
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioconvergent synthesis of chiral fluorenols from racemic secondary alcohols via Pd(II)/chiral norbornene cooperative catalysis†
Bo
Ding
a,
Qilin
Xue
a,
Han
Wei‡
b,
Jiangwei
Chen‡
a,
Ze-Shui
Liu
a,
Hong-Gang
Cheng
 a,
Hengjiang
Cong
a,
Jianting
Tang
c and
Qianghui
Zhou
*ab
a,
Hengjiang
Cong
a,
Jianting
Tang
c and
Qianghui
Zhou
*ab
aEngineering Research Center of Organosilicon Compounds & Materials (Ministry of Education), Hubei Key Lab on Organic and Polymeric OptoElectronic Materials, College of Chemistry and Molecular Sciences, TaiKang Center for Life and Medical Sciences, Wuhan University, Wuhan, 430072, China. E-mail: qhzhou@whu.edu.cn
bThe Institute for Advanced Studies, Wuhan University, Wuhan, 430072, China
cKey Laboratory of Water Environment Evolution and Pollution Control in Three Gorges Reservoir, School of Environmental and Chemical Engineering, Chongqing Three Gorges University, Chongqing, 404100, China
First published on 25th April 2024
Abstract
An efficient protocol for the asymmetric synthesis of fluorenols has been developed through an enantioconvergent process enabled by Pd(II)/chiral norbornene cooperative catalysis. This approach allows facile access to diverse functionalized chiral fluorenols with constantly excellent enantioselectivities, applying readily available racemic secondary ortho-bromobenzyl alcohols and aryl iodides as the starting materials.
Introduction
Racemic secondary alcohols have been recognized as one of the most useful feedstocks for organic transformations owing to their generally stable, nontoxic, and readily available properties.1,2 Therefore, the transformation of racemic secondary alcohols into value-added chiral compounds is an attractive approach in asymmetric synthesis.1a,2–6 Considerable progress has been made in this area so far, culminating in the development of several elegant strategies, including deracemization,3,4 enantioconvergent transformation,1a,2,5 and kinetic resolution (KR).6 For example, two common reaction modes, linear and cyclic redox deracemizations, have been established to prepare enantioenriched secondary alcohols from the corresponding racemates (Fig. 1a).4c–h In particular, Zuo and co-workers reported a photoinduced deracemization of secondary alcohols enabled by asymmetric ligand-to-metal charge transfer (LMCT) catalysis.4i In 2021, Shi and co-workers disclosed the nickel/N-heterocyclic carbene-catalyzed enantioconvergent arylation of racemic secondary alcohols to synthesize enantioenriched tertiary alcohols (Fig. 1b).2a Moreover, enantioconvergent transformations of racemic secondary alcohols into enantioenriched chiral higher-order alcohols, amines, N-heterocycles and ketones through asymmetric borrowing hydrogen catalysis have also been developed (Fig. 1c).1a,2b,5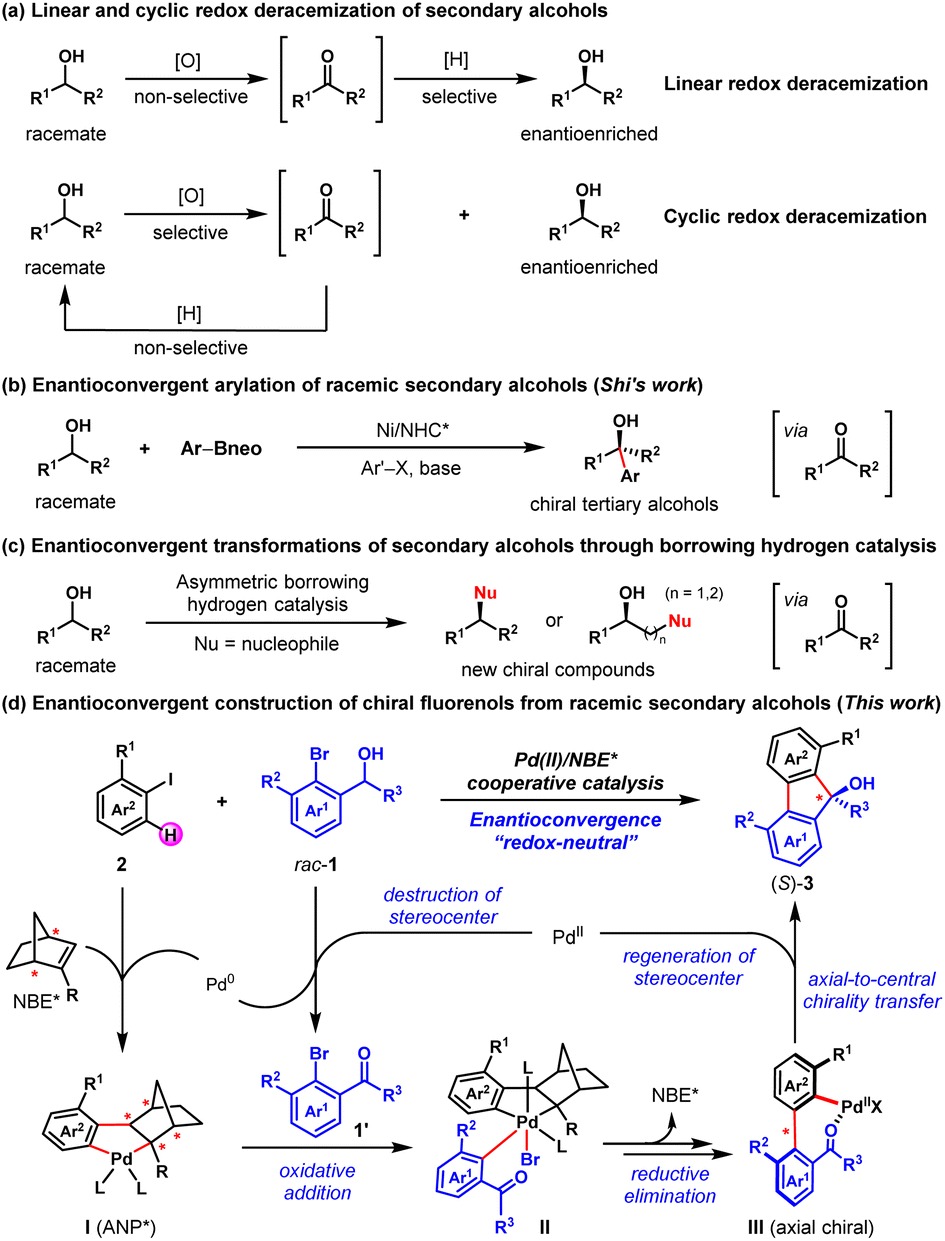 | ||
| Fig. 1 Typical strategies for transforming racemic secondary alcohols into enantioenriched compounds. | ||
Palladium/norbornene (Pd/NBE) cooperative catalysis, firstly reported by Catellani and co-workers in 1997,7 has become a powerful strategy for expeditious synthesis of polysubstituted arenes.8 For instance, the synthesis of racemic fluorenols (FOLs) via Pd/NBE cooperative catalysis has been successfully developed by Lautens9a and Della Ca',9b respectively. Asymmetric palladium/chiral norbornene (Pd/NBE*) cooperative catalysis has been theoretically expected as a potential strategy for asymmetric synthesis. However, exploitation of this asymmetric tactic has rarely been initiated for a long time, due to the associated formidable challenges of this complex process.10 It is only until recently that remarkable breakthroughs have been achieved in this field, owing to the efforts of Yu,11 Dong,12 Song,13 Liang14 and us.15
