
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemoenzymatic synthesis of amino-esters as precursors of ammonium salt-based surfactants from 5-hydroxymethylfurfural (HMF)†‡
Carlos
Moriana Herraiz
 ,
Karen S.
Arias
,
Maria J.
Climent
,
Sara
Iborra
* and
Avelino
Corma
*
,
Karen S.
Arias
,
Maria J.
Climent
,
Sara
Iborra
* and
Avelino
Corma
*
Instituto de Tecnología Química (Universitat Politècnica de València – Agencia Estatal Consejo Superior de Investigaciones Científicas), Avda dels Tarongers s/n, 46022, Valencia, Spain. E-mail: siborra@itq.upv.es; acorma@itq.upv.es; Fax: +34 963877809
First published on 22nd April 2024
Abstract
N-Substituted 5-(hydroxymethyl)-2-furfuryl amines have been obtained through the reductive amination of 5-hydroxymethylfurfural (HMF) with a variety of primary amines using a non-noble metal catalyst based on monodisperse Co nanoparticles covered by a thin carbon layer. The Co@C catalyst was highly active, selective and stable, allowing us to perform the reductive amination of HMF under very mild reaction conditions (60 °C and 4 bar H2) using ethanol as a green solvent and achieve the corresponding amino-alcohol in yields ranging from 80 to 99%. Moreover, the reaction was extended to other furanic aldehydes with excellent success. Furthermore, in order to synthesize amino-ester derivatives, precursors of ammonium salt-based surfactants, the reductive amination of HMF with methylamine was coupled with the selective esterification of the hydroxymethyl group of the furan ring with fatty acids using lipase CALB (Novozym 435) as a biocatalyst in 2-methyltetrahydrofuran as a green and enzyme compatible solvent, achieving practically total conversion to the corresponding amino-esters. The process was implemented in flow reactors by combining two consecutive fixed bed reactors, achieving a global yield of the amino-ester derivative of 85%, which was maintained over 86 h of operation.
1. Introduction
N-Substituted 5-(hydroxymethyl)-2-furfuryl amines constitute an important class of bioactive compounds and also serve as intermediates for the synthesis of a variety of products with pharmacological activities1–3 such as calcium antagonistic activity, cholinergic activity, antimuscarinic activity, and carcinogenesis inhibition (see Scheme 1). Moreover, with the appropriate substituents, they can also serve as precursors of ammonium salt-based surfactants.4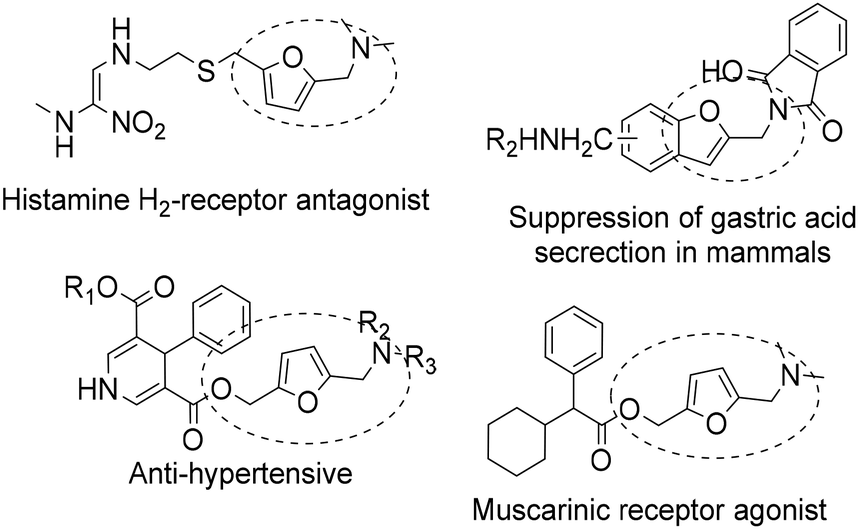 | ||
| Scheme 1 Examples of biomolecules showcasing the aminoalkyl furan functionality.5–11 | ||
N-Substituted-5-(hydroxymethyl)-2-furfuryl amines are typically synthesized through a Mannich-type reaction starting from furfural or furfuryl alcohol, formaldehyde and primary amines1 (Scheme 2). However, the main drawbacks of this process are the harsh reaction conditions that are usually required, leading to low selectivity towards the target compound. Hence, an alternative sustainable method to produce this class of amines could be through the reductive amination of 5-hydroxymethyl furfural (HMF) sourced from lignocellulosic biomass.
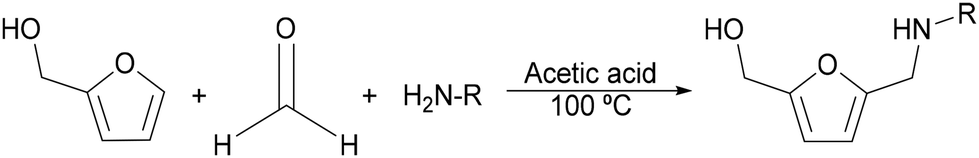 | ||
| Scheme 2 Mannich-type reaction for the synthesis of N-substituted-5-(hydroxymethyl)-2-furfuryl amines. | ||
HMF, produced by the acid catalysed dehydration of hexoses, is a versatile lignocellulosic biomass derived platform molecule which can be used for the production of many different compounds such as bioplastics, polymers, pharmaceutical products, surfactants, adhesives, and liquid fuels.12–16
The direct reductive amination of carbonyl compounds with ammonia or primary (or secondary) amines using molecular hydrogen involving the in situ hydrogenation of the pre-formed imine intermediate has been considered as the best strategy to produce amines (Scheme 3)17,18 in comparison with methods that use other reducing agents such as metal hydrides.19
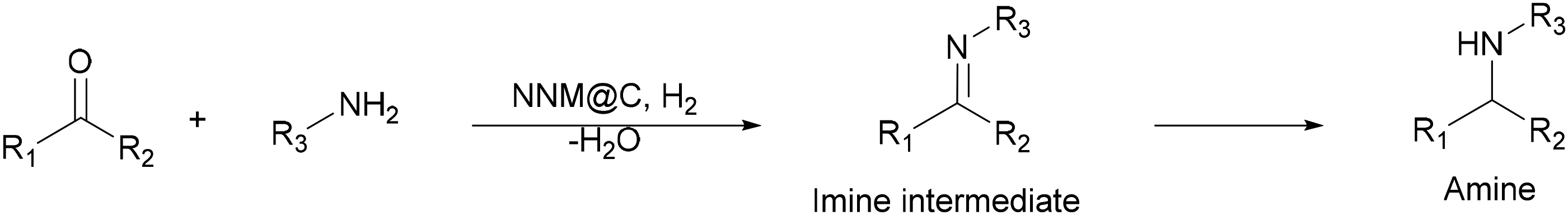 | ||
| Scheme 3 Direct reductive amination of carbonyl compounds. | ||
A variety of homogeneous and heterogeneous catalysts containing noble and non-noble metals such as Ru, Pd, Pt, Fe, Ni or Co have been reported for the reductive amination of different carbonyl compounds employing hydrogen.20 Particularly, the reductive amination of HMF using hydrogen as a reducing agent may involve other competitive reactions, such as the hydrogenation of the formyl group, the hydrogenation of the furanic ring, the removal of hydroxyl or carbonyl groups, and the furan ring opening. Therefore, the selection of the metal catalyst and reaction conditions is crucial to achieving high selectivity to the furfurylamine derivatives. A variety of homogeneous and heterogeneous catalysts based on noble metals such as a homogeneous Ru(II) complex catalyst (Ru(DMP)2Cl2),21 gold supported on titania assisted by CO/H2O,22 Pd nanoparticles supported on carbon23 or immobilized in a porous MOF/polymer composite,24 Pt/Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25 and Ru deposited on Nb2O5·nH2O26 have been recently applied for the reductive amination of HMF with different primary and secondary amines to synthesize N-substituted 5-(hydroxymethyl-)2-furfuryl amines.
25 and Ru deposited on Nb2O5·nH2O26 have been recently applied for the reductive amination of HMF with different primary and secondary amines to synthesize N-substituted 5-(hydroxymethyl-)2-furfuryl amines.
However, from the point of view of a more sustainable chemistry, the use of low-cost and more abundant non-noble metal catalysts represents an interesting alternative strategy to noble metals.27,28 However, to date, studies of the reductive amination of HMF for the synthesis of N-substituted 5-(hydroxymethyl)-2-furfurylamines using non-noble metal-based catalysts are rather limited and most of them are based on Ni catalysts. For instance, Villard et al.29 performed the reductive amination of HMF with alanine sodium salt under hydrogen pressure (5 bar), using Ni RANEY® (20 wt%) as the catalyst, although low yield (38% after 48 h) of the corresponding secondary amine was obtained. The same process was studied by Kirchhecker et al.,30 obtaining similar results. Shi et al.31 performed the reductive amination of HMF with different primary amines, using an optimized Ni6AlOx catalyst. Yields between 76 and 88% of the corresponding secondary amine were achieved by working at 100 °C and 3 bar of hydrogen. Chieffi et al.32 reported the reductive amination of HMF with different amines using a carbonized filter paper supported FeNi alloy as the catalyst. The reactions were performed in a continuous fixed bed reactor (WHSV = 24 h−1) under 10 bar of hydrogen and at temperatures between 100 and 125 °C. Although high conversion of HMF (>99%) was achieved, the selectivity to the secondary amine was limited to 78%. Better results (yields between 55 and 97%) were obtained by Bukhtiyarov et al.33 using CuAlOx in a flow reactor working at 100 °C and 10 bar of hydrogen; however, the possible catalyst deactivation was not studied. More recently, Jagadeesh et al.34 reported the reductive amination of HMF using silica-supported cobalt nanoparticles (prepared by the immobilization and pyrolysis of a Co-terephthalic acid–piperazine MOF template on silica). Reactions were performed at 70 °C under 20 bar of hydrogen and excellent yields of a variety of N-substituted hydroxymethyl furfuryl amines were obtained (85–96%). However, the catalyst experienced a considerable loss of activity after the 3rd recycling run. In Table 1 the results reported with the different catalysts for the reductive amination of HMF with aniline have been compared.
| Entry | Catalyst | Catalyst amount | Solvent | P H2 (bar) | t (h) | T (°C) | Y (%)a | S (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Yield of N-phenyl-5-(hydroxymethyl)-2-furfuryl amine. b Fixed bed reactor. | |||||||||
| 1 | Cu/SiO2–CeO2 | 23.6 mol% | THF | 10 | 9 | 150 | 50 | 50 | 35 |
| 2 | Co-terephthalic acid-piperazine@SiO2-800 | 5 mol% | EtOH | 20 | 16 | 70 | 95 | 95 | 34 |
| H2O | 94 | 94 | |||||||
| 3 | Ni6AlOx | 31.7 wt% | H2O | 3 | 6 | 100 | 85 | 85 | 36 |
| 5b | CuAlOx | 165 mg | MeOH | 10 | 3 | 100 | 97 | 97 | 33 |
| 6 | Co@C | 16.1 mol% | EtOH | 4 | 6 | 60 | 99 | 99 | This work |
Recently, our group has developed mono- and bimetallic catalysts based on non-noble metal nanoparticles (Co@C, CoNi@C, CoW@C, and FeCu@C) which are partially coated by a few layers of carbon. These catalysts displayed excellent activity and selectivity in hydrogenation reactions such as the chemoselective reduction of nitroarenes into anilines, reduction of levulinic acid into γ-valerolactone,37,38 hydrogenation of quinolines,39 and reduction of HMF to bis-(2,5-hydroxymethyl)furfural40 and 3-hydroxymethylcyclopentylamine41
The presence of cracks on the carbon shell allows the diffusion of the reactants to the metal, while the carbon coating easily promotes the in situ reduction of the oxide species on the surface of the metal nanoparticle under the reaction conditions by activating the hydrogen on the metal part of the catalyst. Moreover, the carbon coating preserves the non-noble metal from overoxidation, agglomeration, and metal leaching,37,38 which are the main causes of deactivation of metal nanoparticles.
In view of the high performances and stability of these nanoparticles in hydrogenation reactions, along with their easy and sustainable synthesis method, we have expanded the spectrum of transformations of these nanoparticles, focusing our interest on the reductive amination of HMF. Thus, here we have prepared Ni@C and Co@C nanoparticles that have been studied for the reductive amination of HMF with different primary amines.
