
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly (regio)selective hydroformylation of olefins using self-assembling phosphines†
Pierre
Fablet
 a,
Maria Dolores
Fernandez-Martinez
b,
Anke
Spannenberg
a,
Haijun
Jiao
*a,
Ralf
Jackstell
*a and
Matthias
Beller
*a
a,
Maria Dolores
Fernandez-Martinez
b,
Anke
Spannenberg
a,
Haijun
Jiao
*a,
Ralf
Jackstell
*a and
Matthias
Beller
*a
aLeibniz-Institut für Katalyse e.V. (LIKAT), Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: Ralf.Jackstell@catalysis.de; Matthias.Beller@catalysis.de
bDepartament de Química Física i Inorgánica, Universitat Rovira i Virgili, C/Marceli Domingo 1, 43007 Tarragona, Spain
First published on 6th June 2024
Abstract
New phosphines with self-assembling 6-pyridinone moities were prepared, characterized, and examined in the hydroformylation of diverse olefins. Testing various known and novel ligands in the presence of [Rh(acac)(CO)2] under industrially relevant conditions, the hydroformylation of 1-octene proceeds best with 6,6′-(phenylphosphanediyl)bis(pyridin-2(1H)-one) (DPONP). Control experiments and modelling studies indicate dimerization of this ligand at higher temperatures (>100 °C). The optimal catalyst system is able to conserve high product linearity (>90%) for a broad range of olefins at industrially-employed temperatures at low ligand loading.
Introduction
Hydroformylation or oxo synthesis is unanimously regarded as one of the most important processes in homogeneous catalysis. Using syngas (CO/H2), the volume of alkenes converted to high-value aldehydes is about 10 million metric tons per year.1 Although numerous metals have been investigated,2,3 such as platinum,4,5 palladium,6 ruthenium7 and iridium,8,9 cobalt,10–12 as first discovered by Otto Roelen13 and rhodium14,15 catalysts remain most used by the industry.As such, phosphine ligands have become a powerful and flexible factor to influence the activity, chemo- and regioselectivity of rhodium catalysts.16–18 Indeed, denticity,19,20 electronic21 and steric properties22,23 have been showed to have direct impact on the performance of the catalyst. While catalyst activity and reaction yield matter, an essential aspect of hydroformylation resides in regioselectivity of the reaction. It must be borne in mind that linear aliphatic aldehydes are typically of higher market demand and mixtures of regioisomers can lead to costly separation steps. Important issues for industrial applications are ligand degradation and catalyst deactivation. It is well precedented that phosphorus ligands can react, especially at higher temperatures (>130 °C), in undesired manners with aldehyde products, oxygen, water or hydroperoxides. A common solution to those challenges is to work in industrial processes with high to very high ligand-to-metal ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10–1:100). However, the cost of ligands can then become an important barrier for commercial viability.
:10–1:100). However, the cost of ligands can then become an important barrier for commercial viability.
Therefore, the development of new ligands and their investigation to modulate the catalytic activity continues to attract the interest of many scientists in academia and industry. In this respect, it is desirable to explore novel phosphines that can deliver high n-selectivity at low ligand-to-metal ratio, with minimal catalyst degradation at industrially relevant conditions.
