and J. Talcott, J. Clin. Oncol., 2021, 39, 1274–1305 CrossRef PubMed.
- L. Da Pozzo, C. Cozzarini, A. Briganti, N. Suardi, A. Salonia, R. Bertini, A. Gallina, M. Bianchi, G. Fantini, A. Bolognesi, F. Fazio, F. Montorsi and P. Rigatti, Eur. Urol., 2009, 55, 1003–1011 CrossRef.
- A. V. D'Amico, J. Clin. Oncol., 2014, 32, 362–364 CrossRef PubMed.
- T. M. Beer, E. D. Kwon, C. G. Drake, K. Fizazi, C. Logothetis, G. Gravis, V. Ganju, J. Polikoff, F. Saad, P. Humanski, J. M. Piulats, P. G. Mella, S. S. Ng, D. Jaeger, F. X. Parnis, F. A. Franke, J. Puente, R. Carvajal, L. Sengeløv, M. B. McHenry, A. Varma, A. J. v d Eertwegh and W. Gerritsen, J. Clin. Oncol., 2017, 35, 40–47 CrossRef CAS PubMed.
- D. S. Chen and I. Mellman, Immunity, 2013, 39, 1–10 CrossRef CAS PubMed.
- W. H. Chen, G. F. Luo and X. Z. Zhang, Adv. Mater., 2019, 31, 39 Search PubMed.
- Q. Wei, Y. Su, H. Xin, L. Zhang, J. Ding and X. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 56719–56724 CrossRef CAS PubMed.
- Z. A. Li, J. Huang, T. Du, Y. M. Lai, K. W. Li, M. L. Luo, D. J. Zhu, J. Wu and H. Huang, Chin. Chem. Lett., 2022, 33, 2496–2500 CrossRef CAS.
- Y. Zhang, X. Gao, B. Yan, N. Wen, W. S. V. Lee, X.-J. Liang and X. Liu, ChemMedChem, 2022, 17, e202100656 CrossRef CAS PubMed.
- Z. Wang, C. Sun, H. Wu, J. Xie, T. Zhang, Y. Li, X. Xu, P. Wang and C. Wang, Mater. Today Bio., 2022, 13, 100189 CrossRef CAS PubMed.
- J. Li, S. Zhou, J. Yu, W. Cai, Y. Yang, X. Kuang, H. Liu, Z. He and Y. Wang, J. Controlled Release, 2021, 335, 306–319 CrossRef CAS PubMed.
- F. Y. Huang, J. Lei, Y. Sun, F. Yan, B. Chen, L. M. Zhang, Z. X. Lu, R. Cao, Y. Y. Lin, C. C. Wang and G. H. Tan, OncoImmunology, 2018, 7, e1446720 CrossRef PubMed.
- P. Sengar, P. Juárez, A. Verdugo-Meza, D. L. Arellano, A. Jain, K. Chauhan, G. A. Hirata and P. G. J. Fournier, J. Nanobiotechnol., 2018, 16, 19 CrossRef PubMed.
- S. Yasmin-Karim, B. Ziberi, J. Wirtz, N. Bih, M. Moreau, R. Guthier, V. Ainsworth, J. Hesser, G. M. Makrigiorgos, M. D. Chuong, X. Wei, P. L. Nguyen and W. Ngwa, Int. J. Radiat. Oncol., Biol., Phys., 2022, 112, 475–486 CrossRef PubMed.
- M. A. Islam, J. Rice, E. Reesor, H. Zope, W. Tao, M. Lim, J. X. Ding, Y. H. Chen, D. Aduluso, B. R. Zetter, O. C. Farokhzad and J. J. Shi, Biomaterials, 2021, 266, 120431 CrossRef CAS PubMed.
- W. Lin, C. Li, N. Xu, M. Watanabe, R. Xue, A. Xu, M. Araki, R. Sun, C. Liu, Y. Nasu and P. Huang, Int. J. Nanomed., 2021, 16, 2775–2787 CrossRef PubMed.
- M. H. Teplensky, J. W. Dittmar, L. Qin, S. Y. Wang, M. Evangelopoulos, B. Zhang and C. A. Mirkin, Adv. Healthcare Mater., 2021, 10, 2101262 CrossRef CAS.
- Y. Chao, L. Xu, C. Liang, L. Feng, J. Xu, Z. Dong, L. Tian, X. Yi, K. Yang and Z. Liu, Nat. Biomed. Eng., 2018, 2, 611–621 CrossRef CAS PubMed.
