
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceβ-Terrecyclene synthase constructs the quadrane backbone in terrecyclic acid biosynthesis†
Yongxiang
Song
abcd,
Wengui
Wang
 f,
Jiafan
Yang
abc,
Dewei
Gao
d,
John M.
Billingsley
d,
Songtao
Wang
abc,
Yiguang
Zhu
abcd,
Junfeng
Wang
abcd,
Jianhua
Ju
ac,
Yan
Yan
*abcd and
Yi
Tang
de
f,
Jiafan
Yang
abc,
Dewei
Gao
d,
John M.
Billingsley
d,
Songtao
Wang
abc,
Yiguang
Zhu
abcd,
Junfeng
Wang
abcd,
Jianhua
Ju
ac,
Yan
Yan
*abcd and
Yi
Tang
de
aKey Laboratory of Tropical Marine Bio-resources and Ecology, Guangdong Key Laboratory of Marine Materia Medica, Innovation Academy of South China Sea Ecology and Environmental Engineering, South China Sea Institute of Oceanology, Chinese Academy of Sciences, 164 West Xingang Road, Guangzhou 510301, China. E-mail: yyan@scsio.ac.cn
bSanya Institute of Oceanology Eco-Environmental Engineering Yazhou Scientific Bay, Sanya, 572000, China
cUniversity of Chinese Academy of Science, 19 Yuquan Road, Beijing 100049, China
dDepartment of Chemical and Biomolecular Engineering, University of California, Los Angeles, CA 90095, USA
eDepartment of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095, USA
fSchool of Chemistry and Chemical Engineering, University of Jinan, 336 West Road of Nan Xinzhuang, Jinan 250022, China
First published on 25th April 2024
Abstract
Quadrane sesquiterpenes featuring a distinctive tricyclic skeleton exhibit potent antimicrobial and anticancer activities. Although extensive studies have attempted to reveal the multistep carbocation rearrangement involved in the formation of the tricyclic quadrane scaffold, the exact biosynthetic pathway and chemical logic to generate the quadrane structure remains mysterious. Here we identified a novel sesquiterpene synthase that is capable of generating β-terrecyclene possessing the quadrane scaffold and characterized the biosynthetic pathway of a representative fungal quadrane terrecyclic acid. Further mutagenesis coupled with isotopically sensitive branching studies of this β-terrecyclene synthase provided insight into the mechanism involved in the formation of the quadrane scaffold.
Introduction
Terpenes feature tens of thousands of structurally diverse entities and comprise the largest group of natural products. The structural complexity is generated by terpene cyclases (TCs) using linear oligo-isoprenoid diphosphate precursors including geranyl, farnesyl, geranylgeranyl, and geranylfarnesyl diphosphate.1,2 The TCs are capable of catalyzing the most complex multistep carbocation rearrangement in nature, in which the bonding, hybridization, and stereochemistry of the substrate's carbon atoms are dramatically altered to accomplish the intriguing scaffolds. Among these structurally diversified scaffolds, quadranes belong to a family of naturally occurring sesquiterpenes consisting of a cyclopentane fused with a bicyclo[3.2.1]octane moiety (Fig. 1).3 The first quadrane was discovered as (−)-quadrone (1) from the mold Aspergillus terreus in 1978,4,5 which was shown to be derived from (+)-terrecyclic acid A (2) through an intramolecular oxa-Michael addition reaction.6 In the past two decades, six new quadranoids were isolated from terrestrial fungi, which include (−)-8-hydroxyquadrone (3), terrecyclol (4), (−)-isoquadrone (5), (+)-6-hydroxyisoquadrone (6), (+)-5(6)-dihydro-6-hydroxyterrecyclic acid (7) and (+)-5(6)-dihydro-6-methoxyterrecyclic acid (8).3 Interestingly, molecules possessing this unique quadrane scaffold were also discovered in soft corals as(−)-suberosanone (9), (−)-suberosenol A (10), (−)-suberosenol B (11), (−)-suberosenol A acetate (12), (−)-suberosenol B acetate (13), (+)-suberosenone (14), the intermolecular Michael addition adduct of 14 as (+)-alertenone (15), the 7H-purine-6,8-diol aza-Michael addition adduct 6-(9′-purine-6′,8′-diolyl)-2β-suberosanone (16), ring A opened derivatives (−)-isishippuric acid A (17) and (−)-isishippuric acid A (18).3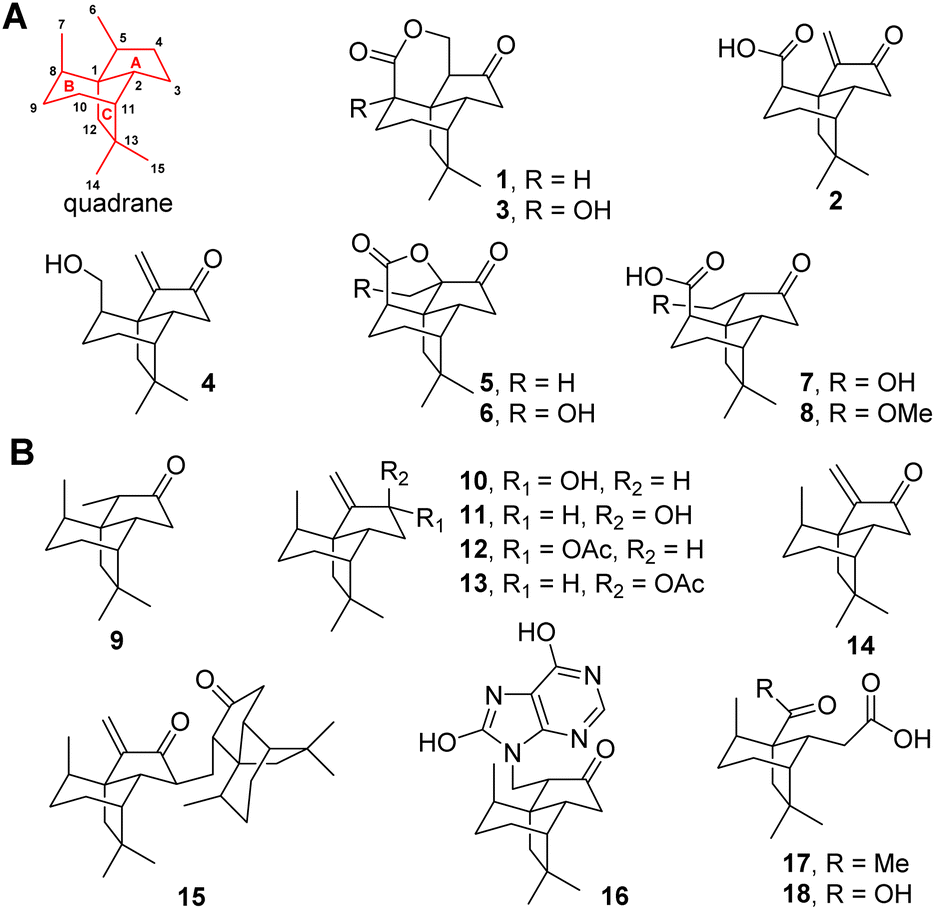 | ||
| Fig. 1 Naturally occurring quadrane sesquiterpenes from fungi (A) and coral (B). The tricyclic quadrane scaffold is shown in red. | ||
Due to the potent antimicrobial and anticancer activities, the challenging construction of the quadrane scaffold has attracted considerable attention from synthetic chemists.3,7,8 Incorporation of isotopically labeled acetate and mevalonate into the biosynthesis of 1 and 2 has revealed that quadranoids are sesquiterpenes resulting from multiple rearrangement reactions.9–11 Based on the altered connectivities of each atom in 1 as compared to the farnesyl diphosphate (FPP) precursor, two distinct possible mechanisms known as Hirota's route12,13 and Coates' route,14 respectively were hypothesized to explain the multistep cascade of carbocation rearrangements involved in the formation of the tricyclic quadrane scaffold (Scheme 1). Hirota's route represents the earliest proposed mechanism involving head-to-tail cyclization of FPP to yield the monocyclic humulyl cation (19), which is subsequently converted to a 5/8-bicyclic carbocation intermediate (20) (Scheme 1A). Then rearrangement of 20 takes place to form a tricyclic intermediate (21), which undergoes a 1,2-hydride shift to generate 22, and then a 1,2-alkyl shift to give 23 (Scheme 1A). Lastly, a 1,3-hydride shift leads 23 to terrecyclanyl cation 24, which is subsequently converted into β-terrecyclene (25) via deprotonation (Scheme 1A).
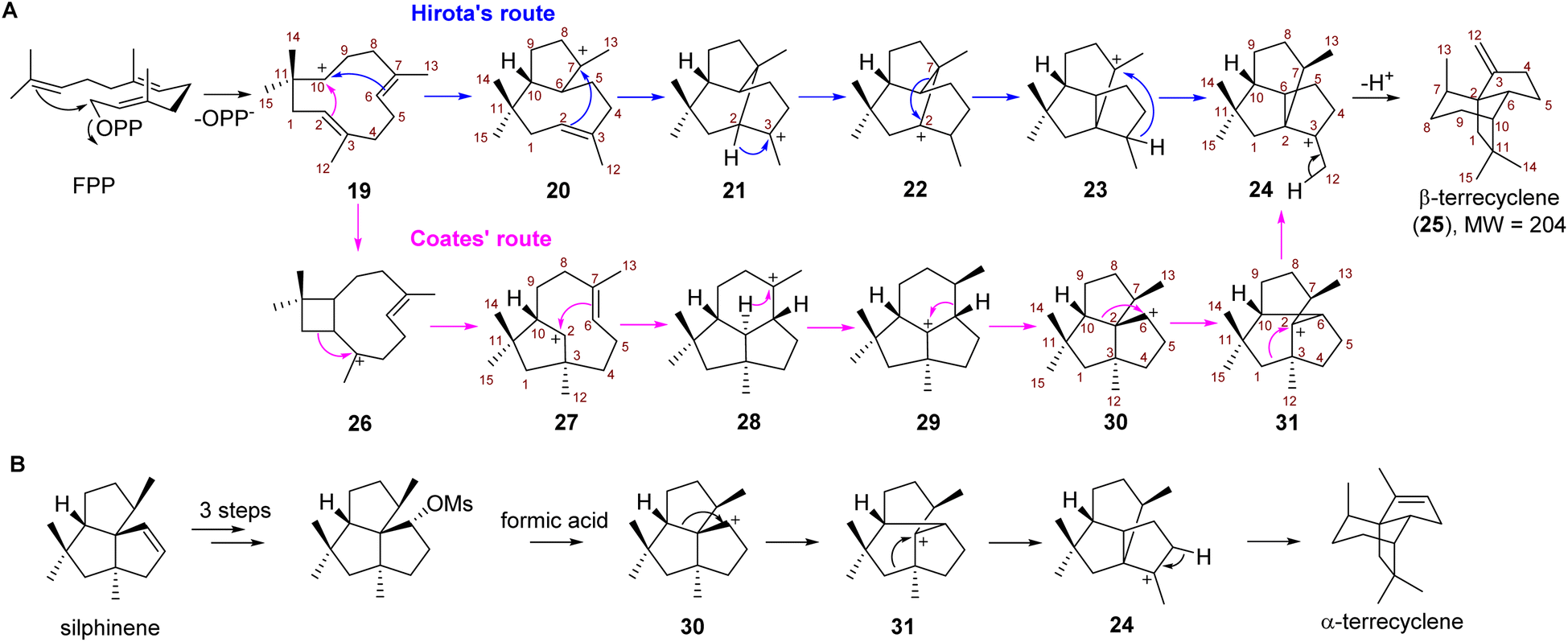 | ||
| Scheme 1 The proposed mechanisms of β-terrecyclene formation. (A) Hirota's route (blue) and Coates' route (pink) to generate β-terrecyclene from farnesyl diphosphate (FPP). (B) Coates' rearrangement to form α-terrecyclene. | ||
In 1992, Coates proposed an alternative route based on the observation that silphinenyl cation (30) was readily transformed into the terrecyclanyl cation (24) which could be further deprotonated to give α-terrecyclene during solvolytic rearrangement (Scheme 1B).15 The difference between these two hypotheses is the rearrangement process during the transformation of 19 into 24 (Scheme 1A). In Coates' route, monocyclic 19 is converted into caryophyllenyl ion 26, which is followed by a 1,2-alkyl shift to generate the 5/8-bicyclic intermediate 27 (Scheme 1A). Then a carbocation rearrangement leads 27 to a tricyclic intermediate 28, which is transformed into presilphiperfolan-8-yl ion (29) via a 1,3-hydride shift. 30 is then formed via a 1,2-alkyl shift of 29, and then another 1,2-alkyl shift takes place to yield 24 (Scheme 1A).