In the past, numerous investigations focused on the variation of electronic properties of P-based ligands to achieve these goals. In contrast, the use of supramolecular chemistry has only been scarcely exploited as a tool to modulate the properties of phosphines.24–29 As an example in hydroformylation catalysis, Breit and co-workers introduced self-assembling monodentate ligands based on the 6-pyridinone moiety.30 Under catalytic conditions, these monodentate phosphines behave like bidentate ligands leading to excellent regioselectivity at low temperature and pressure.31 More recently, the groups of Reek and Breit reported further examples following this ligand design.27,32,33
Based on our long-standing interest in carbonylation reactions and inspired by all those results, we aimed at developing new 6-DPPON analogues33 for the rhodium hydroformylation of terminal olefins. Herein, we report the synthesis and activity assessment of several monodentate self-assembling phosphines bearing the 6-pyridinone moiety. The optimal catalyst system Rh/L6 results in highly regioselective hydroformylations even at temperature >100 °C, where the parent ligand 6-DPPON does not.30
Results and discussion
Ligand synthesis and characterizations
At the start of this work, seven DPPON ligands were prepared in accord with a procedure previously developed by Breit et al.30 Initially, the parent ligand L1 was synthesized by reacting 2,6-dibromopyridine (a) with potassium tert-butoxide to give selectively the mono-alkoxylated bromopyridine at 80 °C in almost quantitative yield. Subsequent lithiation with n-butyl lithium and quenching with the corresponding chlorophosphine at 0 °C provided intermediate c (see ESI†). Finally, the desired DPPON resulted after deprotection via formic acid treatment. It should be noted that no tedious workup/purification procedures are needed for the conversion from a to L1–L7, and final product can be conveniently crystallized from acetone (see below). This general protocol was used for the synthesis of L2–L7, including already described L5,34 and provided the ligands in 43–75% overall yield (see ESI†) (Scheme 1).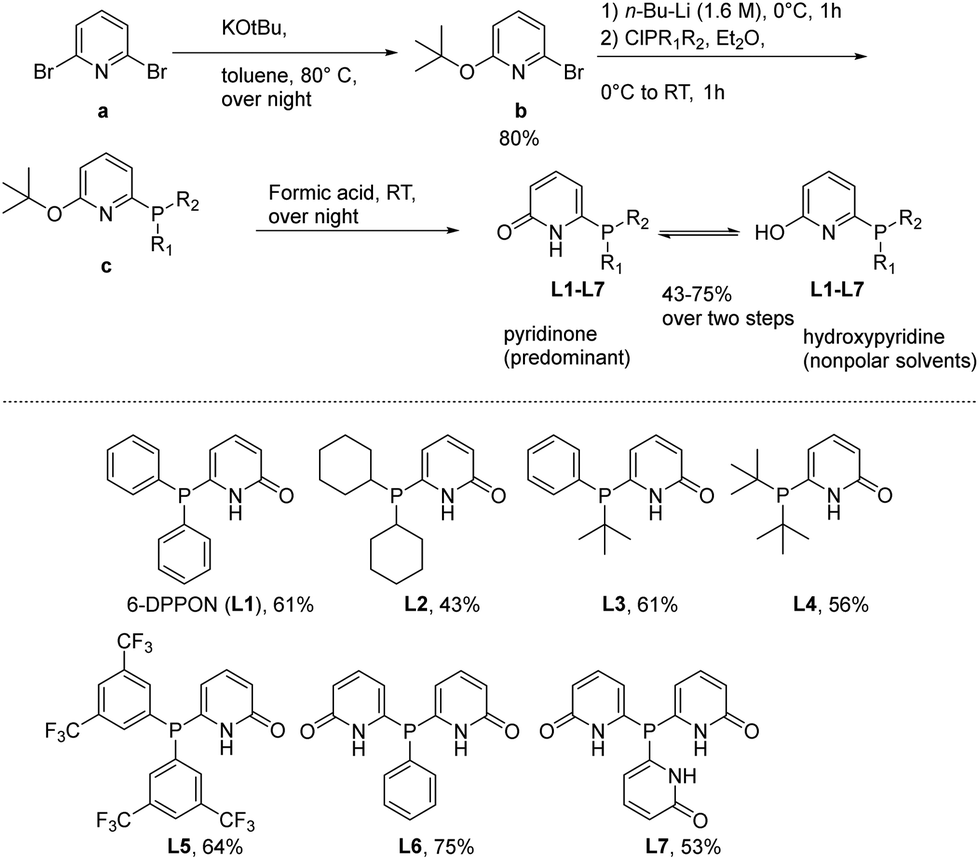 | ||
| Scheme 1 General method for the synthesis of ligands L1–L7 and structures of tertiary phosphines relevant to this work. | ||
In general, all the ligands were characterized by31 P, 1H and 13C NMR and HR-MS. In some cases, crystallisation of the obtained white powders (L2, L4, L5) from acetone yielded crystals suitable for X-ray diffraction. In all the obtained crystal structures, strong intermolecular N–H⋯O hydrogen bonds are observed resulting in the corresponding dimer structures. For L4 and L5 these dimers are further linked by weak intermolecular interactions (L4(C–H⋯O) (Fig. 1) and L5(C–H⋯F)).