This research considers sustainable development goals and the growing demand for surfactants, with a global market size that is expected to reach $58.5 billion by 202742 with a clear increasing interest in designing new renewable and biodegradable surfactants. In this sense, HMF, due to its renewability and biodegradability, has been recognized as an important building block for designing biobased surfactants.4,43 However, because the global production of HMF is still limited compared with other platform chemicals (the global market size of 5-hydroxymethylfurfural reached USD 61 million in 2022 and is expected to reach USD 68.2 million by 2028),44 the use of HMF as a starting compound for the synthesis of surfactants is still at an early stage. Nevertheless, this emerging topic will have an important impact on the design of new biobased surfactants as evidenced by the wide variety of strategies recently developed to combine the polar and non-polar moieties onto the furan ring to produce novel amphiphilic molecules.4,43
As commented above, N-substituted 5-(hydroxymethyl)-2-furfuryl amines may serve as interesting precursors for quaternary ammonium salt surfactants, which have a wide variety of applications such as fabric softeners, antistatic agents, etc., while also possessing high biological activity against microorganisms. Particularly, furan-based ammonium salts have been described as analogues to benzyl quaternary ammonium salt surfactants.45 Moreover, the rate of biodegradation of surfactants can be increased by connecting the polar head and the hydrophobic tail in their structure through a labile bond that can be easily cleaved under chemical conditions or by enzymes.
Considering these aspects, here, we have synthesized precursors of ammonium salt surfactants through a linear chemoenzymatic cascade reaction by coupling the reductive amination of HMF catalysed by non-noble metal nanoparticles with the selective esterification of the hydroxymethyl group of the furan ring with fatty acids using lipase (CALB) as the biocatalyst (Scheme 4). As far as we now, this is a novel approach to obtain precursors of a new class of furan biobased esterquat surfactants, which are defined as surfactants containing cleavable ester bonds as a lipophilic moiety and a polar group containing the quaternary ammonium head group.
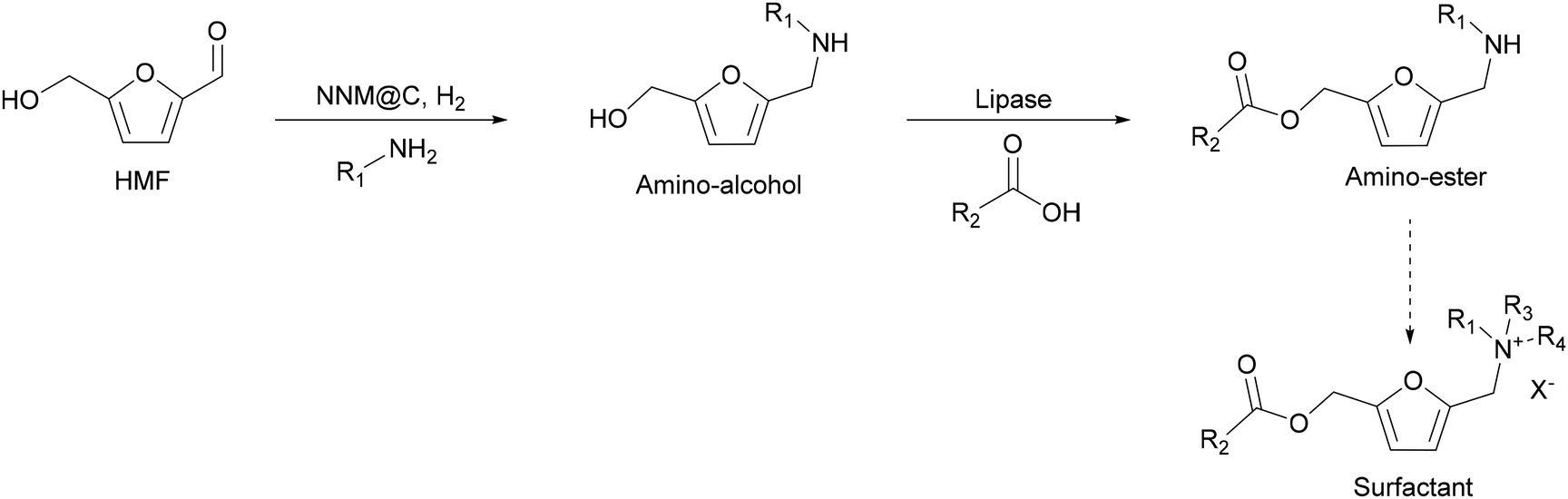 | ||
| Scheme 4 Cascade reaction to produce surfactants coming from HMF. | ||
2. Results and discussion
2.1 Catalyst characterization
Co@C and Ni@C monodisperse nanoparticles were prepared following the hydrothermal method described in the Experimental section using the corresponding oxides as a metal precursor and glucose as a source of carbon.The X-ray diffraction (XRD) patterns of Co@C and Ni@C nanoparticles are presented in Fig. 1, where the diffraction peaks at 44.3, 51.5, and 75.8° corresponding to Co0 (44.2), (51.5), and (75.8) (PDF 00-015-0806) are observed in the Co@C sample (Fig. 1, below), whereas the peaks which appear at 44.5, 51.8, and 76.3° assigned to Ni0 (44.5), (51.8), and (76.3) (PDF 00-004-0850) are observed in the Ni@C sample (Fig. 1, above).
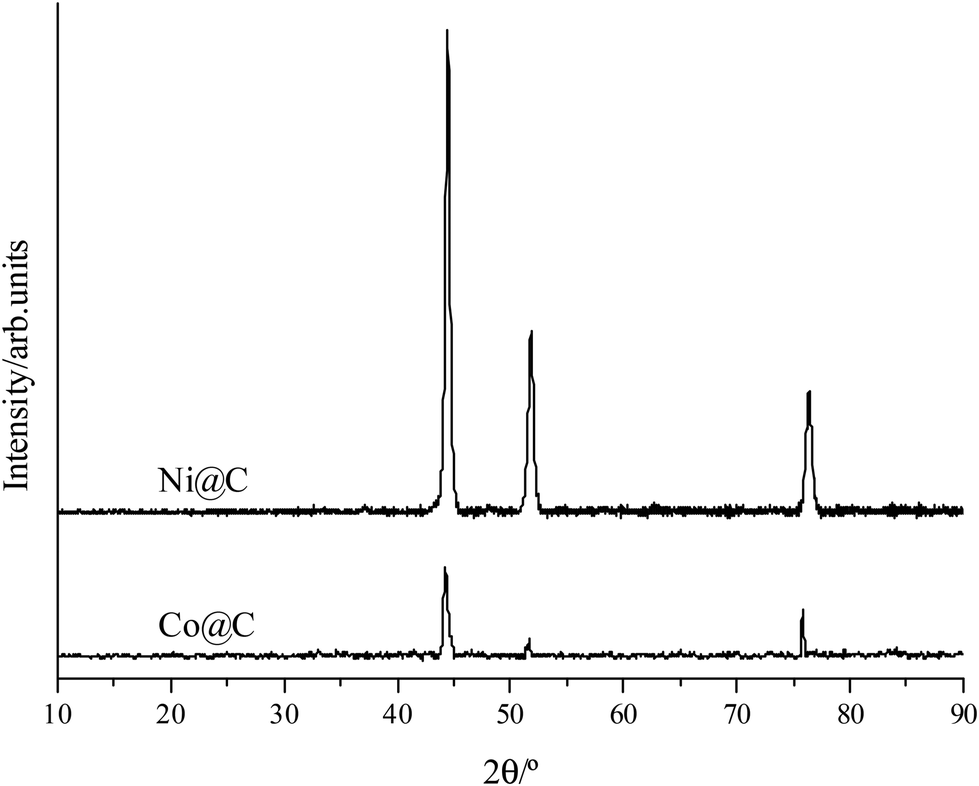 | ||
| Fig. 1 XRD patterns of Co@C and Ni@C nanoparticles. | ||
Furthermore, high-resolution transmission electron microscopy (HRTEM) images shown in Fig. S1,‡ along with energy-dispersive X-ray spectroscopy, revealed that the metal nanoparticles, ranging in size from 10 to 100 nm, exhibit a core–shell structure. This structure comprises a metallic core of either Co or Ni covered by a thin carbon layer serving as the shell. Notably, Fig. S1‡ illustrates that the lattice spacing of both monometallic Co@C and Ni@C catalysts measures 0.20 nm, corresponding to the (111) plane of metallic cobalt and nickel species. Moreover, close examination of the Co@C and Ni@C nanoparticles revealed patches of Co3O4 and NiO adjacent to the carbon shell. These patches likely result from the surface re-oxidation of the metallic species after preparation and exposure to air. Through ICP analysis, it was determined that the metal content within the nanoparticles comprised 94–95%, with the remaining 5–6% constituting the carbon coating. Additionally, a H2 chemisorption experiment of the Co@C catalyst was carried out, revealing 41.76 μmol g−1 (0.04176 mmol g−1) of active Co sites in the catalyst, which corresponds to a metal dispersion of 0.5128%. These comprehensive characterization findings align consistently with our previously documented results regarding the structural attributes of these nanoparticles.37,38,40
2.2 Catalytic activity
As a proof-of-concept investigation, we started our study by performing the reductive amination of HMF with aniline as a model reaction, using Co@C as a catalyst, followed by further optimization of the reaction conditions. The reaction was first accomplished at 100 °C, under 6.5 bar of H2 pressure and using ethanol as a green solvent. In this process, the nucleophilic aniline attacks the formyl group of the HMF, giving imine 1 as an intermediate, which is subsequently hydrogenated to the corresponding secondary amine N-phenyl-5-(hydroxymethyl)-2-furfuryl amine, 2 (Scheme 5).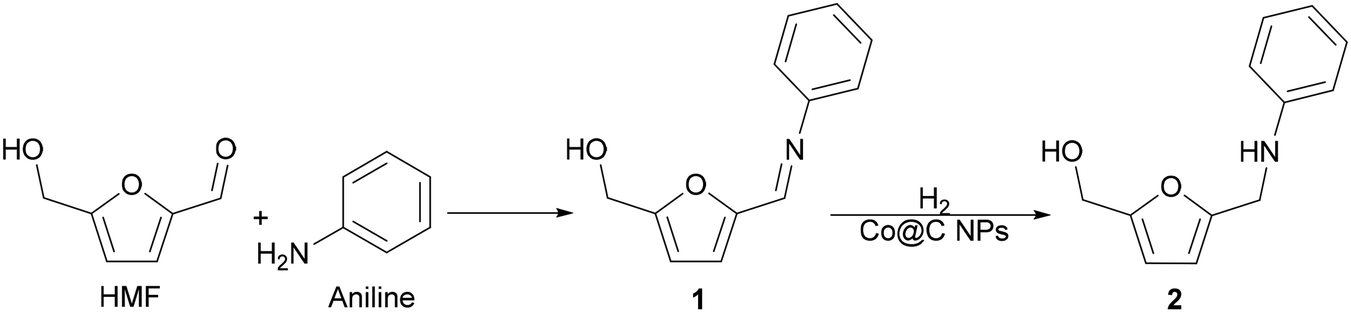 | ||
| Scheme 5 Reductive amination of HMF with aniline. | ||
During the process it was observed that the intermediate imine 1 rapidly formed, in such a way that when introducing H2 in the reactor (see the Experimental section) the complete conversion of HMF into the imine intermediate 1 was already achieved. Additionally, blank experiments performed in the absence of a catalyst or in the absence of hydrogen showed that the imine 1 was the only product observed, indicating that the formation of the intermediate imine does not require the presence of a catalyst.
Under these reaction conditions, 98% conversion of HMF with 92% selectivity to the amine 2 was obtained after 2 h of reaction time, with N-phenyl-5-methyl-2-furfuryl amine (4) being the main byproduct, while N-phenyl-5-methyl-2-furfuryl imine (3) was detected in minor amounts. The results suggest (see Fig. S2‡) that the formation of the amine 4 comes from the over-reduction of the hydroxymethyl group of the imine 1 and amine 2 (Scheme 6). Then, in order to increase selectivity by avoiding hydrodeoxygenation reactions, temperature and H2 pressure were optimized (Table S1‡) (Fig. S2–S6‡). As expected, decreasing the temperature from 100 to 60 °C (entries 1–3, Fig. S2–S4‡) improved the selectivity to 2 up to 97% (at total HMF conversion), while decreasing the H2 pressure up to 3 bar (while working at 60 °C) (Fig. S6‡) practically preserved the selectivity to 2, even though a longer reaction time for the total imine conversion was required (22 h). However, working at 4 bar, the selectivity to 2 was conserved and the reaction time required for the total conversion was maintained at an acceptable value (6 h) (see Fig. S5‡). Furthermore, we observed that upon increasing the reaction time up to 24 h, the amine 2 was stable and no further over-reduction occurred (Fig. 2).
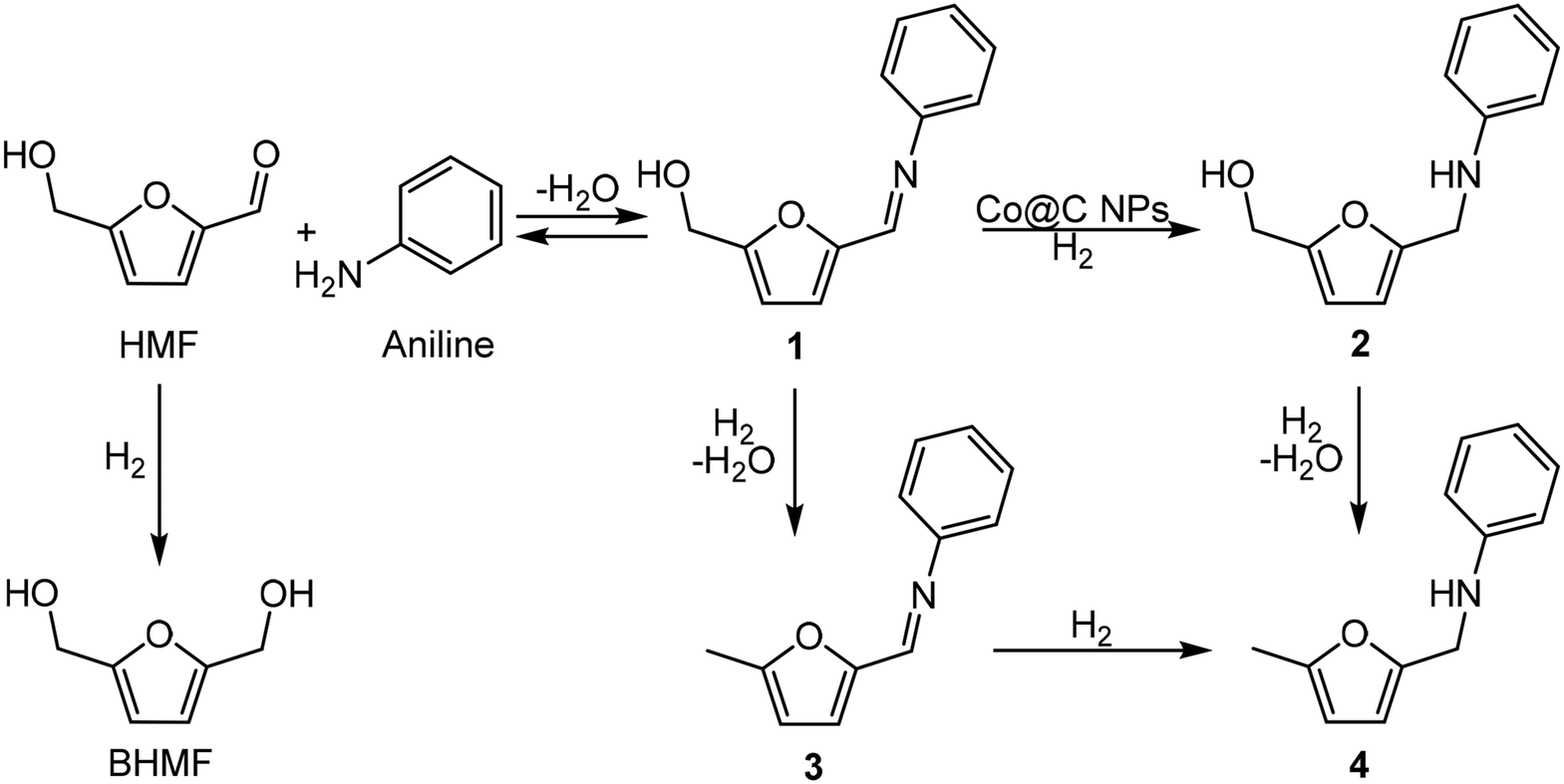 | ||
| Scheme 6 Proposed reaction pathway for the reductive amination of HMF with aniline. | ||
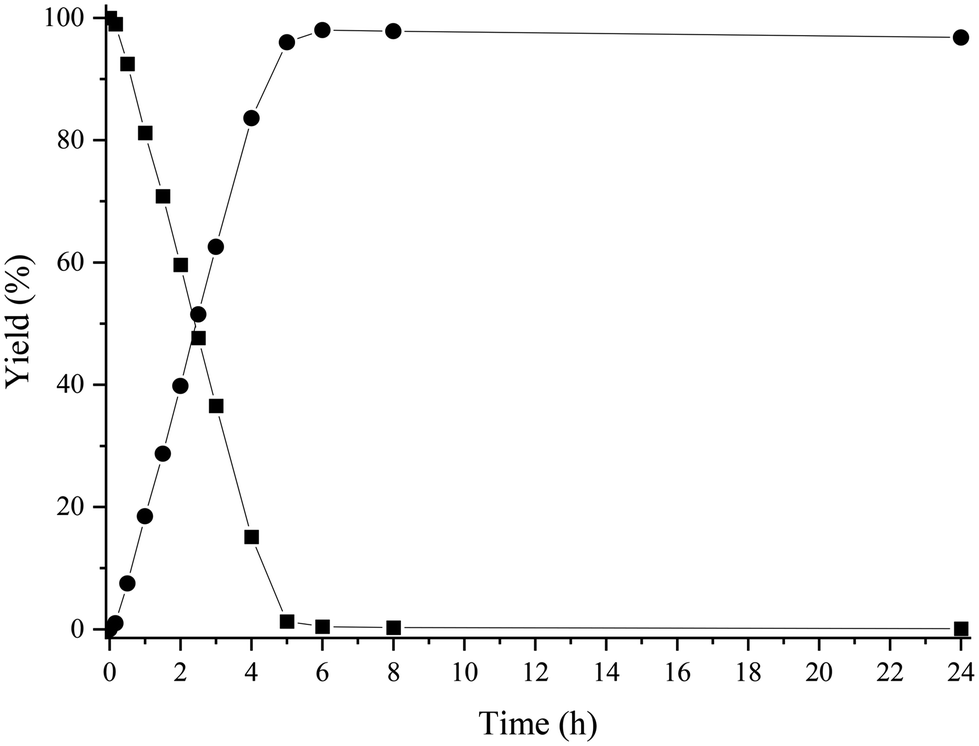 | ||
| Fig. 2 Kinetic study of the reductive amination of HMF with aniline. Reaction conditions: HMF (2 mmol), aniline (2.4 mmol), EtOH (4 mL), 16.1 mol% of Co@C NPs (20 mg), 4 bar of H2 and 60 °C. Imine 1 (■), amine 2 (●). | ||
After optimizing the reaction conditions at 60 °C and 4 bar H2, the efficiency of cobalt on a different support (Co/SiO2) was analysed. However, this catalyst exhibited poor activity and produced only 8% yield after 24 hours of reaction. Moreover, we tested a commercial Co oxide (Co3O4), which was pre-reduced at 450 °C under H2 flow prior to the reaction. In a similar way, after 20 hours of reaction time, the results only showed a 20% conversion (Table 2). These results confirm, as indicated above, the fact that the presence of the carbon shell in the Co@C catalyst not only preserves the Co nanoparticles from the deep oxidation when exposed to air but also allows the easier reduction of the metal species under the mild reaction conditions used in the process. Moreover, and for comparison purposes, we tested the Ni@C sample under the same reaction conditions. The sample showed very good selectivity to the target amine (99%) under the optimized reaction conditions; however, the catalytic activity was considerably lower compared with the Co@C catalyst (see Table 2 and Fig. S7‡), and 20 h of reaction time was required to achieve 98% conversion. The lower catalytic activity of the Ni@C catalyst with respect to the Co@C sample can be attributed to the higher amount of NiO species in the Ni@C sample compared to the amount of Co oxides on Co@C samples, as previously observed by XPS analysis.41
| Entry | Catalyst | Time (h) | Conversion HMF (%) | Yield 2 (%) | Selectivity 2 (%) |
|---|---|---|---|---|---|
| a Reaction conditions: HMF (2 mmol), aniline (2.4 mmol), catalyst (20 mg), EtOH (4 mL) and 4 bar of H2. Reactions performed after a previous pre-reduction of the catalyst. | |||||
| 1 | Co@C NPs | 6 | 99 | 98 | 99 |
| 2 | Co/SiO2 | 24 | 8 | 8 | 100 |
| 3 | Co3O4 commercial | 20 | 20 | 19 | 95 |
| 4 | Ni@C NPs | 22 | 98 | 97 | 99 |
Interestingly, comparing the results obtained with the Co@C sample with those obtained on other non-noble metal catalysts previously reported in the literature for the reductive amination of HMF with aniline (see Table 1), we can conclude that our catalyst is more active being able to operate at lower pressures and temperatures with high yields of the desired product.
2.3 Stability and reusability of the Co@C catalyst
An important feature for the application of any heterogeneous catalyst is stability and recyclability. To evaluate if Co leaching occurs during the reaction, an additional experiment was carried out where the reductive amination of HMF was stopped after 2 h of reaction. At this point, the catalyst was removed with a magnet while hot and the reaction was continued for additional 10 hours. No further conversion was detected during this time (Fig. S8‡). In addition, the reaction mixture was analysed by inductively coupled plasma analysis (ICP-analysis), and the amount of Co was below 0.1 ppm in the solution.The reusability of the catalyst was studied by removing the Co@C catalyst with a magnet after conducting the first run, thoroughly washing it with methanol and reusing the catalyst for subsequent cycles after hydrogenation at 150 °C for 2 h. As can be seen in Fig. S9,‡ the conversion and selectivity were practically maintained after six consecutive runs. However, the imine conversion slightly decreased after the second cycle (see Fig. S9‡). X-ray diffraction (XRD) and electron transmission microscopy (TEM) studies of the used catalyst after the sixth cycle (Fig. S10 and S11‡) indicate that the monodisperse Co nanoparticles are conserved after the recycling process, showing that no agglomeration of the catalyst nanoparticles occurs during the reductive amination of HMF. Therefore, the loss of activity can be attributed to the adsorption of organic material on the catalyst. To check that, after the sixth cycle of reaction, the catalyst was extracted with methanol using a Soxhlet apparatus. The GC analysis of the organic material extracted using Soxhlet (8 wt% organic material with respect to catalyst weight) showed that it was mainly composed of the amino-alcohol. The reused sample extracted using Soxhlet showed that the catalyst had only partially regained its initial activity, indicating the presence of unremoved organic material on the catalyst, causing a loss of activity.