Although Hirota's route seems unlikely since the formation of transition states of two critical steps (22 → 23 → 24) require high energy according to a computational study,16 no TC catalyzing the direct conversion of FPP into 25 or α-terrecyclene had been reported, and the formation of the quadrane scaffold could also result from rearrangement of other terpene scaffolds using tailoring enzymes. Therefore, the biosynthesis of the quadrane scaffold remained unclear. Here, we elucidated the biosynthetic pathway of 2 and demonstrated that a novel TC is capable of directly generating 25 from FPP through Coates' route using isotopically sensitive branching coupled with mutagenesis studies.
Results and discussion
Identification and characterization of the biosynthetic genes of terrecyclic acid
To investigate the biosynthesis of 2, we hypothesized that the biosynthetic genes should include not only a sesquiterpene cyclase as the core gene but also oxidases to generate the highly oxygenated scaffold of 2. To locate these biosynthetic genes, the genome of the producer of 2Aspergillus terreus ATCC 20516 was sequenced and analyzed. Three biosynthetic gene clusters (BGCs) were identified encoding both a TC and oxidative tailoring enzymes such as a cytochrome P450, α-ketoglutarate-dependent oxidase, or flavoenzyme (Fig. S1†). To further identify the BGC of 2, we compared the differential gene expression of A. terreus ATCC 20516 before and after 2 was produced (Fig. S13†). Only one of the BGCs, a four-gene cassette on scaffold 58 was significantly upregulated during the production of 2; this BGC contains a TC (terA), a cytochrome P450 (terB), a short-chain dehydrogenase/reductase (SDR, terC), and a transporter of the major facilitator superfamily (MFS, terD) (Fig. 2A and Table S5†).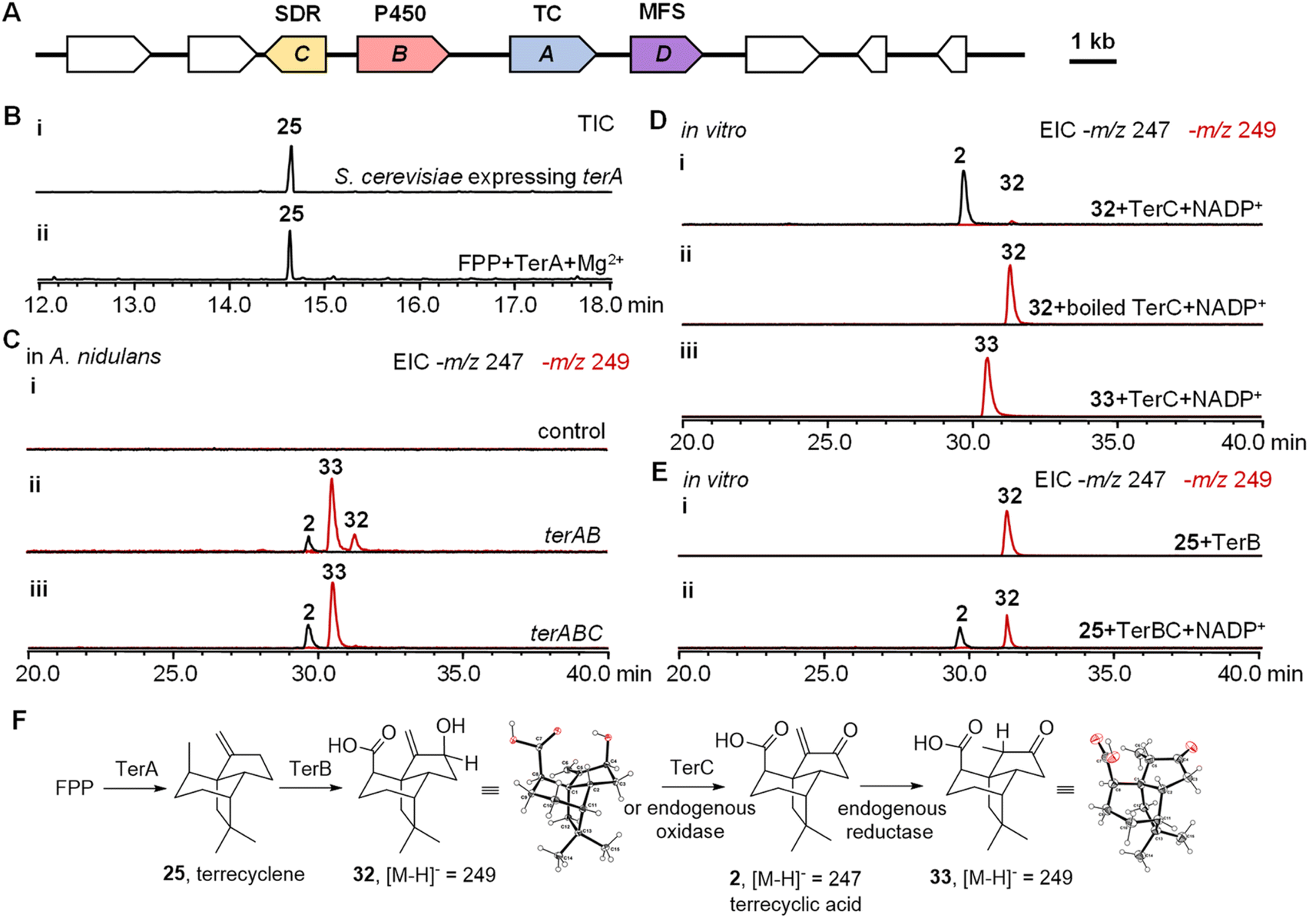 | ||
| Fig. 2 Characterization of the biosynthetic pathway of terrecyclic acid. (A) Biosynthetic gene cluster of terrecyclic acid (2). The hypothetic proteins are in white. (B) Characterization of TerA using heterologous expression in S. cerevisiae (i) and in vitro biochemical analysis (ii). The total ion chromatograms (TICs) were obtained using GC-MS. (C) Heterologous expression of ter biosynthetic genes in A. nidulans: (i) A. nidulans carrying the empty vectors; (ii) A. nidulans carrying terA and terB; (iii) A. nidulans carrying terA, terB, and terC. The extracted ion chromatograms (EICs) were obtained using LC-MS. (D) In vitro characterization of TerC: (i) conversion of 32 into 2 catalyzed by TerC; (ii) boiled TerC abolishes the catalytic activity on substrate 32; (iii) TerC is not able to catalyze the conversion of 33 into 2. The EICs were obtained using LC-MS. (E) In vitro characterization of TerB: (i) conversion of 25 into 32 catalyzed by TerB; (ii) conversion of 25 into 2 catalyzed by TerB and TerC. (F) Proposed biosynthetic pathway of 2 and the related products. The EICs were obtained using LC-MS. The Y-axes of the chromatograms for each experiment are depicted on the same scale, ensuring comparable quantities of the compounds. | ||
To characterize the function of each gene in the BGC, intron-free terA–C were cloned from cDNA and heterologously expressed in Saccharomyces cerevisiae RC01, which was engineered to express the A. terreus electron transfer partner cytochrome P450 reductase.17,18 The yeast cell expressing terA was able to produce a new sesquiterpene, which was determined to be 25 using NMR spectroscopy (Fig. 2B, S2 and Table S6†). When TerA was purified to homogeneity (Fig. S4†), 25 was also observed as the only product in the presence of 1 mM FPP and 5 mM MgCl2 (Fig. 2B). To study the function of terB and terC, we attempted the heterologous expression of terAB and terABC in S. cerevisiae RC01. However, only a trace amount of new products with molecular weights of 250 and 248 were able to be detected possibly due to the low catalytic efficiency of the cytochrome P450 TerB in yeast (Fig. S5†). We then switched to another well-established eukaryotic host Aspergillus nidulans A1145.19,20 When terA and terB were expressed in A. nidulans, the yield of the new products, with identical molecular weights and retention times, was dramatically increased. The new product with a molecular weight of 248 was characterized to be 2, while the fractions with a molecular weight of 250 are 32 and 33, respectively. All these products were characterized using NMR spectroscopy and X-ray crystallography (Fig. 2C, F, S2, S3 and Table S6†). When all three genes terA–C were co-expressed together, the yield of 2 was increased and only compound 33 was accumulated, which implied that 32 was possibly converted into 2 using TerC (Fig. 2C).
To identify the function of TerC, we expressed and purified the short-chain dehydrogenase/reductase in Escherichia coli. Biochemical analysis was performed using compounds 32 and 33 as substrates respectively. The oxidation of 32 into 2 was able to be catalyzed using TerC in the presence of the cofactor nicotinamide adenine dinucleotide phosphate (NADP+), while the activity of boiled TerC was abolished (Fig. 2D). On the other hand, TerC failed to catalyze the oxidation of compound 33 into 2 using the NADP+ cofactor, which indicated that only 32 is the substrate of TerC, while 33 is likely a shunt product derived from 2. The reducing ability of TerC was then assessed in the presence of the cofactor NADPH using 2, resulting in the exclusive formation of product 32. This observation aligns with previous findings that TerC can catalyze the conversion of 32 back to 2 in the presence of NADP+. Thus, it provides further evidence that 32 serves as a precursor to 2 (Fig. S6†). We therefore propose that A. nidulans A1145 possesses an isozyme of TerC which catalyzes the conversion of 32 into 2 at a low efficiency in the absence of TerC. Subsequently, an endogenous reductase in the host likely catalyzes the reduction of 2 to generate 33 (Fig. 2F). To validate our hypothesis, we conducted feeding experiments with compounds 2, 32, and 33 using the heterologous expression host. When 2 was fed to A. nidulans for 4 hours, it underwent conversion into 33 (Fig. S6A†). Conversely, feeding 32 to A. nidulans led to the generation of both 2 and 33 as products. However, no modified product was observed when 33 was fed to A. nidulans (Fig. S6A†). These findings indicate that 32 can be converted into 2 by an endogenous oxidase present in A. nidulans. Subsequently, 2 is further modified through the action of an endogenous reductase in A. nidulans, leading to the generation of 33 (Fig. 2F and S6B†).
To avoid modification of 32 and 2 by the endogenous enzymes, we performed in vitro biochemical analysis using the corresponding microsomal fractions of A. nidulans expressing terB. The sesquiterpene precursor 25 was successfully converted into a single oxidized product 32, which could be further converted into a single product 2 using TerC (Fig. 2E). Thus the biosynthetic pathway of 2 could be proposed. First, TerA catalyzes the cyclization of FPP to give 25, which is then iteratively oxidized by TerB to install the hydroxyl and carboxylic acid in 32. Finally, dehydrogenation of 32 catalyzed by TerC takes place to generate 2 (Fig. 2F).
Mutagenesis studies of TerA
We next investigated the mechanism involved in carbocationic rearrangement during the formation of 25 catalyzed using TerA. To identify the key residues in the active site of TerA that are responsible for guiding the carbocationic intermediates to the quadrane scaffold, we predicted the three-dimensional structure of TerA using the Phyre 2 server.21 Although the identity of the amino acid sequence is not very high between TerA and several representative sesquiterpene synthases including epi-isozizaene synthase,22 selinadiene synthase,23 and pentalenene synthase,24 their overall three-dimensional structures are similar (Fig. S8 and S9†). To approximate the key residues in the active site that are responsible for binding and folding FPP, we aligned the predicted structure of TerA to selinadiene synthase complexed with substrate mimic 2,3-dihydro-FPP and Mg2+ (Fig. S10†). 10 residues lining the FPP binding pocket were identified, which included C111, V114, F115, D116, L120, F189, I219, Q260, W342 and S343 (Fig. 3A). To validate the prediction, alanine scanning was performed on these residues. The corresponding 10 TerA mutants were generated and heterologously produced in S. cerevisiae RC01. Analysis of the sesquiterpene product profile (m/z = 204) of the host expressing each mutant showed the mutations of F115A and W342A lead to almost complete loss of β-terrecyclene synthase activity (Fig. S11†). On the other hand, mutant V114A was capable of producing 25 as a major fraction, together with β-caryophyllene (34) and other unknown sesquiterpenes as minor fractions (Fig. 3B). However, the structure of the other minor fractions was not determined due to low yield. Interestingly, coproduct 34 could be derived via deprotonation of caryophyllenyl ion 26, an intermediate proposed by Coates' route (Fig. 3C). This observation suggested the formation of β-terrecyclene is more likely through Coates' route.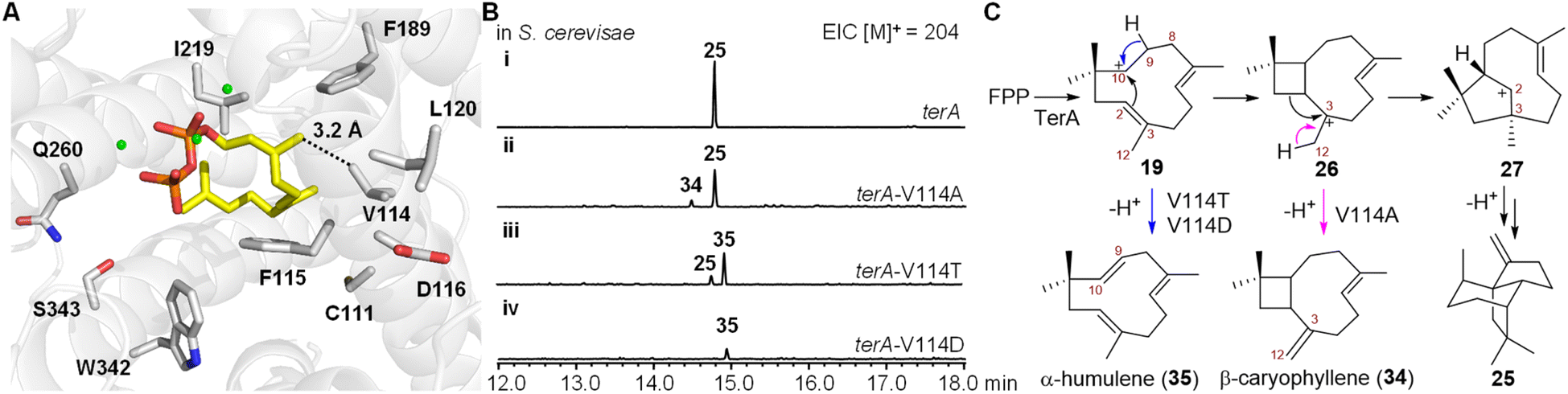 | ||
| Fig. 3 Mutagenesis of TerA. (A) The predicted model of TerA with the key residues (grey) in the active site responsible for terrecyclene formation. The substrate mimic 2,3-dihydro-FPP is in yellow. The magnesium cation is in green. (B) Representative sesquiterpene product profile of S. cerevisiae expressing terA mutants: (i) S. cerevisiae expressing wildtype terA; (ii) S. cerevisiae expressing terA-V114A; (iii) S. cerevisiae expressing terA-V114T; (iv) S. cerevisiae expressing terA-V114D. The EICs were obtained using GC-MS. The Y-axes of the chromatograms for each experiment are depicted on the same scale, ensuring comparable quantities of the compounds. (C) The proposed mechanism of the formation of shunt products catalyzed by TerA mutants. 34 is derived from the key intermediate 26 in Coates' route (pink), while 35 is derived from the intermediate 19 in both Hirota's and Coates' routes (blue). | ||