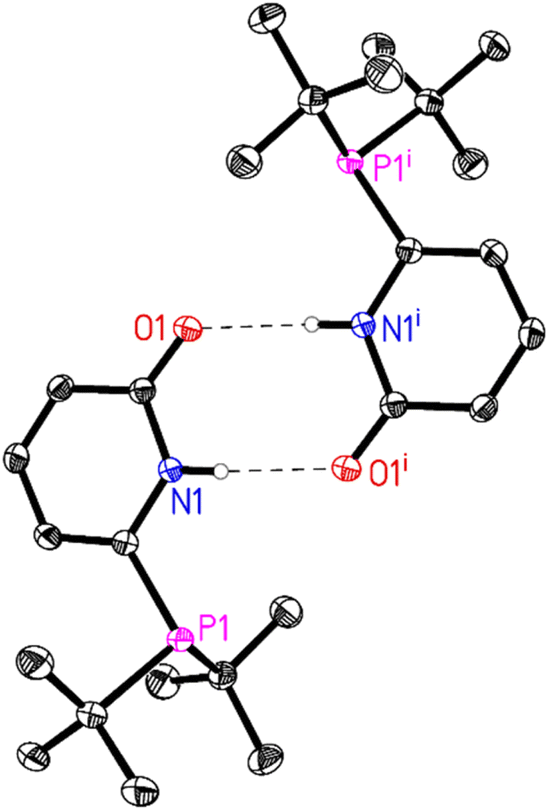 | ||
| Fig. 1 Molecular structure of L4. The thermal ellipsoids correspond to 50% probability. C-Bound hydrogen atoms are omitted for clarity. Only one molecule of the asymmetric unit and its symmetry related molecule (symmetry code (i) 1 − x, 1 − y, 2 − z) is shown. Intermolecular N–H⋯O hydrogen bonds are depicted as dashed lines (N1–H1⋯O1i: N1–H1 0.84(2), H1⋯O1i 2.01(2), N1⋯O1i 2.8437(14) Å, N1–H1⋯O1i 173(2)°). For more details see ESI†e.g. packing diagram and table of hydrogen bonds. | ||
Catalyst testing and reaction conditions optimization
To assess the activity and selectivity of the new phosphines and compare them to selected monodentate ligands including DPPON, the hydroformylation of 1-octene was used as a model system. Conscious of the temperature range (85–130 °C) commonly used with phosphine-modified rhodium catalysts for low-pressure hydroformylation,14,35 experiments at different temperatures were conducted. More specifically, we tested PPh3, L1 (DPPON), and L6 in between 40–140 °C under otherwise similar conditions as described by Breit et al.30 These experiments revealed promising regioselectivity to 2a but minor hydroformylation activity for L6 at low temperature (<80 °C) compared to the two other ligands (Fig. 2). Nonetheless, increasing the temperature Rh/L6 showed a steady increase in hydroformylation activity, reaching near full conversion and the highest yield of 2a at 120 °C. Notably, the excellent linear selectivity for 2a of 98% is kept until 140 °C (Fig. 2 and ESI Table 5†). To the best of our knowledge, this is the first report of a monodentate ligand exhibiting such very good linear selectivity at high temperatures in the rhodium-catalysed hydroformylation of higher aliphatic olefins. In contrast, in the presence of PPh3 and L1 and in agreement with previous literature,21 the regioselectivity decreased at higher temperatures, reaching a plateau at respectively 66 and 80% above 100 °C (ESI Table 5†). Previously, for L1 this effect was explained by the degradation of the pyridinone-hydroxypyridine hydrogen bonding leading to a simple monodentate ligand behaviour.36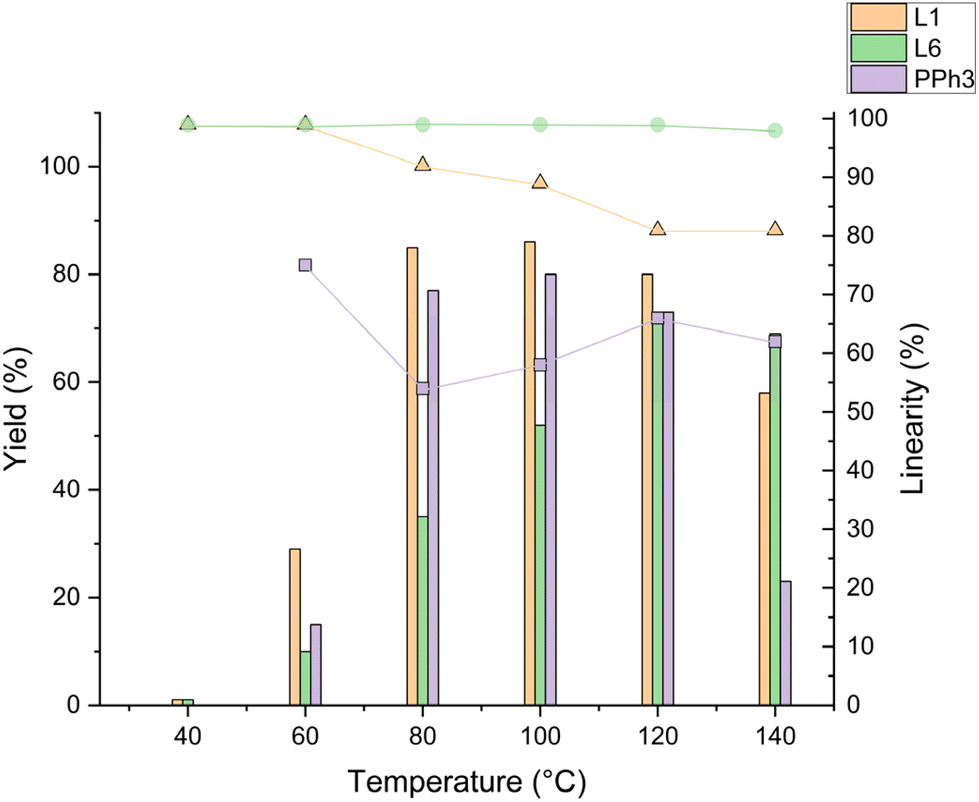 | ||
| Fig. 2 Influence of temperature in hydroformylation of 1a for selected ligands. Conditions: olefin (6.0 mmol), [Rh(CO)2acac] (0.014 mol%), monodentate ligand (0.071 mol%), CO:H2 (10 bar, 1:1), toluene (4.3 mL), 4 h. The yield and regioselectivity of the product were measured by GC analysis using MeOH as solvent and isooctane as internal standard. | ||
Next, experiments at 120 °C were ran at lower ligand loading to make the protocol more economic. In fact, the ligand to metal ratio for L6 could successfully be lowered to 5:1, with no negative impact on regioselectivity for nonanal 2a, providing even higher yields. However, applying ligand to metal ratios below 3:1, led to reproducibility issues (ESI Table 7, entries 4, 6†). Utilizing L1 at lower ligand to metal ratios, e.g. 5:1 showed the necessity to use higher loading of 6-DPPON to preserve the product yield and linear selectivity. Meanwhile, the influence of syngas pressure was studied. In the presence of both ligands, no aldehyde formation was observed at 1 bar of syngas. However, at slightly elevated pressure (5 bar) 2a was obtained in 54% yield and 2a selectivity remained steady for L1 and L6 (ESI Table 8†). Increasing the pressure above 10 bars further increased aldehyde yields, reaching above 90% at 40 bars. Notably, the regioselectivity dropped to 73% applying L1 while remaining virtually unchanged for L6 (ESI Table 7, entries 1 and 2†). This different behaviour can be explained by the increased stability of the ligand dimer in case of L6. This assumption is supported by performing the model reaction in a range of relevant solvents. The choice of solvent media in hydroformylation is challenging as it boils down to a dilemma between transition-state-stabilizing properties and compatibility with the catalyst leading to no inhibition of the system or of the product release.37 Apart from toluene, the use of dioxane, THF, and propylene carbonate (PC) led to moderate to good product yields and above 90% linearity (ESI Table 8, entries 2, 3 and 4†). In agreement with Breit's original report utilizing 6-DPPON, in protic solvents a strong decrease in regioselectivity was observed.30
With regard to green chemistry, it is interesting that the model system performed well under neat conditions leading to 82% yield and 90% linear selectivity, respectively, which could prove superior to other investigated conditions, albeit less regioselective. (ESI Table 9, entry 10†).38
Comparing L2–L7 with triphenylphosphine and 6-DPPON under optimized conditions (Rh:L:octene = 1:5:7000, 10 bar H2:CO = 1:1, toluene, 120 °C, 4 h) showed good to high catalytic activity for most ligands, except for the bis-tert-butyl-substituted phosphine L4. Notably, utilizing L7 no hydroformylation but mainly isomerization and hydrogenation of the olefin occurred. As a ligand, L6 stands out regarding regioselectivity with a linearity of 99% for product 2a (Fig. 3).
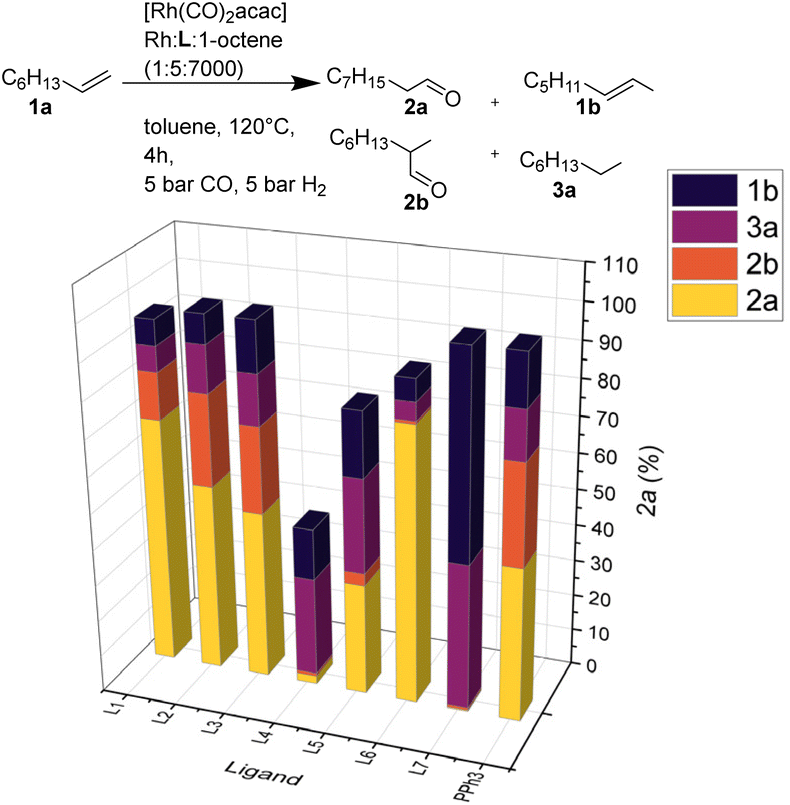 | ||
| Fig. 3 Influence of ligands in hydroformylation of 1a. in optimized conditions. Conditions: 1a (6.0 mmol), [Rh(CO)2acac] (0.014 mol%), monodentate ligand (0.071 mol%), CO:H2 (10 bar, 1:1), toluene (4.3 mL), 120 °C, 4 h. The conversion of 1a and yields of 2a, 2b, 1b, 3a and regioselectivity of the products were measured by GC analysis using MeOH as solvent and isooctane as internal standard. | ||
Mechanistic investigations and structural characterizations
To evaluate the catalyst performance and to gain a deeper understanding on this new catalytic system (Rh/L6) specifically catalyst turnover numbers (TON), turnover frequency (TOF), and gas consumption were measured. Similar experiments were run for 6-DPPON to compare the new ligand with the parent system. To slow down the rate of the catalyst system and allow for establishment of a clear pressure curve, experiments were done under diluted conditions – a rhodium concentration in solvent from 2 × 10−4 to 6 × 10−5 mol L−1 was used – and the pressure of syngas (CO/H2 = 1:1) was increased to 25 bars to monitor gas consumption more accurately. Initial TOF were measured at 1 hour, and TON over 4 hours as both systems reach a plateau in yield and conversions over this time. Good TON and TOF were observed despite the dilution. In general, L1 performed at a much lower rate than previously reported (TOF = 3250 h−1), which is attributed to the difference to the original conditions, e.g. higher temperature, strong dilution, increased pressure, and lower ligand to metal ratio (initially 20:1).39 Gas consumptions analysis were run at the same time and confirmed the faster consumption of gas over 3 hours applying Rh/L6 showed compared to Rh/L1 (Fig. 4a). This observation was further validated by hourly sampling of the reaction mixture (Fig. 4b).
 | ||
| Fig. 4 Kinetic profile of L1 and L6 in the hydroformylation of 1-octene. Conditions: 1a (10.0 mmol), [Rh(CO)2acac] (0.014 mol%), monodentate ligand (0.071 mol%), CO:H2 (25 bar, 1:1), toluene (24 mL), 120 °C, 4 h. The conversion of 1a and, 2b, 1b, 3a (see ESI†) and regioselectivity of the products were measured by GC analysis using MeOH as solvent and isooctane as internal standard. (a) Gas consumption for both systems in the hydroformylation of 1-octene (b) time-yield plot for both systems in the hydroformylation of 1-octene (c) equation of TON and TOF for both systems in the hydroformylation of 1-octene. | ||
To demonstrate the highly (regio)selective hydroformylation in the presence of a monodentate phosphine ligand at higher temperature, the Rh/L6 system was applied to a broader range of olefins. For comparison, we conducted similar tests using 6-DPPON as a reference for typical self-assembling systems.
As shown in Table 1, as a general trend we observed both better aldehyde yields and higher selectivity to the linear aldehydes for all hydroformylated substrates. Although vinyl arenes are known to strongly favour branched aldehydes, through the formation of a η3-allyl intermediate,40 hydroformylation of styrene 6 proceeded to 7 in 82% yield with a linear selectivity of 69%. Similarly, 4-vinylanisole 10 was successfully converted to 11 with a yield of 97% and a linear selectivity of 56%. Dodecene 4 and allyl benzene 8 were converted to 5 and 9, respectively, in good yields and with excellent selectivity above 95% for the linear isomers. Several other substrates were tested for the Rh-L6 system, showcasing potential applications. For example, allyl methacrylate 18 was hydroformylated to 19 in 36% yield with a linear selectivity of 84%. Raising the pressure to 30 bars, yielded 75% of aldehyde but gave lower selectivity (66%) for the linear isomer. Maintaining syngas pressure and raising the ligand to metal ratio from 5:1 to 10:1 led to full conversion with a linear selectivity of 68%. Following standard optimized conditions, eugenol 26 was hydroformylated in good yield (76%) and excellent linear selectivity (93%). Notably, terminal diolefins 20 and 23 could be efficiently and highly selectively (linear selectivity >97%) converted to the corresponding dials 22 and 25. Here, the mono-hydroformylated products 21 and 24 were obtained in minor amounts (16 and 22% yield, respectively), too.
:H2 (10 bar, 1:1), toluene (4.3 mL), 120 °C, 4 h
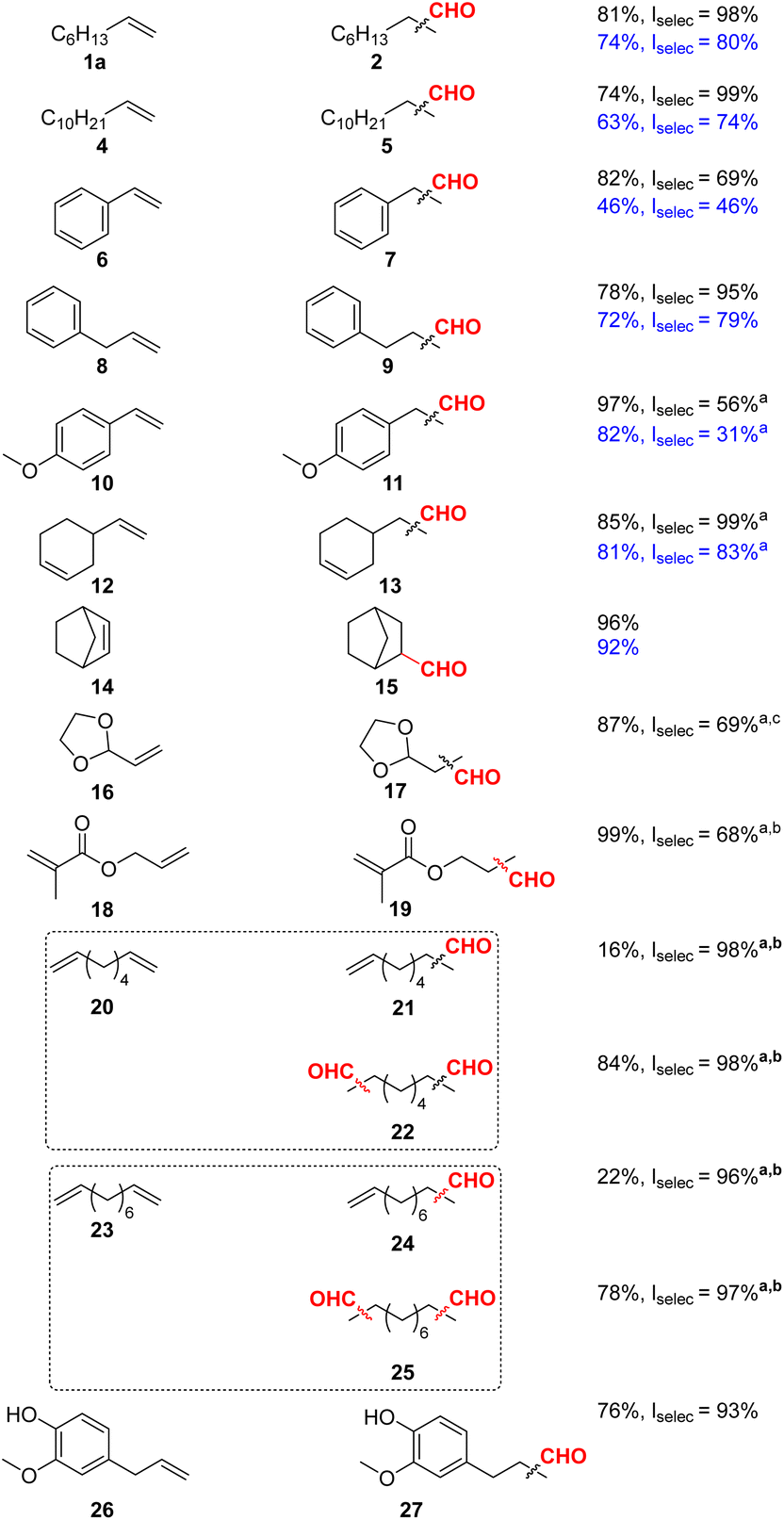
Notably, apart from all these successful examples, the methodology also showed some limitations (see ESI†). For example, both L1 and L6 proved to be inactive for the hydroformylation of cyclic olefins 30 and 32 (ESI Table 18†), for which the starting material was fully recovered. In addition, limonene 34, β-pinene 36 and 2-vinylpiridine 28 did not react in the desired manner, similarly providing the unreacted starting material with minimal isomerization in the case of 34 and 35.
Conclusions
Here, we present the synthesis and characterization of a small library of new self-assembling phosphines related to the known Breit ligand 6-DPPON. After optimizing crucial reaction parameters, e.g. solvents, temperature, and pressure, for the hydroformylation of 1-octene, the activity of all ligands was assessed and compared to the parent 6-DPPON. Upon variation of reaction conditions, it was discovered that Rh-L6 can perform hydroformylations with excellent regioselectivity at higher temperatures (>100 °C). The optimal catalyst results in selective hydroformylation of a diverse set of terminal olefins, including industrially relevant aliphatic substrates as well as vinyl arenes. A special feature of this newly described system is the efficient dicarbonylation of terminal non-conjugated dienes to give linear dials, which are valuable intermediates for various polymers.Author contributions
P. F. performed the experiments and drafted a manuscript; M. D. F. M performed the synthesis of ligands and characterization thereof; A. S. performed XRD analysis; H. J. performed the DFT calculations; R. J. participated in discussions and provided experimental equipment; M. B. planed the project and participated in discussions and revised the manuscript.Data availability
Data for this article, including compound characterizations, calculations, experimental data are available as ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 859910 (CO2PERATE). Additional funding from the Danish Excellence Center CADIAC is acknowledged. Further, we would like to thank the analytic department of LIKAT and Dr Wolfgang Baumann for relevant NMR experiments of synthesized complexes and ligands.References
- A. Börner and R. Franke, Hydroformylation: Fundamentals, Processes, and Applications in Organic Synthesis, Wiley-VCH, 2016 Search PubMed.
- J. Pospech, I. Fleischer, R. Franke, S. Buchholz and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 2852–2872 CrossRef CAS PubMed.
- R. Kumar and S. H. Chikkali, J. Organomet. Chem., 2022, 960, 122231 CrossRef CAS.
- P. Pongracz, L. Kollar and L. T. Mika, Green Chem., 2016, 18, 842–847 RSC.
- R. van Duren, J. I. van der Vlugt, H. Kooijman, A. L. Spek and D. Vogt, Dalton Trans., 2007, 1053–1059 RSC.
- E. Drent and P. H. M. Budzelaar, J. Organomet. Chem., 2000, 593, 211–225 CrossRef.
- L. P. Wu, I. Fleischer, R. Jackstell, I. Profir, R. Franke and M. Beller, J. Am. Chem. Soc., 2013, 135, 14306–14312 CrossRef CAS PubMed.
- W. Huang, X. X. Tian, H. J. Jiao, R. Jackstell and M. Beller, Chem. – Eur. J., 2022, 28, e202104012 CrossRef CAS PubMed.
- I. Piras, R. Jennerjahn, R. Jackstell, A. Spannenberg, R. Franke and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 280–284 CrossRef CAS PubMed.
- L. H. Slaugh and R. D. Mullineaux, J. Organomet. Chem., 1968, 13, 469–477 CrossRef CAS.
- B. X. Zhang, C. Kubis and R. Franke, Science, 2022, 377, 1223 CrossRef CAS PubMed.
- F. G. Delolo, C. Kubis, B. X. Zhang, H. Neumann, E. N. dos Santos, E. V. Gusevskaya and M. Beller, Catal. Sci. Technol., 2024, 14, 1524–1533 RSC.
- O. Roelen, Chemische Verwertungsgesellschaft Oberhausen m.b.H., Ger. Pat849548, US Pat. 2327066, 1943 Search PubMed.
- H. W. Bohnen and B. Cornils, Adv. Catal., 2002, 47, 1–64 CAS.
- M. Kranenburg, Y. E. M. Vanderburgt, P. C. J. Kamer, P. W. N. M. Vanleeuwen, K. Goubitz and J. Fraanje, Organometallics, 1995, 14, 3081–3089 CrossRef CAS.
- G. M. Vieira, A. V. Granato, E. V. Gusevskaya, E. N. dos Santos, P. H. Dixneuf, C. Fischmeister and C. Bruneau, Appl. Catal., A, 2020, 598, 117583 CrossRef CAS.
- Y. J. Yan and X. M. Zhang, J. Am. Chem. Soc., 2006, 128, 7198–7202 CrossRef CAS PubMed.
- T. Tewari, R. Kumar, A. C. Chandanshive and S. H. Chikkali, Chem. Rec., 2021, 21, 1182–1198 CrossRef CAS PubMed.
- C. P. Casey, G. T. Whiteker, M. G. Melville, L. M. Petrovich, J. A. Gavney and D. R. Powell, J. Am. Chem. Soc., 1992, 114, 10680–10680 CrossRef CAS.
- R. L. Pruett and J. A. Smith, Union Carbide, US Pat., 4148830, 1979 Search PubMed; Chem. Abstr. 1971 75 109844 Search PubMed.
- W. R. Moser, C. J. Papile, D. A. Brannon, R. A. Duwell and S. J. Weininger, J. Mol. Catal., 1987, 41, 271–292 CrossRef CAS.
- L. Gonsalvi, A. Guerriero, E. Monflier, F. Hapiot and M. Peruzzini, in Hydroformylation for Organic Synthesis, ed. M. Taddei and A. Mann, Springer, Berlin, Heidelberg, 2013, pp. 1–47 Search PubMed.
- C. Vogl, E. Paetzold, C. Fischer and U. Kragl, J. Mol. Catal. A: Chem., 2005, 232, 41–44 CrossRef CAS.
- V. S. Koshti, A. Sen, D. Shinde and S. H. Chikkali, Dalton Trans., 2017, 46, 13966–13973 RSC.
- D. T. Seidenkranz, J. M. McGrath, L. N. Zakharov and M. D. Pluth, Chem. Commun., 2017, 53, 561–564 RSC.
- P. Dydio, R. J. Detz and J. N. H. Reek, J. Am. Chem. Soc., 2013, 135, 10817–10828 CrossRef CAS PubMed.
- S. S. Nurttila, P. R. Linnebank, T. Krachko and J. N. H. Reek, ACS Catal., 2018, 8, 3469–3488 CrossRef CAS PubMed.
- K. Zhao, X. Z. Wang, D. C. He, H. L. Wang, B. Qian and F. Shi, Catal. Sci. Technol., 2022, 12, 4962–4982 RSC.
- P. R. Linnebank, A. M. Kluwer and J. N. H. Reek, Catal. Sci. Technol., 2024, 14, 1837–1847 RSC.
- B. Breit and W. Seiche, J. Am. Chem. Soc., 2003, 125, 6608–6609 CrossRef CAS PubMed.
- W. Seiche, A. Schuschkowski and B. Breit, Adv. Synth. Catal., 2005, 347, 1488–1494 CrossRef CAS.
- J. N. H. Reek, B. de Bruin, S. Pullen, T. J. Mooibroek, A. M. Kluwer and X. Caumes, Chem. Rev., 2022, 122, 12308–12369 CrossRef CAS PubMed.
- F. Bauer, N. Dierenbach and B. Breit, Asian J. Org. Chem., 2023, 12, e202300244 CrossRef CAS.
- V. Agabekov, W. Seiche and B. Breit, Chem. Sci., 2013, 4, 2418–2422 RSC.
- R. Franke, D. Selent and A. Borner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- U. Gellrich, J. Huang, W. Seiche, M. Keller, M. Meuwly and B. Breit, J. Am. Chem. Soc., 2011, 133, 964–975 CrossRef CAS PubMed.
- F. Jameel and M. Stein, J. Mol. Catal., 2021, 503, 111429 CrossRef CAS.
- A. Loupy, Top. Curr. Chem., 1999, 206, 153–207 CrossRef.
- U. Gellrich, T. Koslowski and B. Breit, Catal. Sci. Technol., 2015, 5, 129–133 RSC.
- I. del Rio, O. Pamies, P. W. N. M. van Leeuwen and C. Claver, J. Organomet. Chem., 2000, 608, 115–121 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of isolated compounds. See DOI: https://doi.org/10.1039/d4ob00714j |
| This journal is © The Royal Society of Chemistry 2024 |