- Q. Xia, C. Gong, F. Gu, Z. Wang, C. Hu, L. Zhang, L. Qiang, X. Ding, S. Gao and Y. Gao, J. Biomed. Nanotechnol., 2018, 14, 1613–1626 CrossRef CAS PubMed.
- A. K. Kozlowska, A. Florczak, M. Smialek, E. Dondajewska, A. Mackiewicz, M. Kortylewski and H. Dams-Kozlowska, Acta Biomater., 2017, 59, 221–233 CrossRef CAS PubMed.
- V. L. Herrmann, D. E. Wieland, D. F. Legler, V. Wittmann and M. Groettrup, Prostate, 2016, 76, 456–468 CrossRef CAS PubMed.
- S. X. Li, X. R. Feng, J. X. Wang, W. G. Xu, M. A. Islam, T. M. Sun, Z. G. Xie, C. X. Wang, J. X. Ding and X. S. Chen, Adv. Funct. Mater., 2019, 29, 1903391 CrossRef CAS.
- F. Rueda, C. Eich, B. Cordobilla, P. Domingo, G. Acosta, F. Albericio, L. J. Cruz and J. C. Domingo, Immunobiology, 2017, 222, 989–997 CrossRef CAS PubMed.
- J. A. Hangasky, W. Chen, S. P. Dubois, A. Daenthanasanmak, J. R. Muller, R. Reid, T. A. Waldmann and D. V. Santi, J. Immunother. Cancer, 2022, 10, e004104 CrossRef PubMed.
- E. E. Hill, J. K. Kim, Y. Jung, C. K. Neeley, K. J. Pienta, R. S. Taichman, J. E. Nor, J. R. Baker, Jr. and P. H. Krebsbach, J. Cell. Biochem., 2018, 119, 8074–8083 CrossRef CAS.
- N. N. Parayath, S. B. Stephan, A. L. Koehne, P. S. Nelson and M. T. Stephan, Nat. Commun., 2020, 11, 6080 CrossRef CAS.
- B. Choi, H. Jung, B. Yu, H. Choi, J. Lee and D. H. Kim, Small, 2019, 15, 1904378 CrossRef CAS PubMed.
- L. Luo, J. Yang, C. Zhu, M. Jiang, X. Guo, W. Li, X. Yin, H. Yin, B. Qin, X. Yuan, Q. Li, Y. Du and J. You, J. Controlled Release, 2018, 278, 87–99 CrossRef CAS PubMed.
- A. M. Gasparri, A. Sacchi, V. Basso, F. Cortesi, M. Freschi, E. Rrapaj, M. Bellone, G. Casorati, P. Dellabona, A. Mondino, A. Corti and F. Curnis, Small, 2019, 15, 1903462 CrossRef CAS PubMed.
- J. H. Lee, J. Song, I. G. Kim, G. You, H. Kim, J.-H. Ahn and H. Mok, Acta Biomater., 2022, 141, 354–363 CrossRef CAS PubMed.
- M. Khoobchandani, A. Khan, K. K. Katti, V. C. Thipe, A. Y. Al-Yasiri, D. K. D. MohanDoss, M. B. Nicholl, A. B. Lugão, C. P. Hans and K. V. Katti, Sci. Rep., 2021, 11, 16797 CrossRef CAS PubMed.
- Y. Liu, W. Shang, H. Liu, H. Hui, J. Wu, W. Zhang, P. Gao, K. Guo, Y. Guo and J. Tian, J. Nanobiotechnol., 2022, 20, 48 CrossRef CAS PubMed.
- Y. X. Lin, Y. Wang, J. X. Ding, A. P. Jiang, J. Wang, M. A. Yu, S. Blake, S. S. Liu, C. J. Bieberich, O. C. Farokhzad, L. Mei, H. Wang and J. J. Shi, Sci. Transl. Med., 2021, 13, eaba9772 CrossRef CAS PubMed.
- L. Kraehenbuehl, C. Weng, S. Eghbali, J. Wolchok and T. Merghoub, Nat. Rev. Clin. Oncol., 2022, 19, 37–50 CrossRef CAS PubMed.
- Y. Zhang, X. Hou, S. Du, Y. Xue, J. Yan, D. D. Kang, Y. Zhong, C. Wang, B. Deng, D. W. McComb and Y. Dong, Nat. Nanotechnol., 2023, 18, 1364–1374 CrossRef CAS PubMed.
- D. Li, S. Liu, Y. Ma, S. Liu, Y. Liu and J. Ding, Small methods, 2023, 7, e2300204 CrossRef PubMed.
