
One-pot synthesized plasmonic black gold nanoparticles for efficient photocatalytic CO oxidation†
Rishi
Verma‡
 ,
Sushma
Kundu‡
and
Vivek
Polshettiwar
*
,
Sushma
Kundu‡
and
Vivek
Polshettiwar
*
Department of Chemical Sciences, Tata Institute of Fundamental Research (TIFR), Mumbai, 400005, India. E-mail: vivekpol@tifr.res.in
First published on 11th September 2024
Abstract
Dendritic plasmonic colloidosome, also known as “black gold”, is a unique plasmonic material with broad-spectrum light absorption extending from visible to near-infrared wavelengths. Black gold has demonstrated remarkable performance in challenging and industrially relevant reactions, such as CO2 reduction, alcohol oxidation, acetylene semi-hydrogenation, and purification of seawater. Unfortunately, one of the major shortcomings of black gold lies in its intricate multi-step synthesis procedure. This involves the step-by-step growth of Au nanoparticles on a dendritic fibrous nanosilica (DFNS) support, a task spanning several days. Furthermore, the need for solid separation through centrifugation after each cycle is an additional bottleneck. Consequently, this complexity poses a challenge for industrial scaling. In this work, we report a simplified and efficient one-pot synthesis of plasmonic black gold. The one-pot method streamlines the synthesis process, significantly reducing the time from days to hours and eliminating the need for solid separation steps, thereby enhancing sustainability. Characterization studies reveal the optimal reaction time of an hour (instead of days) during the growth step, resulting in black gold materials closely resembling those synthesized via the conventional cycle-by-cycle method. We further demonstrated that DPC-60 synthesized via a one-pot protocol efficiently catalyzes CO oxidation under light, achieving 87% CO conversion at 4.0 W cm−2 without external heating. In situ DRIFTS studies revealed key adsorption sites involved in the CO oxidation reaction, with bridged CO adsorbed on Au0 sites reacting faster than linearly adsorbed CO, and CO on high-coordinated Au0 sites exhibiting a faster reaction rate than that on low-coordinated sites. Thus, the one-pot synthesis will facilitate rapid exploration of black gold's applications in a range of fields and also pave the way for scalable industrial deployment.
Introduction
The phenomenon of localized surface plasmon resonance (LSPR) represents a fascinating occurrence stemming from the collective oscillations of conduction-band electrons within metals such as gold, silver, copper, and heavily doped semiconductor nanostructures. This unique property endows plasmonic nanomaterials with the ability to efficiently harvest light energy.1–26 Plasmonic nanoparticles have exhibited potential to catalyze a diverse array of chemical transformations on their surfaces when illuminated with light energy.1–26 However, a notable constraint lies in the tendency of most documented plasmonic catalysts to selectively absorb particular wavelengths of light energy, influenced by factors such as shape, size, and environmental conditions.27 This limitation is significant, given that an optimal photocatalyst should possess the capacity to absorb the entire solar spectrum, spanning from visible to near-infrared (NIR) light.The phenomenon of plasmonic excitation presents a distinct solution to this challenge through the manipulation of plasmonic coupling.28,29 Plasmonic coupling occurs when two or more plasmonic nanoparticles are in close proximity, typically within a few nanometers, resulting in the coupling of their plasmonic modes. This engenders broader absorption bands, as there are now multiple modes of light absorption: the pristine plasmonic mode and the coupled mode with differing excitation energies.28,29 Precisely controlling the distances between nanoparticles to induce plasmonic coupling poses a challenge in colloidal solutions, prompting the development of materials where plasmonic nanoparticles are supported on high-surface-area substrates.
Our recently discovered plasmonic “black gold” also referred to as dendritic plasmonic colloidosomes (DPCs), represents a notable example of one such plasmonic material.30 Its synthesis involves the controlled nucleation and growth of gold nanoparticles (Au NPs) on high-surface-area dendritic fibrous nanosilica (DFNS),31–33 resulting in varied interparticle distances and particle size distributions. This diversity enables black gold to exhibit broad-spectrum light absorption extending from visible to near-infrared (NIR) wavelengths. The presence of different particle sizes and plasmonic coupling among Au NPs creates thermal and electric hotspots within black gold, rendering it an efficient catalyst for various reactions.30
Despite its effectiveness, there exists potential for enhancing the catalytic activity of black gold. As gold nanoparticles do not universally exhibit activity for all chemical transformations, the concept of an antenna reactor has been introduced.34 We loaded different active metals, such as Ni or Ru–Pt, onto black gold to form an antenna–reactor complex where black gold functions as the antenna.35–37 These plasmonic catalysts have demonstrated remarkable performance in challenging and industrially relevant reactions, such as CO2 reduction, alcohol oxidation, hydrosilylation of aldehydes, H2/D2 dissociation, hydrodechlorination, alkene and alkyne hydrogenation, one-electron ferricyanide reduction, and acetylene semi-hydrogenation as well as in protein unfolding and purification of seawater to drinkable water.30,35–37
Unfortunately, one of the biggest drawbacks of black gold is its complex multi-step synthesis process. It involves cycle-by-cycle growth of Au NPs to precisely adjust their size and interparticle distances, a process that requires several days. Moreover, the necessity for solid separation via centrifugation after each cycle presents a considerable bottleneck, rendering its synthesis complex, time-consuming, and consequently unsustainable. This factor also renders it less suitable for industrial scaling.
In this study, we report an innovative one-pot synthesis protocol for black gold that circumvents the cycle-by-cycle method, thereby significantly reducing synthesis time from days to mere hours. This approach not only accelerates the process but also reduces chemical consumption and energy demands, contributing to a more sustainable and efficient synthesis. The simplicity and ease of this method make it a significant advancement over the conventional cycle-by-cycle method, allowing for rapid and scalable production. By streamlining the synthesis process, this approach facilitates the swift exploration of black gold's potential applications across diverse fields and makes the scale-up process more practical and economically viable for industrial purposes. The black gold synthesized through this one-pot protocol was subsequently utilized as a plasmonic photocatalyst, demonstrating its effectiveness in photocatalytic CO oxidation to CO2. This underscores the practical advantages of the new method, highlighting its potential to revolutionize the production and application of black gold in various industries.