2.4 Catalyst scope
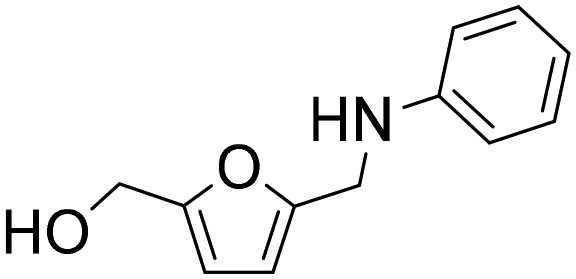
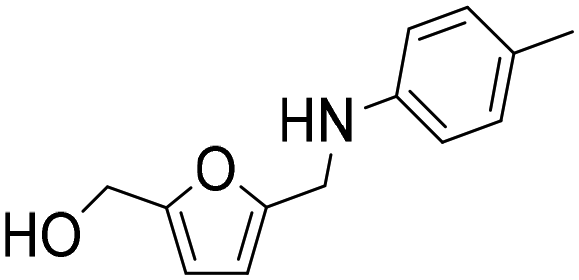
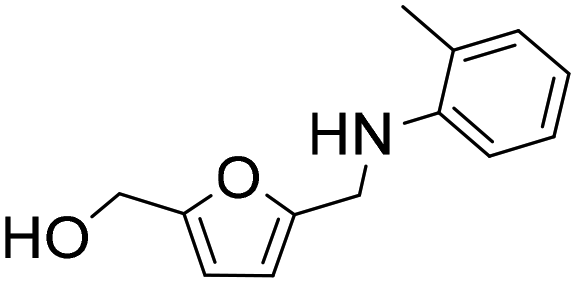
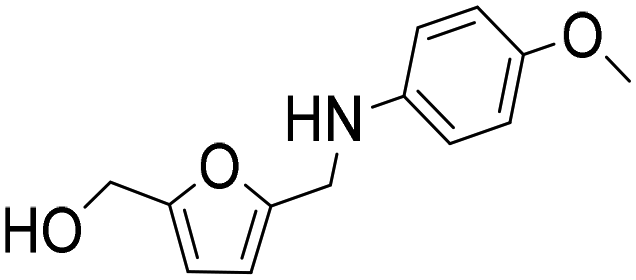
The general applicability of the Co@C NP catalyst in the reductive amination was evaluated by performing the reaction with different furanic aldehydes and amines as starting reagents. In Table 3, results of the reductive amination of HMF with various aromatic and aliphatic amines under the optimal reaction conditions are presented. The influence of the electronic and steric effects of different substituted anilines on the reductive amination of HMF was studied by selecting anilines bearing electron-donating groups (para-CH3, ortho-CH3, and OCH3) (entries 2–4, Table 3) and electron-withdrawing groups (Br and COCH3) (entries 5 and 6, Table 3). As can be observed, substituted anilines with electron-donating groups such –CH3 increase the reaction rate with respect to aniline (entries 2–4), resulting in practically total conversions and high yields of the corresponding amino-alcohol in shorter reaction times, which agrees with previously reported results.23,33 However, the presence of a stronger electron donor such as the methoxy group (entry 4) considerably decreases the reaction rate, and 19 h reaction time was required to achieve high yield of the corresponding amino-alcohol. These results indicate the existence of a strong negative steric effect caused by the presence of a bulkier substituent in the aromatic ring. The introduction of electron-withdrawing groups in the aromatic ring decreases the reaction rate with respect to aniline,23,33 while the negative steric effect is again observed (entries 5 and 6) in the case of 4-acetylaniline (entry 6). Interestingly, in both cases, competitive hydrogenation reactions such as dehydrohalogenation or reduction of the carbonyl group were not observed. Reductive amination with aliphatic amines was also efficiently performed with high selectivity, although steric effects were also observed for amines between 2 and 7 carbon atoms (entries 7–10). The reaction with amino-alcohols such as ethanolamine and 2-methylethanolamine (entries 11 and 12) afforded the corresponding amino-diols in good yields. Secondary amines, such as diethylamine, yielded the corresponding tertiary amine in good yield (entry 13). In this case, since the reaction does not occur through the hydrogenation of the imine intermediate, but through the hydrogenolysis of a hemiaminal, a higher amine concentration was required to shift the equilibrium towards the hemiaminal. Even so, the competitive hydrogenation of HMF into 2,5-bis(hydroxymethyl)furan (BHMF) occurred with 12% yield. Finally, the reductive amination with ammonia (entry 14) was performed using an ammonia solution in methanol and under 10 bar of hydrogen, affording the 5-hydroxymethylfurfuril amine in 85% yield. However, in this case, the molar balance only accounted for 85%, indicating that polymerization of the highly reactive imine intermediate takes places to some extent.|
|
|||||
|---|---|---|---|---|---|
| Entry | Product | Time (h) | Conversion imine (%) | Yield amine (%) | Selectivity amine (%) |
| a Reaction conditions: HMF (2 mmol), amine (2.4 mmol), Co@C NPs (20 mg), EtOH (4 mL), 60 °C, 4 bar H2. b Using 2 mmol of the primary amine. c The only by-product detected was BHMF produced from the hydrolysis of the imine and subsequent hydrogenation. d Silylated samples with N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) before being measured in GC. e Using 4 mmol of the secondary amine. f HMF conversion. g Reaction conditions: HMF (2 mmol), 4 mL of NH3 (2 M in MeOH, 8 mmol) and 10 bar of H2. h Reaction conditions: HMF (2 mmol), nitrobenzene (2.8 mmol), EtOH (4 mL), 120 °C, and 20 bar of H2. | |||||
| 1 |
|
1 | 21 | 21 | 100 |
| 6 | 99 | 99 | 100 | ||
| 2b |
|
1 | 28 | 28 | 100 |
| 4 | 99 | 99 | 100 | ||
| 3 |
|
1 | 28 | 26 | 93 |
| 5 | 99 | 93 | 94c | ||
| 4b |
|
1 | 10 | 9 | 99 |
| 19 | 99 | 98 | 99 | ||
| 5 |
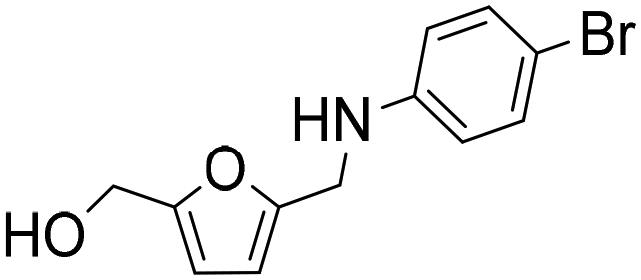
|
1 | 29 | 27 | 94c |
| 7 | 100 | 95 | 95c | ||
| 6b |
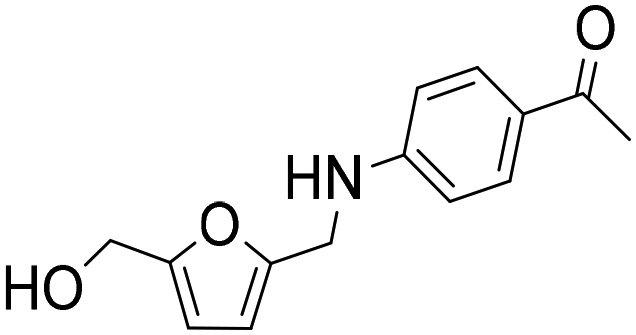
|
1 | 6 | 5 | 84c |
| 20 | 93 | 84 | 90c | ||
| 7 |
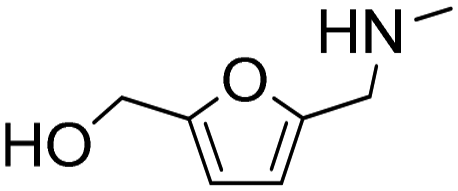
|
1 | 14 | 14 | 100 |
| 7 | 99 | 99 | 100 | ||
| 8 |
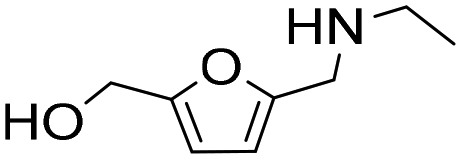
|
1 | 2 | 2 | 100 |
| 24 | 97 | 97 | 100 | ||
| 9 |
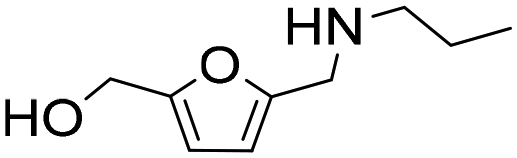
|
1 | 2 | 2 | 100 |
| 22 | 81 | 81 | 100 | ||
| 10 |
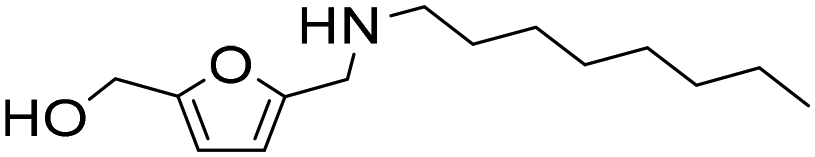
|
1 | 11 | 10 | 98c |
| 23 | 98 | 96 | 98c | ||
| 11d |
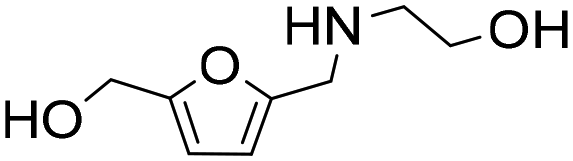
|
1 | 14 | 14 | 100 |
| 24 | 97 | 97 | 100 | ||
| 12d |
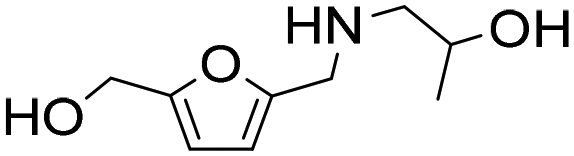
|
1 | 6 | 6 | 99 |
| 24 | 99 | 97 | 98c | ||
| 13e |
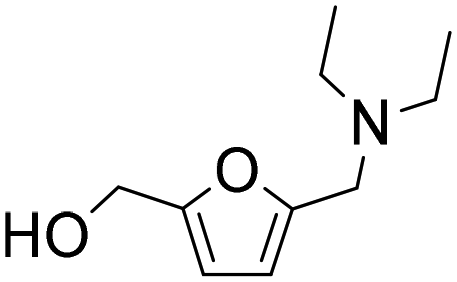
|
1 | 79f | 68 | 86c |
| 4 | 100f | 88 | 88c | ||
| 14g |
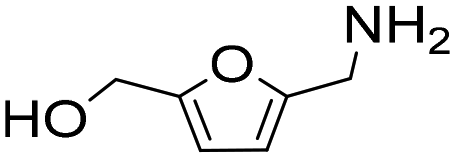
|
1 | 100 | 29 | 29 |
| 3 | 100 | 85 | 85 | ||
| 15h |
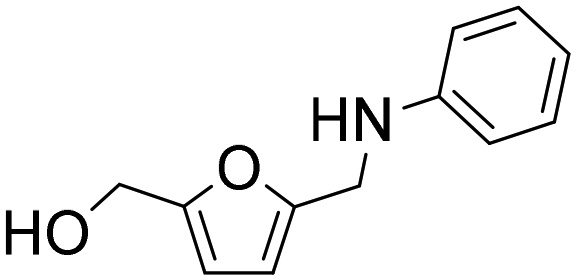
|
3 | 100 | 90 | 90c |
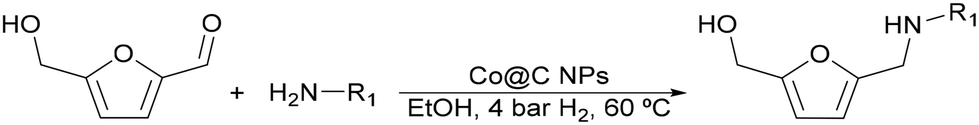
Finally, the possibility to obtain furfuryl amine-alcohol derivatives starting from nitro-compounds through a cascade one pot reaction was studied using nitrobenzene as the aniline precursor. In this case, a nitrone intermediate46 and traces of the hydroxylamine derivative were detected in the reaction media (Fig. S25‡) and the target amine 2 was obtained in 90% yield. The results showed that the cascade one-pot process starting from the most inexpensive and readily available nitrobenzene is fully feasible, although the selectivity to the amino-alcohol was slightly lower than that obtained when starting directly from aniline. This is due to the formation of 2,5-bishydroxymethylfuran (BHMF) (detected as a byproduct in 10% yield). The secondary character of BHMF indicates that it should be produced by the hydrolysis of the imine intermediate into HMF, which is subsequently hydrogenated into BHMF.
The Co@C catalyst was also applied to the reductive amination of furfural and 2-methyl furfural with different aromatic and aliphatic amines. As can be observed from Table S2,‡ both furanic aldehydes afforded the corresponding secondary amines in good to excellent yields and selectivities, while an important negative steric effect was observed particularly in the case of 2-methylfurfural. This could be associated with a different adsorption mode of this substrate (with respect to HMF and furfural) on the catalyst surface caused by the presence of the methyl group in the furan ring that affects the hydrogenation rate of the imine intermediate. Note that even with the smaller methylamine, the reaction rate was very low (entry 7, Table S2‡).
2.5 Combining chemocatalytic reductive amination of HMF with selective biocatalyzed esterification
Following our interest to synthesize 5-(N-substituted aminomethyl)-2-furanmethanol ester derivatives, precursors of ammonium salt surfactants, we studied the second step, i.e., the selective esterification of the hydroxymethyl group in the N-substituted-5-(hydroxymethyl-)2-furfuryl amine substrate. At this point, it is interesting to note that the conventional chemical acylation suffers from significant limitations, such as the requirement of hazardous reagents and solvents and high reaction temperatures. Moreover, in the case of amino-alcohols, one of the most serious problems is to control the chemoselectivity towards esterification or amidation and, therefore, additional steps for alcohol or amino group protection and deprotection are required, thus increasing the production of wastes. Contrarily, enzymes are environmentally friendly catalytic systems able to work under mild reaction conditions and can give high selectivity to the desired product.In this sense, lipases are excellent biocatalysts in O-acylation, transesterification and N-acylation reactions, and in the case of amino-alcohols the chemoselectivity toward the hydroxy or amino group has been found to depend on factors such as solvent, substrate structure,47–50 and acyl donor.51,52 Interestingly, among the different lipases used in organic synthesis, Candida antarctica lipase B (CALB) has been reported to selectively acylate alcohols when competing with amines.50,53,54 Therefore, commercially available lipase B from Candida antarctica immobilized on microporous acrylic resin (Novozym 435) was selected as the biocatalyst for our purpose. Moreover, Novozym 435 has been successfully used as the biocatalyst for the esterification and transesterification of HMF and its derivatives.40,55,56 In these studies, it was found that the bioesterifications of HMF and its derivatives with fatty acids can be successfully performed using a variety of solvents such as 2-methyltetrahydrofuran (2-MTHF), 1,4-dioxane or acetone in the presence of molecular sieves. In fact, the elimination of the water released during the esterification by the molecular sieves was critical to obtain the target esters in high yields.
Then, taking into account these findings, we first tested the bioesterification of N-methyl-(5-hydroxymethyl)-2-furfuryl amine (5), with hexanoic acid using Novozym 435, at 35 °C, and 2-MTHF as a lipase compatible solvent in the presence of molecular sieves (Scheme 7). The optimization of the molar ratio of substrates and the amount of the biocatalyst was performed under these reaction conditions (see Table S3‡). As can be observed, a molar ratio between the amino-alcohol and hexanoic acid of 1:3 (entry 3) was required to shift the equilibrium toward practically complete conversion (99%) into the amino-ester (6). Moreover, it is interesting to note that when the carboxylic acid was in excess (entries 2 and 3) the selectivity to the amino-ester was also 100%, indicating the high selectivity of Novozym 435 towards esterification. In addition, when the amount of enzyme was decreased to half, as expected, the process required a longer reaction time to achieve practically total conversion (entry 5).
 | ||
| Scheme 7 Enzymatic esterification reaction of the amino-alcohol 5 with carboxylic acids in the presence of Novozym 435. | ||
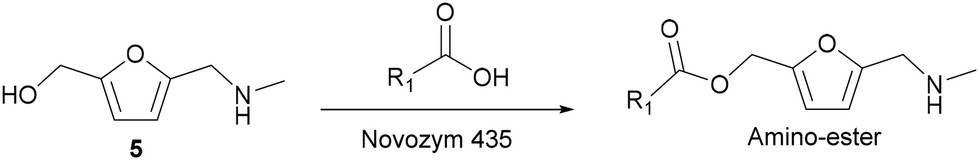
Then, we selected a molar ratio of amino-alcohol to carboxylic acid of 1:3 and 62 mg of Novozym 435 as the optimal reaction conditions (see Fig. 3). Under these conditions, the reuse and stability of the lipase were studied by performing the enzymatic process during 6 consecutive cycles. As can be seen in Fig. 4, lipase deactivation was not observed, achieving similar conversion and 100% selectivity to the amino-ester (6) in all cases. In addition, an experiment without the use of molecular sieves was carried out, resulting in a maximum yield of 40% after 0.5 h of reaction.
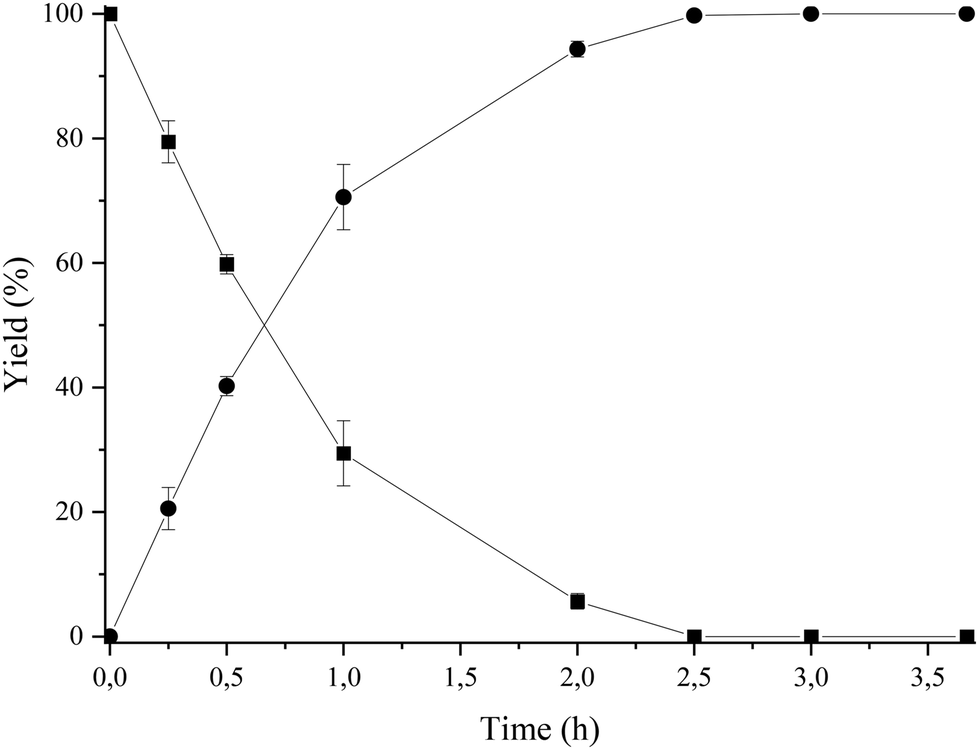 | ||
| Fig. 3 Kinetic study of the esterification of amino-alcohol 5 (0.1 mmol) with hexanoic acid (0.3 mmol), using Novozym 435 (62.5 mg) in 2-MTHF (2.5 mL) and molecular sieves (500 mg) at 35 °C. Amine-alcohol 5 (■); amino-ester 6 (●). | ||
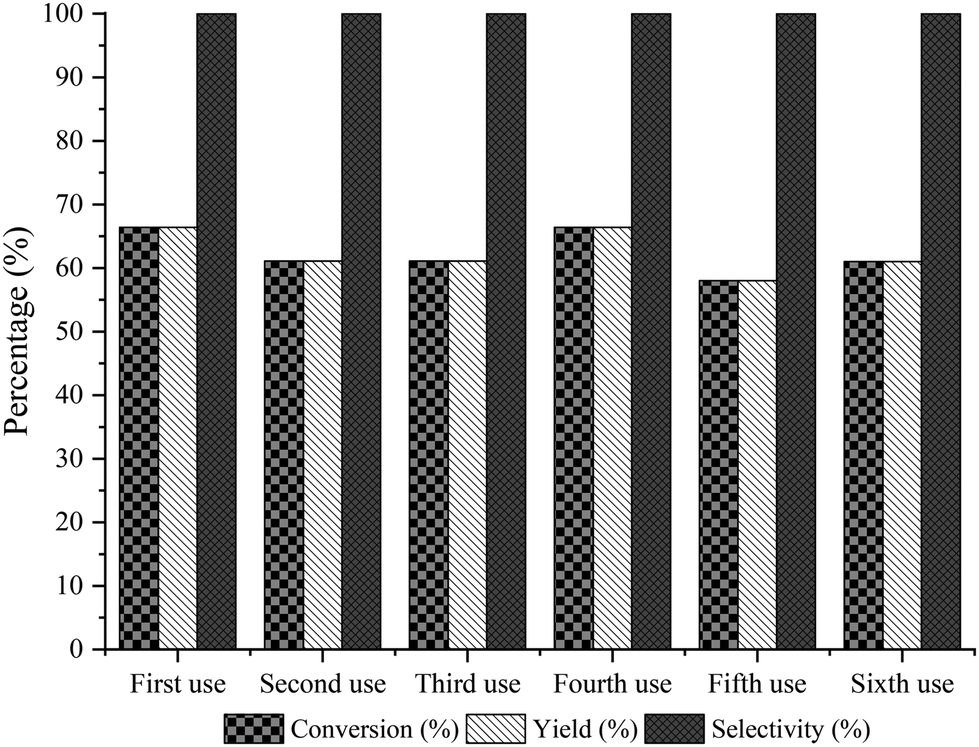 | ||
| Fig. 4 Recyclability of Novozym 435 for the esterification reaction of the amino-alcohol 5 with hexanoic acid at 1 h of reaction. | ||
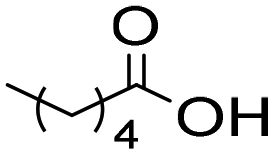
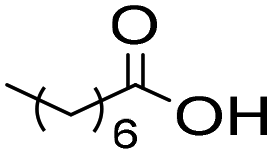
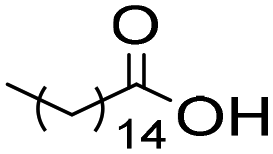
The general applicability of the chemoenzymatic process was evaluated using different carboxylic acids of different chain lengths as the acyl donor (Table 4). As can be observed, besides hexanoic acid, excellent yields and selectivities to the corresponding amino-ester were obtained when using butanoic, octanoic and palmitic acids (entries 1, 3 and 4, Table 4).
| Entry | Acyl donor | Amino-ester | Time (h) | Conversion (%) | Yield (%) | Selectivity (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: 5 (0.1 mmol, 14.1 mg), acyl donor (0.3 mmol, 3 equivalents), Novozym 435 (62.5 mg), molecular sieves (500 mg), 2.5 mL of 2-MTHF, 35 °C and 800 rpm. b Without molecular sieves. | ||||||
| 1 |
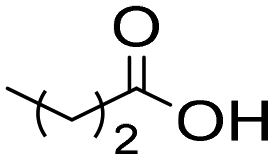
|
6a | 7 | 100 | 99 | 100 |
| 2 |
|
6 | 2.5 | 100 | 99 | 100 |
| 3 |
|
6b | 3 | 100 | 99 | 100 |
| 4 |
|
6c | 5 | 100 | 99 | 100 |
| 5b |
|
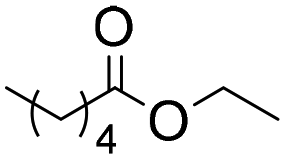
6 | 0.25 | 30 | 30 | 100 |
| 6b |
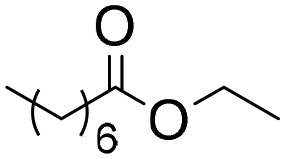
|
6b | 0.25 | 40 | 40 | 100 |
In order to avoid using molecular sieves, the reaction was performed using ethyl esters instead of carboxylic acids (entries 6 and 7, Table 4). In this case, although the selectivity to the amino-ester was high (100%), the afforded conversion was much lower. This could be due to the inactivation of the enzyme by the ethanol released as a byproduct.40,57
On the other hand, to show the advantage of the use of Novozym 435 as a biocatalyst, in the selective preparation of the amino-esters, the reaction was carried out using a conventional acylation method in which the amino-alcohol 5 was reacted with hexanoyl chloride in the presence of triethylamine and dichloromethane as the solvent.58 In this way, after 3.5 h stirring at room temperature, the major product detected was the corresponding amido-ester.
Based on these findings, we designed a sequential two-step one reactor process to transform HMF into an N-substituted amino-ester, a precursor of an ammonium salt surfactant. In this process, we combined the first chemocatalytic pathway, involving the reductive amination of HMF using the Co@C catalyst, with the selective biocatalyzed esterification of the produced amino-alcohol with fatty acids in the presence of immobilized lipase (Novozym 435) (Scheme 4).
The global process starting from HMF was performed in batch mode taking the reductive amination of HMF with methylamine in the presence of the Co@C catalyst at 60 °C as the reaction model, according to entry 7 of Table 3, and using 2-MTHF as a lipase compatible solvent. The reductive amination of HMF with methylamine was satisfactorily performed in 2-MTHF, achieving the target N-methyl-(5-hydroxymethyl)furfurylamine (5) in 98% yield after 5 h (Fig. S39‡). Next, the Co@C catalyst was removed with a magnet, the amino-alcohol produced was diluted with 2-MTHF to 0.04 M, and 6 mmol of hexanoic acid, 125 mg of the lipase Novozym 435, and 500 mg of molecular sieves were added to the reactor while the temperature was kept at 35 °C. Complete conversion into the amino ester was achieved within 19 h.
2.6 Chemo-enzymatic process in a continuous flow reactor
We recently presented the possibility of combining chemo-enzymatic reactions in separate flow reactors. This method allows for operative compartmentalization of chemo- and bio-catalysts, as well as reaction conditions that avoid incompatibility problems.40,59–61Then, with the aim to implement the chemoenzymatic process in a flow mode, the continuous reductive amination of HMF with methylamine using the Co@C catalyst and 2-MTHF as a solvent was first studied using a continuous fixed bed reactor. The contact time (CT) was optimized by varying the HMF concentration from 3.5 wt% (contact time = 7.7 h) (WHSV of 0.13 h−1) to 6.5 wt% (CT = 4 h) (WHSV of 0.25 h−1) (see Fig. 5). As can be seen, a CT of 7.7 h provided an excellent conversion and selectivity to the corresponding amino-alcohol 5. The continuous flow reactor was able to operate for 95 h, showing excellent activity as well as achieving an average yield of 95% (Fig. 6).
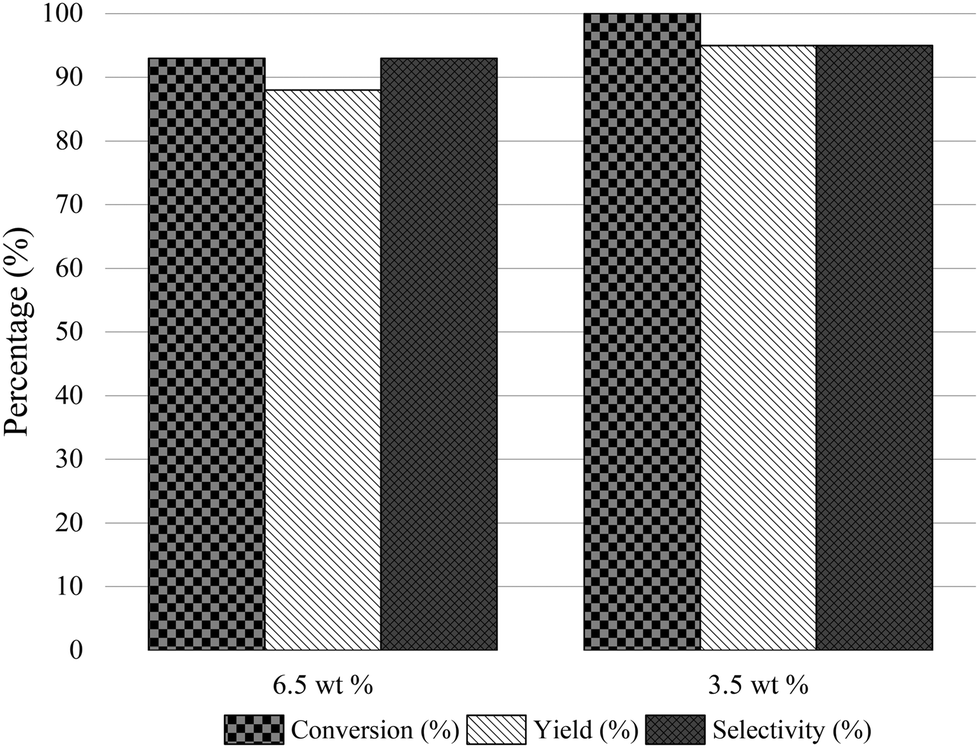 | ||
| Fig. 5 Optimization of the contact time in the flow reactor for the reductive amination of HMF with methylamine. Reaction conditions: HMF in 2-MTHF feed (6.5 and 3.5 wt%, respectively), at 60 °C, at a flow rate of 1.32 mL h−1, at 4 bar of H2 and at a hydrogen flow of 29 mL h−1. | ||
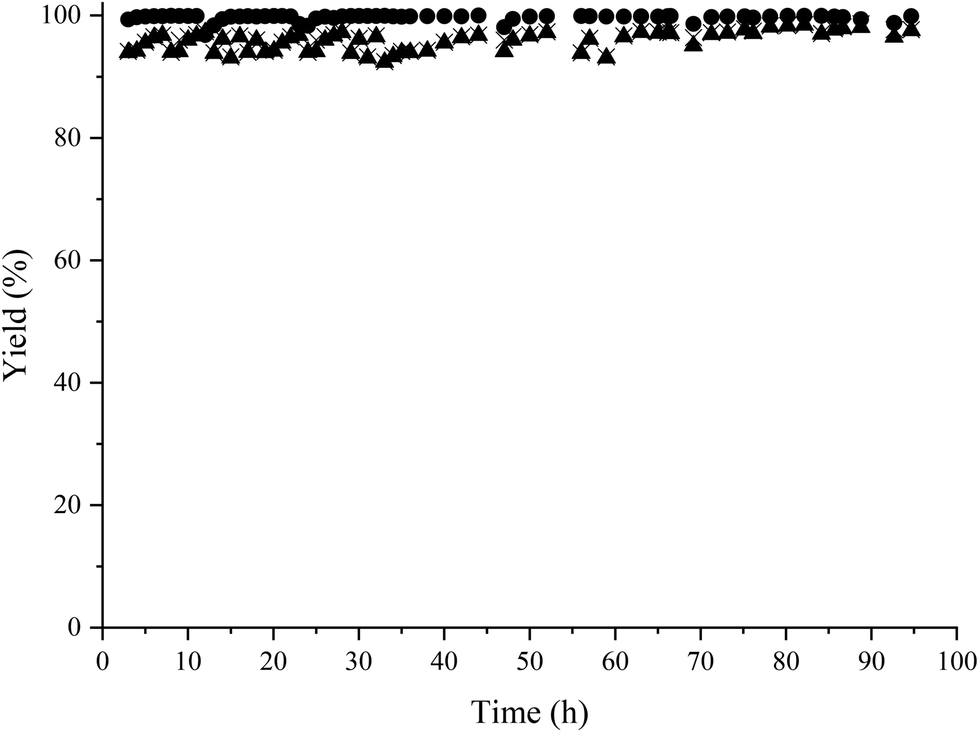 | ||
| Fig. 6 Results of the reductive amination of HMF with methylamine in a continuous-flow reactor using Co@C NPs as a catalyst. Reaction conditions: HMF in 2-MTHF (3.5 wt%), methylamine (2 M in THF) (1.2 equivalents regarding HMF), 300 mg of catalyst, flow rate 1.32 mL h−1 (WHSV: 0.13 h−1) at 60 °C and 4 bar of H2, hydrogen flow 29 mL h−1. HMF conversion (▲); amino-alcohol 5 yield (●); amino-alcohol selectivity (×). | ||
The catalyst stability under flow conditions was studied by maintaining the process at lower conversion (30%) through decreasing the catalyst loading to 150 mg and increasing the feed flow to 0.2 mL min−1, corresponding to a contact time of 0.45 h, while the other parameters were kept constant. In Fig. 7 it is shown that the catalyst maintains its activity and selectivity during 50 h of operation, indicating the high stability of the Co@C nanoparticles under these reaction conditions.
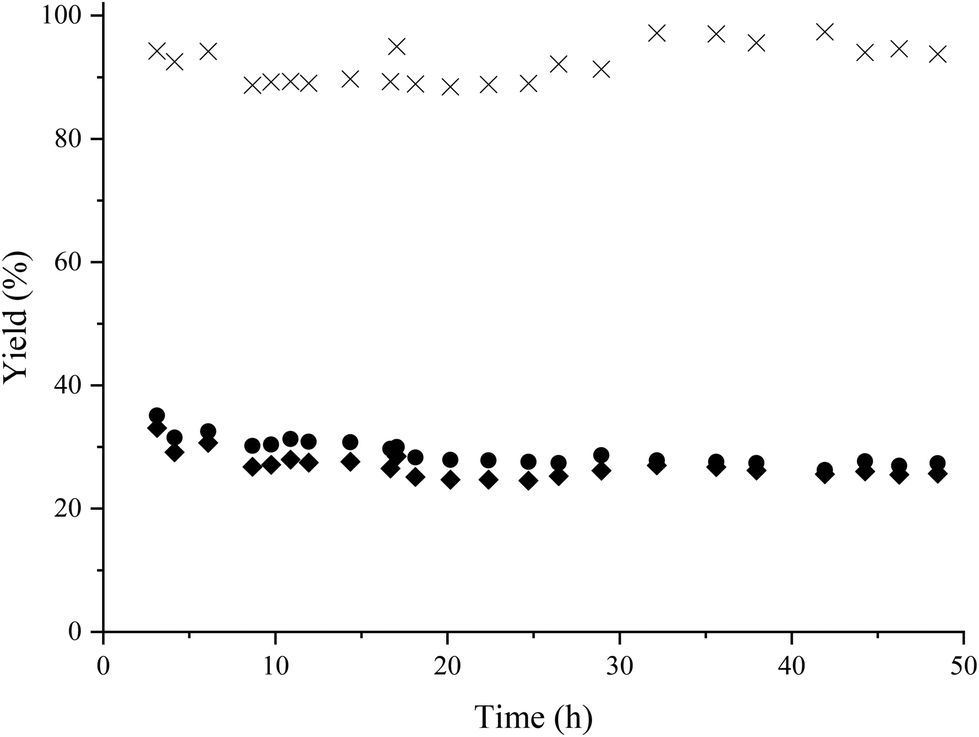 | ||
| Fig. 7 Study of the catalyst stability in the reductive amination of HMF with methylamine in a continuous-flow reactor using Co@C NPs as a catalyst. Reaction conditions: HMF in 2-MTHF (3.5 wt%), 150 mg of catalyst, flow rate 11.1 mL h−1 (WHSV: 2.22 h−1) at 60 °C and 4 bar of H2, hydrogen flow 29 mL h−1. HMF conversion (▲); amino-alcohol 5 yield (●); amino-alcohol selectivity (×). | ||
Then, the second step, the bioesterification reaction of the amino-alcohol 5, was optimized in a flow reactor. For that, Novozym 435 and molecular sieves were placed in the flow reactor that was fed with a mixture of the amino-alcohol 5 and hexanoic acid diluted with 2-MTHF. The contact time was optimized by varying the concentration of 5 (Table 5). As can be seen in Table 5, the control of contact time was key in order to achieve high yields of the corresponding amino-ester, and 90% yield of the amino-ester could be obtained. The effect of the reaction temperature was also studied by increasing the temperature to 60 °C because it has been reported that Novozym-435 can retain 95% activity at 60 °C in diphenyl ether.62 As can be seen, an increase of temperature from 35 °C to 60 °C has an impact on the amino-ester yield (see entries 2 and 4 in Table 5) that was maintained at 90% for 18 h; however, after this time a strong deactivation of the biocatalyst was observed (Fig. S45‡).
| Entry | Amino-alcohol (5) (wt%) | T (°C) | CT (h) | Yield (%) | Working time (h) |
|---|---|---|---|---|---|
|
a Reaction conditions: amino-alcohol 5:hexanoic acid molar ratio = 4 in MTHF as fed, flow rate of 1 mL h−1, 2.4 g of molecular sieves 4 A 0.4–0.6 mm and 50 mg of Novozym 435.
|
|||||
| 1 | 1 | 35 | 6 | 70 | 25 |
| 2 | 0.5 | 35 | 11 | 75 | 35 |
| 3 | 0.28 | 35 | 22 | 85 | 86 |
| 4 | 0.5 | 60 | 11 | 90 | 18 |
Finally, the two continuous flow reactors were coupled is such a way that the amino-alcohol was first produced from HMF and methylamine using the Co@C catalyst under optimized reaction conditions. Then, the amino-alcohol 5 coming from the first flow reactor was directly diluted with 2-MTHF to a concentration of 0.28 wt%, hexanoic acid was added (0.9 wt%) and the mixture was fed to the second reactor where the lipase and molecular sieves were placed. The bioesterification proceeded at 35 °C with a flow rate of 1 mL h−1. Under these reaction conditions, the global yield of the target amino-ester was maintained at around 85% during 86 h of operation (see Fig. 8).
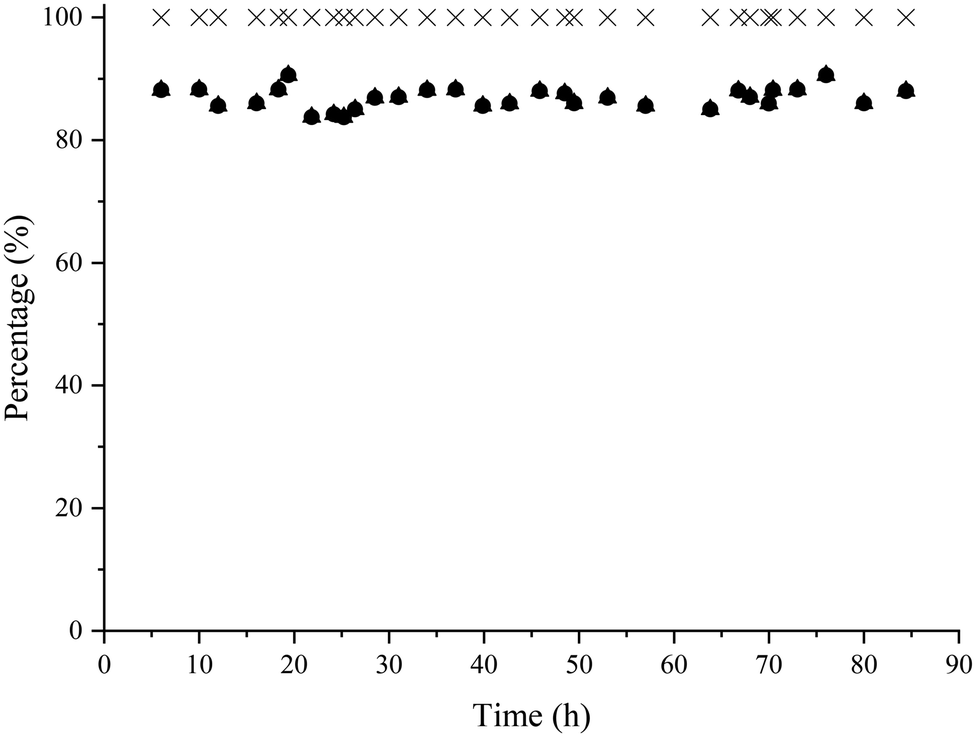 | ||
| Fig. 8 Results of the bioesterification of the amino-alcohol 5 with hexanoic acid in a continuous-flow reactor using Novozym 435 as a catalyst and molecular sieves. Reaction conditions: amino-alcohol 5 in 2-MTHF feed (0.28 wt%), hexanoic acid (0.9 wt%), Novozym 435 (50 mg), molecular sieves 4 A 0.4–0.6 mm (2.4 g), flow rate 1 mL h−1 (WHSV: 0.045 h−1) at 35 °C. Amino-alcohol 5 conversion (▲); amino-ester 6 yield (●); selectivity to 6 (×). | ||
3. Conclusion
Monodisperse Co nanoparticles covered by a thin carbon layer (Co@C) resulted in a highly active, selective and stable catalyst for the reductive amination of HMF with a variety of primary and secondary amines, as well as nitro-compounds, achieving excellent yields of the corresponding N-substituted 5-(hydroxymethyl)-2-furfuryl amines under very mild reaction conditions (60 °C and 4 bar H2) using ethanol as a green solvent. Furthermore, amino-ester derivatives, precursors of ammonium salt-based surfactants, were successfully prepared for the first time through a chemo-enzymatic process by coupling the reductive amination of HMF with methylamine with the selective esterification of the amino-alcohol intermediate with fatty acids, using lipase CALB (Novozym 435) as a biocatalyst in 2-methyltetrahydrofuran as a green and enzyme compatible solvent. The process was implemented in continuous flow reactors, achieving a global yield of the amino-ester derivative of 85%, which was maintained over 86 h of operation. The process represents a new chemo-enzymatic strategy to valorise HMF into highly valuable surfactant precursors.4. Experimental
4.1 Materials
All reagents used were of analytical grade, purchased from Sigma-Aldrich, ABCR or ChemPur, and used without any purification unless otherwise specified.HMF (>98% purity) was obtained from ABCR and purified before its use. Lipase acrylic resin from Candida antarctica (Novozym 435), molecular sieves 4 A powder, 2-MTHF (anhydrous, >99% purity), acetic, butyric, hexanoic, octanoic acid and palmitic acids (>99% purity), aniline (>99% purity), methylamine (2 M in THF), cobalt(II,III) oxide (<10 μm), cobalt(II) nitrate hexahydrate (>98% purity), cobalt(II) acetate tetrahydrate (reagent grade), nickel(II) acetate tetrahydrate (98% purity), ethylene glycol (>99% purity), sodium carbonate (>99.5% purity) and D-(+)-glucose (<99.5% purity) were purchased from Sigma-Aldrich. Silicon oxide catalyst support 250 m2 g−1 (SiO2) was purchased from ChemPur.
4.2 Catalyst preparation
The Co@C NPs were prepared by the hydrothermal treatment of metal oxide nanoparticles with aqueous glucose following the below-mentioned process reported by Liu et al.37Initially, Co3O4 NPs were prepared by dissolving 4.94 g of Co(Ac)2 in 100 mL of ethylene glycol and heating the solution to 160 °C while stirring. Once Co(Ac)2 was completely dissolved and a purple solution was formed, an aqueous solution of Na2CO3 (4.24 g of Na2CO3 dissolved in 160 mL of distilled water) was added dropwise over a duration of 1.5–2 hours. The resulting suspension was further aged for an hour before cooling. The obtained purple solid, corresponding to Co oxide, was filtered, followed by washing with water and acetone. Subsequently, the solid product was dried in an oven at 60 °C for 16 hours and calcined in static air at 450 °C for 3 hours with a ramp rate of 1 °C min−1 from room temperature to 450 °C.
In the subsequent step, 0.5 g of the previously synthesized Co3O4 NPs were dispersed in a glucose aqueous solution (360 mg of glucose dissolved in 20 mL of distilled water) under ultrasonic treatment for approximately 1 hour. The resulting black suspension was transferred into an autoclave and maintained at 175 °C for 18 hours. After cooling to room temperature, the solid product was filtered, washed with water and acetone, and dried in an oven at 60 °C, yielding dark-green solid two-dimensional Co(OH)2/C composites. The Co@C NPs were then obtained by annealing the Co(OH)2/C composites in N2 at 600 °C for 2 hours, employing a ramp rate of 10 °C min−1 from room temperature to 600 °C. After being held at 600 °C for 2 hours, the sample was gradually cooled down in a N2 flow to room temperature and subsequently stored in a glass vial in the ambient environment.
The Ni@C nanoparticles were prepared in a similar way starting from Ni(Ac)2.37
The catalysts were characterized by inductively coupled plasma atomic emission spectroscopy (ICP-AES) using a Varian 715-ES, X-ray diffraction (XRD) was performed by using a PAnalytical CubiX diffractometer with CuKα, and electron microscopy studies were performed using a JEOL 2100F microscope operating at 200 kV both in transmission (TEM) and in scanning transmission modes (STEM). STEM images were obtained using a high angle annular dark-field detector (HAADF), which allows Z-contrast imaging. Cobalt active sites were determined by H2-chemisorption at 100 °C on a Quantachrome Autosorb-1C apparatus by extrapolating the total gas uptake in the adsorption isotherm at zero pressure. Prior to adsorption, 0.3 g of Co@C catalyst samples were pre-treated in flowing He at 100 °C for 1 h and then reduced in situ by flowing pure H2 at 150 °C for 2 h (heating rate of 10 °C min−1). After reduction, the samples were degassed and cooled to 100 °C to record the adsorption isotherm. The active area of the catalyst and its Co monolayer was calculated from the amount of irreversibly adsorbed H2, assuming a Co/H2 stoichiometry of 1:2.
The Co/SiO2 catalyst was prepared via incipient wetness impregnation in a similar way to that reported by Gould et al.63 According to this method, the support (SiO2 250 m2 g−1) was calcined in static air at 540 °C for 3 hours with a ramp rate of 5 °C min−1. Then, 136.2 mg of cobalt nitrate hexahydrate and 500 mg of the support were mixed and stirred in 20 g of extra pure water (quantities corresponding to a cobalt loading of 5 wt%) for 3 h. After evaporating the water under reduced pressure, the resulting violet solid was calcined again under the same conditions used for the support. Finally, the resulting grey solid was reduced at 450 °C for 3 hours with a ramp rate of 5 °C min−1 and a hydrogen flow of 50 mL min−1.
4.3 General reaction procedure for reductive amination
Before use, the commercial HMF was purified to remove possible traces of contaminants following a simple procedure using silica and basic alumina. Thus, a solution of HMF in ethyl acetate (around 1–2 g in 10–20 mL) is passed through a filter plate filled with silica and basic alumina. The resulting yellow liquid solution was analyzed by GC and after evaporation of the solvent, HMF was stored in a glass vial at 4 °C in a fridge until its use.Typically, to 20 mg of the catalyst (pre-reduced under 7 bar of hydrogen at 150 °C for 2 h), placed in a 12 mL stainless autoclave reactor provided with a magnetic stirrer, a solution of 2 mmol of HMF and 2.4 mmol of aniline in 4 mL of EtOH was added. Then, the mixture was pressurized with 4 bar of H2 at 60 °C and stirred at 1000 rpm for the required time.
Three diffusion experiments using 500, 1000 and 1500 rpm were carried out to check the best conditions for the reaction. Thus, it was determined that 1000 rpm was the best condition in terms of the external diffusion of the reaction (this condition was better than that of 500 rpm and almost the same as that of 1500 rpm).
During the reaction, aliquots of ∼25 μL were taken from the reactor at different reaction times. After the addition of dodecane as the external standard, the products were analyzed using an Agilent GC-7980A gas chromatograph equipped with a capillary column HP-5 (30 m × 0.25 μm × 0.25 mm) and an FID detector. The identification of the products was performed by using a GC-MS spectrometer (Agilent 6890N). The molar balance was >95%.
4.4 Stability and recyclability of the Co catalyst
To study if the catalytic process could involve possible Co leaching, the reductive amination of HMF with aniline using 20 mg of Co@C NPs was stopped after 2 h; then, the catalyst was removed with a magnet and the reaction was continued until 22 h.The reusability of the catalyst was evaluated under a hydrogen pressure of 4 bar and at 60 °C. Thus, after the first run, the catalyst was removed using a magnet and washed thoroughly with ethanol and then with acetone at 60 °C. Then, the catalyst was dried in the oven at 60 °C and reused in the second cycle. The same process was repeated for the subsequent cycles, and for the sixth cycle, a Soxhlet extraction with ethanol was carried out.
4.5 General reaction procedure for the enzymatic esterification
Typically, a solution of N-methyl-5-(hydroxymethyl)-2-furfurylamine (5) (0.1 mmol) and carboxylic acid (0.3 mmol), in 2-MTHF (2.5 mL), powder 4 A molecular sieves (500 mg), and Novozym 435 (62.5 mg) were placed in a 5 mL glass batch reactor. After sealing the reactor, the mixture was stirred (800 rpm) at 35 °C for 2.5 h. During the experiment, aliquots of ∼100 μL were taken from the reactor at different reaction times. After the addition of dodecane as the external standard, the products were analyzed using an Agilent GC-7980A gas chromatograph equipped with a capillary column HP-5 (30 m × 0.25 μm × 0.25 mm) and an FID detector. The identification of the products was performed by using a GC-MS spectrometer. The molar balance was >95%.4.6 Chemo-enzymatic one-pot reaction in a batch reactor
The reductive amination of HMF with methylamine, 2 M in THF, was first performed following the procedure described above, but in this case using 2-MTHF as a solvent compatible with the enzyme. After complete conversion of the imine into the target amino-alcohol, the heterogeneous catalyst was removed with a magnet, and the amino-alcohol 5 was diluted with 2-MTHF to 0.04 M for the enzymatic process. Then 6 mmol of hexanoic acid, 125 mg of Novozym 435 and 500 mg of molecular sieves were placed. The solution was stirred (800 rpm) at 35 °C, achieving a complete conversion within 19 h. The molar balance was >95%.4.7 General procedure for the reductive amination of HMF with methylamine in a flow reactor
The reductive amination of HMF with methylamine using Co@C NPs in a continuous fixed bed reactor was carried out using a tubular stainless-steel reactor of 22 cm, with an internal diameter of 3.5 mm and an external diameter of 6 mm. In a standard experiment, 300 mg of Co@C pelletized and sieved to a particle size of 0.2–0.4 mm and diluted by silicon carbide (0.4–0.6 nm, 5.8 g) were introduced in the reactor. Before the reaction, the Co@C catalyst was pre-reduced at 150 °C and 4 bar of H2, under a hydrogen flow of 30 mL min−1 for 2 h. Then, the reactor was cooled down to 60 °C, and a solution of HMF (3.5 wt%) and methylamine (1.2 equivalents) in 2-MTHF was fed into the reactor by using a syringe pump and a Hamilton syringe of 10 mL at a rate of 1.32 mL h−1, which corresponds to a WHSV of 0.13 h−1 and a contact time of 7.5 h. WHSV (weight of feed per hour per unit weight of catalyst loaded in the reactor) and contact time (CT, 1/WHSV) were calculated considering the flow (1.32 mL h−1), density (0.86 g mL−1) and percentage of reagent (3.5 wt%) of the feed and the amount of the catalyst (0.3 g) in the reactor: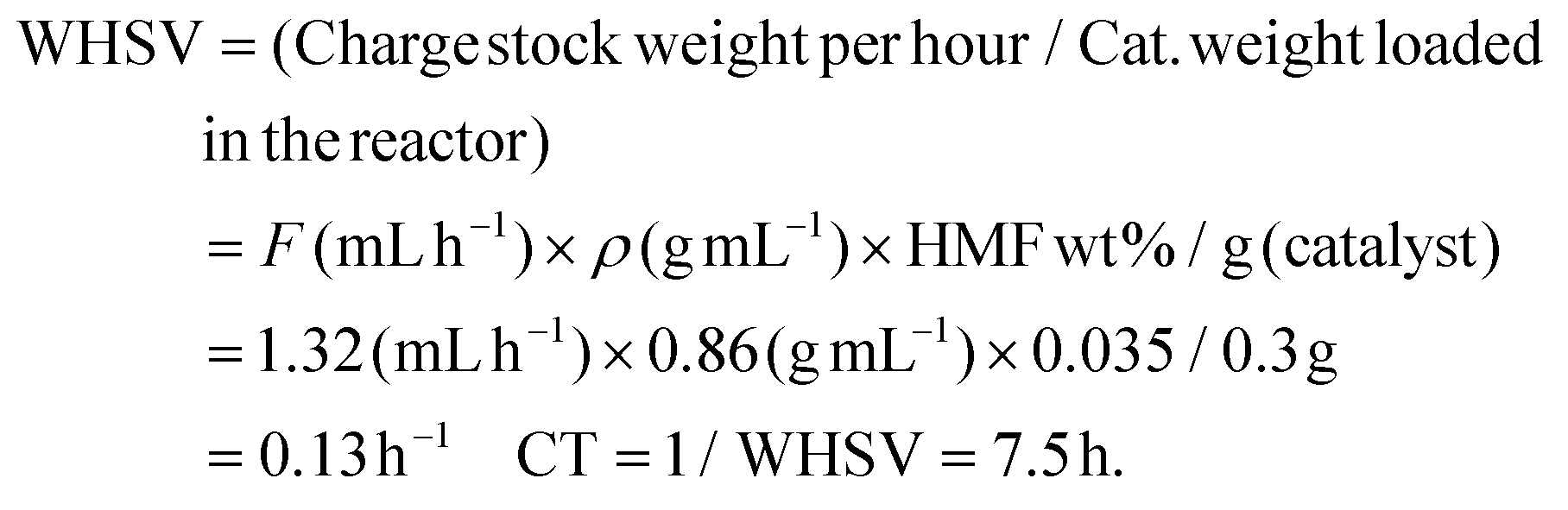
The product was collected in a cooled bottle and fractions were taken and analyzed by gas chromatography using dodecane as the external standard. The molar balance was >95%.
4.8 General procedure for the enzymatic esterification of N-methyl-5-(hydroxymethyl)-2-furfurylamine in a flow reactor
The esterification reaction of the amino-alcohol 5 with hexanoic acid using Novozym 435 in a continuous fixed bed reactor was carried out using a tubular stainless-steel reactor of 30 cm, with an internal diameter of 3.5 mm and an external diameter of 6 mm. In a typical experiment, Novozym 435 (50.5 mg) was diluted with powder 4 A molecular sieves pelletized and sieved to a particle size of 0.4–0.6 mm and packed in the reactor, which was maintained at 35 °C. Then, the reactor was fed with a solution of the amino-alcohol 5 (0.28 wt%) and 4 equivalents of hexanoic acid (0.91 wt%) in 2-MTHF, by using a syringe pump with a Hamilton syringe of 20 mL, at a rate of 1 mL h−1, which corresponds to a WHSV of 0.04 h−1 and a contact time of 22.3 h (calculated in the same way as explained before). Fractions were taken and analyzed by gas chromatography using dodecane as the internal standard. The molar balance was >95%.4.9 Chemo-enzymatic process by combining two fixed bed flow reactors
The chemo-enzymatic process was performed by combining both flow reactors under the conditions indicated above. In this case, the amino-alcohol 5 obtained from the first bed was used, after dilution with 2-MTHF to the required concentration, as feed for the second reactor where the bioesterification occurs.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work has been funded by the Spanish Ministry of Science and Innovation through the “Severo Ochoa Program” (CEX-2021-001230-S) and PID2021-125897OB-I00 (MCIU/AEI/) 10.13039/501100011033 by “ERDF A way of making Europe project”. C. M. thanks the Severo Ochoa program for predoctoral fellowships. The authors also thank the Microscopy Service of UPV for the TEM measurements.References
- A. Feriani, G. Gaviraghi, G. Toson, M. Mor, A. Barbieri, E. Grana, C. Boselli, M. Guarneri, D. Simoni and S. Manfredini, J. Med. Chem., 1994, 37, 4278–4287 CrossRef CAS PubMed.
- B. Plitta, E. Adamska, M. Giel-Pietraszuk, A. Fedoruk-Wyszomirska, M. Naskręt-Barciszewska, W. T. Markiewicz and J. Barciszewski, Eur. J. Med. Chem., 2012, 55, 243–254 CrossRef CAS PubMed.
- S. F. Michardy and J. A. Lowe III, WO2008065500, 2008.
- A. Velty, S. Iborra and A. Corma, ChemSusChem, 2022, 15, e2202200181 CrossRef PubMed.
- K. S. K. Murthy, G. Weeratunga, B. K. Radatus and K. P. S. Sidhu, US Pat., 5750714, 1995 Search PubMed.
- B. J. Price, J. W. Clitherow and J. Bradshaw, US Pat., 4128658, 1978 Search PubMed.
- J. Cooper and D. V. Lee, US Pat., 4347191, 1981 Search PubMed.
- J. Bradshaw, US Pat., 4399294, 1981 Search PubMed.
- B. Alhede and F. P. Clausen, EP0219225A1, 1986.
- L. D. Vicentiis, EP0164040A2, 1985.
- L. H. Schlager, US Pat., 5017586, 1989 Search PubMed.
- R. J. Van Putten, J. C. Van Der Waal, E. De Jong, C. B. Rasrendra, H. J. Heeres and J. G. De Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC.
- A. Messori, A. Fasolini and R. Mazzoni, ChemSusChem, 2022, 15, e202201044 CrossRef.
- Y. Wang, H. Wang, X. Kong and Y. Zhu, ChemSusChem, 2022, 15, e202200421 CrossRef CAS PubMed.
- J. He, Q. Qiang, S. Liu, K. Song, X. Zhou, J. Guo, B. Zhang and C. Li, Fuel, 2021, 306, 121765 CrossRef CAS.
- T. Stemmler, A. E. Surkus, M. M. Pohl, K. Junge and M. Beller, ChemSusChem, 2014, 7, 3012–3016 CrossRef CAS PubMed.
- S. Pisiewicz, T. Stemmler, A. E. Surkus, K. Junge and M. Beller, ChemCatChem, 2015, 7, 62–64 CrossRef CAS.
- J. He, L. Chen, S. Liu, K. Song, S. Yang and A. Riisager, Green Chem., 2020, 22, 6714–6747 RSC.
- T. Irrgang and R. Kempe, Chem. Rev., 2020, 120, 9583–9674 CrossRef CAS PubMed.
- Z. Xu, P. Yan, W. Xu, S. Jia, Z. Xia, B. Chung and Z. C. Zhang, RSC Adv., 2014, 4, 59083–59087 RSC.
- M. M. Zhu, L. Tao, Q. Zhang, J. Dong, Y. M. Liu, H. Y. He and Y. Cao, Green Chem., 2017, 19, 3880–3887 RSC.
- A. García-Ortiz, J. D. Vidal, M. J. Climent, P. Concepción, A. Corma and S. Iborra, ACS Sustainable Chem. Eng., 2019, 7, 6243–6250 CrossRef.
- V. V. Karve, D. T. Sun, O. Trukhina, S. Yang, E. Oveisi, J. Luterbacher and W. L. Queen, Green Chem., 2020, 22, 368–378 RSC.
- A. L. Nuzhdin, P. A. Simonov and V. I. Bukhtiyarov, Kinet. Catal., 2021, 62, 507–512 CrossRef CAS.
- D. Deng, Y. Kita, K. Kamata and M. Hara, ACS Sustainable Chem. Eng., 2019, 7, 4692–4698 CrossRef CAS.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS.
- J. Liu, Y. Song and L. Ma, Chem. – Asian J., 2021, 16, 2371–2391 CrossRef CAS.
- R. Villard, F. Robert, I. Blank, G. Bernardinelli, T. Soldo and T. Hofmann, J. Agric. Food Chem., 2003, 51, 4040–4045 CrossRef CAS PubMed.
- S. Kirchhecker, S. Tröger-Müller, S. Bake, M. Antonietti, A. Taubert and D. Esposito, Green Chem., 2015, 17, 4151–4156 RSC.
- H. Yuan, J. P. Li, F. Su, Z. Yan, B. T. Kusema, S. Streiff, Y. Huang, M. Pera-Titus and F. Shi, ACS Omega, 2019, 2510–2516 CrossRef CAS PubMed.
- G. Chieffi, M. Braun and D. Esposito, ChemSusChem, 2015, 8, 3590–3594 CrossRef CAS PubMed.
- A. L. Nuzhdin, M. V. Bukhtiyarova and V. I. Bukhtiyarov, Molecules, 2020, 25, 4771 CrossRef CAS PubMed.
- V. G. Chandrashekhar, K. Natte, A. M. Alenad, A. S. Alshammari, C. Kreyenschulte and R. V. Jagadeesh, ChemCatChem, 2022, e202101234 CrossRef CAS.
- E. Redina, N. Arkhipova, G. Kapustin, O. Kirichenko, I. Mishin and L. Kustov, ChemCatChem, 2023, e202300294 CrossRef CAS.
- (a) H. Yuan, J.-P. Li, F. Su, Z. Yan, B. T. Kusema, S. Streiff, Y. Huang, M. Pera-Titus and F. Shi, ACS Omega, 2019, 4, 2510–2516 CrossRef CAS PubMed; (b) P. Li, A. T. Liebens, H. Yuan, F. Su and F. Shi, WO2019174221A1, 2018.
- L. Liu, F. Gao, P. Concepción and A. Corma, J. Catal., 2017, 350, 218–225 CrossRef CAS.
- L. Liu, P. Concepción and A. Corma, J. Catal., 2016, 340, 1–9 CrossRef CAS.
- M. Puche, L. Liu, P. Concepción, I. Sorribes and A. Corma, ACS Catal., 2021, 11, 8197–8210 CrossRef CAS PubMed.
- K. S. Arias, J. M. Carceller, M. J. Climent, A. Corma and S. Iborra, ChemSusChem, 2020, 13, 1864–1875 CrossRef CAS PubMed.
- B. Hurtado, K. S. Arias, M. J. Climent, P. Concepción, A. Corma and S. Iborra, ChemSusChem, 2022, e202200194 CrossRef CAS PubMed.
- Market Research Company, Market Research Company offers Syndicate & Custom Market Research Reports with Consulting Services – Allied Market Research, https://www.alliedmarketresearch.com/.
- X. Yue and Y. Queneau, ChemSusChem, 2022, 15, e202102660 CrossRef CAS PubMed.
- 5-Hydroxymethylfurfural Market Report by Grade (Industrial Grade, Food Grade), End User (Flavor and Fragrance Industry, Pharmaceutical Industry, and Others), and Region 2024–2032.
- P. Galletti, A. Montecavalli, F. Moretti, A. Pasteris, C. Samorì and E. Tagliavini, New J. Chem., 2009, 33, 1859–1868 RSC.
- L. Cisneros, P. Serna and A. Corma, Angew. Chem., Int. Ed., 2014, 53, 9306–9310 CrossRef CAS PubMed.
- E. Husson, C. Humeau, C. Paris, R. Vanderesse, X. Framboisier, I. Marc and I. Chevalot, Process Biochem., 2009, 428–434 CrossRef CAS.
- F. Le Joubioux, N. Bridiau, Y. Ben Henda, O. Achour, M. Graber and T. Maugard, J. Mol. Catal. B: Enzym., 2013, 95, 99–110 CrossRef CAS.
- F. Le Joubioux, Y. Ben Henda, N. Bridiau, O. Achour, M. Graber and T. Maugard, J. Mol. Catal. B: Enzym., 2013, 85–86, 193–199 CrossRef CAS.
- P. O. Syrén, F. LeJoubioux, Y. BenHenda, T. Maugard, K. Hult and M. Graber, ChemCatChem, 2013, 5, 1842–1853 CrossRef.
- N. Chinsky, A. L. Margolin and A. M. Klibanov, J. Am. Chem. Soc., 1989, 111, 386–388 CrossRef CAS.
- A. Pintor, I. Lavandera, A. Volkov and V. Gotor-Fernández, ACS Sustainable Chem. Eng., 2023, 11(28), 10284–10292 CrossRef CAS PubMed.
- F. Le Joubioux, O. Achour, N. Bridiau, M. Graber and T. Maugard, J. Mol. Catal. B: Enzym., 2011, 70, 108–113 CrossRef CAS.
- N. Öhrner, C. Orrenius, A. Mattson, T. Norin and K. Hult, Enzyme Microb. Technol., 1996, 328–331 CrossRef.
- M. A. Lǎcǎtuş, L. C. Bencze, M. I. Toşa, C. Paizs and F. D. Irimie, ACS Sustainable Chem. Eng., 2018, 6, 11353–11359 CrossRef.
- M. Krystof, M. Pérez-Sánchez and P. Domínguez de María, ChemSusChem, 2013, 6, 630–634 CrossRef CAS PubMed.
- C. José, R. D. Bonetto, L. A. Gambaro, M. D. P. Guauque Torres, M. L. Foresti, M. L. Ferreira and L. E. Briand, J. Mol. Catal. B: Enzym., 2011, 95–107 CrossRef.
- H. L. Chan, L. Lyu, J. Aw, W. Zhang, J. Li, H.-H. Yang, H. Hayashi, S. Chiba and B. Xing, ACS Chem. Biol., 2018, 13, 1890–1896 CrossRef CAS PubMed.
- D. Kracher and R. Kourist, Curr. Opin. Green Sustainable Chem., 2021, 32, 100538 CrossRef CAS.
- J. M. Carceller, M. Mifsud, M. J. Climent, S. Iborra and A. Corma, Green Chem., 2020, 22, 2767–2777 RSC.
- S. Strompen, M. Weiß, H. Gröger, L. Hilterhaus and A. Liese, Adv. Synth. Catal., 2013, 355, 2391–2399 CrossRef CAS.
- Y. Poojari and S. J. Clarson, Biocatal. Agric. Biotechnol., 2013, 2, 7–11 CrossRef.
- N. S. Gould, H. Landfield, B. Dinkelacker, C. Brady, X. Yang and B. Xu, ChemCatChem, 2020, 12, 2106–2115 CrossRef CAS.
Footnotes |
| † Dedicated to Professor G. Hutchings on his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc00425f |
| This journal is © The Royal Society of Chemistry 2024 |