- D. V. Krysko, A. D. Garg, A. Kaczmarek, O. Krysko, P. Agostinis and P. Vandenabeele, Nat. Rev. Cancer, 2012, 12, 860–875 CrossRef CAS PubMed.
- A. Rastinehad, H. Anastos, E. Wajswol, J. Winoker, J. Sfakianos, S. Doppalapudi, M. Carrick, C. Knauer, B. Taouli, S. Lewis, A. Tewari, J. Schwartz, S. Canfield, A. George, J. West and N. Halas, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18590–18596 CrossRef CAS PubMed.
- S. Shalapour, J. Font-Burgada, G. Di Caro, Z. Y. Zhong, E. Sanchez-Lopez, D. Dhar, G. Willimsky, M. Ammirante, A. Strasner, D. E. Hansel, C. Jamieson, C. J. Kane, T. Klatte, P. Birner, L. Kenner and M. Karin, Nature, 2015, 521, 94–U235 CrossRef CAS PubMed.
- P. De Silva, M. A. Saad, H. C. Thomsen, S. Bano, S. Ashraf and T. Hasan, J. Porphyr. Phthalocyanines, 2020, 24, 1320–1360 CrossRef CAS PubMed.
- L. Li, B. Zhou, H. Xu, H. Shi, L. Gao and B. Ge, Front. Endocrinol., 2022, 13, 872411 CrossRef PubMed.
- Y. Philippou, H. T. Sjoberg, E. Murphy, S. Alyacoubi, K. I. Jones, A. N. Gordon-Weeks, S. Phyu, E. E. Parkes, W. Gillies McKenna, A. D. Lamb, U. Gileadi, V. Cerundolo, D. A. Scheiblin, S. J. Lockett, D. A. Wink, I. G. Mills, F. C. Hamdy, R. J. Muschel and R. J. Bryant, Br. J. Cancer, 2020, 123, 1089–1100 CrossRef CAS PubMed.
- S. Go, M. Jung, S. Lee, S. Moon, J. Hong, C. Kim, Y. Chung and B. Kim, Adv. Mater., 2023, 35, 2303979 CrossRef CAS PubMed.
- J. Han, R. Bhatta, Y. Liu, Y. Bo, A. Elosegui-Artola and H. Wang, Nat. Commun., 2023, 14, 5049 CrossRef CAS PubMed.
- L. Li, S. Yang, L. Song, Y. Zeng, T. He, N. Wang, C. Yu, T. Yin, L. Liu, X. Wei, Q. Wu, Y. Wei, L. Yang and C. Gong, Theranostics, 2018, 8, 860–873 CrossRef CAS PubMed.
- Z. A. Lopez-Bujanda, A. Obradovic, T. R. Nirschl, L. Crowley, R. Macedo, A. Papachristodoulou, T. O’Donnell, U. Laserson, J. C. Zarif, R. Reshef, T. Yuan, M. K. Soni, E. S. Antonarakis, M. C. Haffner, H. B. Larman, M. M. Shen, P. Muranski and C. G. Drake, J. Immunother. Cancer, 2021, 9, e001649 CrossRef PubMed.
- W. G. Xu, Y. Z. Su, Y. Ma, Q. Wei, J. Z. Yang, X. L. Zhuang, J. X. Ding and X. S. Chen, Sci. China: Chem., 2023, 66, 1150–1160 CrossRef CAS.
- Y. Z. Su, W. G. Xu, Q. Wei, Y. Ma, J. X. Ding and X. S. Chen, Sci. Bull., 2023, 68, 284–294 CrossRef CAS PubMed.
- E. Antonarakis, J. Piulats, M. Gross-Goupil, J. Goh, K. Ojamaa, C. Hoimes, U. Vaishampayan, R. Berger, A. Sezer, T. Alanko, R. de Wit, C. Li, A. Omlin, G. Procopio, S. Fukasawa, K. Tabata, S. Park, S. Feyerabend, C. Drake, H. Wu, P. Qiu, J. Kim, C. Poehlein and J. de Bono, J. Clin. Oncol., 2020, 38, 395–405 CrossRef CAS PubMed.
- T. Thambi, Y. Li and D. S. Lee, J. Controlled Release, 2017, 267, 57–66 CrossRef CAS PubMed.
- A. Palucka and L. Coussens, Cell, 2016, 164, 1233–1247 CrossRef CAS PubMed.
