 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarnessing conformational dynamics in enzyme catalysis to achieve nature-like catalytic efficiencies: the shortest path map tool for computational enzyme redesign†
Cristina
Duran
 a,
Guillem
Casadevall
a and
Sílvia
Osuna
*ab
a,
Guillem
Casadevall
a and
Sílvia
Osuna
*ab
aDepartament de Química, Institut de Química Computacional i Catàlisi, Universitat de Girona, c/Maria Aurèlia Capmany 69, 17003, Girona, Spain. E-mail: silvia.osuna@udg.edu
bICREA, Pg. Lluís Companys 23, 08010, Barcelona, Spain
First published on 18th March 2024
Abstract
Enzymes exhibit diverse conformations, as represented in the free energy landscape (FEL). Such conformational diversity provides enzymes with the ability to evolve towards novel functions. The challenge lies in identifying mutations that enhance specific conformational changes, especially if located in distal sites from the active site cavity. The shortest path map (SPM) method, which we developed to address this challenge, constructs a graph based on the distances and correlated motions of residues observed in nanosecond timescale molecular dynamics (MD) simulations. We recently introduced a template based AlphaFold2 (tAF2) approach coupled with 10 nanosecond MD simulations to quickly estimate the conformational landscape of enzymes and assess how the FEL is shifted after mutation. In this study, we evaluate the potential of SPM when coupled with tAF2-MD in estimating conformational heterogeneity and identifying key conformationally-relevant positions. The selected model system is the beta subunit of tryptophan synthase (TrpB). We compare how the SPM pathways differ when integrating tAF2 with different MD simulation lengths from as short as 10 ns until 50 ns and considering two distinct Amber forcefield and water models (ff14SB/TIP3P versus ff19SB/OPC). The new methodology can more effectively capture the distal mutations found in laboratory evolution, thus showcasing the efficacy of tAF2-MD-SPM in rapidly estimating enzyme dynamics and identifying the key conformationally relevant hotspots for computational enzyme engineering.
Introduction
Enzymes do not have a single, rigid structure, instead they adopt multiple conformations, which enable the enzyme to evolve towards novel functions.1 Evolution leverages pre-existing diversities, thereby granting proteins the capacity for functional innovation.1–5 These ensembles of pre-existing conformations in thermal equilibrium with the native state, provide enzymes with the ability to evolve and also explains the existing divergence from only a few common ancestors,6 as well as the promiscuous side activities (both in substrate and in reactivity) that many enzymes exhibit.6 However, the ability of enzymes to adopt diverse conformations may initially appear to conflict with their traits of efficiency, precision, and specificity. The high catalytic activity of enzymes is largely attributed to their well-structured active site pockets, which hold the catalytic machinery in place for efficiently stabilising the transition state(s) of the reactions.7,8 Still, proficient catalysis necessitates a fine balance between active site pre-organization for transition state stabilisation and optimisation of the conformational ensemble along the catalytic pathway.Tracing an enzymatic cycle in detail, the core steps within a generic catalytic pathway include: (i) substrate binding in the catalytic pocket, often involving exploration of additional conformational states with properly positioned loops and flexible domains facilitating active site access;9,10 (ii) substrate(s) activation for enzyme–substrate (ES) formation; (iii) transition state stabilisation leading to the formation of reaction intermediates and products; (iv) product release, often accompanied by conformational alterations resetting the catalytic cycle. Each of these steps are pivotal for enhanced catalytic activity.
The experimental and computational (or combined) studies exploring the conformational landscape of natural and laboratory-evolved enzymes highlight the key role of mutations at the active site but also at distal sites, in changing the stabilities of the pre-existing conformations.1,3–5,16 This can be, for instance, shown experimentally through NMR and room-temperature X-ray crystallography as shown for some Kemp eliminases,17,18 or by the evolution of B-factors in static X-ray structures of the multiple enzyme variants generated via directed evolution (DE) along an evolutionary trajectory.2,16 From a computational perspective, molecular dynamics (MD) simulations and enhanced sampling methods can be applied to estimate this ensemble of conformations, i.e., the free energy landscape (FEL, see Fig. 1).4,13,19,20 In the reconstructed FEL, the relative stability of thermally accessible conformations, as well as the kinetic barriers separating them are displayed. The barrier height separating a specific pair of conformational states dictates the timescale of the associated transition. Conformational changes directly influencing catalytic function encompass side-chain conformational shifts, loop motions often crucial for substrate binding/product release, and in some cases allosteric transitions.11
 | ||
| Fig. 1 Shortest path map (SPM) construction, workflow and applications. SPM is a correlation-based tool that can be used to identify conformationally relevant positions of importance for inducing a population shift in the FEL. The first step requires the estimation of the conformational heterogeneity of the studied system (FEL estimation) using for instance MD simulations. From these simulations the distance and correlation matrices are constructed, which are used to generate a first complex graph (shown in step 2, middle panel) that contains all protein residues represented as spheres and some edges of different length (weighted according to the correlation value) that link those positions situated, on average, less than 6 Å along the MD run. This complex graph is further simplified by finding the edges that are shorter, i.e., more correlated, providing the final SPM graph than can be plotted on top of the 3D structure (step 3). In the SPM graph, the sphere size and edges are weighted according to the number of times the pair of residues have been included in the shortest paths (i.e., the size qualitatively represents the importance of the identified positions for the conformational dynamics of the enzyme). SPM has been used to rationalise DE mutations,11 to study and understand the allosteric regulation within monomers in multimers,12,13 and more recently to design new enzyme variants starting from natural scaffolds.14,15 SPM is a strategy to reduce the number of potential hotspots, thus reducing the sequence space for enzyme engineering. The figure has been generated with a model system, the SPM graph, and displayed structure correspond to the beta subunit of tryptophan synthase. | ||
The evaluation of the FEL and understanding how it is altered after mutation provides crucial insights for comprehending and engineering enzyme function.4 The mutations introduced at the active site and often at distal sites induce a long-range conformational effect impacting enzymatic catalysis. Triggered by the introduced mutations, catalytically productive conformational states are stabilised, while the non-beneficial ones for the new functionality are disfavoured, thus transforming computational enzyme redesign into a population shift problem.11 These discoveries boosted the exploration of enzyme conformational dynamics for enzyme redesign.3,4,16 The reconstruction of ancestral enzymes displaying higher flexibility compared to modern counterparts, and their utilization as initial scaffolds for enzyme redesign, brought interesting new insights.21 Increased flexibility in many ancestral variants proved crucial for attaining superior levels of catalytic activity with only a few mutations at the active site. Various ancestrally-reconstructed enzymes have since been employed as starting points for enzyme design, for example, to enhance some residual catalytic promiscuity within an enzyme family, or to alter the allosteric regulation of some heterodimeric enzymes, among others.21–23
Although the above-mentioned examples highlight the importance of the enzyme conformational heterogeneity for function, what remains challenging is the identification of which mutations should be introduced to enhance a given conformational change, especially if those involve mutating distal sites.11 In this line, we developed the shortest path map (SPM) tool that relies on the construction of a graph based on the computed mean distances and correlation values obtained from MD simulations, following a similar strategy as the protocol reported by Sethi et al. for investigating allosterically-regulated enzymes (see Fig. 1).20 SPM instead of identifying communities in this first original complex graph (see step 2 in Fig. 1), focuses on the identification of the shortest path lengths.24 SPM therefore reduces the sequence space to a smaller number of conformationally-relevant positions, and of importance is the fact that SPM has the potential to identify the challenging distal activity-enhancing positions.11 Indeed, we successfully applied SPM for identifying DE mutations in the retro-aldolase, monoamine oxidase and tryptophan synthase enzymes, suggesting its potential application for the rational design of enzyme variants (Fig. 1).11 Our SPM tool was also applied by the Mulholland lab to evaluate the changes in the dynamical networks at the transition-state ensemble along DE of a computationally designed Kemp eliminase.25 We have also used SPM to investigate the allosteric communication within monomers, and have recently found that SPM is highly complementary to distance fluctuation analysis and dynamical non-equilibrium MD simulations for investigating allosteric systems.12,13 Despite the successes in identifying key positions targeted in DE, the application of SPM in computational enzyme design is not direct, as it identifies multiple conformationally relevant positions (usually around 50–70 residues are included) and, most importantly, it does not provide which specific amino-acid substitution should be introduced for achieving the desired conformational change.11 We combined SPM and ancestral sequence reconstruction to mitigate some of these limitations and design new stand-alone tryptophan synthase B (TrpB) variants.15 As the ancestral LBCA TrpB was known to display stand-alone activity, our approach focused on including the LBCA amino acid in those non-conserved SPM positions. The stand-alone activity of the new SPM6-TrpB variant was increased 7-fold (in terms of kcat). Still, it is worth highlighting that by testing only one single variant the fold increase in kcat was similar to the 9-fold obtained by DE that required the generation and screening of more than 3000 variants. In a recent pre-print, we have further demonstrated the power of our SPM methodology for redesigning natural scaffolds and boosting an existing side-activity into nature-like catalytic activities. In particular, we were able to increase the esterase catalytic efficiency of a hydroxynitrile lyase (HNL) more than 1300-fold, and we actually surpassed the activity of the esterase taken as reference.14 These studies therefore provide further evidence for the potential of our SPM methodology for computational enzyme redesign.