Inspired by these elegant works, we envisioned an asymmetric method to access enantioenriched FOLs through an enantioconvergent process enabled by Pd/NBE* cooperative catalysis, with widely available racemic secondary benzyl alcohols and aryl iodides as the reactants. As depicted in Fig. 1d, this method includes two sequential steps. Firstly, a Pd(II)-initiated oxidation of the racemic secondary ortho-bromobenzyl alcohol (1) will afford the achiral ortho-bromoacetophenone (1′) and the corresponding Pd(0) species.9b,16 Secondly, the Pd(0)/NBE* co-catalyzed asymmetric Catellani-type reaction between the in situ generated ortho-bromoacetophenone (1′) and aryl iodide (2) will lead to enantioenriched FOL (3) via Pd(IV) intermediate (II) and axial chiral Pd(II) intermediate (III), and meanwhile regenerating the Pd(II) species9,15a (see Fig. S5 in ESI† for more details of the proposed catalytic cycle). The potential features of this protocol include readily available starting materials, redox-neutral process (no external redox reagent is needed), the intriguing asymmetric strategy for destruction/reconstruction of stereoelement and the unique axial-to-central chirality transfer mode. From a practical perspective, the FOL products are known for their biological activity and applications in material.17 Until now, extensive efforts have been devoted to the synthesis of racemic FOLs and a number of effective strategies have been developed.9,18 However, only three examples of the asymmetric synthesis of FOLs have been reported,15a,19 to the best of our knowledge. Thus, the development of new strategies for their asymmetric synthesis is highly desirable. Nevertheless, there are three foreseeable main challenges regarding this proposal: (1) the identification of an appropriate catalyst combination of Pd(II) species, ligand and NBE* to ensure that the oxidation of racemic benzyl alcohol and the Catellani-type reaction connect well to each other and progress in an overall redox-neutral manner; (2) since NBE* is the only chiral source of this complex asymmetric transformation, the employment of a versatile NBE* cocatalyst is pivotal to achieve both good reactivity and enantioselectivity of this method; (3) multiple competitive side reactions, for example, the direct annulation of racemic secondary benzyl alcohol with aryl iodide to afford the benzo[c]chromene product,15d,20 premature ipso-protonation of the transient ary-Pd(II) species (e.g., III)21 and etc. (see Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Pd] | Ligand | Solvent | Yieldb (%) | E.e.c (%) |
| a All reactions were performed on a 0.1 mmol scale. b GC yield with biphenyl as an internal standard and isolated yield shown in parentheses. c Determined by HPLC analysis on a chiral stationary phase. d 20 mol% TFP. e 110 °C instead of 105 °C. f 25 mol% NBE* was applied. g 1.5 equiv. of K2CO3 was applied. —: not detected. TFP: tri(2-furyl)phosphine. DCE: 1,2-dichloroethane. | |||||
| 1 | Pd(OAc)2 | TFPd | Toluene | 24 | 98 |
| 2 | Pd(OAc)2 | DPEPhos | Toluene | 65 | — |
| 3 | Pd(OAc)2 | DPPE | Toluene | 66 | — |
| 4 | Pd(OAc)2 | DPPP | Toluene | 78 | 98 |
| 5 | Pd(OAc)2 | DPPPe | Toluene | 73 | — |
| 6 | PdI2 | DPPP | Toluene | 23 | — |
| 7 | Pd(PPh3)4 | DPPP | Toluene | Trace | — |
| 8e | Pd(OAc)2 | DPPP | Toluene | 83 | — |
| 9e | Pd(OAc)2 | DPPP | DCE | 89 | — |
| 10e,f | Pd(OAc)2 | DPPP | DCE | 88 | 98 |
| 11e,f,g | Pd(OAc)2 | DPPP | DCE | 88 (82) | 98 |
|
|||||
Results and discussion
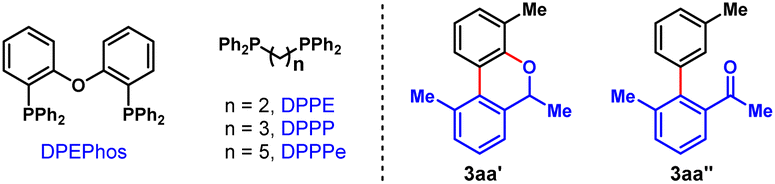
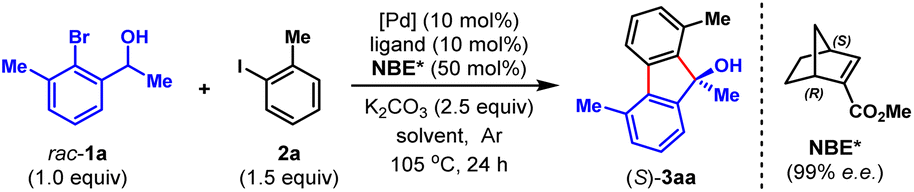
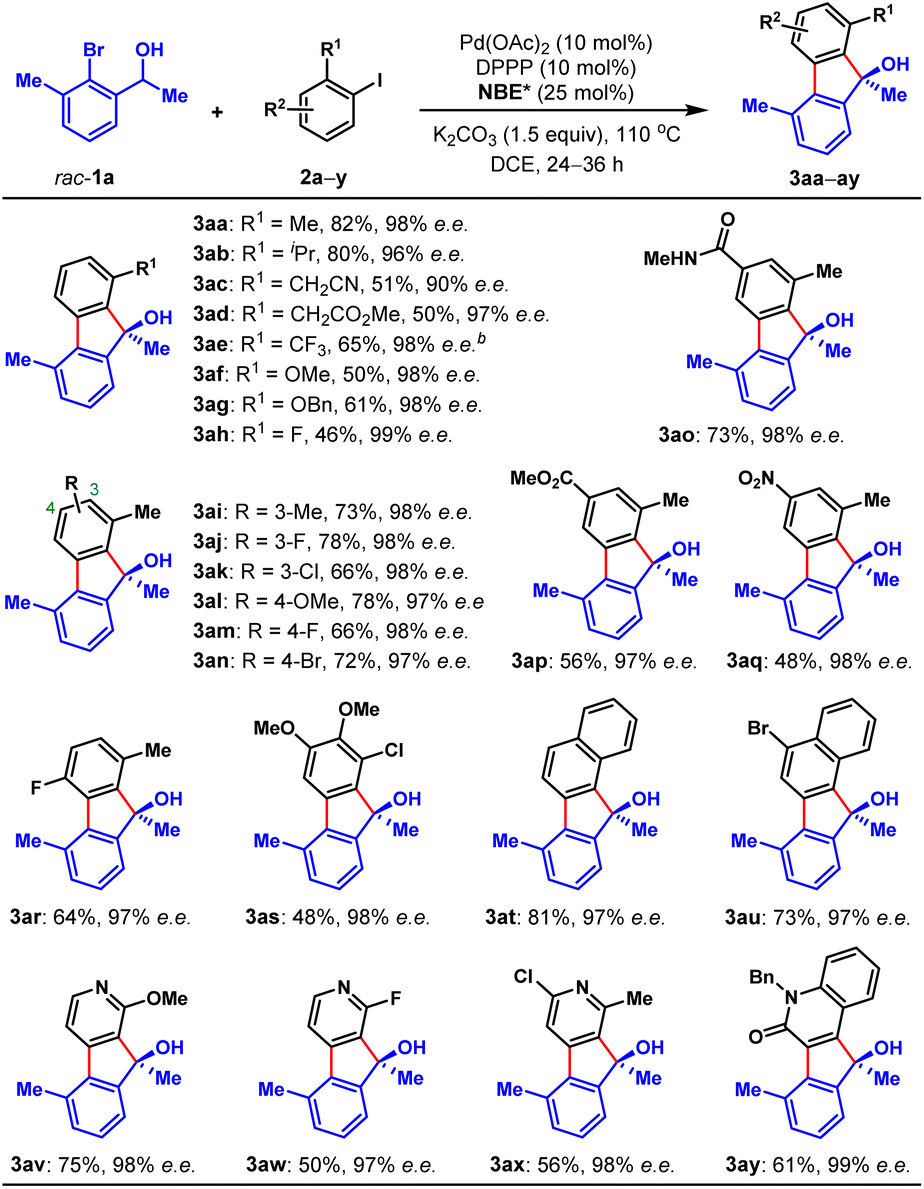
To validate our hypothesis, we performed a model reaction using racemic secondary ortho-bromobenzyl alcohol (rac-1a) and 2-iodotoluene (2a) as the reactants (Table 1). To our delight, under our previously reported reaction conditions (Pd(OAc)2 (10 mol%), tri(2-furyl)phosphine (TFP) (20 mol%), (1R,4S)-2-methyl ester-substituted NBE (NBE*, 99% e.e., 50 mol%) and heating at 105 °C),15d the desired chiral FOL 3aa was obtained with excellent enantioselectivity (98% e.e.), albeit in only 24% yield (entry 1). Meanwhile, two main expected side products were isolated, which were the benzo[c]chromene 3aa′ (12% isolated yield) formed through intramolecular ipso-etherification and the ipso-hydrogen-terminated one 3aa′′ (8% isolated yield). Encouraged by these preliminary results, we next focused on optimization of the reaction parameters to increase the reaction efficiency and chemical selectivity. Gratifyingly, the phosphine ligand proved to take a critical role for increasing the yield and chemical selectivity. The unique bidentate phosphine ligands with a flexible backbone,22 such as DPEPhos, DPPE, DPPP and DPPPe, are generally superior to monodentate phosphine ligands, to deliver 3aa in significantly improved yields (65–78%, entries 2–5). DPPP is identified as the optimal one (entry 4). Further screening of palladium catalyst, solvent, and reaction temperature indicated that Pd(OAc)2, 1,2-dichloroethane (DCE) and 110 °C was the optimal combination (entries 6–9), which provided 3aa in a substantially improved yield (89%, entry 9). It is worth mentioning that the use of DCE can reduce the production of side product 3aa′′. Importantly, the loading of NBE* could be reduced to a truly catalytic amount (25 mol%) without any erosion of the yield and enantiopurity of 3aa (entry 10). Moreover, the amount of K2CO3 could also be reduced to 1.5 equiv. without any deleterious effects on the reaction (entry 11). Additional optimization of other reaction parameters (e.g., base, concentration and etc.) did not improve the efficiency further (see Tables S1–S5 in ESI† for more details). Thus, the optimal reaction conditions were identified to be entry 11 of Table 1, which delivered 3aa in 82% isolated yield and 98% e.e.With the optimal reaction conditions on hand, we first examined the scope of aryl iodides (2), using rac-1a as the reaction partner. As shown in Table 2, a wide variety of ortho-substituted aryl iodides were suitable substrates, including sterically hindered isopropyl (3ab), functionalized alkyl (3ac and 3ad), electron-withdrawing trifluoromethyl (3ae), electron donating methoxy and benzyloxy (3af and 3ag), fluoro (3ah) and chloro (3as) groups, and the corresponding products were produced in 46–82% yields. In addition, the substituents at either the 3-, 4- or 5-positions of aryl iodides were well tolerated, which included alkyl (3ai), halogen (3aj, 3ak, 3am, 3an and 3ar), methoxy (3al), amide (3ao), ester (3ap), and nitro (3aq) groups. Moreover, 1-iodonaphtalene derivatives with extended π system were also suitable substrates for this reaction and afforded the benzo[a]fluorenol products in good yields (3at and 3au). More importantly, this method could be extended to heteroaryl iodides (2v–x) and 4-iodo-2-quinolone (2y), delivering the enantioenriched heterocyclic FOLs in 50–75% yields (3av–ay). These heterocyclic chiral FOLs may have potential applications in the development of new antimycobacterial and antiprotozoal agents.17d,g,18d Remarkably, all the products (3aa–ay) were obtained with excellent enantioselectivities (90–99% e.e.s), and those compatible functional groups would provide handles for further manipulation of the obtained FOLs.
Next, the scope of racemic secondary ortho-bromobenzyl alcohols (1) was investigated, with 2-iodotoluene (2a) as the reaction partner (Table 3). Various alkyl substituents other than methyl group at the R2 position were applicable for this protocol, including butyl (3ba), branched alkyl (3ca and 3da), and cycloalkyl (3ea and 3fa). It is worth mentioning that the alcohol substrate with a sterically very hindered tert-butyl substitution was able to provide the corresponding FOL in 76% yield after extending the reaction time to 36 h (3da). Additionally, the scope of R2 substitution of racemic alcohols could be extended to a wide range of aryl groups, and the corresponding enantioenriched FOLs (3ga–oa) were obtained in 77–87% yields. For example, besides the simple phenyl group (3ga), other functionalized aryl groups with various substitution including methyl (3ha–ja), electron-withdrawing trifluoromethyl (3la), electron-donating methoxy (3ma), fluoro (3na) and chloro (3oa) groups were tolerated. Notably, secondary benzyl alcohols with a heteroaryl motif were also suitable substrates to deliver corresponding products in moderate yields (3pa and 3qa). Finally, we probed the scope of ortho-substituent of the aryl bromide moiety of racemic alcohols, and found sterically hindered isopropyl (3ra), phenyl (3sa), methoxy (3ta and 3xa), chloro (3ua), morpholino-methyl (3wa) and naphthyl (3ya) were well tolerated (3ra–ya), delivering the desired products in good yields (60–89%). In particular, the protocol was applicable for densely functionalized secondary ortho-bromobenzyl alcohol, providing polysubstituted chiral FOL 3xa in 64% yield. Similar to Table 2, all the products in Table 3 were obtained with constantly excellent enantioselectivities (93–98% e.e.s). The absolute configuration of 3ha was unambiguously confirmed to be (S) by X-ray crystallographic analysis and those of other products were assigned by analogy.
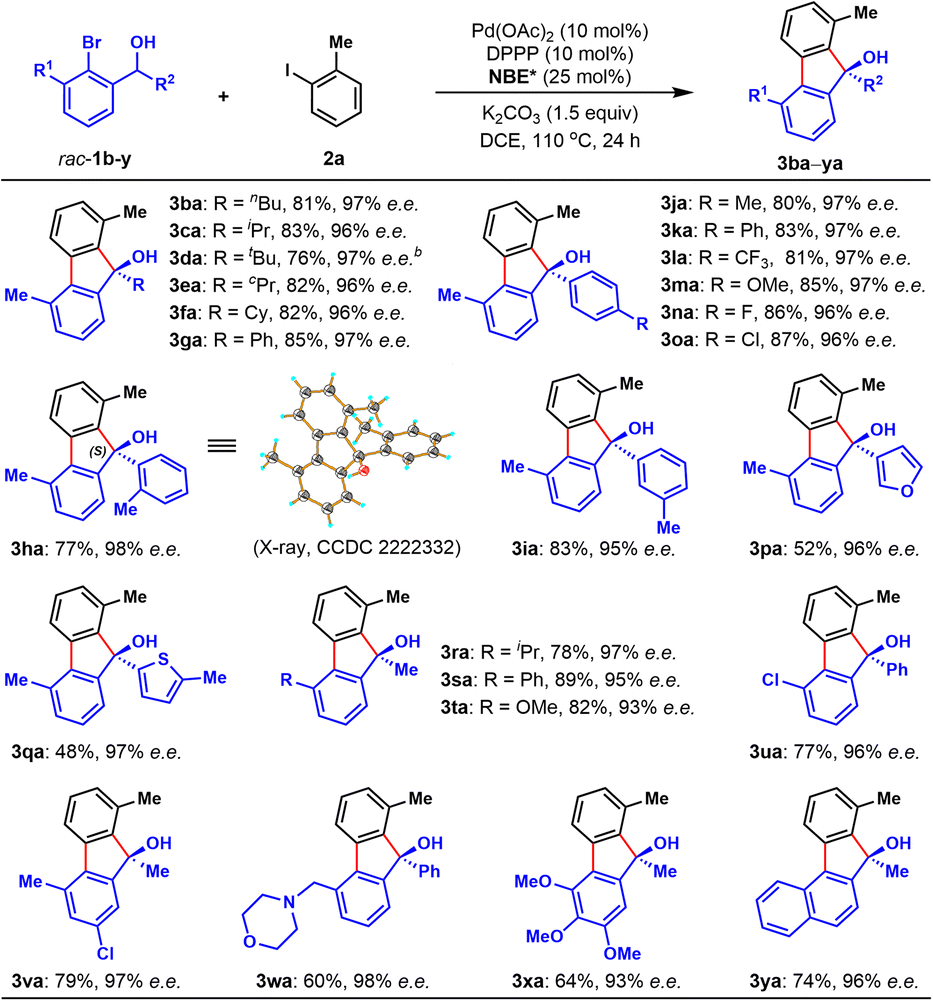
To demonstrate the synthetic practicality of this method, a larger scale (4.3 mmol) reaction was performed, which proceeded smoothly to give 1.09 gram of the desired product 3oa in 79% yield and 98% e.e. (Scheme 1a), alongside the recovery of 64% of NBE* (99% e.e.) after work-up. Furthermore, based on this method, we developed an intriguing desymmetrization strategy possessing the ability to generate three chiral products in just one operation (Scheme 1b). For instance, by using readily available symmetric secondary dialcohols meso-1z as the reactant, the reaction with 2a under standard conditions delivered three separable chiral products (R,S)-3za (20%, 92% e.e.), (S,S)-3za (14%, >99% e.e.) and 3Aa (43%, 89% e.e.) (Scheme 1b, eqn (1)). The structure of (R,S)-3za was unambiguously confirmed by X-ray crystallographic analysis. The ratio of (R,S)-3za, (S,S)-3za and 3Aa can be simply tuned by the reaction time (see Fig. S1b in ESI† for details). Similar results were obtained from the reaction of racemic secondary dialcohols rac-1z′ with 2a under standard conditions (Scheme 1b, eqn (2)). We reasoned that both meso-1z and rac-1z′ could be partially oxidized to generate the same intermediate rac-1A, which was the real arylating reagent to react with 2a. Interestingly, a control experiment involving the reaction of fully oxidized symmetric diketone intermediate 1A′ with 2a resulted in almost racemic product 3Aa in a poor yield (Scheme 1c), which indicated that product 3Aa of Scheme 1b was mainly formed through in situ oxidation of the preformed products (R,S)-3za and (S,S)-3za.
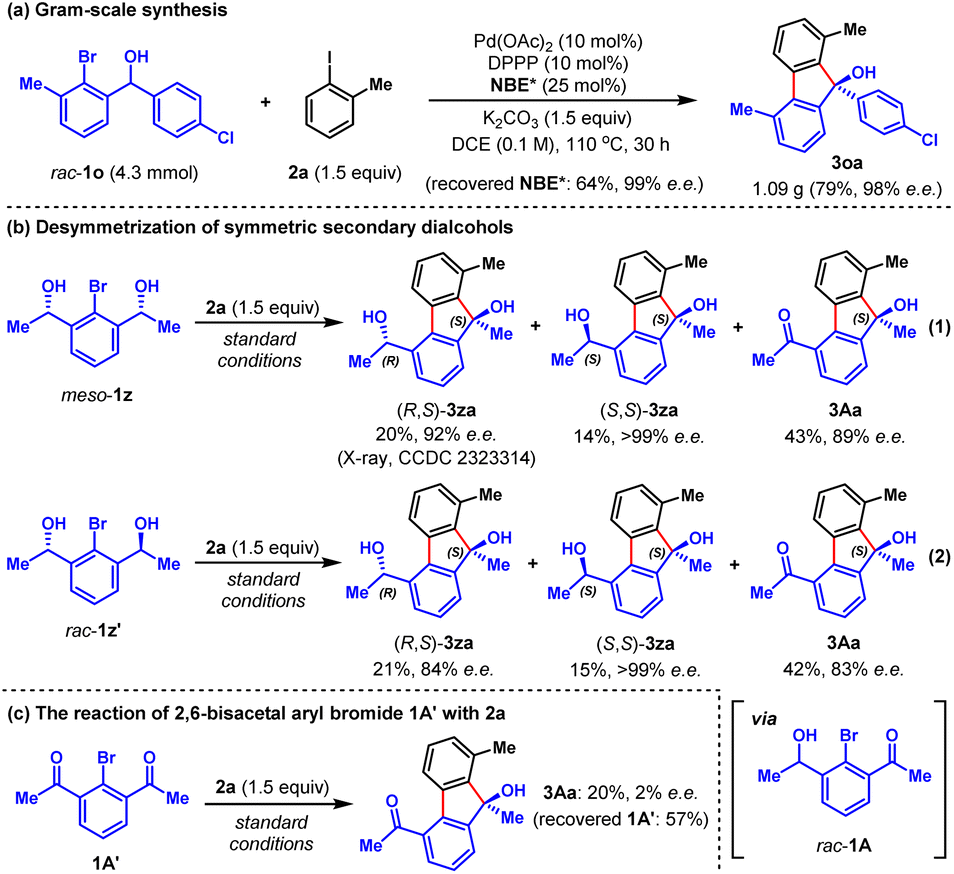 | ||
| Scheme 1 (a) Gram-scale synthesis; (b) desymmetrization of symmetric secondary dialcohols; (c) the reaction of 2,6-bisacetal aryl bromide with 2a. | ||
Lastly, to probe the reaction mechanism9b,23 and the origin of enantioselectivity, density functional theory (DFT) calculations were performed. As revealed in Fig. 2a (the detailed free energy profiles of the generation of both (S)- and (R)-3aa are included in Fig. S3 and S4†), the reductive elimination step for ortho-C–H arylation is the stereoselectivity-determining step of this reaction. The corresponding transition state TS2′ leading to (R)-3aa is disfavored because of the steric repulsions between the methyl substituent and the NBE* fragment (Fig. 2b). In contrast, such steric repulsions are absent in the favored transition state TS2, which leads to (S)-3aavia a stereospecific axial-to-central chirality transfer process. The free energy difference between the two competing reductive elimination processes is calculated to be 7.5 kcal mol−1. These DFT calculations are in good agreement with the observed experimental results and the determined absolute configuration of 3ha by X-ray crystallographic analysis. Based on these experimental and calculated results, a possible catalytic cycle for this reaction was proposed in Fig. S5.†
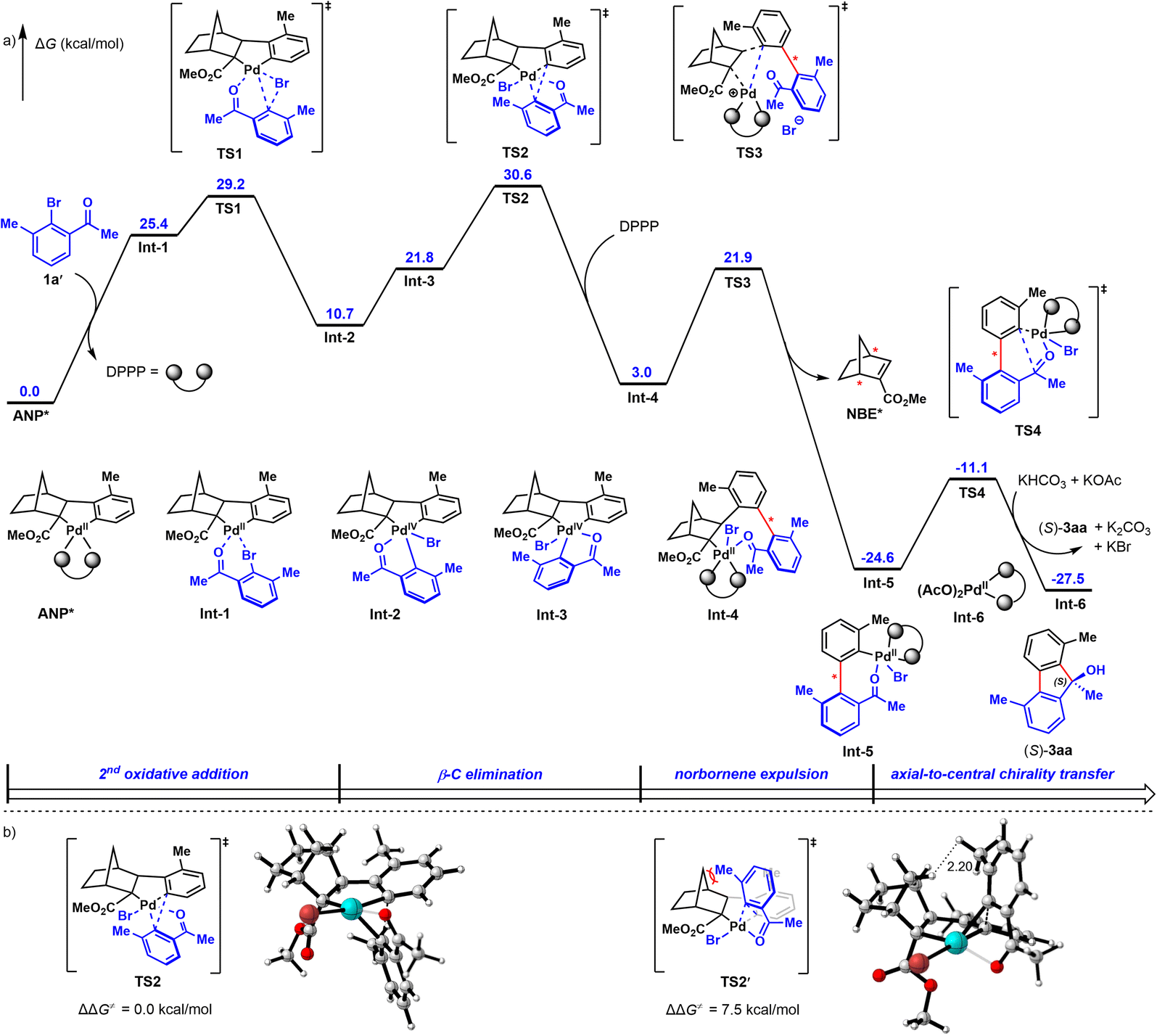 | ||
| Fig. 2 (a) DFT-computed free energy profile for the formations of (S)-3aavia the computational method of M06L/6-311+G(d,p)-SDD/SMD(DCE)//B3LYP-D3(BJ)/6-31G(d)-LANL2DZ. (b) The optimized structures and relative free energies of the transition states of the stereoselectivity-determining reductive elimination step. | ||
Conclusions
In summary, we have developed an enantioconvergent strategy for the synthesis of enantioenriched fluorenols from racemic secondary benzyl alcohols and aryl iodides via Pd(II)/chiral norbornene cooperative catalysis. This is an overall redox-neutral transformation involving sequential steps of destroying and regenerating stereoelements. A wide range of readily available aryl iodides and racemic secondary benzyl alcohols are compatible with this protocol, affording a diverse of chiral fluorenols bearing various functional groups with constantly excellent enantioselectivities (49 examples, 90–99% e.e.s). Based on this method, an intriguing desymmetrization strategy possessing the ability to generate three chiral fluorenols in one operation is developed. This work provides a valuable addition to the toolbox of enantioconvergent transformations. Preliminary DFT calculations were carried out to elucidate the reaction mechanism and the origin of enantioselectivity. Lastly, we believe this work will also promote the research and application of chiral fluorenols.Data availability
All experimental procedures, characterisation data, mechanistic investigations, NMR spectra and HPLC spectra can be found in the ESI.†Author contributions
Q. Z. conceived the idea and directed the project. B. D., Q. X., H. W., J. C., Z.-S. L. and J. T. performed the experiments under the supervision of H.-G. C. and Q. Z. Q. X. performed the density functional theory calculations. H. C. performed the X-ray crystallographic analysis. B. D. and Q. Z. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2022YFA1503703) and National Natural Science Foundation of China (No. 22325106). The DFT calculations in this paper have been done on the supercomputing system in the Supercomputing Center of Wuhan University.Notes and references
- (a) Y. Gao, G. Hong, B.-M. Yang and Y. Zhao, Chem. Soc. Rev., 2023, 52, 5541–5562 RSC; (b) S. Bähn, S. Imm, L. Neubert, M. J. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1853–1864 CrossRef; (c) S. Thiyagarajan and C. Gunanathan, J. Am. Chem. Soc., 2019, 141, 3822–3827 CrossRef CAS PubMed.
- (a) Y. Cai and S.-L. Shi, J. Am. Chem. Soc., 2021, 143, 11963–11968 CrossRef CAS PubMed; (b) D.-Y. Yang, H. Wang and C.-R. Chang, Adv. Synth. Catal., 2022, 364, 3100–3121 CrossRef CAS.
- Deracemization is used in this context in its strict sense. For selected reviews on deracemization of secondary alcohols, see: (a) C. C. Gruber, I. Lavandera, K. Faber and W. Kroutil, Adv. Synth. Catal., 2006, 348, 1789–1805 CrossRef CAS; (b) C. V. Voss, C. C. Gruber and W. Kroutil, Synlett, 2010, 2010, 991–998 CrossRef; (c) C. Aranda, G. Oksdath-Mansilla, F. R. Bisogno and G. de Gonzalo, Adv. Synth. Catal., 2020, 362, 1233–1257 CrossRef CAS; (d) M. Huang, T. Pan, X. Jiang and S. Luo, J. Am. Chem. Soc., 2023, 145, 10917–10929 CrossRef CAS PubMed; (e) K.-K. Qiao, G.-S. Feng and L. Shi, J. Catal., 2023, 422, 99–116 CrossRef CAS.
- For selected examples on deracemization of secondary alcohols, see: (a) C. V. Voss, C. C. Gruber and W. Kroutil, Angew. Chem., Int. Ed., 2008, 47, 741–745 CrossRef CAS PubMed; (b) C. V. Voss, C. C. Gruber, K. Faber, T. Knaus, P. Macheroux and W. Kroutil, J. Am. Chem. Soc., 2008, 130, 13969–13972 CrossRef CAS PubMed; (c) C. Magallanes-Noguera, M. M. Ferrari, M. Kurina-Sanz and A. A. Orden, J. Biotechnol., 2012, 160, 189–194 CrossRef CAS PubMed; (d) I. Karume, M. Takahashi, S. M. Hamdan and M. M. Musa, ChemCatChem, 2016, 8, 1459–1463 CrossRef CAS; (e) P. Qu, M. Kuepfert, S. Jockusch and M. Weck, ACS Catal., 2019, 9, 2701–2706 CrossRef CAS; (f) B. Yang, P. Cui, Y. Chen, Q. Liu and H. Zhou, Tetrahedron Lett., 2020, 61, 152530–152532 CrossRef CAS; (g) Z. Zhao, C. Wang, Q. Chen, Y. Wang, R. Xiao, C. Tan and G. Liu, ChemCatChem, 2021, 13, 4055–4063 CrossRef CAS; (h) Z. Zhang and X. Hu, Angew. Chem., Int. Ed., 2021, 60, 22833–22838 CrossRef CAS PubMed; (i) L. Wen, J. Ding, L. Duan, S. Wang, Q. An, H. Wang and Z. Zuo, Science, 2023, 382, 458–464 CrossRef CAS PubMed.