- D. Dangaj, M. Bruand, A. J. Grimm, C. Ronet, D. Barras, P. A. Duttagupta, E. Lanitis, J. Duraiswamy, J. L. Tanyi, F. Benencia, J. Conejo-Garcia, H. R. Ramay, K. T. Montone, D. J. Powell, Jr., P. A. Gimotty, A. Facciabene, D. G. Jackson, J. S. Weber, S. J. Rodig, S. F. Hodi, L. E. Kandalaft, M. Irving, L. Zhang, P. Foukas, S. Rusakiewicz, M. Delorenzi and G. Coukos, Cancer Cell, 2019, 35, 885–900 CrossRef CAS PubMed.
- X. Han, Q. Wei, Y. Lv, L. Weng, H. Huang, Q. Wei, M. Li, Y. Mao, D. Hua, X. Cai, M. Cao and P. Cao, Mol. Ther., 2022, 30, 327–340 CrossRef CAS PubMed.
- G. Motz, S. Santoro, L. Wang, T. Garrabrant, R. Lastra, I. Hagemann, P. Lal, M. Feldman, F. Benencia and G. Coukos, Nat. Med., 2014, 20, 607–615 CrossRef CAS PubMed.
- S. Barzilai, S. Yadav, S. Morrell, F. Roncato, E. Klein, L. Stoler-Barak, O. Golani, S. Feigelson, A. Zemel, S. Nourshargh and R. Alon, Cell Rep., 2017, 18, 685–699 CrossRef CAS PubMed.
- G. T. Motz and G. Coukos, Immunity, 2013, 39, 61–73 CrossRef CAS PubMed.
- C. Li, A. F. Teixeira, H.-J. Zhu and P. ten Dijke, Mol. Cancer, 2021, 20, 154 CrossRef CAS PubMed.
- J. Zhang, Y. Lin, Z. Lin, Q. Wei, J. Qian, R. Ruan, X. Jiang, L. Hou, J. Song, J. Ding and H. Yang, Adv. Sci., 2022, 9, 2103444 CrossRef CAS PubMed.
- P. Sharma and J. P. Allison, Science, 2015, 348, 56–61 CrossRef CAS PubMed.
- K. Choucair, J. R. Duff, C. S. Cassidy, M. T. Albrethsen, J. D. Kelso, A. Lenhard, H. Staats, R. Patel, F. C. Brunicardi, L. Dworkin and J. Nemunaitis, Future Oncol., 2019, 15, 3053–3069 CrossRef CAS PubMed.
- D. A. A. Vignali and V. K. Kuchroo, Nat. Immunol., 2012, 13, 722–728 CrossRef CAS PubMed.
- C. R. Glassman, Y. K. Mathiharan, K. M. Jude, L. Su, O. Panova, P. J. Lupardus, J. B. Spangler, L. K. Ely, C. Thomas, G. Skiniotis and K. C. Garcia, Cell, 2021, 184, 983–999 CrossRef CAS PubMed.
- Z. Ma and B. Damania, Cell Host Microbe, 2016, 19, 150–158 CrossRef CAS PubMed.
- Q. Chen, L. Sun and Z. J. Chen, Nat. Immunol., 2016, 17, 1142–1149 CrossRef CAS PubMed.
- Z. Hu, Y. Yang, L. Fang, J. Zhou and H. Zhang, Immunopharmacol. Immunotoxicol., 2021, 43, 126–137 CrossRef CAS PubMed.
- A. M. Esteves, E. Papaevangelou, P. Dasgupta and C. Galustian, Front. Oncol., 2021, 11, 621550 CrossRef CAS PubMed.
- E. Conde, E. Vercher, M. Soria-Castellano, J. Suarez-Olmos, U. Mancheño, E. Elizalde, M. L. Rodriguez, J. Glez-Vaz, N. Casares, E. Rodríguez-García, M. Hommel, G. González-Aseguinolaza, I. Uranga-Murillo, J. Pardo, G. Alkorta, I. Melero, J. Lasarte and S. Hervas-Stubbs, J. Immunother. Cancer, 2021, 9, e003351 CrossRef PubMed.
- J. Guo and L. Huang, Trends Pharmacol. Sci., 2022, 43, 957–972 CrossRef CAS PubMed.
- X. Lu, L. Miao, W. Gao, Z. Chen, K. J. McHugh, Y. Sun, Z. Tochka, S. Tomasic, K. Sadtler, A. Hyacinthe, Y. Huang, T. Graf, Q. Hu, M. Sarmadi, R. Langer, D. G. Anderson and A. Jaklenec, Sci. Transl. Med., 2020, 12, eaaz6606 CrossRef CAS PubMed.