The neural network Alphafold2 (AF2) revolutionized the structural biology and protein design field as it is able to predict the folded structure of proteins and enzymes from the primary sequence with high levels of precision.26–29 The innovative AF2 neuronal network exploits data on the evolutionary, physical and geometric constraints of existing protein structures. AF2 is acknowledged as a pivotal milestone in protein structure prediction and has boosted the utilization of deep-learning techniques for numerous other applications.29 Despite the remarkable efficacy of AF2 algorithms in predicting the native lowest energy structure of proteins, using AF2 for understanding and engineering function directly from the acquired single static structure is not straightforward. However, some recent research has indicated that AF2 can also be used to predict multiple conformations of the same protein, thereby potentially being utilized to explore the conformational adaptability of biological systems.30–32 In this regard, we have recently developed a template-based AF2 approach to estimate the conformational landscape of enzymes, and quickly assess how it is shifted by mutations.33,34 This is promising as it suggests that AF2 could be employed for evaluating the impact of introduced mutations on the conformational landscape at a significantly reduced computational cost: in hours instead of days/weeks of simulation time, thus accelerating the creation of conformationally-driven enzyme design protocols.4,11
In this study, we evaluate the potential of our developed SPM tool and template based AF2 (tAF2) approach coupled to MD simulations for estimating the conformational heterogeneity and identifying key active site and distal conformationally-relevant positions. We first compare the reconstructed FEL from tAF2 coupled to 10–50 nanosecond timescale MD simulations employing different forcefields and water models. Second, the predictions from tAF2-MD are compared to the previously reconstructed FELs from multiple replica well-tempered metadynamics simulations.15 Finally, we generate the SPM maps using the tAF2-MD data and considering different conformational states. Our results show the potential of SPM and tAF2-MD, for estimating the conformational heterogeneity and identifying the key conformationally-relevant hotspots. The developed approach has a reduced computational cost (results in a few hours instead of days/weeks or even months compared to metadynamics) and can be used to computationally generate and screen multiple enzyme variants, thus potentially giving access to nature-like activities.