- For selected recent examples, see: (a) Y. Liu, R. Tao, Z.-K. Lin, G. Yang and Y. Zhao, Nat. Commun., 2021, 12, 5035 CrossRef CAS PubMed; (b) F. Li, L. Long, Y.-M. He, Z. Li, H. Chen and Q.-H. Fan, Angew. Chem., Int. Ed., 2022, 61, e202202972 CrossRef CAS PubMed; (c) X. Zhang, W. Ma, J. Zhang, W. Tang, D. Xue, J. Xiao, H. Sun and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202203244 CrossRef CAS PubMed; (d) X. Chang, X. Cheng, X.-T. Liu, C. Fu, W.-Y. Wang and C.-J. Wang, Angew. Chem., Int. Ed., 2022, 61, e202206517 CrossRef CAS PubMed; (e) W. Miao, J. Zhang, Y. Yang, W. Tang, D. Xue, J. Xiao, H. Sun and C. Wang, Angew. Chem., Int. Ed., 2023, 62, e202306015 CrossRef CAS PubMed; (f) X. Q. Ng, C. S. Lim, M. W. Liaw, T. T. Quach, B.-M. Yang, V. Isoni, J. Wu and Y. Zhao, Nat. Synth., 2023, 2, 572–580 CrossRef; (g) Y. Liu, H. Diao, G. Hong, J. Edward, T. Zhang, G. Yang, B.-M. Yang and Y. Zhao, J. Am. Chem. Soc., 2023, 145, 5007–5016 CrossRef CAS PubMed.
- For selected reviews on kinetic resolution of secondary alcohols, see: (a) S. France, D. J. Guerin, S. J. Miller and T. Lectka, Chem. Rev., 2003, 103, 2985–3012 CrossRef CAS PubMed; (b) C. E. Müller and P. R. Schreiner, Angew. Chem., Int. Ed., 2011, 50, 6012–6042 CrossRef PubMed; (c) H. Pellissier, Adv. Synth. Catal., 2011, 353, 1613–1666 CrossRef CAS; (d) I. Ahmad, Shagufta and A. R. AlMallah, Chirality, 2017, 29, 798–810 CrossRef CAS PubMed; (e) H. Pellissier, Tetrahedron, 2018, 74, 3459–3468 CrossRef CAS; (f) Z. Wang, D. Pan, T. Li and Z. Jin, Chem.–Asian J., 2018, 13, 2149–2163 CrossRef CAS PubMed; (g) S. Chen, Y.-H. Shi and M. Wang, Chem.–Asian J., 2018, 13, 2184–2194 CrossRef CAS PubMed; (h) H. Yang and W.-H. Zheng, Tetrahedron Lett., 2018, 59, 583–591 CrossRef CAS; (i) W. Liu and X. Yang, Asian J. Org. Chem., 2021, 10, 692–710 CrossRef CAS.
- M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed., 1997, 36, 119–122 CrossRef CAS.
- For selected reviews, see: (a) J. Ye and M. Lautens, Nat. Chem., 2015, 7, 863–870 CrossRef CAS PubMed; (b) H. Zhu, C. Ye and Z. Chen, Chin. J. Org. Chem., 2015, 35, 2291–2300 CrossRef CAS; (c) N. Della Ca', M. Fontana, E. Motti and M. Catellani, Acc. Chem. Res., 2016, 49, 1389–1400 CrossRef PubMed; (d) J. Wang and G. Dong, Chem. Rev., 2019, 119, 7478–7528 CrossRef CAS PubMed; (e) R. Li and G. Dong, J. Am. Chem. Soc., 2020, 142, 17859–17875 CrossRef CAS PubMed; (f) S. Dong and X. Luan, Chin. J. Chem., 2021, 39, 1690–1705 CrossRef CAS; (g) H.-G. Cheng, S. Jia and Q. Zhou, Acc. Chem. Res., 2023, 56, 573–591 CrossRef CAS PubMed.
- (a) Y.-B. Zhao, B. Mariampillai, D. A. Candito, B. Laleu, M. Li and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 1849–1852 CrossRef CAS PubMed; (b) A. Casnati, M. Fontana, E. Motti and N. Della Ca', Org. Biomol. Chem., 2019, 17, 6165–6173 RSC.
- (a) K. Zhao, S. Xu, C. Pan, X. Sui and Z. Gu, Org. Lett., 2016, 18, 3782–3785 CrossRef CAS PubMed; (b) L. Ding, X. Sui and Z. Gu, ACS Catal., 2018, 8, 5630–5635 CrossRef CAS; (c) Z.-S. Liu, G. Qian, Q. Gao, P. Wang, H.-G. Cheng, Q. Wei, Q. Liu and Q. Zhou, ACS Catal., 2018, 8, 4783–4788 CrossRef CAS; (d) Q. Gao, Z.-S. Liu, Y. Hua, L. Li, H.-G. Cheng, H. Cong and Q. Zhou, Chem. Commun., 2019, 55, 8816–8819 RSC; (e) Z. Zhang, B.-S. Zhang, K.-L. Li, Y. An, C. Liu, X.-Y. Gou and Y.-M. Liang, J. Org. Chem., 2020, 85, 7817–7839 CrossRef CAS PubMed; (f) X.-M. Chen, L. Zhu, D.-F. Chen and L.-Z. Gong, Angew. Chem., Int. Ed., 2021, 60, 24844–24848 CrossRef CAS PubMed; (g) L. Zhou, H.-G. Cheng, L. Li, K. Wu, J. Hou, C. Jiao, S. Deng, Z. Liu, J.-Q. Yu and Q. Zhou, Nat. Chem., 2023, 15, 815–823 CrossRef CAS PubMed.
- (a) H. Shi, A. N. Herron, Y. Shao, Q. Shao and J.-Q. Yu, Nature, 2018, 558, 581–586 CrossRef CAS PubMed; (b) J.-J. Li, J.-H. Zhao, H.-C. Shen, K. Wu, X. Kuang, P. Wang and J.-Q. Yu, Chem, 2023, 9, 1452–1463 CrossRef CAS.
- R. Li, F. Liu and G. Dong, Org. Chem. Front., 2018, 5, 3108–3112 RSC.
- Q. Feng, X. Ma, W. Bao, S.-J. Li, Y. Lan and Q. Song, CCS Chem., 2021, 3, 377–387 CrossRef CAS.
- Y. An, X.-Y. Zhang, Y.-N. Ding, Y. Li, X.-Y. Liu and Y.-M. Liang, Org. Lett., 2022, 24, 7294–7299 CrossRef CAS PubMed.