- Q. Li, Q. Chen, X. Yang, Y. Zhang, L. Lv, Z. Zhang, S. Zeng, J. Lv, S. Liu and B. Fu, J. Nanobiotechnol., 2022, 20, 84 CrossRef CAS PubMed.
- Y. Komohara, M. Jinushi and M. Takeya, Cancer Sci., 2014, 105, 1–8 CrossRef CAS PubMed.
- L. Barile and G. Vassalli, Pharmacol. Ther., 2017, 174, 63–78 CrossRef CAS PubMed.
- C. P. Chang, Y. C. Su, C. W. Hu and H. Y. Lei, Cell Death Differ., 2013, 20, 515–523 CrossRef CAS PubMed.
- R. Noy and J. W. Pollard, Immunity, 2014, 41, 49–61 CrossRef CAS PubMed.
- M. Chang, C. Dong, H. Huang, L. Ding, W. Feng and Y. Chen, Adv. Funct. Mater., 2022, 32, 2204791 CrossRef CAS.
- Y. Togashi, K. Shitara and H. Nishikawa, Nat. Rev. Clin. Oncol., 2019, 16, 356–371 CrossRef CAS PubMed.
- V. Karpisheh, S. M. Mousavi, P. Naghavi Sheykholeslami, M. Fathi, M. Mohammadpour Saray, L. Aghebati-Maleki, R. Jafari, N. Majidi Zolbanin and F. Jadidi-Niaragh, Life Sci., 2021, 284, 119132 CrossRef CAS PubMed.
- M. D. Sharma, R. Shinde, T. L. McGaha, L. Huang, R. B. Holmgaard, J. D. Wolchok, M. R. Mautino, E. Celis, A. H. Sharpe, L. M. Francisco, J. D. Powell, H. Yagita, A. L. Mellor, B. R. Blazar and D. H. Munn, Sci. Adv., 2015, 1, e1500845 CrossRef PubMed.
- M. Zhao, R. Wang, K. Yang, Y. Jiang, Y. Peng, Y. Li, Z. Zhang, J. Ding and S. Shi, Acta Pharm. Sin. B, 2023, 13, 916–941 CrossRef CAS PubMed.
- A. Calcinotto, C. Spataro, E. Zagato, D. Di Mitri, V. Gil, M. Crespo, G. De Bernardis, M. Losa, M. Mirenda, E. Pasquini, A. Rinaldi, S. Sumanasuriya, M. B. Lambros, A. Neeb, R. Lucianò, C. A. Bravi, D. Nava-Rodrigues, D. Dolling, T. Prayer-Galetti, A. Ferreira, A. Briganti, A. Esposito, S. Barry, W. Yuan, A. Sharp, J. de Bono and A. Alimonti, Nature, 2018, 559, 363–369 CrossRef CAS PubMed.
- T. A. Wynn, Nat. Immunol., 2013, 14, 197–199 CrossRef CAS PubMed.
- I. Bah, A. Kumbhare, L. Nguyen, C. E. McCall and M. El Gazzar, Cell. Immunol., 2018, 332, 32–38 CrossRef CAS PubMed.
- D. Vasquez-Dunddel, F. Pan, Q. Zeng, M. Gorbounov, E. Albesiano, J. Fu, R. L. Blosser, A. J. Tam, T. Bruno, H. Zhang, D. Pardoll and Y. Kim, J. Clin. Invest., 2013, 123, 1580–1589 CrossRef CAS PubMed.
- X. Lu, J. W. Horner, E. Paul, X. Shang, P. Troncoso, P. Deng, S. Jiang, Q. Chang, D. J. Spring, P. Sharma, J. A. Zebala, D. Y. Maeda, Y. A. Wang and R. A. DePinho, Nature, 2017, 543, 728–732 CrossRef CAS PubMed.
- S. Yasmin-Karim, J. Wood, J. Wirtz, M. Moreau, N. Bih, W. Swanson, A. Muflam, V. Ainsworth, B. Ziberi and W. Ngwa, Front. Oncol., 2021, 11, 711018 Search PubMed.
- X. Wang, T. Yang, Z. Yu, T. Liu, R. Jin, L. Weng, Y. Bai, J. J. Gooding, Y. Zhang and X. Chen, Adv. Mater., 2022, 34, 2110219 CrossRef CAS PubMed.
- B. Mirlekar and Y. Pylayeva-Gupta, Cancers, 2021, 13, 167 CrossRef CAS PubMed.
- Y. Cheng, X.-l Ma, Y.-q Wei and X.-W. Wei, Biochim. Biophys. Acta, Rev. Cancer, 2019, 1871, 289–312 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |