
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile modular synthesis of jasmonoyl-L-isoleucine analogs possessing a pyrazolidin-3-one core†
Samuel Vizcaíno Páez ab,
Diego Durangob,
Christian Jürgen Müllerc,
Matthias Breuning*c and
Wiston Quiñones Fletcher*a
ab,
Diego Durangob,
Christian Jürgen Müllerc,
Matthias Breuning*c and
Wiston Quiñones Fletcher*a
aQuímica Orgánica de Productos Naturales, Universidad de Antioquia, Medellín, 050010, Antioquia, Colombia. E-mail: wiston.quinones@udea.edu.co
bQuímica de los Productos Naturales y los Alimentos, Universidad Nacional de Colombia, Medellín, 050034, Antioquia, Colombia
cDepartment of Chemistry, University of Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany. E-mail: matthias.breuning@uni-bayreuth.de
First published on 25th January 2024
Abstract
A short and flexible route to pyrazolidin-3-one analogs of the phytohormone (+)-7-iso-jasmonoyl-L-isoleucine is presented. The compounds were assembled from four basic building blocks, namely a pyrazolidin-3-one core, alkyl chain, linker and amino ester or acid. The efficacy of this approach was demonstrated in the synthesis of 11 analogs with variations in all parts of the molecule.
Introduction
The jasmonates (JAs), which constitute a family of more than 30 secondary metabolites derived from jasmonic acid (1) (Fig. 1), fulfill multiple physiological roles throughout the plant kingdom.1 The most remarkable molecule from this group is (+)-7-iso-jasmonoyl-L-isoleucine (JA-Ile, 2), identified as the cornerstone for plant responses to biotic stress.2 Other jasmonate-induced physiological responses observed in higher plants include inhibition of cytokinin-induced growth,3 stimulation of ethylene production, induction of proteinase inhibitor II,4,5 tuber inducing,6 accumulation of secondary metabolites,7 tendril coiling,8 odor production,9,10 JIP-23 gene expression,11 phytoalexin production,12 and stomatal opening.13 The importance of these jasmonate-induced responses has triggered the interest in developing synthetic routes to JA analogs.14 The prepared analogs permitted structure activity relationship (SAR) studies through which the main structural features required to keep or to potentiate such activities were identified. SAR studies have been done on activities in plants such as tendril coiling15 or secondary metabolites accumulation,16 and even in humans, such as anti-inflammatory,17 neoplastic,18 or anticancer,19 because of the structural similarity of these compounds to prostaglandins in mammals. Notwithstanding, all existing routes to JAs analogs are mainly linear, including de novo syntheses20–22 and partial syntheses starting from commercially available methyl jasmonate or cis-jasmone.23–25 In an interesting approach to prostaglandin E2 analogs, the cyclopentanone moiety was replaced by a pyrazolidin-3-one, yielding several potent and selective in vitro EP2 and EP4 receptor agonists.26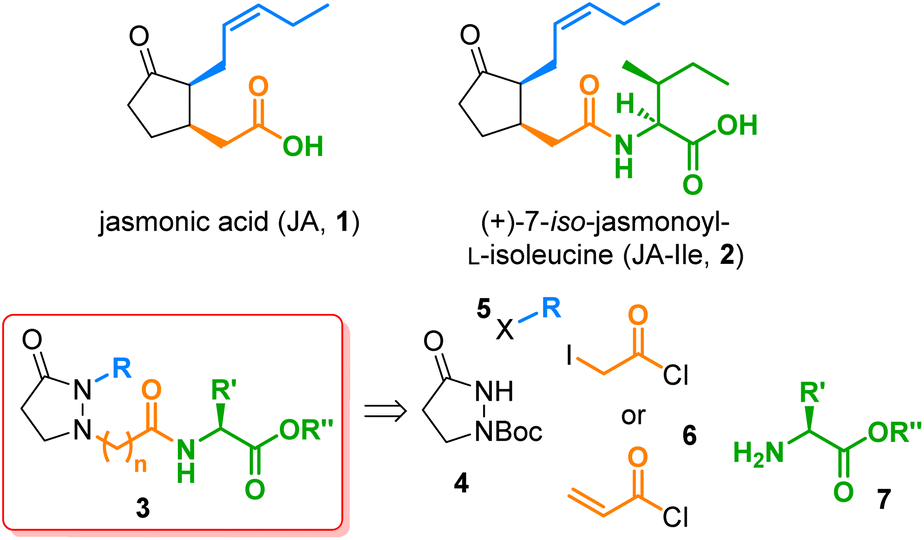 | ||
| Fig. 1 The phytohormones jasmonic acid (1) and (+)-7-iso-jasmonoyl-L-isoleucine (JA-Ile, 2), the new pyrazolidin-3-one based JA-Ile analogs 3, and their four building blocks 4–7. | ||
A similar substitution in the field of JA-Ile (2), viz. the replacement of the chiral cyclopentanone core by a simple, achiral pyrazolidine-3-one, would lead to analogs of type 3, which should be by far more easily accessible since all stereocenters are omitted and the nitrogen atoms provide versatile attachment points for side chains. Consequently, a quick preparation of a structurally broad library of JA-Ile derivatives, as required for in-depth SAR studies, would be possible. In this work, we realized this idea and synthesized a couple of JA-Ile analogs 3. We developed a convergent route based on four different building blocks, the pyrazolidin-3-one core 4, the alkyl side chain 5, the acid linker 6, and the terminal amino acid 7. This strategy is highly efficient and permits access to a wide range of structural modifications, because all building blocks can be varied independently and prior to the final coupling stage.
Results and discussion
The syntheses of the core building blocks 10 commenced with methyl acrylate (8), which was cyclized with hydrazine hydrate. After N-Boc protection of the amino function, which is required in order to direct the following alkylation to the less nucleophilic amide nitrogen, the pyrazolidin-3-one 4 was obtained in good 72% yield. Deprotonation of the amide with NaH in DMF at 0 °C and subsequent electrophilic quench with (Z)-pent-2-en-1-yl methanesulfonate (5a), 1-bromopentane (5b), 1-bromohexane (5c), and 1-bromobutane (5d), respectively, introduced the first variable substituent R and provided the protected pyrazolidin-3-ones 9a–d in reasonable yields (55–63%). As side products, minor quantities (<15%) of the corresponding O-alkylated imidates were observed, which was characterized in the case of that formed together with 9b. We also explored an alternative route to 9 based on an inversed reaction sequence, namely the alkylation of tert-butyl carbazate followed by cyclization with 3-chloropropionyl chloride or ethyl acrylate (8),26 which, however, was met with low success and provided only traces of 9a. Final deprotection of 9a–d with TFA afforded the core building blocks 10a–d in excellent yield (>97%) and overall just four steps from methyl acrylate (Table 1).
|
|||
|---|---|---|---|
| Entry | R | Cmpdb (Yield [%]) | Cmpdb (Yield [%]) |
| a Reaction conditions: (i) 1. H2NNH2·H2O, mol sieves 3 Å, EtOH, 0 °C to reflux, 22.5 h; 2. Boc2O, MeOH, rt, 5 h, 72%; (ii) NaH, DMF, 0 °C, 30 min, then R–X (5a–d), rt, 18 h; (iii) TFA, CHCl3, rt, 18 h.b Isolated yields. | |||
| 1 | (Z)-2-Pentenyl | 9a (55) | 10a (99) |
| 2 | nPent | 9b (67) | 10b (99) |
| 3 | nHex | 9c (57) | 10c (99) |
| 4 | nBu | 9d (63) | 10d (97) |

The side chain connected to the amino group of 3 consists of an acid linker and an amino acid, which both were independently varied. In a first set, the methyl esters of L-isoleucine (7a), L-leucine (7b), L-valine (7c), and L-alanine (7d), which all possess aliphatic and hydrophobic side chains as found in JA-Ile (2), were chosen as models for the amino acid part. Note that the esters are eligible bio-precursors for the corresponding amino acids in the final products because saponification will readily occur in a living environment.2 According to a known procedure,27,28 7a–d were treated with chloroacetyl chloride (11) and NEt3, which introduced the ‘natural’ two-carbon linker of JA-Ile (2) and furnished the known chlorides 12aa–ad in excellent yields above 90%. It should be stressed that, under slightly modified conditions,29 this procedure is also applicable to unprotected amino acids. The chloride 12ae was obtained in good 80% yield from 11 and two equivalents of L-isoleucine (7e) in MeCN at −20 °C. Cl/I-exchange in 12aa–ae under Finkelstein conditions, in order to improve the quality of the leaving group for the final coupling, delivered the desired iodides 6aa–6ae in high 74–94% yield. Note that compounds of type 6ae and 12ae, which possess a free acid group and a leaving group like chloride or iodine, are prone to undergo intramolecular cyclization to morpholin-2,5-diones.30 In our case, however, only traces of such by-products were observed. Finally, a precursor for a side chain with a longer three-carbon linker was prepared, too. Treatment of acryloyl chloride (13) with L-isoleucine methyl ester (7a) furnished the enamide 6ba in 77% yield (Table 2).
|
||||
|---|---|---|---|---|
| Entry | Cmpd | R′b | R′′ | Yieldc [%] |
| a Reaction conditions: (i) for 12aa–ad and 6ba: Et3N, CHCl3, 0 °C, 1 h, 62–96%.31,32 for 12ae: Et3N, MeCN, −20 °C, 48 h, 92%; (ii) KI, acetone, rt, 24 h.b In brackets: corresponding L-amino acid.c Isolated yields. | ||||
| 1 | 6aa | sBu (Ile) | Me | 80 |
| 2 | 6ab | iBu (Leu) | Me | 94 |
| 3 | 6ac | iPr (Val) | Me | 74 |
| 4 | 6ad | Me (Ala) | Me | 77 |
| 5 | 6ae | sBu (Ile) | H | 89 |
| 6 | 6ba | sBu (Ile) | Me | 77 |

The final coupling step between the pyrazolidin-3-one cores 10a–d and the side chains 6aa–ad occurred smoothly under SN2-conditions (Cs2CO3, DMF) at room temperature, giving the desired JA-Ile analogs 3 in good to high yields (48–83%). Mol sieves 4 Å was added to remove traces of water, as formed by the carbonate, to prevent any yield-lowering solvolysis of the iodide. An aza-Michael addition was intended for the linkage of the α,β-unsaturated amide 6ba to the core 10a. Under standard conditions with MeOH as the solvent, only an oxo-Michael addition of MeOH was observed. We therefore changed to more forcing conditions in a polar, but low-nucleophilic solvent (TFE/H2O 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 100 °C),31,32 under which the desired aza-Michael product 3aba was formed in acceptable 58% yield (Table 3).