To further investigate the catalytic functions of residues in the active site of TerA, we performed additional mutagenesis on these residues, substituting them with various amino acids other than alanine (Fig. S11†). We generated 21 TerA mutants and analyzed their product profiles. While most of the mutations decreased the terpene cyclase activity, they did not yield any new products. Their activities were similar to either the wild-type TerA or the corresponding alanine mutants. For instance, mutations of C111 to serine, tyrosine, and aspartate resulted in a reduction in the yield of 25 as the sole product. Mutation of F115 to other aromatic amino acids, such as tyrosine and tryptophan, resulted in a reduced yield of 25 compared to F115A. The F115W mutant also exhibited very low production of a few other sesquiterpenes, possibly due to improper folding of the substrate FPP. Substituting the acidic residue aspartate at position 116 with glutamate or asparagine led to a decrease in the yield of 25. The mutation of L120 to a smaller amino acid did not affect the yield of 25, whereas the L120I mutation decreased production. Mutating F198 to aromatic residues, such as tyrosine and tryptophan, resulted in a decreased yield of 25. Similarly, mutations of W342 to aromatic residues, such as tyrosine and phenylalanine, also led to a decrease in the production of 25, and mutations of Q260 to asparagine and glutamate reduced the activity of TerA. When S343 was mutated to threonine, the production of 25 decreased. However, mutating S343 to phenylalanine completely abolished the catalytic activity of TerA. The hydrophobic residue V114 was mutated to a larger hydrophobic residue (leucine), an aromatic residue (phenylalanine), a polar residue (threonine), and an acidic residue (aspartate), respectively. Compared to V114A, both V114L and V114F mutants completely lost terpene cyclase activity, suggesting a strict size limitation for the residue at this position. On the other hand, mutants V114T and V114D produced a new compound, 35, identified as α-humulene (Fig. 3B and S11†).
These results indicated that the structure of each residue located in the active sites of TerA is not strictly required for the generation of 25. Except for V114, mutations of these residues do not dramatically switch the cyclization pathway to yield other shunt products. Intriguingly, 35 could be derived from intermediate 19, forming a shunt product for both Hirota's and Coates' pathways (Fig. 3C). In the predicted model of the TerA active sites, residue V114 is close to the C12 of substrate mimic 2,3-dihydro-FPP (Fig. 3A). Therefore, we propose that the mutation of V114 to the smaller residue alanine enlarges the substrate binding pocket, leading to reduced selectivity of the 1,2-alkyl shift of 26, and the concurrent generation of the C12 proton elimination product 34 (Fig. 3C). Additionally, the mutation of V114 to hydrogen bonding acceptor-containing residues may facilitate hydrogen abstraction at C9 of intermediate 19, resulting in the formation of another shunt product 35 (Fig. 3C).
Isotopically sensitive branching reveals that the formation of β-terrecyclene follows Coates' route
Although the coproduction of 34 resulting from mutant V114A suggested the formation of β-terrecyclene is more likely through Coates' route, additional evidence would be needed to conclusively demonstrate that 26 is on the direct pathway to 25. As a main distinction between Hirota's and Coates' routes, the formation of 26 could be validated using the well-established isotopically sensitive branching approach.25–27 Therefore, we speculated that the substitution of all three protons on C12 of FPP by deuterium ([12-2H3]-FPP) would result in the decrease of the ratio of 34 to 25 produced by V114A mutant if the carbocation rearrangement is via the Coates' route (Scheme 1A and Fig. 4A). The production of 34 would be suppressed due to the primary deuterium kinetic isotope effect (KIE) on the deprotonation of 26 to yield 34 (Fig. 4A). On the other hand, the distribution of 34 and 25 would not be affected while using [12-2H3]-FPP as substrate, on the condition that cyclization follows Hirota's route (Fig. 4A).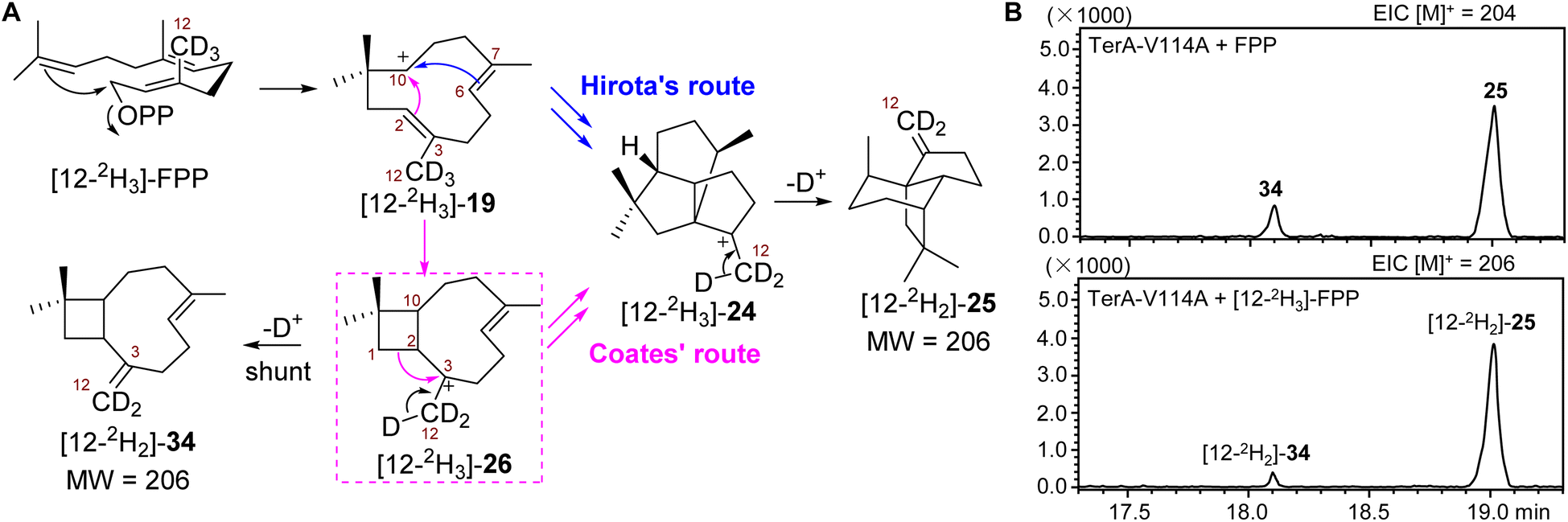 | ||