- (a) Z.-S. Liu, Y. Hua, Q. Gao, Y. Ma, H. Tang, Y. Shang, H.-G. Cheng and Q. Zhou, Nat. Catal., 2020, 3, 727–733 CrossRef CAS; (b) Q. Gao, C. Wu, S. Deng, L. Li, Z.-S. Liu, Y. Hua, J. Ye, C. Liu, H.-G. Cheng, H. Cong, Y. Jiao and Q. Zhou, J. Am. Chem. Soc., 2021, 143, 7253–7260 CrossRef CAS PubMed; (c) Z.-S. Liu, P.-P. Xie, Y. Hua, C. Wu, Y. Ma, J. Chen, H.-G. Cheng, X. Hong and Q. Zhou, Chem, 2021, 7, 1917–1932 CrossRef CAS; (d) Y. Hua, Z.-S. Liu, P.-P. Xie, B. Ding, H.-G. Cheng, X. Hong and Q. Zhou, Angew. Chem., Int. Ed., 2021, 60, 12824–12828 CrossRef CAS PubMed; (e) J. Ye, L. Li, Y. You, C. Jiao, Z. Cui, Y. Zhang, S. Jia, H. Cong, S. Liu, H.-G. Cheng and Q. Zhou, JACS Au, 2023, 3, 384–390 CrossRef CAS PubMed; (f) Z.-S. Liu, S. Deng, Q. Gao, Y. Hua, H.-G. Cheng, X. Qi and Q. Zhou, ACS Catal., 2023, 13, 2968–2980 CrossRef CAS; (g) C. Wu, Z.-S. Liu, Y. Shang, C. Liu, S. Deng, H.-G. Cheng, H. Cong, Y. Jiao and Q. Zhou, Chin. J. Chem., 2024, 42, 699–704 CrossRef CAS.
- J. Muzart, Tetrahedron, 2003, 59, 5789–5816 CrossRef CAS.
- (a) J. A. McCubbin, X. Tong, R. Wang, Y. Zhao, V. Snieckus and R. P. Lemieux, J. Am. Chem. Soc., 2004, 126, 1161–1167 CrossRef CAS PubMed; (b) J. Wang, W. Wan, H. Jiang, Y. Gao, X. Jiang, H. Lin, W. Zhao and J. Hao, Org. Lett., 2010, 12, 3874–3877 CrossRef CAS PubMed; (c) A. Shimizu and Y. Tobe, Angew. Chem., Int. Ed., 2011, 50, 6906–6910 CrossRef CAS PubMed; (d) R. S. Upadhayaya, P. D. Shinde, S. A. Kadam, A. N. Bawane, A. Y. Sayyed, R. A. Kardile, P. N. Gitay, S. V. Lahore, S. S. Dixit, A. Földesi and J. Chattopadhyaya, Eur. J. Med. Chem., 2011, 46, 1306–1324 CrossRef CAS PubMed; (e) I. V. Kurdyukova and A. A. Ishchenko, Russ. Chem. Rev., 2012, 81, 258–290 CrossRef CAS; (f) D. Dunn, G. Hostetler, M. Iqbal, V. R. Marcy, Y. G. Lin, B. Jones, L. D. Aimone, J. Gruner, M. A. Ator, E. R. Bacon and S. Chatterjee, Bioorg. Med. Chem. Lett., 2012, 22, 3751–3753 CrossRef CAS PubMed; (g) R. S. Upadhayaya, S. S. Dixit, A. Földesi and J. Chattopadhyaya, Bioorg. Med. Chem. Lett., 2013, 23, 2750–2758 CrossRef CAS PubMed; (h) X. Sun, Q. Xue, Z. Zhu, Q. Xiao, K. Jiang, H.-L. Yip, H. Yan and Z. Li, Chem. Sci., 2018, 9, 2698–2704 RSC.
- For selected examples, see: (a) L. Liu and J. Zhang, Angew. Chem., Int. Ed., 2009, 48, 6093–6096 CrossRef CAS PubMed; (b) Z. Chen, M. Zeng, J. Yuan, Q. Yang and Y. Peng, Org. Lett., 2012, 14, 3588–3591 CrossRef CAS PubMed; (c) M. Itoh, K. Hirano, T. Satoh, Y. Shibata, K. Tanaka and M. Miura, J. Org. Chem., 2013, 78, 1365–1370 CrossRef CAS PubMed; (d) Y. Kurimoto, K. Mitsudo and S. Suga, Chem. Lett., 2021, 50, 378–381 CrossRef CAS; (e) X. Zhao, X. Yu, M. Liu, Y. Huo, S. Ji, X. Li and Q. Chen, J. Org. Chem., 2023, 88, 2612–2620 CrossRef CAS PubMed; (f) B. S. Gore, L.-W. Pan, J.-H. Lin, Y.-C. Luo and J.-J. Wang, Green Chem., 2024, 26, 513–519 RSC.
- (a) Z. Chao, N. Li, B. Hong, M. Ma and Z. Gu, Org. Lett., 2021, 23, 7759–7764 CrossRef CAS PubMed; (b) J. Xi, H. Yang, L. Li, X. Zhang, C. Li and Z. Gu, Org. Lett., 2022, 24, 2387–2392 CrossRef CAS PubMed.
- E. Motti, N. Della Ca’, D. Xu, A. Piersimoni, E. Bedogni, Z.-M. Zhou and M. Catellani, Org. Lett., 2012, 14, 5792–5795 CrossRef CAS PubMed.
- (a) S. Deledda, E. Motti and M. Catellani, Can. J. Chem., 2005, 83, 741–747 CrossRef CAS; (b) A. Martins, D. A. Candito and M. Lautens, Org. Lett., 2010, 12, 5186–5188 CrossRef CAS PubMed.
- (a) M.-N. Birkholz, Z. Freixa and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099–1118 RSC; (b) Z. Dong, G. Lu, J. Wang, P. Liu and G. Dong, J. Am. Chem. Soc., 2018, 140, 8551–8562 CrossRef CAS PubMed.
- (a) G. Maestri, E. Motti, N. Della Ca', M. Malacria, E. Derat and M. Catellani, J. Am. Chem. Soc., 2011, 133, 8574–8585 CrossRef CAS PubMed; (b) D. I. Chai, P. Thansandote and M. Lautens, Chem. - Eur. J., 2011, 17, 8175–8188 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2222332 and 2323314. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01004c |
| ‡ These authors contributed equally: Han Wei and Jiangwei Chen. |
| This journal is © The Royal Society of Chemistry 2024 |