We evaluated the one-pot synthesis protocol by characterizing the resulting black gold material using high-resolution transmission electron microscopy (HRTEM), scanning electron microscopy (SEM), scanning transmission electron microscopy-energy dispersive spectroscopy (STEM-EDS), powder X-ray diffraction (pXRD), ultraviolet-visible (UV-vis) absorption spectroscopy, and N2 sorption measurement. We also used in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) under light and in the dark to explore the binding nature of CO on Au sites and the reaction mechanism of CO oxidation to CO2.
Results and discussion
One-pot synthesis of black gold
DFNS was first functionalized with (3-aminopropyl)triethoxysilane (APTS) using the previously optimized protocol to synthesize DFNS-APTS.31,32 DFNS-APTS (100 mg) was crushed into a fine powder using a mortar and pestle to ensure homogeneity. The resulting powder was then transferred into a 250 mL conical flask, followed by the addition of 50 mL of deionized (DI) water. The mixture underwent sonication for 15 minutes at room temperature (23–25 °C) to promote dispersion. Subsequently, 10 mg of HAuCl4·3H2O (from a stock solution in DI water with a concentration of 100 mg mL−1) was introduced into the conical flask, and the mixture was sonicated for an additional 15 minutes at room temperature. Stirring at 400 rpm was then initiated and maintained for 2 minutes at room temperature. A freshly prepared solution of NaBH4 was prepared by dissolving 40 mg in 1 mL of DI water. This solution was then added dropwise in 30 seconds using a dropper into the DFNS-APTS and gold mixture under stirring at room temperature. A noticeable color change from light golden to brown was observed. The solution was continuously stirred at 400 rpm for 2 hours. This 2-hour stirring duration ensured the complete reduction of Au3+ ions and the full decomposition of NaBH4, which was crucial to prevent any residual NaBH4 from interfering with further Au loading and potentially initiating unintended nucleation instead of growth.During this step, the utilization of a strong reducing agent, NaBH4, induced a rapid increase in the concentration of Au0, surpassing the critical concentration necessary for the formation and nucleation of small Au NPs. These formed Au0 nuclei served as sites for the subsequent growth of Au NPs upon further reduction of Au3+ ions. The material obtained at this stage was denoted as DPC-N, where ‘N’ signifies the nucleation step. Subsequently, the loading of Au was performed on DPC-N within the same reaction mixture without isolation.
In the subsequent step, the existing Au nuclei were allowed to grow into larger NPs with varied sizes and interparticle distances. For this, in a 50 mL falcon tube, HAuCl4·3H2O (150 mg) and K2CO3 (280 mg) were dissolved in 45 mL of DI water, and the solution was vortexed for 15 minutes. The amount of gold precursor was carefully calculated to achieve a 43 wt% Au loading on the silica spheres. This reaction between the Au precursor and K2CO3 served to passivate the Au3+ ions, ensuring their slow reduction and favoring the growth of Au nanoparticles rather than new nucleation.
The resulting solution, termed K–Au solution, was then added dropwise to the reaction mixture containing DPC-N in 1.5 minutes using a 10 mL pipette under stirring. Subsequently, ammonium hydroxide solution in water (1 mL, 25% v/v) was added dropwise in 30 seconds into the reaction mixture, followed by the dropwise addition of formaldehyde solution in 1 minute (20 mL, 37 wt% in H2O). The ammonium hydroxide solution provided a basic environment in the reaction mixture, while formaldehyde acted as a weak reducing agent under these basic conditions. Over the course of 5–10 minutes, the color of the solution gradually transitioned from brown to black, indicating the progression of the reaction. The reaction mixture was stirred at 400 rpm for different durations: 30 minutes, 60 minutes, and 120 minutes, and denoted as DPC-30, DPC-60, and DPC-120, respectively. Following the selected stirring period, the solid product was isolated by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 minutes. The isolated product was then washed with ethanol (20 mL, three times) and water (20 mL, three times). Finally, the resulting black gold powder was obtained by drying the isolated and washed product in an oven at 80 °C for 10 hours, ensuring complete removal of the solvent and moisture.
000 rpm for 10 minutes. The isolated product was then washed with ethanol (20 mL, three times) and water (20 mL, three times). Finally, the resulting black gold powder was obtained by drying the isolated and washed product in an oven at 80 °C for 10 hours, ensuring complete removal of the solvent and moisture.
Additionally, we synthesized black gold using the conventional cycle-by-cycle approach by conducting 4 cycles of Au loading.30 This was done to enable comparative analysis between the outcomes of our new protocol and the standard black gold, also known as DPC-C4.
Characterization of black gold
The black coloration of the final product post-one-pot synthesis served as an initial indicator of black gold formation. However, the distinct catalytic potential of black gold hinges not only on its light absorption capabilities but also on its chemical properties, which are intricately linked to various physicochemical attributes such as Au nanoparticle size, surface area, pore volume, crystallinity, etc. As these attributes evolve during the synthesis process, we systematically characterized the material at different stages to ensure the synthesis of black gold with the desired physicochemical properties.In the case of the DPC-N sample, Au NPs were not readily visible in the SEM images, attributed to the small sizes of the Au NPs (Fig. 1a and S1†). Subsequent HRTEM analysis revealed the presence of small Au NPs distributed on the DFNS sphere (Fig. 1b and c). Particle size distribution analysis indicated the presence of small Au NPs ranging from 1 to 6 nm (Fig. 1d). Following the addition of formaldehyde solution, more Au NPs were loaded onto the DFNS sphere. SEM and HRTEM images demonstrated the uniform presence of Au NPs (Fig. 1e–g and S2†). Varying the reaction time from 30 minutes to 120 minutes showed that 30 minutes of reaction was sufficient to facilitate the growth of Au NPs on existing nuclei. Particle size distribution analysis of DPC-30 revealed slightly larger Au NPs, with sizes ranging from 2 to 16 nm and an average size of 6.7 ± 2.6 nm. Increasing the reaction time to 60 minutes maintained a similar morphology to DPC-30 (Fig. 1i–k and S3†), albeit with some observation of slightly larger Au NPs. Particle size distribution analysis of DPC-60 indicated sizes ranging from 2 to 19 nm with an average size of 7.8 ± 2.7 nm (Fig. 1l). Further extension of reaction time to 120 minutes revealed some Au NP overgrowth on the silica sphere, observed through SEM and HRTEM analysis (Fig. 1m–o and S4†). Particle size distribution of DPC-120 showed sizes ranging from 2 to 23 nm with an average size of 9.5 ± 3.5 nm (Fig. 1p). In comparison, standard black gold prepared through the cycle-by-cycle approach exhibited uniformly loaded Au NPs on the silica sphere (Fig. 1q–s and S5†), with particle sizes ranging from 2 to 18 nm and an average size of 8.3 ± 3.3 nm (Fig. 1t). Electron microscopy analysis suggested that a reaction time of 60 minutes represents the optimal condition for growing existing Au nuclei in DPC-N toward the formation of black gold. Despite similar morphologies and particle size distributions across all three stirring times, the product obtained after 60 minutes of stirring closely resembled the standard black gold sample.