| Fig. 4 Isotopically sensitive branching study of TerA-V114A mutant. (A) Distinguishing between Hirota's and Coates' routes through an isotopically sensitive branching experiment using [12-2H3]-FPP as substrate. (B) Effect of deuteration at C12 of FPP on the ratio of products generated by TerA-V114A. GC-MS chromatograms of reaction mixtures using FPP (top) and [12-2H3]-FPP (bottom). The EICs were obtained using GC-MS. | ||
Thus, recombinant TerA-V114A mutant was obtained upon heterologous expression in S. cerevisiae RC01 to perform the following in vitro analysis (Fig. S4†). In the presence of 1 mM FPP and 5 mM MgCl2, TerA-V114A was able to convert FPP to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 mixture of coproducts 34 and 25 (Fig. 4B and S11†). When using [12-2H3]-FPP as a substrate at the same reaction condition, the ratio of 34 to 25 was significantly shifted to 1:10, with the ∼53% decreased yield of 34 and ∼18% increased production of 25 (Fig. 4B and S12†). The decreased ratio of 34 to 25 from 1:4 to 1:10 resulting from isotopically sensitive branching demonstrated that 26 is a common carbocation precursor to form both 34 and 25 (Fig. 4B). This indicated the cyclization of FPP to construct 25 is through Coates' route. Based on the shift of product distribution, the observed primary KIE on deprotonation of 26 to give 34 was determined to be kH/kD = 2.5. The magnitude of KIE is consistent with primary deuterium KIEs on similar terpene synthase-promoted methyl deprotonation, kH/kD = 2–6.27–31
:4 mixture of coproducts 34 and 25 (Fig. 4B and S11†). When using [12-2H3]-FPP as a substrate at the same reaction condition, the ratio of 34 to 25 was significantly shifted to 1:10, with the ∼53% decreased yield of 34 and ∼18% increased production of 25 (Fig. 4B and S12†). The decreased ratio of 34 to 25 from 1:4 to 1:10 resulting from isotopically sensitive branching demonstrated that 26 is a common carbocation precursor to form both 34 and 25 (Fig. 4B). This indicated the cyclization of FPP to construct 25 is through Coates' route. Based on the shift of product distribution, the observed primary KIE on deprotonation of 26 to give 34 was determined to be kH/kD = 2.5. The magnitude of KIE is consistent with primary deuterium KIEs on similar terpene synthase-promoted methyl deprotonation, kH/kD = 2–6.27–31
Conclusions
In summary, we elucidated the biosynthetic pathway of 2via quadrane scaffold formation using the β-terrecyclene synthase and oxidative tailoring enzymes. The mechanism of enzymatic quadrane scaffold formation was demonstrated through mutagenesis and isotopically sensitive branching experiments, settling a decades-old mystery in the field of natural product biosynthesis. We propose that more natural products of the quadrane family can be identified using ter biosynthetic genes as a query to search in the public fungal genome database.Data availability
Crystallographic data for compound 32 and 33 have been deposited at the CCDC under 2271713 and 2271714 respectively. The other datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Y. Y. and Y. T. designed the research. Y. S., W. W., J. Y., and S. W. performed the experiments. Y. S., Y. Y., Y. Z., J. B., D. G., J. W., and J. J. analyzed the data. Y. Y., Y. S., and J. B. wrote the manuscript. All the authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFC2805000, 2023YFA0914200); Hainan Provincial Joint Project of Sanya Yazhou Bay Science and Technology City (2021CXLH0013); the National Natural Science Foundation of China (32000044); Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2019BT02Y262); K. C. Wong Education Foundation (GJTD-2020-12); Hainan Provincial Natural Science Foundation of China (823CXTD393); Key Science and Technology Plan Projects in Nansha District (2023ZD010). We are grateful to the analytical facilities in SCSIO for recording mass, NMR, and X-ray crystallography data.Notes and references
- D. W. Christianson, Chem. Rev., 2017, 117, 11570–11648 CrossRef CAS PubMed.
- J. D. Rudolf and C.-Y. Chang, Nat. Prod. Rep., 2020, 37, 425–463 RSC.
- M. Presset, Y. Coquerel and J. Rodriguez, Eur. J. Org Chem., 2010, 2010, 2247–2260 CrossRef.