:1, 100 °C),31,32 under which the desired aza-Michael product 3aba was formed in acceptable 58% yield (Table 3).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cmpd | R | n | R′ | R′′ | Yieldb [%] |
| a Reaction conditions: (i) for 6aa–ad: Cs2CO3, DMF, mol sieves 4 Å, rt, 16–48 h; for 6ba: TFE/H2O 1:1, 100 °C, 24 h.b Isolated yields. |
||||||
| 1 | 3aaa | (Z)-2-Pentenyl | 1 | sBu | Me | 70 |
| 2 | 3aab | (Z)-2-Pentenyl | 1 | iBu | Me | 63 |
| 3 | 3aac | (Z)-2-Pentenyl | 1 | iPr | Me | 77 |
| 4 | 3aad | (Z)-2-Pentenyl | 1 | Me | Me | 48 |
| 5 | 3aae | (Z)-2-Pentenyl | 1 | sBu | H | 77 |
| 6 | 3baa | nPent | 1 | sBu | Me | 83 |
| 7 | 3caa | nHex | 1 | sBu | Me | 66 |
| 8 | 3daa | nBu | 1 | sBu | Me | 74 |
| 9 | 3aba | (Z)-2-Pentenyl | 2 | sBu | Me | 58 |
| 10 | 3bab | nPent | 1 | iBu | Me | 52 |

Conclusions
A modular and convergent strategy for the synthesis of pyrazolidin-3-one analogs 3 of the key phytohormone involved in the plant chemical responses to biotic or abiotic stress JA-Ile has been developed. The four basic building blocks, the achiral pyrazolidin-3-one core 4, the alkyl side chain 5, the acid linker 6, and the terminal amino acid ester 7, can be independently varied and combined without restrictions, thus permitting a facile access to a structurally broad array of compounds in just a few steps. The high potential and efficacy of this approach was proven in the synthesis of eleven different JA-Ile analogs 3, which include variations at all possible positions. Any epimerization in the amino acid side chain was excluded. By using this approach, large libraries of JA-Ile analogs 3 are now easily accessible, which can be used for in-depth SAR studies on JA (1) or JA-Ile (2) triggered biological processes. Investigations in this direction are in progress.Experimental
General
All reactions with moisture-sensitive reagents were carried out under an Ar atmosphere in anhydrous solvents, which were prepared using standard procedures. Commercially available reagents were used as received. Reaction monitoring was done by thin-layer chromatography (TLC) on precoated silica gel plates (Merck TLC Silica gel 60 F254). Spots were visualized by UV light (254 nm or 366 nm) or by staining with aqueous KMnO4, vanillin, or phosphomolybdic acid (PMA). Silica gel (40–63 μm) was used for column chromatography. Melting points were measured on a Büchi M-565 melting point apparatus and are uncorrected. Optical rotations were recorded through 5 continuous repetitions on a Jasco P-2000 polarimeter (1 dm cell) at 589 nm and concentrations close to 1.0% in chloroform or methanol. Infrared spectra were recorded on a PerkinElmer Spectrum 100 FT-IR spectrometer. NMR spectra were measured on Bruker Avance III HD 300 and 500 instruments and calibrated using the residual non-deuterated solvent as an internal reference. High resolution mass spectrometry (HRMS) was performed on a ThermoFischer Scientific Q-Extractive (Orbitrap) mass spectrometer using electrospray ionization (ESI). Compounds 12aa–ad were synthesized in 92–96% yield according to literature.27,28Synthesis
:2 → 90:10) delivered product 4 (2.70 g, 14.4 mmol, 72%) as white needles. Mp. 107–110 °C. FTIR (ATR): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) max/cm−1 3301 (NH), 2978, 2933, 1686 (C
max/cm−1 3301 (NH), 2978, 2933, 1686 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).·1H NMR (500 MHz, CDCl3): δ (ppm) 8.56 (s, 1H), 3.95 (t, J = 8.6 Hz, 2H), 2.67 (t, J = 8.6 Hz, 2H), 1.49 (s, 9H). 13C NMR (125 MHz, CDCl3): δ (ppm) 170.72 (CO), 153.78 (CO), 82.75 (Cq), 44.82 (CH2), 31.09 (CH2), 28.36 (CH3).:15) provided the 9a (66.0 mg, 0.263 mmol, 55%) as a colorless oil. FTIR (ATR): max/cm−1 3009 (CCH), 2973, 2935, 2875, 1704 (CO), 1640 (CC), 1314, 1153. 1H NMR (500 MHz, CDCl3): δ (ppm) 5.66–5.58 (m, 1H), 5.36–5.28 (m, 1H), 4.34 (d, J = 7.1 Hz, 2H), 3.94 (t, J = 7.7 Hz, 2H), 2.54 (t, J = 7.7 Hz, 2H), 2.11 (pd, J = 7.5, 1.2 Hz, 2H), 1.50 (s, 9H), 0.97 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.02 (CO), 156.93 (CO), 137.16 (CH), 122.01 (CH), 82.77 (Cq), 47.80 (CH2), 42.13 (CH2), 31.44 (CH2), 28.27 (CH3), 20.86 (CH2), 14.39 (CH3). HRMS (+ESI): m/z calcd for C13H23N2O3+ [M + H]+: 255.1703; found: 255.1698.max/cm−1 2958, 2933, 2874, 1702 (CO), 1315, 1155. 1H NMR (500 MHz, CDCl3): δ (ppm) 3.95 (t, J = 7.7 Hz, 2H), 3.69 (t, J = 7.2 Hz, 2H), 2.55 (t, J = 7.7 Hz, 2H), 1.57 (p, J = 7.4 Hz, 2H), 1.50 (s, 9H), 1.38–1.28 (m, 2H), 1.28–1.20 (m, 2H), 0.88 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 171.57 (CO), 157.05 (CO), 82.83 (Cq), 47.63 (CH2), 45.18 (CH2), 31.65 (CH2), 28.99 (CH2), 28.30 (CH3), 26.16 (CH2), 22.44 (CH2), 14.11 (CH3). HRMS (+ESI): m/z calcd for C13H25N2O3+ [M + H]+: 257.1860; found: 257.1855.max/cm−1 2959, 2931, 2860, 1703 (CO), 1314, 1155. 1H NMR (500 MHz, CDCl3): δ (ppm) 3.95 (t, J = 7.7 Hz, 2H), 3.69 (t, J = 7.2 Hz, 2H), 2.55 (t, J = 7.7 Hz, 2H), 1.56 (p, J = 7.2 Hz, 2H), 1.50 (s, 9H), 1.33–1.22 (m, 6H), 0.87 (t, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 171.58 (CO), 157.06 (CO), 82.85 (Cq), 47.64 (CH2), 45.22 (CH2), 31.66 (CH2), 31.55 (CH2), 28.31 (CH3), 26.51 (CH2), 26.44 (CH2), 22.65 (CH2), 14.14 (CH3). HRMS (+ESI): m/z calcd for C14H27N2O3+ [M + H]+: 271.2016; found: 271.2010.max/cm−1 2960, 2934, 2875, 1701 (CO), 1316, 1155. 1H NMR (500 MHz, CDCl3): δ (ppm) 3.94 (t, J = 7.8 Hz, 2H), 3.68 (t, J = 7.2 Hz, 2H), 2.53 (t, J = 7.8 Hz, 2H), 1.54 (p, 7.4 Hz, 2H), 1.48 (s, 9H), 1.27 (sext, J = 7.5 Hz, 2H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 171.54 (CO), 157.01 (CO), 82.79 (Cq), 47.59 (CH2), 44.85 (CH2), 31.60 (CH2), 28.52 (CH3), 28.26 (CH2), 20.02 (CH2), 13.83 (CH3). HRMS (+ESI): m/z calcd for C12H23N2O3+ [M + H]+: 243.1703; found: 243.1699.:2.6:0.3). Traces of TFA were removed by treatment with 3 M KOH (1.0 mL), followed by extraction with EtOAc (3 × 10 mL). Evaporation of the solvent delivered analytically pure amine 10a (688 mg, 4.46 mmol, 99%) as an orange oil. FTIR (ATR): max/cm−1 3218 (NH), 3020 (CCH), 2966, 2936, 2878, 1666 (CO). 1H NMR (500 MHz, CDCl3): δ (ppm) 5.68–5.61 (m, 1H), 5.38–5.31 (m, 1H), 4.37 (s, 1H, N–H), 4.08 (d, J = 7.0 Hz, 2H), 3.37 (t, J = 7.8 Hz, 2H), 2.54 (t, J = 7.7 Hz, 2H), 2.15 (pd, J = 7.5, 1.3 Hz, 2H), 0.99 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 171.88 (CO), 137.17 (CH), 122.17 (CH), 43.83 (CH2), 40.67 (CH2), 33.35 (CH2), 20.80 (CH2), 14.27 (CH3). HRMS (+ESI): m/z calcd for C8H15N2O+ [M + H]+: 155.1179; found: 155.1175.max/cm−1 3216 (NH), 2957, 2930, 2872, 1663 (CO). 1H NMR (500 MHz, CDCl3): δ (ppm) 4.31 (s, 1H, N–H), 3.41 (t, J = 7.2 Hz, 2H), 3.36 (t, J = 7.7 Hz, 2H), 2.55 (t, J = 7.6 Hz, 2H), 1.58 (p, J = 7.4 Hz, 2H), 1.38–1.23 (m, 4H), 0.89 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.27 (CO), 44.18 (CH2), 44.12 (CH2), 33.67 (CH2), 29.07 (CH2), 27.06 (CH2), 22.46 (CH2), 14.13 (CH3). HRMS (+ESI): m/z calcd for C8H17N2O+ [M + H]+: 157.1335; found: 157.1330.max/cm−1 3215 (NH), 2959, 2929, 2858, 1663 (CO). 1H NMR (500 MHz, CDCl3): δ (ppm) 4.35 (s, 1H, N–H), 3.41 (t, J = 7.2 Hz, 2H), 3.36 (t, J = 7.7 Hz, 2H), 2.54 (t, J = 7.5 Hz, 2H), 1.57 (p, J = 7.1 Hz, 2H), 1.33–1.25 (m, 6H), 0.87 (t, J = 6.7 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.26 (CO), 44.18 (CH2), 44.15 (CH2), 33.67 (CH2), 31.58 (CH2), 27.33 (CH2), 26.59 (CH2), 22.67 (CH2), 14.15 (CH3). HRMS (+ESI): m/z calcd for C9H19N2O+ [M + H]+: 171.1492; found: 171.1487.max/cm−1 3216 (NH), 2960, 2934, 2875, 1662 (CO). 1H NMR (500 MHz, CDCl3): δ (ppm) 4.36 (s, 1H, N–H), 3.41 (t, J = 7.2 Hz, 2H), 3.35 (t, J = 7.7 Hz, 2H), 2.53 (t, J = 7.6 Hz, 2H), 1.55 (p, J = 7.4 Hz, 2H), 1.32 (sext, J = 7.5 Hz, 2H), 0.92 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.27 (CO), 44.16 (CH2), 43.82 (CH2), 33.64 (CH2), 29.40 (CH2), 20.11 (CH2), 13.84 (CH2). HRMS (+ESI): m/z calcd for C7H15N2O+ [M + H]+: 143.1179; found: 143.1175.max/cm−1 3500–2500 (COOH), 3364 (NH), 2964, 2925, 2878, 1709 (CO), 1622 (CO), 1533 (NH), 1235. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.06 (d, J = 8.4 Hz, 1H, N–H), 4.63 (dd, J = 8.6, 4.6 Hz, 1H), 4.12 (s, 2H, CH2Cl), 2.07–1.98 (m, 2H), 1.57–1.47 (m, 1H), 1.33–1.20 (m, 1H), 0.99 (d, J = 6.8 Hz, 3H), 0.97 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.85 (CO), 166.36 (CO), 56.81 (CH), 42.67 (CH2), 37.74 (CH), 25.14 (CH2), 15.58 (CH3), 11.73 (CH3). HRMS (+ESI): m/z calcd for C8H15NClO3+ [M + H]+: 210.0735; found: 208.0734.max/cm−1 3317 (NH), 2969, 2936, 2879, 1736 (CO), 1644 (CO), 1536 (NH). 1H NMR (500 MHz, CDCl3): δ (ppm) 6.49 (d, J = 7.6 Hz, 1H, N–H), 4.57 (dd, J = 8.6, 4.8 Hz, 1H), 3.75 (s, 3H, OCH3), 3.75 (d, J = 11.5 Hz, 1H), 3.70 (d, J = 11.5 Hz, 1H), 1.99–1.90 (m, 1H), 1.50–1.40 (m, 1H), 1.27–1.15 (m, 1H), 0.94 (t, J = 7.4 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.19 (CO), 166.81 (CO), 57.23 (CH), 52.43 (OCH3), 38.18 (CH), 25.22 (CH2), 15.52 (CH3), 11.73 (CH3), −0.90 (ICH2). HRMS (+ESI): m/z calcd for C9H17NIO3+ [M + H]+: 314.0248; found: 314.0243.max/cm−1 3287 (NH), 2956, 2871, 1742 (CO), 1648 (CO), 1539 (NH). 1H NMR (500 MHz, CDCl3): δ (ppm) 6.37 (d, J = 7.3 Hz, 1H, N–H), 4.61 (td, J = 8.6, 4.9 Hz, 1H), 3.75 (s, 3H, OCH3), 3.74 (d, J = 11.1 Hz, 1H, H–CHI), 3.70 (d, J = 11.5 Hz, 1H, H–CHI), 1.73–1.63 (m, 2H), 1.63–1.53 (m, 1H), 0.95 (d, J = 6.1 Hz, 3H), 0.95 (d, J = 6.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 173.26 (CO), 166.92 (CO), 57.62 (CH), 51.59 (OCH3), 41.63 (CH2), 24.97 (CH), 22.94 (CH3), 22.10 (CH3), −1.13 (ICH2). HRMS (+ESI): m/z calcd for C9H17NIO3+ [M + H]+: 314.0248; found: 314.0243.max/cm−1 3279 (NH), 2965, 2902, 2875, 1723 (CO), 1640 (CO), 1548 (NH). 1H NMR (500 MHz, CDCl3): δ (ppm) 6.49 (d, J = 7.4 Hz, 1H, N–H), 4.53 (dd, J = 8.8, 4.8 Hz, 1H), 3.76 (d, J = 11.4 Hz, 1H, H–CHI), 3.76 (s, 3H, OCH3), 3.71 (d, J = 11.4 Hz, 1H, H–CHI), 2.22 (septd, J = 6.9, 4.8 Hz, 1H), 0.96 (d, J = 6.9 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.23 (CO), 167.05 (CO), 57.82 (CH), 52.49 (OCH3), 31.65 (CH), 19.01 (CH3), 17.78 (CH3), −0.98 (ICH2). HRMS (+ESI): m/z calcd for C8H15NIO3+ [M + H]+: 300.0091; found: 300.0086.max/cm−1 3285 (NH), 2966, 2952, 2928, 1722 (CO), 1644 (CO), 1542 (NH). 1H NMR (500 MHz, CDCl3): δ (ppm) 6.58 (s, 1H, N–H), 4.57 (p, J = 7.2 Hz, 1H), 3.77 (s, 3H, OCH3), 3.71 (s, 2H, CH2I), 1.43 (d, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 173.20 (CO), 166.55 (CO), 52.80 (OCH3), 48.98 (CH), 18.27 (CH3), −1.09 (ICH2). HRMS (+ESI): m/z calcd for C6H11NIO3+ [M + H]+: 271.9778; found: 271.9776.max/cm−1 3292 (NH), 3200–2700 (COOH), 2968, 2957, 2877, 1704 (CO), 1623 (CO), 1557 (NH), 1255. 1H NMR (500 MHz, CD3OD): δ (ppm) 4.33 (d, J = 5.8 Hz, 1H), 3.86 (d, J = 9.8 Hz, 1H, H–CHI), 3.75 (d, J = 9.8 Hz, 1H, H–CHI), 1.99–1.89 (m, 1H), 1.58–1.48 (m, 1H), 1.33–1.22 (m, 1H), 0.97 (d, J = 6.9 Hz, 3H), 0.93 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CD3OD): δ (ppm) 174.47 (CO), 171.38 (CO), 58.57 (CH), 38.35 (CH), 26.03 (CH2), 16.01 (CH3), 11.76 (CH3), −2.05 (ICH2). HRMS (+ESI): m/z calcd for C8H15NIO3+ [M + H]+: 300.0091; found: 300.0089.:30) to give 6ba (154 mg, 0.770 mmol, 77%) as a pale yellow oil. [α]26D + 30.8 (c 1.1, HCCl3). FTIR (ATR): max/cm−1 3286 (NH), 2965, 2937, 2879, 1741 (CO), 1657 (CO), 1532 (NH). 1H NMR (500 MHz, CDCl3): δ (ppm) 6.31 (dd, J = 17.0, 1.3 Hz, 1H), 6.15 (dd, J = 17.0, 10.3 Hz, 1H), 6.11 (s, 1H, N–H), 5.68 (dd, J = 10.3, 1.0 Hz, 1H), 4.70 (dd, J = 8.6, 5.0 Hz, 1H), 3.75 (s, 3H, OCH3), 1.97–1.88 (m, 1H), 1.51–1.41 (m, 1H), 1.27–1.14 (m, 1H), 0.93 (t, J = 7.4 Hz, 3H), 0.91 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 172.65 (CO), 165.21 (CO), 130.55 (CH), 127.35 (CH2), 56.48 (CH), 52.32 (OMe), 38.31 (CH), 25.41 (CH2), 15.52 (CH3), 11.72 (CH3). HRMS (+ESI): m/z calcd for C10H18NO3+ [M + H]+: 200.1281; found: 200.1279.max/cm−1 3310 (NH), 3021 (CCH), 2964, 2935, 2878, 1740 (CO), 1683 (CO), 1669 (CO), 1513 (NH), 1204. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.22 (d, J = 8.9 Hz, 1H, N–H), 5.67–5.59 (m, 1H), 5.43–5.35 (m, 1H), 4.61 (dd, J = 9.0, 4.8 Hz, 1H), 4.20–3.97 (m, 2H), 3.74 (s, 3H, OCH3), 3.53 (d, J = 15.8 Hz, 1H), 3.46 (d, J = 15.9 Hz, 1H), 3.48–3.20 (m, 2H), 2.74–2.47 (m, 2H), 2.14 (pd, J = 7.5, 0.8 Hz, 2H), 1.99–1.89 (m, 1H), 1.48–1.38 (m, 1H), 1.21–1.10 (m, 1H), 0.98 (t, J = 7.5 Hz, 3H), 0.93 (t, J = 7.4 Hz, 3H), 0.92 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.19 (CO), 171.23 (CO), 168.23 (CO), 136.44 (CH), 122.75 (CH), 58.66 (CH2), 56.14 (CH), 52.32 (OCH3), 51.47 (CH2), 39.61 (CH2), 37.79 (CH), 29.03 (CH2), 25.23 (CH2), 20.85 (CH2), 15.74 (CH3), 14.21 (CH3), 11.68 (CH3). HRMS (+ESI): m/z calcd for C17H30N3O4+ [M + H]+: 340.2231; found: 340.2224.max/cm−1 3303 (NH), 3019 (CCH), 2958, 2935, 2873, 1744 (CO), 1689 (CO), 1667 (CO), 1516 (NH), 1204. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.05 (d, J = 8.9 Hz, 1H, N–H), 5.67–5.58 (m, 1H), 5.43–5.35 (m, 1H), 4.67 (td, J = 8.9, 5.0 Hz, 1H), 4.20–3.95 (m, 2H), 3.74 (s, 3H, OCH3), 3.52 (d, J = 15.8 Hz, 1H), 3.46 (d, J = 15.8 Hz, 1H), 3.47–3.20 (m, 2H), 2.75–2.46 (m, 2H), 2.12 (p, J = 7.5 Hz, 2H), 1.74–1.53 (m, 3H), 0.99 (t, J = 7.5 Hz, 3H), 0.95 (d, J = 6.3 Hz, 6H). 13C NMR (125 MHz, CDCl3): δ (ppm) 173.25 (CO), 171.29 (CO), 168.31 (CO), 136.46 (CH), 122.82 (CH), 58.72 (CH2), 52.53 (OCH3), 51.51 (CH2), 50.38 (CH), 41.46 (CH2), 39.62 (CH2), 29.06 (CH2), 25.17 (CH), 23.00 (CH3), 21.94 (CH3), 20.88 (CH2), 14.24 (CH3). HRMS (+ESI): m/z calcd for C17H30N3O4+ [M + H]+: 340.2231; found: 340.2226.max/cm−1 3308 (NH), 3020 (CCH), 2964, 2935, 2876, 1741 (CO), 1683 (CO), 1667 (CO), 1513 (NH), 1206. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.21 (d, J = 9.0 Hz, 1H, N–H), 5.67–5.60 (m, 1H), 5.43–5.36 (m, 1H), 4.57 (dd, J = 9.2, 4.7 Hz, 1H), 4.21–3.97 (m, 2H), 3.74 (s, 3H, OCH3), 3.54 (d, J = 15.8 Hz, 1H), 3.46 (d, J = 15.9 Hz, 1H), 3.47–3.20 (m, 2H), 2.74–2.47 (m, 2H), 2.22 (septd, J = 6.9, 4.9 Hz, 1H), 2.12 (pd, J = 7.5, 1.0 Hz, 2H), 0.98 (t, J = 7.5 Hz, 3H), 0.95 (d, J = 6.9 Hz, 3H), 0.90 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.21 (CO), 171.24 (CO), 168.37 (CO), 136.48 (CH), 122.75 (CH), 58.66 (CH2), 56.72 (CH), 52.39 (OCH3), 51.49 (CH2), 39.61 (CH2), 31.14 (CH), 29.04 (CH2), 20.87 (CH2), 19.23 (CH3), 17.78 (CH3), 14.24 (CH3). HRMS (+ESI): m/z calcd for C16H28N3O4+ [M + H]+: 326.2074; found: 326.2069.max/cm−1 3307 (NH), 3062 (CCH), 2956, 2941, 2876, 1743 (CO), 1683 (CO), 1663 (CO), 1520 (NH), 1209. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.20 (d, J = 7.7 Hz, 1H, N–H), 5.58–5.48 (m, 1H), 5.34–5.22 (m, 1H), 4.59–4.46 (m, 1H), 4.08–3.87 (m, 2H), 3.66 (s, 3H, OCH3), 3.44 (d, J = 15.9 Hz, 1H), 3.37 (d, J = 16.0 Hz, 1H), 3.35–3.14 (m, 2H), 2.64–2.38 (m, 2H), 2.02 (p, J = 7.3 Hz, 2H), 1.33 (d, J = 7.2 Hz, 3H), 0.89 (t, J = 7.5 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) 172.84 (CO), 171.17 (CO), 167.93 (CO), 136.12 (CH), 122.53 (CH), 58.62 (CH2), 52.39 (OCH3), 51.36 (CH2), 47.47 (CH), 39.36 (CH2), 28.87 (CH2), 20.57 (CH2), 18.03 (CH3), 13.92 (CH3). HRMS (+ESI): m/z calcd for C14H23N3O4+ [M + H]+: 298.1761; found: 298.1758.:4.8) to give the acid 3aae (42.0 mg, 0.129 mmol, 58%) as an orange oil. [α]23D + 1.0 (c 0.7, HCCl3). FTIR (ATR): max/cm−1 3300–2700 (COOH), 3290 (NH), 3061 (CCH), 2964, 2934, 2877, 1730 (CO), 1653 (CO), 1526 (NH). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.26 (d, J = 8.4 Hz, 1H, N–H), 5.68–5.58 (m, 1H), 5.43–5.33 (m, 1H), 4.65–4.49 (m, 1H), 4.21–3.94 (m, 2H), 3.56 (d, J = 15.8 Hz, 1H), 3.49 (d, J = 16.0 Hz, 1H), 3.47–3.24 (m, 2H), 2.78–2.50 (m, 2H), 2.11 (p, J = 7.5 Hz, 2H), 2.05–1.91 (m, 2H), 1.56–1.43 (m, 1H), 1.32–1.13 (m, 1H), 1.01–0.88 (m, 9H). 13C NMR (125 MHz, CDCl3): δ (ppm) 174.49 (CO), 171.72 (CO), 168.81 (CO), 136.57 (CH), 122.55 (CH), 58.44 (CH2), 56.44 (CH), 51.47 (CH2), 39.69 (CH2), 37.44 (CH), 29.13 (CH2), 25.12 (CH2), 20.86 (CH2), 15.79 (CH3), 14.20 (CH3), 11.74 (CH3). HRMS (+ESI): m/z calcd for C16H28N3O4+ [M + H]+: 326.2074; found: 326.2070.max/cm−1 3308 (NH), 2960, 2934, 2875, 1741 (CO), 1681 (CO), 1665 (CO), 1513 (NH), 1204. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.27 (d, J = 8.9 Hz, 1H, N–H), 4.63 (dd, J = 9.0, 4.7 Hz, 1H), 3.75 (s, 3H, OCH3), 3.48 (d, J = 16.1 Hz, 1H), 3.43 (d, J = 16.1 Hz, 1H), 3.43–3.25 (m, 4H), 2.71–2.51 (m, 2H), 2.01–1.91 (m, 1H), 1.68–1.58 (m, 2H), 1.50–1.39 (m, 1H), 1.39–1.23 (m, 4H), 1.23–1.11 (m, 1H), 0.94 (t, J = 7.4 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H), 0.90 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.23 (CO), 171.22 (CO), 168.28 (CO), 58.35 (CH2), 56.15 (CH), 52.37 (OCH3), 51.56 (CH2), 41.94 (CH2), 37.83 (CH), 29.30 (CH2), 28.98 (CH2), 27.12 (CH2), 25.27 (CH2), 22.45 (CH2), 15.80 (CH3), 14.09 (CH3), 11.71 (CH3). HRMS (+ESI): m/z calcd for C17H32N3O4+ [M + H]+: 342.2387; found: 342.2381.max/cm−1 3308 (NH), 2959, 2933, 2858, 1741 (CO), 1681 (CO), 1665 (CO), 1513 (NH), 1205. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.27 (d, J = 9.3 Hz, 1H, N–H), 4.63 (dd, J = 9.0, 4.7 Hz, 1H), 3.75 (s, 3H, OCH3), 3.48 (d, J = 16.1 Hz, 1H), 3.43 (d, J = 16.1 Hz, 1H), 3.44–3.23 (m, 4H), 2.71–2.52 (m, 2H), 2.01–1.92 (m, 1H), 1.66–1.57 (m, 2H), 1.49–1.39 (m, 1H), 1.36–1.24 (m, 6H), 1.22–1.11 (m, 1H), 0.94 (t, J = 7.4 Hz, 3H), 0.94 (d, J = 6.9 Hz, 3H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.24 (CO), 171.22 (CO), 168.29 (CO), 58.36 (CH2), 56.15 (CH), 52.37 (OCH3), 51.57 (CH2), 41.99 (CH2), 37.82 (CH), 31.57 (CH2), 29.31 (CH2), 27.40 (CH2), 26.51 (CH2), 25.27 (CH2), 22.64 (CH2), 15.81 (CH3), 14.14 (CH3), 11.73 (CH3). HRMS (+ESI): m/z calcd for C18H34N3O4+ [M + H]+: 356.2544; found: 356.2534.max/cm−1 3304 (NH), 2961, 2935, 2876, 1741 (CO), 1683 (CO), 1663 (CO), 1513 (NH), 1204. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.28 (d, J = 8.9 Hz, 1H, N–H), 4.63 (dd, J = 9.0, 4.7 Hz, 1H), 3.75 (s, 3H, OCH3), 3.49 (d, J = 16.1 Hz, 1H), 3.43 (d, J = 16.1 Hz, 1H), 3.45–3.23 (m, 4H), 2.71–2.49 (m, 2H), 2.01–1.91 (m, 1H), 1.65–1.57 (m, 2H), 1.50–1.40 (m, 1H), 1.35 (sext, J = 7.5 Hz, 2H), 1.22–1.12 (m, 1H), 0.94 (t, J = 6.8 Hz, 6H), 0.93 (d, J = 6.8 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.25 (CO), 171.23 (CO), 168.27 (CO), 58.36 (CH2), 56.15 (CH), 52.38 (OCH3), 51.57 (CH2), 41.67 (CH2), 37.85 (CH), 29.53 (CH2), 29.30 (CH2), 25.29 (CH2), 20.08 (CH2), 15.80 (CH3), 13.87 (CH3), 11.73 (CH3). HRMS (+ESI): m/z calcd for C16H30N3O4+ [M + H]+: 328.2231; found: 328.2224.:1H2O/trifluoroethanol (TFE) biphasic system (1.0 mL). This mixture was heated at 100 °C for 24 h. At rt, acetone was added (5 mL) and the resulting solution was concentrated under reduced pressure to give an orange oil. The Michael adduct was purified by column chromatography (silica gel, DCM/MeOH/NH3 (aq., 25%) 97.1:2.6:0.3) providing 3aba (40.0 mg, 0.113 mmol, 58%) as a brown oil. [α]25D + 15.5 (c 1.0, HCCl3). FTIR (ATR): max/cm−1 3293 (NH), 3021 (CCH), 2964, 2935, 2878, 1742 (CO), 1667 (CO), 1652 (CO), 1535 (NH), 1201. 1H NMR (500 MHz, CDCl3): δ (ppm) 6.77 (d, J = 8.0 Hz, 1H, H–N), 5.60–5.52 (m, 1H), 5.43–5.36 (m, 1H), 4.59 (dd, J = 8.5, 5.0 Hz, 1H), 4.26–3.95 (m, 2H), 3.73 (s, 3H, OCH3), 3.40–3.00 (m, 4H), 2.71–2.45 (m, 2H), 2.44 (t, J = 6.8 Hz, 2H), 2.13 (pd, J = 7.5, 0.8 Hz, 2H), 1.93–1.83 (m, 1H), 1.48–1.38 (m, 1H), 1.22–1.10 (m, 1H), 0.98 (t, J = 7.5 Hz, 3H), 0.92 (t, J = 7.3 Hz, 3H), 0.90 (d, J = 6.8 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.61 (CO), 171.50 (CO), 170.61 (CO), 135.68 (CH), 123.32 (CH), 56.54 (CH), 52.26 (OCH3), 51.39 (CH2), 49.16 (CH2), 40.08 (CH2), 37.99 (CH), 33.84 (CH2), 29.66 (CH2), 25.45 (CH2), 20.96 (CH2), 15.61 (CH3), 14.20 (CH3), 11.73 (CH3). HRMS (+ESI): m/z calcd for C18H32N3O4+ [M + H]+: 354.2387; found: 354.2378.max/cm−1 3297 (NH), 2955, 2933, 2869, 1743 (CO), 1661 (CO), 1519 (NH), 1204. 1H NMR (500 MHz, CDCl3): δ 7.09 (d, J = 8.6 Hz, 1H), 4.71–4.64 (m, 1H), 3.74 (s, 3H), 3.47 (d, J = 16.0 Hz, 1H), 3.43 (d, J = 16.1 Hz, 1H), 3.47–3.20 (m, 4H), 2.72–2.47 (m, 2H), 1.80–1.53 (m, 5H), 1.39–1.22 (m, 4H), 0.95 (d, J = 6.3 Hz, 6H), 0.90 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 173.26 (CO), 171.27 (CO), 168.33 (CO), 58.41 (CH2), 52.54 (OCH3), 51.55 (CH2), 50.42 (CH), 42.00 (CH2), 41.47 (CH2), 29.29 (CH2), 28.97 (CH2), 27.16 (CH2), 25.21 (CH), 22.96 (CH3), 22.43 (CH2), 22.01 (CH3), 14.09 (CH3). HRMS (+ESI): m/z calcd for C17H32N3O4+ [M + H]+: 342.2387; found: 342.2388.max/cm−1 3305 (NH), 2958, 2933, 2874, 1741 (CO), 1684 (CO), 1664 (CO), 1515 (NH), 1208. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.24 (d, J = 9.1 Hz, 1H), 4.53 (dd, J = 9.1, 4.7 Hz, 1H), 3.70 (s, 3H), 3.46 (d, J = 16.1 Hz, 1H), 3.39 (d, J = 16.2 Hz, 1H), 3.41–3.22 (m, 2H), 2.65–2.47 (m, 2H), 2.27–2.10 (m, 1H), 1.58 (p, J = 7.0 Hz, 2H), 1.35–1.18 (m, 4H), 0.92 (d, J = 6.9 Hz, 3H), 0.87 (d, J = 7.0 Hz, 3H), 0.85 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 172.10 (CO), 171.15 (CO), 168.33 (CO), 58.20 (CH2), 56.61 (CH), 52.28 (OCH3), 51.43 (CH2), 41.82 (CH2), 31.03 (CH), 29.19 (CH2), 28.86 (CH2), 26.97 (CH2), 22.31 (CH2), 19.14 (CH3), 17.65 (CH3), 13.97 (CH3). HRMS (+ESI): m/z calcd for C16H30N3O4+ [M + H]+: 328.2231; found: 328.2237.
O).·1H NMR (500 MHz, CDCl3): δ (ppm) 8.56 (s, 1H), 3.95 (t, J = 8.6 Hz, 2H), 2.67 (t, J = 8.6 Hz, 2H), 1.49 (s, 9H). 13C NMR (125 MHz, CDCl3): δ (ppm) 170.72 (CO), 153.78 (CO), 82.75 (Cq), 44.82 (CH2), 31.09 (CH2), 28.36 (CH3).:15) provided the 9a (66.0 mg, 0.263 mmol, 55%) as a colorless oil. FTIR (ATR): max/cm−1 3009 (CCH), 2973, 2935, 2875, 1704 (CO), 1640 (CC), 1314, 1153. 1H NMR (500 MHz, CDCl3): δ (ppm) 5.66–5.58 (m, 1H), 5.36–5.28 (m, 1H), 4.34 (d, J = 7.1 Hz, 2H), 3.94 (t, J = 7.7 Hz, 2H), 2.54 (t, J = 7.7 Hz, 2H), 2.11 (pd, J = 7.5, 1.2 Hz, 2H), 1.50 (s, 9H), 0.97 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.02 (CO), 156.93 (CO), 137.16 (CH), 122.01 (CH), 82.77 (Cq), 47.80 (CH2), 42.13 (CH2), 31.44 (CH2), 28.27 (CH3), 20.86 (CH2), 14.39 (CH3). HRMS (+ESI): m/z calcd for C13H23N2O3+ [M + H]+: 255.1703; found: 255.1698.max/cm−1 2958, 2933, 2874, 1702 (CO), 1315, 1155. 1H NMR (500 MHz, CDCl3): δ (ppm) 3.95 (t, J = 7.7 Hz, 2H), 3.69 (t, J = 7.2 Hz, 2H), 2.55 (t, J = 7.7 Hz, 2H), 1.57 (p, J = 7.4 Hz, 2H), 1.50 (s, 9H), 1.38–1.28 (m, 2H), 1.28–1.20 (m, 2H), 0.88 (t, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 171.57 (CO), 157.05 (CO), 82.83 (Cq), 47.63 (CH2), 45.18 (CH2), 31.65 (CH2), 28.99 (CH2), 28.30 (CH3), 26.16 (CH2), 22.44 (CH2), 14.11 (CH3). HRMS (+ESI): m/z calcd for C13H25N2O3+ [M + H]+: 257.1860; found: 257.1855.max/cm−1 2959, 2931, 2860, 1703 (CO), 1314, 1155. 1H NMR (500 MHz, CDCl3): δ (ppm) 3.95 (t, J = 7.7 Hz, 2H), 3.69 (t, J = 7.2 Hz, 2H), 2.55 (t, J = 7.7 Hz, 2H), 1.56 (p, J = 7.2 Hz, 2H), 1.50 (s, 9H), 1.33–1.22 (m, 6H), 0.87 (t, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 171.58 (CO), 157.06 (CO), 82.85 (Cq), 47.64 (CH2), 45.22 (CH2), 31.66 (CH2), 31.55 (CH2), 28.31 (CH3), 26.51 (CH2), 26.44 (CH2), 22.65 (CH2), 14.14 (CH3). HRMS (+ESI): m/z calcd for C14H27N2O3+ [M + H]+: 271.2016; found: 271.2010.max/cm−1 2960, 2934, 2875, 1701 (CO), 1316, 1155. 1H NMR (500 MHz, CDCl3): δ (ppm) 3.94 (t, J = 7.8 Hz, 2H), 3.68 (t, J = 7.2 Hz, 2H), 2.53 (t, J = 7.8 Hz, 2H), 1.54 (p, 7.4 Hz, 2H), 1.48 (s, 9H), 1.27 (sext, J = 7.5 Hz, 2H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 171.54 (CO), 157.01 (CO), 82.79 (Cq), 47.59 (CH2), 44.85 (CH2), 31.60 (CH2), 28.52 (CH3), 28.26 (CH2), 20.02 (CH2), 13.83 (CH3). HRMS (+ESI): m/z calcd for C12H23N2O3+ [M + H]+: 243.1703; found: 243.1699.:2.6:0.3). Traces of TFA were removed by treatment with 3 M KOH (1.0 mL), followed by extraction with EtOAc (3 × 10 mL). Evaporation of the solvent delivered analytically pure amine 10a (688 mg, 4.46 mmol, 99%) as an orange oil. FTIR (ATR): max/cm−1 3218 (NH), 3020 (CCH), 2966, 2936, 2878, 1666 (CO). 1H NMR (500 MHz, CDCl3): δ (ppm) 5.68–5.61 (m, 1H), 5.38–5.31 (m, 1H), 4.37 (s, 1H, N–H), 4.08 (d, J = 7.0 Hz, 2H), 3.37 (t, J = 7.8 Hz, 2H), 2.54 (t, J = 7.7 Hz, 2H), 2.15 (pd, J = 7.5, 1.3 Hz, 2H), 0.99 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 171.88 (CO), 137.17 (CH), 122.17 (CH), 43.83 (CH2), 40.67 (CH2), 33.35 (CH2), 20.80 (CH2), 14.27 (CH3). HRMS (+ESI): m/z calcd for C8H15N2O+ [M + H]+: 155.1179; found: 155.1175.max/cm−1 3216 (NH), 2957, 2930, 2872, 1663 (CO). 1H NMR (500 MHz, CDCl3): δ (ppm) 4.31 (s, 1H, N–H), 3.41 (t, J = 7.2 Hz, 2H), 3.36 (t, J = 7.7 Hz, 2H), 2.55 (t, J = 7.6 Hz, 2H), 1.58 (p, J = 7.4 Hz, 2H), 1.38–1.23 (m, 4H), 0.89 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.27 (CO), 44.18 (CH2), 44.12 (CH2), 33.67 (CH2), 29.07 (CH2), 27.06 (CH2), 22.46 (CH2), 14.13 (CH3). HRMS (+ESI): m/z calcd for C8H17N2O+ [M + H]+: 157.1335; found: 157.1330.max/cm−1 3215 (NH), 2959, 2929, 2858, 1663 (CO). 1H NMR (500 MHz, CDCl3): δ (ppm) 4.35 (s, 1H, N–H), 3.41 (t, J = 7.2 Hz, 2H), 3.36 (t, J = 7.7 Hz, 2H), 2.54 (t, J = 7.5 Hz, 2H), 1.57 (p, J = 7.1 Hz, 2H), 1.33–1.25 (m, 6H), 0.87 (t, J = 6.7 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.26 (CO), 44.18 (CH2), 44.15 (CH2), 33.67 (CH2), 31.58 (CH2), 27.33 (CH2), 26.59 (CH2), 22.67 (CH2), 14.15 (CH3). HRMS (+ESI): m/z calcd for C9H19N2O+ [M + H]+: 171.1492; found: 171.1487.max/cm−1 3216 (NH), 2960, 2934, 2875, 1662 (CO). 1H NMR (500 MHz, CDCl3): δ (ppm) 4.36 (s, 1H, N–H), 3.41 (t, J = 7.2 Hz, 2H), 3.35 (t, J = 7.7 Hz, 2H), 2.53 (t, J = 7.6 Hz, 2H), 1.55 (p, J = 7.4 Hz, 2H), 1.32 (sext, J = 7.5 Hz, 2H), 0.92 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.27 (CO), 44.16 (CH2), 43.82 (CH2), 33.64 (CH2), 29.40 (CH2), 20.11 (CH2), 13.84 (CH2). HRMS (+ESI): m/z calcd for C7H15N2O+ [M + H]+: 143.1179; found: 143.1175.max/cm−1 3500–2500 (COOH), 3364 (NH), 2964, 2925, 2878, 1709 (CO), 1622 (CO), 1533 (NH), 1235. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.06 (d, J = 8.4 Hz, 1H, N–H), 4.63 (dd, J = 8.6, 4.6 Hz, 1H), 4.12 (s, 2H, CH2Cl), 2.07–1.98 (m, 2H), 1.57–1.47 (m, 1H), 1.33–1.20 (m, 1H), 0.99 (d, J = 6.8 Hz, 3H), 0.97 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.85 (CO), 166.36 (CO), 56.81 (CH), 42.67 (CH2), 37.74 (CH), 25.14 (CH2), 15.58 (CH3), 11.73 (CH3). HRMS (+ESI): m/z calcd for C8H15NClO3+ [M + H]+: 210.0735; found: 208.0734.max/cm−1 3317 (NH), 2969, 2936, 2879, 1736 (CO), 1644 (CO), 1536 (NH). 1H NMR (500 MHz, CDCl3): δ (ppm) 6.49 (d, J = 7.6 Hz, 1H, N–H), 4.57 (dd, J = 8.6, 4.8 Hz, 1H), 3.75 (s, 3H, OCH3), 3.75 (d, J = 11.5 Hz, 1H), 3.70 (d, J = 11.5 Hz, 1H), 1.99–1.90 (m, 1H), 1.50–1.40 (m, 1H), 1.27–1.15 (m, 1H), 0.94 (t, J = 7.4 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.19 (CO), 166.81 (CO), 57.23 (CH), 52.43 (OCH3), 38.18 (CH), 25.22 (CH2), 15.52 (CH3), 11.73 (CH3), −0.90 (ICH2). HRMS (+ESI): m/z calcd for C9H17NIO3+ [M + H]+: 314.0248; found: 314.0243.max/cm−1 3287 (NH), 2956, 2871, 1742 (CO), 1648 (CO), 1539 (NH). 1H NMR (500 MHz, CDCl3): δ (ppm) 6.37 (d, J = 7.3 Hz, 1H, N–H), 4.61 (td, J = 8.6, 4.9 Hz, 1H), 3.75 (s, 3H, OCH3), 3.74 (d, J = 11.1 Hz, 1H, H–CHI), 3.70 (d, J = 11.5 Hz, 1H, H–CHI), 1.73–1.63 (m, 2H), 1.63–1.53 (m, 1H), 0.95 (d, J = 6.1 Hz, 3H), 0.95 (d, J = 6.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 173.26 (CO), 166.92 (CO), 57.62 (CH), 51.59 (OCH3), 41.63 (CH2), 24.97 (CH), 22.94 (CH3), 22.10 (CH3), −1.13 (ICH2). HRMS (+ESI): m/z calcd for C9H17NIO3+ [M + H]+: 314.0248; found: 314.0243.max/cm−1 3279 (NH), 2965, 2902, 2875, 1723 (CO), 1640 (CO), 1548 (NH). 1H NMR (500 MHz, CDCl3): δ (ppm) 6.49 (d, J = 7.4 Hz, 1H, N–H), 4.53 (dd, J = 8.8, 4.8 Hz, 1H), 3.76 (d, J = 11.4 Hz, 1H, H–CHI), 3.76 (s, 3H, OCH3), 3.71 (d, J = 11.4 Hz, 1H, H–CHI), 2.22 (septd, J = 6.9, 4.8 Hz, 1H), 0.96 (d, J = 6.9 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.23 (CO), 167.05 (CO), 57.82 (CH), 52.49 (OCH3), 31.65 (CH), 19.01 (CH3), 17.78 (CH3), −0.98 (ICH2). HRMS (+ESI): m/z calcd for C8H15NIO3+ [M + H]+: 300.0091; found: 300.0086.max/cm−1 3285 (NH), 2966, 2952, 2928, 1722 (CO), 1644 (CO), 1542 (NH). 1H NMR (500 MHz, CDCl3): δ (ppm) 6.58 (s, 1H, N–H), 4.57 (p, J = 7.2 Hz, 1H), 3.77 (s, 3H, OCH3), 3.71 (s, 2H, CH2I), 1.43 (d, J = 7.2 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 173.20 (CO), 166.55 (CO), 52.80 (OCH3), 48.98 (CH), 18.27 (CH3), −1.09 (ICH2). HRMS (+ESI): m/z calcd for C6H11NIO3+ [M + H]+: 271.9778; found: 271.9776.max/cm−1 3292 (NH), 3200–2700 (COOH), 2968, 2957, 2877, 1704 (CO), 1623 (CO), 1557 (NH), 1255. 1H NMR (500 MHz, CD3OD): δ (ppm) 4.33 (d, J = 5.8 Hz, 1H), 3.86 (d, J = 9.8 Hz, 1H, H–CHI), 3.75 (d, J = 9.8 Hz, 1H, H–CHI), 1.99–1.89 (m, 1H), 1.58–1.48 (m, 1H), 1.33–1.22 (m, 1H), 0.97 (d, J = 6.9 Hz, 3H), 0.93 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CD3OD): δ (ppm) 174.47 (CO), 171.38 (CO), 58.57 (CH), 38.35 (CH), 26.03 (CH2), 16.01 (CH3), 11.76 (CH3), −2.05 (ICH2). HRMS (+ESI): m/z calcd for C8H15NIO3+ [M + H]+: 300.0091; found: 300.0089.:30) to give 6ba (154 mg, 0.770 mmol, 77%) as a pale yellow oil. [α]26D + 30.8 (c 1.1, HCCl3). FTIR (ATR): max/cm−1 3286 (NH), 2965, 2937, 2879, 1741 (CO), 1657 (CO), 1532 (NH). 1H NMR (500 MHz, CDCl3): δ (ppm) 6.31 (dd, J = 17.0, 1.3 Hz, 1H), 6.15 (dd, J = 17.0, 10.3 Hz, 1H), 6.11 (s, 1H, N–H), 5.68 (dd, J = 10.3, 1.0 Hz, 1H), 4.70 (dd, J = 8.6, 5.0 Hz, 1H), 3.75 (s, 3H, OCH3), 1.97–1.88 (m, 1H), 1.51–1.41 (m, 1H), 1.27–1.14 (m, 1H), 0.93 (t, J = 7.4 Hz, 3H), 0.91 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 172.65 (CO), 165.21 (CO), 130.55 (CH), 127.35 (CH2), 56.48 (CH), 52.32 (OMe), 38.31 (CH), 25.41 (CH2), 15.52 (CH3), 11.72 (CH3). HRMS (+ESI): m/z calcd for C10H18NO3+ [M + H]+: 200.1281; found: 200.1279.max/cm−1 3310 (NH), 3021 (CCH), 2964, 2935, 2878, 1740 (CO), 1683 (CO), 1669 (CO), 1513 (NH), 1204. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.22 (d, J = 8.9 Hz, 1H, N–H), 5.67–5.59 (m, 1H), 5.43–5.35 (m, 1H), 4.61 (dd, J = 9.0, 4.8 Hz, 1H), 4.20–3.97 (m, 2H), 3.74 (s, 3H, OCH3), 3.53 (d, J = 15.8 Hz, 1H), 3.46 (d, J = 15.9 Hz, 1H), 3.48–3.20 (m, 2H), 2.74–2.47 (m, 2H), 2.14 (pd, J = 7.5, 0.8 Hz, 2H), 1.99–1.89 (m, 1H), 1.48–1.38 (m, 1H), 1.21–1.10 (m, 1H), 0.98 (t, J = 7.5 Hz, 3H), 0.93 (t, J = 7.4 Hz, 3H), 0.92 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.19 (CO), 171.23 (CO), 168.23 (CO), 136.44 (CH), 122.75 (CH), 58.66 (CH2), 56.14 (CH), 52.32 (OCH3), 51.47 (CH2), 39.61 (CH2), 37.79 (CH), 29.03 (CH2), 25.23 (CH2), 20.85 (CH2), 15.74 (CH3), 14.21 (CH3), 11.68 (CH3). HRMS (+ESI): m/z calcd for C17H30N3O4+ [M + H]+: 340.2231; found: 340.2224.max/cm−1 3303 (NH), 3019 (CCH), 2958, 2935, 2873, 1744 (CO), 1689 (CO), 1667 (CO), 1516 (NH), 1204. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.05 (d, J = 8.9 Hz, 1H, N–H), 5.67–5.58 (m, 1H), 5.43–5.35 (m, 1H), 4.67 (td, J = 8.9, 5.0 Hz, 1H), 4.20–3.95 (m, 2H), 3.74 (s, 3H, OCH3), 3.52 (d, J = 15.8 Hz, 1H), 3.46 (d, J = 15.8 Hz, 1H), 3.47–3.20 (m, 2H), 2.75–2.46 (m, 2H), 2.12 (p, J = 7.5 Hz, 2H), 1.74–1.53 (m, 3H), 0.99 (t, J = 7.5 Hz, 3H), 0.95 (d, J = 6.3 Hz, 6H). 13C NMR (125 MHz, CDCl3): δ (ppm) 173.25 (CO), 171.29 (CO), 168.31 (CO), 136.46 (CH), 122.82 (CH), 58.72 (CH2), 52.53 (OCH3), 51.51 (CH2), 50.38 (CH), 41.46 (CH2), 39.62 (CH2), 29.06 (CH2), 25.17 (CH), 23.00 (CH3), 21.94 (CH3), 20.88 (CH2), 14.24 (CH3). HRMS (+ESI): m/z calcd for C17H30N3O4+ [M + H]+: 340.2231; found: 340.2226.max/cm−1 3308 (NH), 3020 (CCH), 2964, 2935, 2876, 1741 (CO), 1683 (CO), 1667 (CO), 1513 (NH), 1206. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.21 (d, J = 9.0 Hz, 1H, N–H), 5.67–5.60 (m, 1H), 5.43–5.36 (m, 1H), 4.57 (dd, J = 9.2, 4.7 Hz, 1H), 4.21–3.97 (m, 2H), 3.74 (s, 3H, OCH3), 3.54 (d, J = 15.8 Hz, 1H), 3.46 (d, J = 15.9 Hz, 1H), 3.47–3.20 (m, 2H), 2.74–2.47 (m, 2H), 2.22 (septd, J = 6.9, 4.9 Hz, 1H), 2.12 (pd, J = 7.5, 1.0 Hz, 2H), 0.98 (t, J = 7.5 Hz, 3H), 0.95 (d, J = 6.9 Hz, 3H), 0.90 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.21 (CO), 171.24 (CO), 168.37 (CO), 136.48 (CH), 122.75 (CH), 58.66 (CH2), 56.72 (CH), 52.39 (OCH3), 51.49 (CH2), 39.61 (CH2), 31.14 (CH), 29.04 (CH2), 20.87 (CH2), 19.23 (CH3), 17.78 (CH3), 14.24 (CH3). HRMS (+ESI): m/z calcd for C16H28N3O4+ [M + H]+: 326.2074; found: 326.2069.max/cm−1 3307 (NH), 3062 (CCH), 2956, 2941, 2876, 1743 (CO), 1683 (CO), 1663 (CO), 1520 (NH), 1209. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.20 (d, J = 7.7 Hz, 1H, N–H), 5.58–5.48 (m, 1H), 5.34–5.22 (m, 1H), 4.59–4.46 (m, 1H), 4.08–3.87 (m, 2H), 3.66 (s, 3H, OCH3), 3.44 (d, J = 15.9 Hz, 1H), 3.37 (d, J = 16.0 Hz, 1H), 3.35–3.14 (m, 2H), 2.64–2.38 (m, 2H), 2.02 (p, J = 7.3 Hz, 2H), 1.33 (d, J = 7.2 Hz, 3H), 0.89 (t, J = 7.5 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) 172.84 (CO), 171.17 (CO), 167.93 (CO), 136.12 (CH), 122.53 (CH), 58.62 (CH2), 52.39 (OCH3), 51.36 (CH2), 47.47 (CH), 39.36 (CH2), 28.87 (CH2), 20.57 (CH2), 18.03 (CH3), 13.92 (CH3). HRMS (+ESI): m/z calcd for C14H23N3O4+ [M + H]+: 298.1761; found: 298.1758.:4.8) to give the acid 3aae (42.0 mg, 0.129 mmol, 58%) as an orange oil. [α]23D + 1.0 (c 0.7, HCCl3). FTIR (ATR): max/cm−1 3300–2700 (COOH), 3290 (NH), 3061 (CCH), 2964, 2934, 2877, 1730 (CO), 1653 (CO), 1526 (NH). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.26 (d, J = 8.4 Hz, 1H, N–H), 5.68–5.58 (m, 1H), 5.43–5.33 (m, 1H), 4.65–4.49 (m, 1H), 4.21–3.94 (m, 2H), 3.56 (d, J = 15.8 Hz, 1H), 3.49 (d, J = 16.0 Hz, 1H), 3.47–3.24 (m, 2H), 2.78–2.50 (m, 2H), 2.11 (p, J = 7.5 Hz, 2H), 2.05–1.91 (m, 2H), 1.56–1.43 (m, 1H), 1.32–1.13 (m, 1H), 1.01–0.88 (m, 9H). 13C NMR (125 MHz, CDCl3): δ (ppm) 174.49 (CO), 171.72 (CO), 168.81 (CO), 136.57 (CH), 122.55 (CH), 58.44 (CH2), 56.44 (CH), 51.47 (CH2), 39.69 (CH2), 37.44 (CH), 29.13 (CH2), 25.12 (CH2), 20.86 (CH2), 15.79 (CH3), 14.20 (CH3), 11.74 (CH3). HRMS (+ESI): m/z calcd for C16H28N3O4+ [M + H]+: 326.2074; found: 326.2070.max/cm−1 3308 (NH), 2960, 2934, 2875, 1741 (CO), 1681 (CO), 1665 (CO), 1513 (NH), 1204. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.27 (d, J = 8.9 Hz, 1H, N–H), 4.63 (dd, J = 9.0, 4.7 Hz, 1H), 3.75 (s, 3H, OCH3), 3.48 (d, J = 16.1 Hz, 1H), 3.43 (d, J = 16.1 Hz, 1H), 3.43–3.25 (m, 4H), 2.71–2.51 (m, 2H), 2.01–1.91 (m, 1H), 1.68–1.58 (m, 2H), 1.50–1.39 (m, 1H), 1.39–1.23 (m, 4H), 1.23–1.11 (m, 1H), 0.94 (t, J = 7.4 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H), 0.90 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.23 (CO), 171.22 (CO), 168.28 (CO), 58.35 (CH2), 56.15 (CH), 52.37 (OCH3), 51.56 (CH2), 41.94 (CH2), 37.83 (CH), 29.30 (CH2), 28.98 (CH2), 27.12 (CH2), 25.27 (CH2), 22.45 (CH2), 15.80 (CH3), 14.09 (CH3), 11.71 (CH3). HRMS (+ESI): m/z calcd for C17H32N3O4+ [M + H]+: 342.2387; found: 342.2381.max/cm−1 3308 (NH), 2959, 2933, 2858, 1741 (CO), 1681 (CO), 1665 (CO), 1513 (NH), 1205. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.27 (d, J = 9.3 Hz, 1H, N–H), 4.63 (dd, J = 9.0, 4.7 Hz, 1H), 3.75 (s, 3H, OCH3), 3.48 (d, J = 16.1 Hz, 1H), 3.43 (d, J = 16.1 Hz, 1H), 3.44–3.23 (m, 4H), 2.71–2.52 (m, 2H), 2.01–1.92 (m, 1H), 1.66–1.57 (m, 2H), 1.49–1.39 (m, 1H), 1.36–1.24 (m, 6H), 1.22–1.11 (m, 1H), 0.94 (t, J = 7.4 Hz, 3H), 0.94 (d, J = 6.9 Hz, 3H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.24 (CO), 171.22 (CO), 168.29 (CO), 58.36 (CH2), 56.15 (CH), 52.37 (OCH3), 51.57 (CH2), 41.99 (CH2), 37.82 (CH), 31.57 (CH2), 29.31 (CH2), 27.40 (CH2), 26.51 (CH2), 25.27 (CH2), 22.64 (CH2), 15.81 (CH3), 14.14 (CH3), 11.73 (CH3). HRMS (+ESI): m/z calcd for C18H34N3O4+ [M + H]+: 356.2544; found: 356.2534.max/cm−1 3304 (NH), 2961, 2935, 2876, 1741 (CO), 1683 (CO), 1663 (CO), 1513 (NH), 1204. 1H NMR (500 MHz, CDCl3): δ (ppm) 7.28 (d, J = 8.9 Hz, 1H, N–H), 4.63 (dd, J = 9.0, 4.7 Hz, 1H), 3.75 (s, 3H, OCH3), 3.49 (d, J = 16.1 Hz, 1H), 3.43 (d, J = 16.1 Hz, 1H), 3.45–3.23 (m, 4H), 2.71–2.49 (m, 2H), 2.01–1.91 (m, 1H), 1.65–1.57 (m, 2H), 1.50–1.40 (m, 1H), 1.35 (sext, J = 7.5 Hz, 2H), 1.22–1.12 (m, 1H), 0.94 (t, J = 6.8 Hz, 6H), 0.93 (d, J = 6.8 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.25 (CO), 171.23 (CO), 168.27 (CO), 58.36 (CH2), 56.15 (CH), 52.38 (OCH3), 51.57 (CH2), 41.67 (CH2), 37.85 (CH), 29.53 (CH2), 29.30 (CH2), 25.29 (CH2), 20.08 (CH2), 15.80 (CH3), 13.87 (CH3), 11.73 (CH3). HRMS (+ESI): m/z calcd for C16H30N3O4+ [M + H]+: 328.2231; found: 328.2224.:1H2O/trifluoroethanol (TFE) biphasic system (1.0 mL). This mixture was heated at 100 °C for 24 h. At rt, acetone was added (5 mL) and the resulting solution was concentrated under reduced pressure to give an orange oil. The Michael adduct was purified by column chromatography (silica gel, DCM/MeOH/NH3 (aq., 25%) 97.1:2.6:0.3) providing 3aba (40.0 mg, 0.113 mmol, 58%) as a brown oil. [α]25D + 15.5 (c 1.0, HCCl3). FTIR (ATR): max/cm−1 3293 (NH), 3021 (CCH), 2964, 2935, 2878, 1742 (CO), 1667 (CO), 1652 (CO), 1535 (NH), 1201. 1H NMR (500 MHz, CDCl3): δ (ppm) 6.77 (d, J = 8.0 Hz, 1H, H–N), 5.60–5.52 (m, 1H), 5.43–5.36 (m, 1H), 4.59 (dd, J = 8.5, 5.0 Hz, 1H), 4.26–3.95 (m, 2H), 3.73 (s, 3H, OCH3), 3.40–3.00 (m, 4H), 2.71–2.45 (m, 2H), 2.44 (t, J = 6.8 Hz, 2H), 2.13 (pd, J = 7.5, 0.8 Hz, 2H), 1.93–1.83 (m, 1H), 1.48–1.38 (m, 1H), 1.22–1.10 (m, 1H), 0.98 (t, J = 7.5 Hz, 3H), 0.92 (t, J = 7.3 Hz, 3H), 0.90 (d, J = 6.8 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.61 (CO), 171.50 (CO), 170.61 (CO), 135.68 (CH), 123.32 (CH), 56.54 (CH), 52.26 (OCH3), 51.39 (CH2), 49.16 (CH2), 40.08 (CH2), 37.99 (CH), 33.84 (CH2), 29.66 (CH2), 25.45 (CH2), 20.96 (CH2), 15.61 (CH3), 14.20 (CH3), 11.73 (CH3). HRMS (+ESI): m/z calcd for C18H32N3O4+ [M + H]+: 354.2387; found: 354.2378.max/cm−1 3297 (NH), 2955, 2933, 2869, 1743 (CO), 1661 (CO), 1519 (NH), 1204. 1H NMR (500 MHz, CDCl3): δ 7.09 (d, J = 8.6 Hz, 1H), 4.71–4.64 (m, 1H), 3.74 (s, 3H), 3.47 (d, J = 16.0 Hz, 1H), 3.43 (d, J = 16.1 Hz, 1H), 3.47–3.20 (m, 4H), 2.72–2.47 (m, 2H), 1.80–1.53 (m, 5H), 1.39–1.22 (m, 4H), 0.95 (d, J = 6.3 Hz, 6H), 0.90 (t, J = 7.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 173.26 (CO), 171.27 (CO), 168.33 (CO), 58.41 (CH2), 52.54 (OCH3), 51.55 (CH2), 50.42 (CH), 42.00 (CH2), 41.47 (CH2), 29.29 (CH2), 28.97 (CH2), 27.16 (CH2), 25.21 (CH), 22.96 (CH3), 22.43 (CH2), 22.01 (CH3), 14.09 (CH3). HRMS (+ESI): m/z calcd for C17H32N3O4+ [M + H]+: 342.2387; found: 342.2388.max/cm−1 3305 (NH), 2958, 2933, 2874, 1741 (CO), 1684 (CO), 1664 (CO), 1515 (NH), 1208. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.24 (d, J = 9.1 Hz, 1H), 4.53 (dd, J = 9.1, 4.7 Hz, 1H), 3.70 (s, 3H), 3.46 (d, J = 16.1 Hz, 1H), 3.39 (d, J = 16.2 Hz, 1H), 3.41–3.22 (m, 2H), 2.65–2.47 (m, 2H), 2.27–2.10 (m, 1H), 1.58 (p, J = 7.0 Hz, 2H), 1.35–1.18 (m, 4H), 0.92 (d, J = 6.9 Hz, 3H), 0.87 (d, J = 7.0 Hz, 3H), 0.85 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 172.10 (CO), 171.15 (CO), 168.33 (CO), 58.20 (CH2), 56.61 (CH), 52.28 (OCH3), 51.43 (CH2), 41.82 (CH2), 31.03 (CH), 29.19 (CH2), 28.86 (CH2), 26.97 (CH2), 22.31 (CH2), 19.14 (CH3), 17.65 (CH3), 13.97 (CH3). HRMS (+ESI): m/z calcd for C16H30N3O4+ [M + H]+: 328.2231; found: 328.2237.Author contributions
Concept: W. Q. F. and D. D.; experiments: S. V. P, D. D., and C. J. M.; supervision and resources: M. B. and W. Q. F.; writing: S. V. P, M. B. and W. Q. F.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author S. V. P. thanks the DAAD (German Academic Exchange Service) for financial support and a scholarship, and Minciencias (Ministerio de Ciencia Tecnología e Innovación de Colombia) for financial support.Notes and references
- Y. Yan, E. Borrego and M. V Kolomiets, in Lipid Metabolism, ed. R. V. Baez, IntechOpen, 2013, pp. 393–442 Search PubMed.
- S. Fonseca, A. Chini, M. Hamberg, B. Adie, A. Porzel, R. Kramell, O. Miersch, C. Wasternack and R. Solano, Nat. Chem. Biol., 2009, 5, 344–350 CrossRef CAS PubMed.
- J. Ueda and J. Kato, Physiol. Plant., 1982, 54, 249–252 CrossRef CAS.
- E. E. Farmer and C. A. Ryan, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 7713–7716 CrossRef CAS PubMed.
- E. E. Farmer and C. A. Ryan, Plant Cell, 1992, 4, 129–134 CrossRef CAS.
- Y. Koda, Y. Kikuta, H. Tazaki, Y. Tsujino, S. Sakamura and T. Yoshihara, Phytochemistry, 1991, 30, 1435–1438 CrossRef CAS.
- H. Gundlach, M. J. Müller, T. M. Kutchan and M. H. Zenk, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 2389–2393 CrossRef CAS PubMed.
- E. W. Weiler, T. Albrecht, B. Groth, Z.-Q. Xia, M. Luxem, H. Liß, L. Andert and P. Spengler, Phytochemistry, 1993, 32, 591–600 CrossRef CAS.
- W. Boland, J. Hopke, J. Donath, J. Nüske and F. Bublitz, Angew Chem. Int. Ed. Engl., 1995, 34, 1600–1602 CrossRef CAS.
- J. Hopke, J. Donath, S. Blechert and W. Boland, FEBS Lett., 1994, 352, 146–150 CrossRef CAS PubMed.
- R. Kramell, O. Miersch, B. Hause, B. Ortel, B. Parthier and C. Wasternack, FEBS Lett., 1997, 414, 197–202 CrossRef PubMed.
- S. Tamogami and O. Kodama, Phytochemistry, 2000, 54, 689–694 CrossRef CAS PubMed.
- M. Okada, S. Ito, A. Matsubara, I. Iwakura, S. Egoshi and M. Ueda, Org. Biomol. Chem., 2009, 7, 3065–3073 RSC.
- C. Chapuis, D. Skuy and C.-A. Richard, Chimia, 2019, 73, 194–204 CrossRef CAS PubMed.
- S. Blechert, C. Bockelmann, M. Füßlein, T. v. Schrader, B. Stelmach, U. Niesel and E. W. Weiler, Planta, 1999, 207, 470–479 CrossRef CAS.
- G. Haider, T. von Schrader, M. Füßlein, S. Blechert and T. M. Kutchan, Biol. Chem., 2000, 381, 741 CAS.
- H. T. Dang, H. J. Lee, E. S. Yoo, J. Hong, B. Bao, J. S. Choi and J. H. Jung, Bioorg. Med. Chem., 2008, 16, 10228–10235 CrossRef CAS PubMed.
- I. Jarocka-Karpowicz and A. Markowska, Molecules, 2021, 26, 2901–2915 CrossRef CAS PubMed.
- D. Uludağ, S. Bay, B. O. Sucu, Ö. Şavluğİpek, T. Mohr, M. Güzel and N. Karakaş, Front. Pharmacol, 2022, 13, 828400 CrossRef PubMed.
- T.-L. Ho, Synth. Commun., 1974, 4, 265–287 CrossRef CAS.
- B. M. Trost and A. B. Pinkerton, J. Org. Chem., 2001, 66, 7714–7722 CrossRef CAS PubMed.
- A. Pawełczyk and L. Zaprutko, Eur. J. Med. Chem., 2006, 41, 586–591 CrossRef PubMed.
- I. Monte, M. Hamberg, A. Chini, S. Gimenez-Ibanez, G. García-Casado, A. Porzel, F. Pazos, M. Boter and R. Solano, Nat. Chem. Biol., 2014, 10, 671 CrossRef CAS PubMed.
- H. Kiyota, Y. Yoneta and T. Oritani, Phytochemistry, 1997, 46, 983–986 CrossRef CAS.
- J. L. Ward and M. H. Beale, J. Chem. Soc., Perkin Trans. 1, 1993, 2379–2381 RSC.
- Z. Zhao, G. L. Araldi, Y. Xiao, A. P. Reddy, Y. Liao, S. Karra, N. Brugger, D. Fischer and E. Palmer, Bioorg. Med. Chem. Lett., 2007, 17, 6572–6575 CrossRef CAS PubMed.
- S. Y. Han and Y.-D. Gong, Synth. Commun., 2019, 49, 3426–3434 CrossRef CAS.
- B. D. White, J. Mallen, K. A. Arnold, F. R. Fronczek, R. D. Gandour, L. M. B. Gehrig and G. W. Gokel, J. Org. Chem., 1989, 54, 937–947 CrossRef CAS.
- A. E. Fedorov, A. M. Shestopalov and P. A. Belyakov, Russ. Chem. Bull., 2003, 52, 2197–2202 CrossRef CAS.
- X. Liang, M. Behl and A. Lendlein, Eur. Polym. J., 2021, 143, 110189 CrossRef CAS.
- K. De, J. Legros, B. Crousse and D. Bonnet-Delpon, J. Org. Chem., 2009, 74, 6260–6265 CrossRef CAS PubMed.
- A. Fedotova, E. Kondrashov, J. Legros, J. Maddaluno and A. Y. Rulev, C. R. Chim., 2018, 21, 639–643 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07887f |
| This journal is © The Royal Society of Chemistry 2024 |