 | ||
| Fig. 1 Characterization of black gold (DPC). SEM, TEM, and HRTEM images, and particle size distribution (PSD) of Au NPs in (a–d) DPC-N, (e–h) DPC-30, (i–l) DPC-60, (m–p) DPC-120, and (q–t) DPC-C4. In the particle size distribution plots, Dp is the mean particle size of Au NPs. The particle size distribution was estimated by measuring the size of 200 Au NPs from different silica spheres. | ||
The elemental composition of the materials was evaluated using SEM-EDS. The gold weight percentage loading increased from 4.4 ± 1.0% in DPC-N during the nucleation step to 42 ± 5% in the growth step. Interestingly, the gold loading remained consistent in the case of DPC-30, DPC-60, and DPC-120, suggesting that prolonged reaction times did not necessarily yield a significant increase in gold loading (Table 1). The obtained gold weight percentage closely matched the theoretical loading, which was calculated to be 43.2 wt% based on the amount of gold precursor introduced during synthesis. Additionally, the achieved gold loading closely resembled that of standard DPC-C4 (Table 1). However, it was noted that the weight percentage of additionally grown gold nanostructures outside the silica sphere in DPC-120 was predominantly composed of gold (∼84 wt% gold). This observation indicated that optimal reaction time was crucial to prevent the undesired growth of gold nanostructures outside the silica sphere.
| Si (wt%) | O (wt%) | Au (wt%) | |
|---|---|---|---|
| DPC-N | 44 ± 0.5 | 51 ± 0.5 | 4.4 ± 1 |
| DPC-30 | 25 ± 0.5 | 34 ± 0.5 | 41 ± 1 |
| DPC-60 | 24 ± 3 | 33 ± 3 | 42 ± 5 |
| DPC-120 | 23 ± 6 | 32 ± 5 | 47 ± 11 |
| DPC-C4 | 24 ± 3 | 31 ± 4 | 45 ± 5 |
Further characterization was conducted through STEM-EDS elemental mapping to visualize the distribution of gold NPs in DPCs. The mapping revealed that the gold NPs were uniformly distributed over the silica sphere (Fig. S6†). In the case of DPC-N, due to the low counts resulting from the low concentration of gold NPs, some signals were also observed from outside the sphere in addition to the bright spots observed from the location of gold NPs (Fig. S6a†). However, in the case of DPC-30, DPC-60, and DPC-120, the gold signals originated from all over the silica sphere, indicating uniform loading of gold NPs (Fig. S6b–d†). The distribution of gold NPs closely matched that of DPC-C4 prepared using a standard cycle-by-cycle approach (Fig. S6e†). Overall, the morphological and compositional characterization indicated that the black gold or DPC formed using the new one-pot synthesis closely resembled DPC-C4 made by the cycle-by-cycle approach.
We further investigated the light absorption properties of the DPCs using UV-vis absorption spectroscopy. The DPC-N powder was brown in color and demonstrated very weak absorption in the visible region (Fig. 2a and b). This weak absorption was attributed to the lower concentration of gold NPs on the silica sphere during the nucleation step. In the case of growth samples, DPC-30 appeared dark red to purple rather than completely black. Its absorption spectrum displayed a broadband absorption with a minor hump at around 460 nm (Fig. 2a and b). This coloration could be attributed to the presence of a larger number of smaller Au NPs, which tend to absorb in the 450 nm region, resulting in the material appearing dark red to purple instead of completely black.
 | ||
| Fig. 2 Optical and crystallographic characterization of black gold (DPC). (a) Optical images, (b) UV-vis absorption spectra, and (c) pXRD patterns of DPC-N, DPC-30, DPC-60, DPC-120, and DPC-C4. | ||
Upon further increase in reaction time for DPC-60 and DPC-120, the solid powder achieved a dark black color with broadband absorption across the entire visible range (Fig. 2a and b). This phenomenon occurred because sufficient stirring time allowed for the growth of Au NPs, leading to heterogeneity in particle sizes and distances between adjacent Au NPs. This ensured optimal plasmonic coupling between the Au NPs, facilitated by the appropriate distances between them and the overlapping of multiple plasmon modes due to particle size heterogeneity, thus imparting the characteristic black color and broadband absorbance in the visible range to the black gold material (Fig. 2b).30 These findings suggested that achieving a completely black gold material necessitates adequate reaction time during the growth step to facilitate Au NP growth. Moreover, the absorption profiles of DPC-60 and DPC-120 closely resembled those of DPC-C4 synthesized using the cycle-by-cycle approach (Fig. 2b).
The pXRD patterns of the samples revealed peaks corresponding to the diffraction pattern of the gold cubic lattice and a hump at around 22° due to the amorphous DFNS support (Fig. 2c). In the case of DPC-N, characterized by small sizes and low concentration of Au nanoparticles (NPs), the diffraction peak intensities were low and the peaks were broader (Fig. 2c). Following the growth step, all samples exhibited increased peak intensities due to larger particle sizes and a higher concentration of Au NPs (Fig. 2c). Notably, the diffraction pattern closely matched that of DPC-C4 synthesized using the standard cycle-by-cycle approach (Fig. 2c).