- G. J. Calton, R. L. Ranieri and M. A. Espenshade, J. Antibiot., 1978, 31, 38–42 CrossRef CAS PubMed.
- R. L. Ranieri and G. J. Calton, Tetrahedron Lett., 1978, 19, 499–502 CrossRef.
- M. Nakagawa, A. Hirota, H. Sakai and A. Isogai, J. Antibiot., 1982, 35, 778–782 CrossRef CAS PubMed.
- C. Peter, P. Geoffroy and M. Miesch, Org. Biomol. Chem., 2018, 16, 1381–1389 RSC.
- Y. Ren, M. Presset, J. Godemert, N. Vanthuyne, J.-V. Naubron, M. Giorgi, J. Rodriguez and Y. Coquerel, Chem. Commun., 2016, 52, 6565–6568 RSC.
- D. E. Cane, Y. G. Whittle and T.-C. Liang, Tetrahedron Lett., 1984, 25, 1119–1122 CrossRef CAS.
- A. Hirota, M. Nakagawa, H. Sakai and A. Isogai, Agric. Biol. Chem., 1984, 48, 835–837 CAS.
- J. J. M. Beale, R. L. Chapman and J. P. N. Rosazza, J. Antibiot., 1984, 37, 1376–1381 CrossRef CAS PubMed.
- A. Hirota, M. Nakagawa, H. Sakai, A. Isogai, K. Furihata and H. Seto, Tetrahedron Lett., 1985, 26, 3845–3848 CrossRef CAS.
- D. E. Cane, Y. G. Whittle and T.-C. Liang, Bioorg. Chem., 1986, 14, 417–428 CrossRef CAS.
- R. M. Coates, J. Z. Ho, M. Klobus and L. Zhu, J. Org. Chem., 1998, 63, 9166–9176 CrossRef CAS.
- M. Klobus, L. Zhu and R. M. Coates, J. Org. Chem., 1992, 57, 4327–4329 CrossRef CAS.
- J. E. Barquera-Lozada and G. Cuevas, J. Org. Chem., 2011, 76, 1572–1577 CrossRef CAS PubMed.
- M.-C. Tang, H.-C. Lin, D. Li, Y. Zou, J. Li, W. Xu, R. A. Cacho, M. E. Hillenmeyer, N. K. Garg and Y. Tang, J. Am. Chem. Soc., 2015, 137, 13724–13727 CrossRef CAS PubMed.
- D. A. Yee, T. B. Kakule, W. Cheng, M. Chen, C. T. Y. Chong, Y. Hai, L. F. Hang, Y.-S. Hung, N. Liu, M. Ohashi, I. C. Okorafor, Y. Song, M. Tang, Z. Zhang and Y. Tang, J. Am. Chem. Soc., 2020, 142, 710–714 CrossRef CAS PubMed.
- N. Liu, Y.-S. Hung, S.-S. Gao, L. Hang, Y. Zou, Y.-H. Chooi and Y. Tang, Org. Lett., 2017, 19, 3560–3563 CrossRef CAS PubMed.
- Z. Sun, C. S. Jamieson, M. Ohashi, K. N. Houk and Y. Tang, Nat. Commun., 2022, 13, 2568 CrossRef CAS PubMed.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. E. Sternberg, Nat. Protoc., 2015, 10, 845–858 CrossRef CAS PubMed.
- P. N. Blank, G. H. Barrow, W. K. W. Chou, L. Duan, D. E. Cane and D. W. Christianson, Biochemistry, 2017, 56, 5798–5811 CrossRef CAS PubMed.
- P. Baer, P. Rabe, K. Fischer, C. A. Citron, T. A. Klapschinski, M. Groll and J. S. Dickschat, Angew. Chem., Int. Ed., 2014, 53, 7652–7656 CrossRef CAS PubMed.
- C. A. Lesburg, G. Zhai, D. E. Cane and D. W. Christianson, Science, 1997, 277, 1820–1824 CrossRef CAS PubMed.
- L. Zu, M. Xu, M. W. Lodewyk, D. E. Cane, R. J. Peters and D. J. Tantillo, J. Am. Chem. Soc., 2012, 134, 11369–11371 CrossRef CAS PubMed.
- X. He and D. E. Cane, J. Am. Chem. Soc., 2004, 126, 2678–2679 CrossRef CAS PubMed.
- D. J. Schenk, C. M. Starks, K. R. Manna, J. Chappell, J. P. Noel and R. M. Coates, Arch. Biochem. Biophys., 2006, 448, 31–44 CrossRef CAS PubMed.
- K. Wagschal, T. J. Savage and R. Croteau, Tetrahedron, 1991, 47, 5933–5944 CrossRef CAS.
- R. B. Croteau, C. J. Wheeler, D. E. Cane, R. Ebert and H. J. Ha, Biochemistry, 1987, 26, 5383–5389 CrossRef CAS PubMed.
- H. J. Pyun, R. M. Coates, K. C. Wagschal, P. McGeady and R. B. Croteau, J. Org. Chem., 1993, 58, 3998–4009 CrossRef CAS.
- K. C. Wagschal, H. J. Pyun, R. M. Coates and R. Croteau, Arch. Biochem. Biophys., 1994, 308, 477–487 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details regarding general materials and methods, construction of strains S. cerevisiae and A. nidulans, fermentation, isolation, structure elucidation, synthetic procedures, biochemical analysis, X-ray crystallography, NMR data and NMR spectra. CCDC 2271713 and 2271714. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01208a |
| This journal is © The Royal Society of Chemistry 2024 |