The extensive applicability of black gold in plasmonic catalysis, particularly as a plasmonic support for anchoring active sites, is not only due to its optical properties but also due to its exceptional textural characteristics. These include high surface area and pore volume, which ensure effective dispersion of active sites, efficient adsorption–desorption of reactant molecules, and facile diffusion of reactants within the nanostructure. To evaluate these textural properties, nitrogen sorption measurements were conducted on the black gold prepared using the one-pot synthesis method. The nitrogen adsorption–desorption isotherms of DFNS-APTS, utilized as a support for loading Au NPs, exhibited type-II curves with hysteresis (Fig. 3a). The Brunauer–Emmett–Teller (BET) surface area was determined to be 298 m2 g−1, with a Barrett–Joyner–Halenda (BJH) pore volume of 0.34 cm3 g−1 (Fig. 3a). Upon loading of Au NPs during the nucleation step, a reduction in the BET surface area to 218 m2 g−1 was observed, accompanied by a BJH pore volume of 0.33 cm3 g−1 (Fig. 3b). Notably, weak type-II hysteresis persisted throughout this phase (Fig. 3b).
 | ||
| Fig. 3 Textural characterization of black gold (DPC). Nitrogen adsorption–desorption isotherms; (inset): BJH adsorption pore-size distributions of (a) DFNS-APTS, (b) DPC-N, (c) DPC-30, (d) DPC-60, (e) DPC-120, and (f) DPC-C4. The BET surface area and BJH pore volume are written in the plots. The error in the BET surface area was ±10% and the BJH pore volume was 0.08 cm3 g−1. | ||
During the subsequent growth steps, a further decrease in surface area and pore volume was observed in DPC-30 (169 m2 g−1 and 0.23 cm3 g−1), DPC-60 (165 m2 g−1 and 0.19 cm3 g−1), and DPC-120 (149 m2 g—1 and 0.21 cm3 g−1), accompanied by the loss of hysteresis (Fig. 3c–e). Despite increasing reaction time, surface areas and pore volumes remained relatively unchanged, indicating that additional growth of initially formed Au nanoparticles did not significantly impact these parameters. The primary reduction in surface area and pore volume occurred during the initial growth phase (Fig. 3c–e). Comparatively, black gold prepared using the standard cycle-by-cycle approach exhibited surface area (188 m2 g−1) and pore volume (0.32 cm3 g−1) within a similar range to that of DPCs formed using the one-pot synthesis after the growth step (Fig. 3f). Overall, the morphological, optical, and textural characterization corroborated the successful synthesis of black gold using the one-pot synthesis protocol, with 60 minutes as the optimum reaction time.
Photocatalytic CO oxidation by plasmonic black gold
Photocatalytic carbon monoxide (CO) oxidation is a crucial area of research due to the significant environmental and health hazards posed by CO, a colorless, odorless, and toxic gas.38–42 Originating primarily from the incomplete combustion of fossil fuels, CO is a persistent pollutant in the atmosphere, stemming from sources such as industrial flue gas and motor vehicle exhaust. Conventional CO removal methods include thermal catalysis and photocatalysis, and while thermocatalysis is a widely studied and effective method for CO oxidation, it requires high energy inputs due to the need for high temperatures. Photocatalysis, on the other hand, provides a low-temperature alternative by utilizing sunlight to drive the CO oxidation process. This method not only facilitates the removal of CO but also enables its conversion into valuable chemicals and fuels, such as alkanes, light olefins, and oxygenates, through reactions with H2 and other agents. Despite its promise, the conversion efficiency of photocatalytic technology remains below the threshold for industrial application, necessitating further research to enhance its effectiveness.38–42We investigated the photocatalytic CO oxidation capabilities of black gold (DPC-60). Plasmonic CO oxidation using O2 was carried out in a pike flow reactor with a catalyst bed thickness of approximately 1 mm, allowing light photons to penetrate each DPC-60 sphere throughout the catalyst bed (Fig. S7†). A xenon lamp (240–1100 nm) was used as the light source, and the reaction was monitored by using online microgas chromatography (GC) (Fig. S7†). The reactant gas feed consisted of CO (10% in helium), O2 (100%), and argon (Ar, 100%). Initially, we optimized the total flow of the reactant feed while maintaining an O2:CO:Ar ratio of 1:1:1 under light exposure (240–1100 nm, 1.6 W cm−2). CO2 productivity increased from 54 mmol g−1 h−1 to 454 mmol g−1 h−1 as the total flow increased from 6 mL min−1 to 300 mL min−1, though CO conversion decreased from 73% to 11% (Fig. 4a). This decrease was attributed to the reduced residence time of the reactant gases on the catalyst surface at higher gas flows. We determined that a 6 mL min−1 flow rate was optimal. At this optimum flow, we varied the ratio of the reactant gases to optimize CO conversion and CO2 productivity. While CO conversion remained nearly constant (∼71%) with varying gas ratios, CO2 productivity increased to 78 mmol g−1 h−1 with a higher CO ratio in the feed (Fig. 4b). Thus, the optimum conditions were an O2:CO:Ar ratio of 1:2:0 and total flow of 6 mL min−1.
 | ||
| Fig. 4 Photocatalytic CO oxidation by black gold (DPC-60). Photocatalytic CO oxidation by DPC-60 as a function of (a) total flow with an O2:CO:Ar ratio of 1:1:1 under light exposure (240–1100 nm, 1.6 W cm−2) and (b) ratio of O2:CO:Ar at a total flow rate of 6 mL min−1 under light exposure (240–1100 nm, 1.6 W cm−2). (c) Photocatalytic CO oxidation by DPC-60 and catalyst bed temperature as a function of light intensity at a total flow rate of 6 mL min−1 with an O2:CO:Ar ratio of 1:2:0. (d) CO oxidation by DPC-60 in the dark at various catalyst bed temperatures at a total flow rate of 6 mL min−1 with an O2:CO:Ar ratio of 1:2:0. | ||
Under these optimized conditions, we then conducted CO oxidation at different light intensities (without external heating) and in the dark (with external heating) (Fig. 4c). A thin thermocouple inserted directly into the catalyst powder bed measured the catalyst bed temperature (Fig. S8†). CO conversion increased with light intensity, increasing from 27% at 0.29 W cm−2 to 77% at 2.4 W cm−2, and further increases in light intensity led to a saturation of conversion rates at 3.3 W cm−2 (Fig. 4c). Catalyst bed temperature increased from 45.5 °C to 140 °C with an increase in light intensity from 0.29 to 4 W cm−2 (Fig. 4c and S8†). CO oxidation in the dark using external heating showed that CO conversion increased from 29% to 66% as the catalyst temperature increased from 45 °C to 141 °C (Fig. 4d).
In the case of DPC, following the decay of LSPR, the initial few femtoseconds (fs) involve the dephasing of LSPR, leading to the generation of excited electron–hole pairs via Landau damping.2,37 During this short period, these excited electron–hole pairs exhibit a non-thermal distribution and decay either through radiative processes (photon re-emission) or non-radiative processes (electron–electron interactions). Subsequently, over a timespan ranging from 100 fs to several picoseconds, the excited carriers transfer their energy to lower-energy electrons through electron–electron interactions, resulting in the formation of a quasi-Fermi–Dirac distribution for electron energy. Eventually, the electron–hole pairs relax, releasing thermal energy via electron–phonon interactions. This relaxation process occurs over a relatively extended timescale, from hundreds of picoseconds to nanoseconds.37 Plasmon decay generates sufficient local temperatures, and this local heat has a sufficient lifetime to drive the reaction. Consequently, the generation of local heat following LSPR decay and the utilization of this local heat for catalysis are feasible due to the longer lifetime of the local heat compared to the extraction of short-lived hot charge carriers. In the case of photocatalytic CO oxidation, the similarity in CO conversion under both light and dark conditions indicated that DPC-60 catalyzed CO oxidation through photothermal pathways.
Additionally, we performed photocatalytic CO oxidation using conventional DPC-C4 under optimized conditions at a total flow of 6 mL min−1 with an O2:CO:Ar ratio of 1:2:0 at a light intensity of 3.3 W cm−2. DPC-C4 achieved a CO conversion of 75%, comparable to that of DPC-60 under the same reaction conditions (Fig. S9†). This suggested that DPC-60 synthesized via the one-pot method possessed not only structural similarity but also similar chemical properties to the conventionally synthesized DPC-C4.
Mechanism of CO oxidation using in situ DRIFTS studies
We carried out in situ DRIFTS studies under light and dark conditions to elucidate the molecular mechanism of photocatalytic CO oxidation on DPC-60 (Fig. S10†). Initially, the reactant gases, CO, O2, and Ar were introduced in the dark at 25 °C to study the adsorption behavior of molecules on the Au surface. The adsorption of CO on DPC-60 resulted in multiple peaks in the 1900–2300 cm−1 region. Peaks at 2172 and 2119 cm−1 were identified as gaseous CO molecules in the reaction feed (Fig. 5a).11 Two sharp peaks at 2033 and 2013 cm−1 were attributed to CO linearly adsorbed on Au0 and low-coordinated Au0, respectively (Fig. 5a and b).43–47 A shoulder at 1990 cm−1 indicated CO adsorbed in a bridge configuration on Au0 (Fig. 5a and b).43–47 Additionally, the 2119 cm−1 peak exhibited a shoulder towards lower wavenumbers, corresponding to CO linearly adsorbed on Auδ+, formed due to partial oxidation of Au0 sites in the presence of O2 in the reaction feed (Fig. 5a).47 Upon exposure to light, no new peaks appeared in the 1900–2300 cm−1 region, but the intensities of the 2033 and 2013 cm−1 peaks changed relatively (Fig. 5a). The shoulder at 1990 cm−1, associated with bridged CO on Au0, disappeared (Fig. 5a).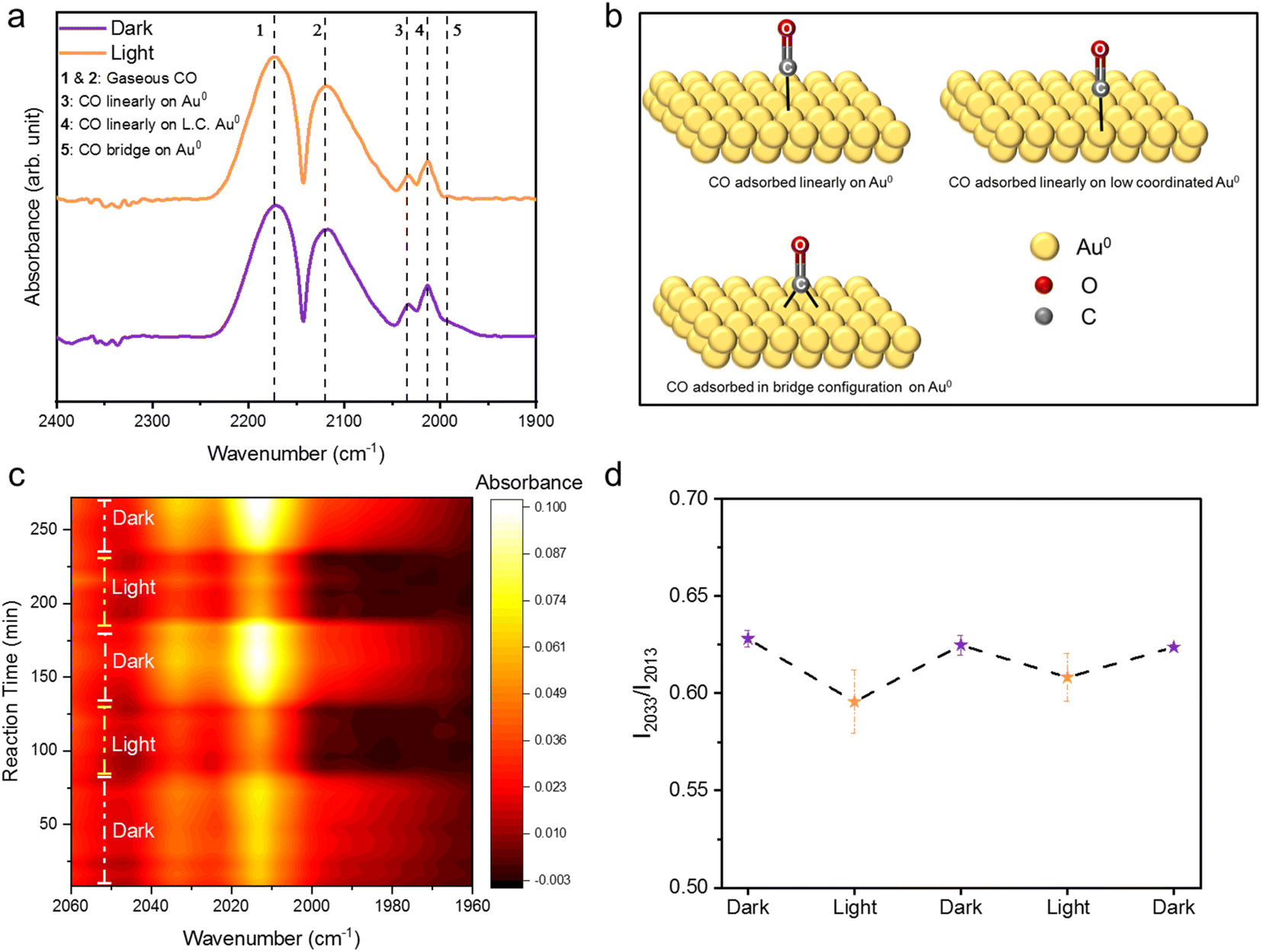 | ||
| Fig. 5 In situ DRIFTS of photocatalytic CO oxidation by black gold (DPC-60). (a) In situ DRIFTS spectra of CO adsorbed on Au sites in the dark and under light, with a total flow rate of 44 mL min−1 with zero air (2 mL min−1), CO (100%, 2 mL min−1), and Ar (100%, 40 mL min−1). For dark, the temperature was 25 °C and for light, exposure was provided by xenon light (240–1100 nm, 2.6 W cm−2). L. C. means low coordinated; (b) schematic of CO adsorbed on various Au sites; (c) in situ DRIFTS spectra of CO oxidation by DPC-60 in the light on–off cycle, with a total flow rate of 44 mL min−1 with zero air (2 mL min−1), CO (100%, 2 mL min−1), and Ar (100%, 40 mL min−1). For dark, the temperature was 25 °C and for light, exposure was provided by xenon light (240–1100 nm, 2.6 W cm−2); (d) ratio of intensities of the peaks at 2033 and 2013 cm−1 in various light on–off cycles. | ||
This dynamic behavior was further probed using in situ DRIFTS under light on–off cycles, recording spectra in the dark–light–dark–light–dark sequence (Fig. 5c and S11†). In each cycle, the 1990 cm−1 shoulder rapidly disappeared (Fig. 5c and S11†), and the relative intensities of the 2033 and 2013 cm−1 peaks changed (Fig. 5c, d and Table S1†). The faster disappearance of the bridged CO peak suggests quicker reaction kinetics for this adsorbed species due to a more polarized carbon atom, which is bound to two gold sites. The decrease in the ratio of the intensity of the 2033 peak to the 2013 peak (I2033/I2013) indicated that, under light, CO adsorbed on high-coordinated Au0 reacts faster than CO on low-coordinated Au0 (Fig. 5d and Table S1†). This phenomenon may be attributed to the presence of more thermal hotspots around highly coordinated Au0, facilitating faster reaction kinetics (Fig. 5d and Table S1†).
Conclusions
In conclusion, we achieved a successful synthesis of a plasmonic black gold material using a one-pot synthesis approach. Effective loading of Au was attained, as evidenced by the close agreement between the observed Au weight percentage in SEM-EDS analysis and the theoretically calculated loading based on the amount of Au precursor added. Optimization of the reaction time during the growth step revealed that 60 minutes was the optimum duration. The one-pot synthesized black gold material was compared with black gold synthesized using the conventional multi-step cycle-by-cycle approach, revealing close resemblance in terms of morphology, absorption properties, textural characteristics, and crystallinity. A shorter reaction time of 30 minutes in the growth step led to a uniform distribution of Au NPs on silica, but the final product exhibited a color ranging from dark red to purple rather than being completely black. Conversely, an excessive reaction time of 120 minutes resulted in the growth of unwanted Au nanostructures outside the silica sphere. Thus, detailed morphological, optical, and textural characterization confirmed that a reaction time of 60 minutes in the growth step resulted in optimal outcomes. Importantly, we were able to significantly reduce the synthesis time of black gold from several days to 3.5 hours using the one-pot synthesis approach.We also demonstrated that plasmonic black gold (DPC-60) synthesized using the one-pot protocol efficiently catalyzes CO oxidation under light. The reaction took place without external heating. It showed a CO conversion of 87% at 4.0 W cm−2 under the optimized conditions. The similarity in CO conversion under both light and dark conditions indicates that DPC-60 primarily operates via photothermal pathways. The in situ DRIFTS provided important insights into the reaction mechanism. In situ DRIFTS showed that the CO adsorbed in a bridged manner reacts faster as compared to linearly adsorbed CO on Au0 sites. Additionally, the relative peak intensities of the CO adsorbed on low and high-coordinated Au0 sites indicated the faster kinetics of the CO adsorbed on high-coordinated Au0 sites.
Overall, this one-pot synthesis approach not only optimizes the synthesis of black gold but also enhances its practical application potential, making it a valuable method for industrial-scale production and diverse catalytic applications. Additionally, the ability to rapidly produce high-quality black gold materials allows for quicker experimentation and development of new applications, ranging from environmental remediation to advanced chemical reactions.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and ESI.†Author contributions
V. P. proposed the research direction, designed the project, and guided the project. R. V. and V. P. designed various experiments. R. V. performed the experiments (synthesis, characterization, and in situ DRIFTS). S. K. performed photocatalytic CO oxidation assisted by R. V. Data were analyzed by R. V. S. K. and V. P. R. V. and V. P. wrote the overall manuscript. Everyone commented on the MS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the funding support of the Department of Atomic Energy, Government of India, project no. 12-R&D-TFR-RTI4003. We acknowledge the EM and XRD facility of TIFR, Mumbai.References
- U. Aslam, V. G. Rao, S. Chavez and S. Linic, Catalytic conversion of solar to chemical energy on plasmonic metal nanostructures, Nat. Catal., 2018, 1, 656–665 CrossRef.
- R. Verma, R. Belgamwar and V. Polshettiwar, Plasmonic photocatalysis for CO2 conversion to chemicals and fuels, ACS Mater. Lett., 2021, 3, 574–598 CrossRef CAS.
- G. Baffou and R. Quidant, Nanoplasmonics for chemistry, Chem. Soc. Rev., 2014, 43, 3898–3907 RSC.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Plasmon-induced hot carrier science and technology, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- S. Y. Ding, E. M. You, Z. Q. Tian and M. Moskovits, Electromagnetic theories of surface-enhanced Raman spectroscopy, Chem. Soc. Rev., 2017, 46, 4042–4076 RSC.
- C. Zhan, X.-J. Chen, J. Yi, J.-F. Li, D.-Y. Wu and Z.-Q. Tian, From plasmon-enhanced molecular spectroscopy to plasmon-mediated chemical reactions, Nat. Rev. Chem, 2018, 2, 216–230 CrossRef.
- E. Cortés, W. Xie, J. Cambiasso, A. S. Jermyn, R. Sundararaman, P. Narang, S. Schlücker and S. A. Maier, Plasmonic hot electron transport drives nano-localized chemistry, Nat. Commun., 2017, 8, 14880 CrossRef PubMed.
- J. Zhang, B. Guan, X. Wu, Y. Chen, J. Guo, Z. Ma, S. Bao, X. Jiang, L. Chen, K. Shu, H. Dang, Z. Guo, Z. Li and Z. Huang, Research on photocatalytic CO2 conversion to renewable synthetic fuels based on localized surface plasmon resonance: current progress and future perspectives, Catal. Sci. Technol., 2023, 13, 1932–1975 RSC.
- J. Li, Y. Zhang, Y. Huang, B. Luo, L. Jing and D. Jing, Noble-metal free plasmonic nanomaterials for enhanced photocatalytic applications-a review, Nano Res., 2022, 15, 10268–10291 CrossRef CAS.
- V. Jain, R. K. Kashyap and P. P. Pillai, Plasmonic photocatalysis: activating chemical bonds through light and plasmon, Adv. Opt. Mater., 2022, 10, 2200463 CrossRef CAS.
- S. Singh, R. Verma, N. Kaul, J. Sa, A. Punjal, S. Prabhu and V. Polshettiwar, Surface plasmon-enhanced photo-driven CO2 hydrogenation by hydroxy-terminated nickel nitride nanosheets, Nat. Commun., 2023, 14, 2551 CrossRef CAS PubMed.
- J. Gargiulo, R. Berté, Y. Li, S. A. Maier and E. Cortés, From optical to chemical hot spots in plasmonics, Acc. Chem. Res., 2019, 52, 2525–2535 CrossRef CAS PubMed.
- A. Stefancu, J. Gargiulo, G. Laufersky, B. Auguié, V. Chiş, E. C. L. Ru, M. Liu, N. Leopold and E. Cortés, Interface-dependent selectivity in plasmon- driven chemical reactions, ACS Nano, 2023, 17, 3119–3127 CrossRef CAS PubMed.
- M. Herran, S. Juergensen, M. Kessens, D. Hoeing, A. Köppen, A. Sousa-Castillo, W. J. Parak, H. Lange, S. Reich, F. Schulz and E. Cortés, Plasmonic bimetallic two-dimensional supercrystals for H2 generation, Nat. Catal., 2023, 6, 1205–1214 CrossRef CAS.
- E. Peiris, S. Hanauer, T. Le, J. Wang, T. Salavati-fard, P. Brasseur, E. V. Formo, B. Wang and P. H. C. Camargo, Controlling selectivity in plasmonic catalysis: switching reaction pathway from hydrogenation to homocoupling under visible-light irradiation, Angew. Chem., Int. Ed., 2022, 62, e202216398 CrossRef PubMed.
- R. Kamarudheen, G. W. Catellanos, L. J. P. Kamp, H. J. H. Clercx and A. Baldi, Quantifying photothermal and hot charge carrier effects in plasmon-driven nanoparticle syntheses, ACS Nano, 2018, 12, 8447–8455 CrossRef CAS PubMed.
- A. G. M. da Silva, T. S. Rodrigues, J. Wang and P. H. C. Camargo, Plasmonic catalysis with designer nanoparticles, Chem. Commun., 2022, 58, 2055–2074 RSC.
- R. K. Kashyap, I. Dwivedi, S. Roy, S. Roy, A. Rao, C. Subramaniam and P. P. Pillai, Insights into the utilization and quantification of thermoplasmonic properties in gold nanorod arrays, Chem. Mater., 2022, 34, 7369–7378 CrossRef CAS.
- D. Mittal, M. Ahlawat and V. G. Rao, Recent progress and challenges in plasmon-mediated reduction of CO2 to chemicals and fuels, Adv. Mater. Interfaces, 2022, 9, 2102383 CrossRef CAS.
- S. Swaminathan, J. K. Bera and M. Chandra, Simultaneous harvesting of multiple hot holes via visible-light excitation of plasmonic gold nanospheres for selective oxidative bond scission of olefins to carbonyls, Angew. Chem., Int. Ed., 2023, 62, e202215933 CrossRef CAS PubMed.
- G. K. Joshi, R. Kashyap, K. Patrikar, A. Mondal and S. Khatua, Ligand-mediated electron transport channels enhance photocatalytic activity of plasmonic nanoparticles, Nanoscale, 2023, 15, 16552–16560 RSC.
- B. Su, M. Zheng, W. Lin, X. F. Lu, D. Luan, S. Wang and X. W. Lou, S-Scheme Co9S8@Cd0.8Zn0.2S-DETA hierarchical nanocages bearing organic CO2 activators for photocatalytic Syngas production, Adv. Energy Mater., 2023, 13, 2203290 CrossRef CAS.
- H. Liang, Q. Chen, Q.-L. Mo, Y. Wu and F.-X. Xiao, Atomically precise thiolate-protected gold nanoclusters: current advances in solar-powered photoredox catalysis, J. Mater. Chem. A, 2023, 11, 9401–9426 RSC.
- Y. Liu, W. Xue, X. Liu, F. Wei, X. Lin, X. F. Lu, W. Lin, Y. Hou, G. Zhang and S. Wang, Ultrafine Pt nanoparticles on defective tungsten oxide for photocatalytic ethylene synthesis, Small, 2024, 20, 2402004 CrossRef PubMed.
- A. Kumar, P. Choudhary, T. Chhabra, H. Kaur, A. Kumar, M. Qamar and V. Krishnan, Frontier nanoarchitectonics of graphitic carbon nitride based plasmonic photocatalysts and photoelectrocatalysts for energy, environment and organic reactions, Mater. Chem. Front., 2023, 7, 1197–1247 RSC.
- A. Kumar, P. Choudhary, A. Kumar, P. H. C. Camargo and V. Krishnan, Recent advances in plasmonic photocatalysis based on TiO2 and noble metal nanoparticles for energy conversion, environmental remediation, and organic synthesis, Small, 2022, 18, 2101638 CrossRef CAS PubMed.
- S. Link and M. A. El-Sayed, Size and temperature dependence of the plasmon absorption of colloidal gold nanoparticles, J. Phys. Chem. B, 1999, 103, 4212–4217 CrossRef CAS.
- P. K. Jain and M. A. El-Sayed, Plasmonic coupling in noble metal nanostructures, Chem. Phys. Lett., 2010, 487, 153–164 CrossRef CAS.
- P. K. Jain, W. Huang and M. A. El-Sayed, On the universal scaling behavior of the distance decay of plasmon coupling in metal nanoparticle pairs: a plasmon ruler equation, Nano Lett., 2007, 7, 2080–2088 CrossRef CAS.
- M. Dhiman, A. Maity, A. Das, R. Belgamwar, B. Chalke, Y. Lee, K. Sim, J.-M. Nam and V. Polshettiwar, Plasmonic colloidosomes of black gold for solar energy harvesting and hotspots directed catalysis for CO2 to fuel conversion, Chem. Sci., 2019, 10, 6594–6603 RSC.
- A. Maity and V. Polshettiwar, Dendritic fibrous nanosilica for catalysis, energy harvesting, carbon dioxide mitigation, drug delivery, and sensing, ChemSusChem, 2017, 10, 3866–3913 CrossRef CAS PubMed.
- A. Maity, R. Belgamwar and V. Polshettiwar, Facile synthesis protocol to tune size, textural properties & fiber density of dendritic fibrous nanosilica (DFNS): applications in catalysis and CO2 capture, Nat. Protoc., 2019, 14, 2177–2204 CrossRef CAS PubMed.
- V. Polshettiwar, Dendritic fibrous nano-silica (DFNS): discovery, synthesis, formation mechanism, catalysis, and CO2 capture-conversion, Acc. Chem. Res., 2022, 55, 1395–1410 CrossRef CAS PubMed.
- F. D. Swearer, H. Zhao, L. Zhou, C. Zhang, H. Robatjazi, P. M. J. Martirez, M. C. Krauter, S. Yazdi, J. M. McClain, E. Ringe, A. E. Carter, P. Nordlander and J. N. Halas, Heterometallic antenna–reactor complexes for photocatalysis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8916–8920 CrossRef PubMed.
- G. Sharma, R. Verma, S. Masuda, K. Badawy, N. Singh, T. Tsukuda and V. Polshettiwar, Pt-doped Ru nanoparticles loaded on ‘black gold’ plasmonic nanoreactors as air stable reduction catalysts, Nat. Commun., 2024, 15, 713 CrossRef CAS PubMed.
- R. Verma, R. Tyagi, V. K. Voora and V. Polshettiwar, Black gold-based “antenna–reactor” to activate non-plasmonic nickel: photocatalytic hydrodechlorination and hydrogenation reactions, ACS Catal., 2023, 13, 7395–7406 CrossRef CAS.
- R. Verma, R. Belgamwar, P. Chatterjee, R. B. Vadell, J. Sa and V. Polshettiwar, Nickel-laden dendritic plasmonic colloidosomes of black gold: forced plasmon mediated photocatalytic CO2 hydrogenation, ACS Nano, 2023, 17, 4526–4538 CrossRef CAS PubMed.
- X. Wu, J. Lang, Z. Sun, F. Jin and Y. H. Hu, Photocatalytic conversion of carbon monoxide: from pollutant removal to fuel production, Appl. Catal., B, 2021, 295, 120312 CrossRef CAS.
- J. Zhang, M. Shu, Y. Niu, L. Yi, H. Yi, Y. Zhou, S. Zhao, X. Tang and F. Gao, Advances in CO catalytic oxidation on typical noble metal catalysts: Mechanism, performance and optimization, Chem. Eng. J., 2024, 495, 153523 CrossRef CAS.
- N. K. Soliman, Factors affecting CO oxidation reaction over nanosized materials: A review, J. Mater. Res. Technol., 2019, 8, 2395–2407 CrossRef CAS.
- K. Eid, A. Gamal and A. M. Abdullah, Graphitic carbon nitride-based nanostructures as emergent catalysts for carbon monoxide (CO) oxidation, Green Chem., 2023, 25, 1276–1310 RSC.
- J. P. N. Kembo, J. Wang, N. Luo, F. Gao, H. Yi, S. Zhao, Y. Zhou and X. Tang, A review of catalytic oxidation of carbon monoxide over different catalysts with an emphasis on hopcalite catalysts, New J. Chem., 2023, 47, 20222–20247 RSC.
- J. Raskó and J. Kiss, CO oxidation in the presence of hydrogen on Au/TiO2 catalyst: an FTIR-MS study, Catal. Lett., 2006, 111, 87–95 CrossRef.
- R. Li, J. Zhao, Z. Gan, W. Jia, C. Wu and D. Han, Gold promotion of MCM-41 supported ruthenium catalysts for selective hydrogenation of α,β-unsaturated aldehydes and ketones, Catal. Lett., 2018, 148, 267–276 CrossRef CAS.
- J. Luo, Y. Liu, Y. Niu, Q. Jiang, R. Huang, B. Zhang and D. Su, Insight into the chemical adsorption properties of CO molecules supported on Au or Cu and hybridized Au–CuO nanoparticles, Nanoscale, 2017, 9, 15033–15043 RSC.
- X. Wang, A. Rosspeintner, A. Ziarati, J. Zhao and T. Bürgi, Insight into the transient inactivation effect on Au/TiO2 catalyst by in situ DRIFT and UV-vis spectroscopy, Nat. Commun., 2022, 13, 5458 CrossRef CAS PubMed.
- O. S. Bezkrovnyi, D. Blaumeiser, M. Vorokhta, P. Kraszkiewicz, M. Pawlyta, T. Bauer, J. Libuda and L. Kepinski, NAP-XPS and in situ DRIFTS of the interaction of CO with Au nanoparticles supported by Ce1−xEuxO2 nanocubes, J. Phys. Chem. C, 2020, 124, 5647–5656 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, Fig. S1–S11, and Table S1 are available for this paper. See DOI: https://doi.org/10.1039/d4ta05117c |
| ‡ Shared 1st authors. |
| This journal is © The Royal Society of Chemistry 2024 |