Results
Coupling AF2 with molecular dynamics simulations for exploring the conformational heterogeneity
We use tryptophan synthase (TrpS) as the model system, which is an allosterically regulated enzyme composed of two subunits: TrpA and TrpB. The allosteric interaction between TrpA and TrpB sustains appropriate conformation throughout the catalytic cycle enhancing the catalytic steps, hence the lack of protein counterpart results in an inadequate conformational landscape that hampers catalysis.19 In the case of TrpB, this entails the conformational change of the so-called COMM domain covering the active site, recognized to adopt closed, semi-closed, and open conformations (see Fig. 2).19,35,36 As we found in a previous study, the inability of PfTrpB to operate efficiently in the absence of PfTrpA is mostly due to the restricted conformational heterogeneity and the non-productive closure of the COMM domain.19 The activating distal mutations introduced in the laboratory-evolved 0B2-PfTrpB variant37,38 recover the conformational ensemble of the allosterically-regulated complex, thus enhancing its stand-alone activity.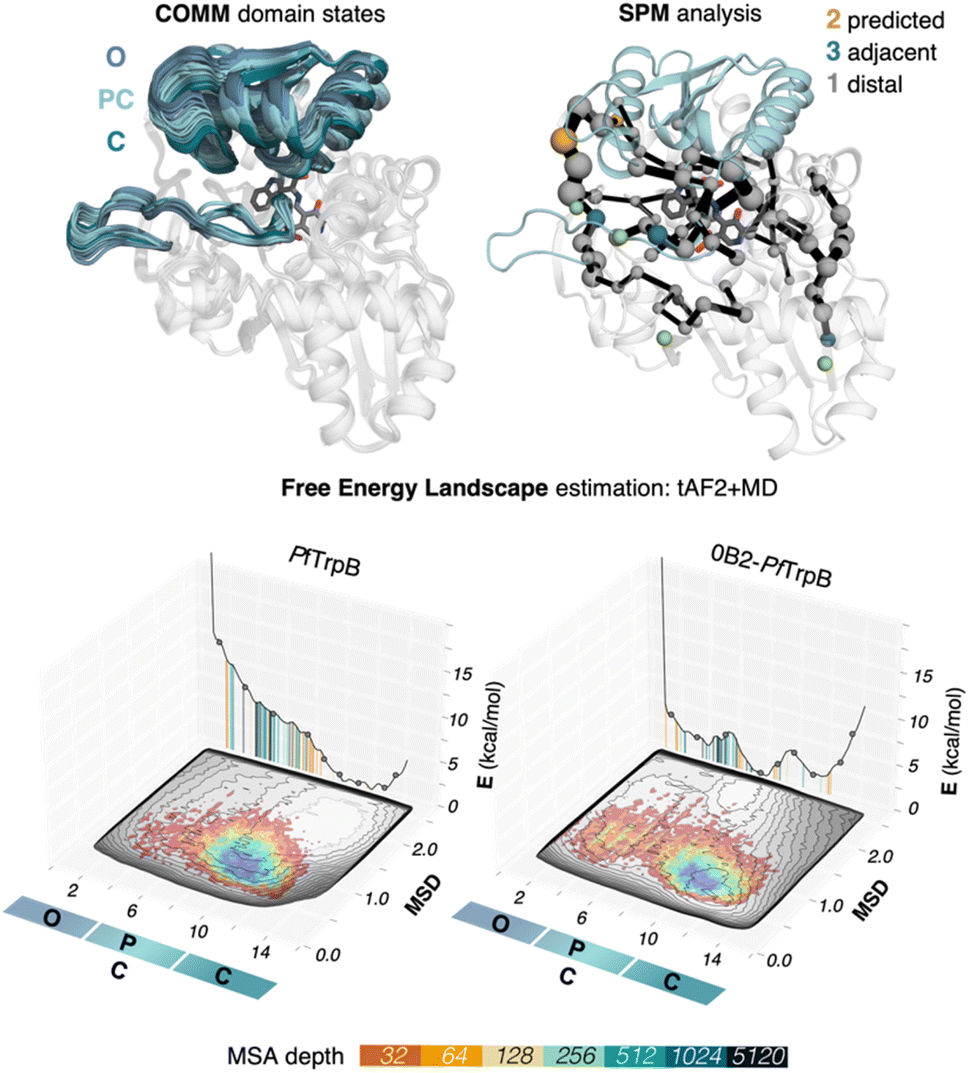 | ||
| Fig. 2 SPM and reconstructed free energy landscapes (FELs) of PfTrpB and the evolved variant 0B2-PfTrpB. The catalytically relevant COMM domain that covers the active site of the enzyme (shown in teal) can adopt different conformations along the catalytic cycle: open (O) states are adopted in the resting state E(Ain), partially closed (PC) at the reaction intermediates E(Aex1) and E(A–A) and closed (C) at E(Q2) states. Representation of the computed SPM for PfTrpB with DE mutations highlighted: 2 mutations are predicted (shown in orange), three are adjacent to SPM residues (teal) and one is distal (grey).19 The pyridoxal phosphate cofactor is shown in dark grey. The previously reconstructed FELs from the developed tAF2-MD protocol in the red-blue colourmap (blue for the most stable conformations, red for the least stable ones)33 are shown on top of the FELs computed from the multiple walker well-tempered metadynamics simulations (shown in grey).19 The predictions of AF2 are represented on the 2D-FEL representation using vertical lines coloured from orange to dark blue depending on the MSA depth: AF2 predictions obtained with a 32 MSA depth are shown with a vertical orange line, 64 in light orange, 128 in light brown, 256 in light cyan, 512 in cyan, 1024 in teal, and 5120 in dark blue. | ||
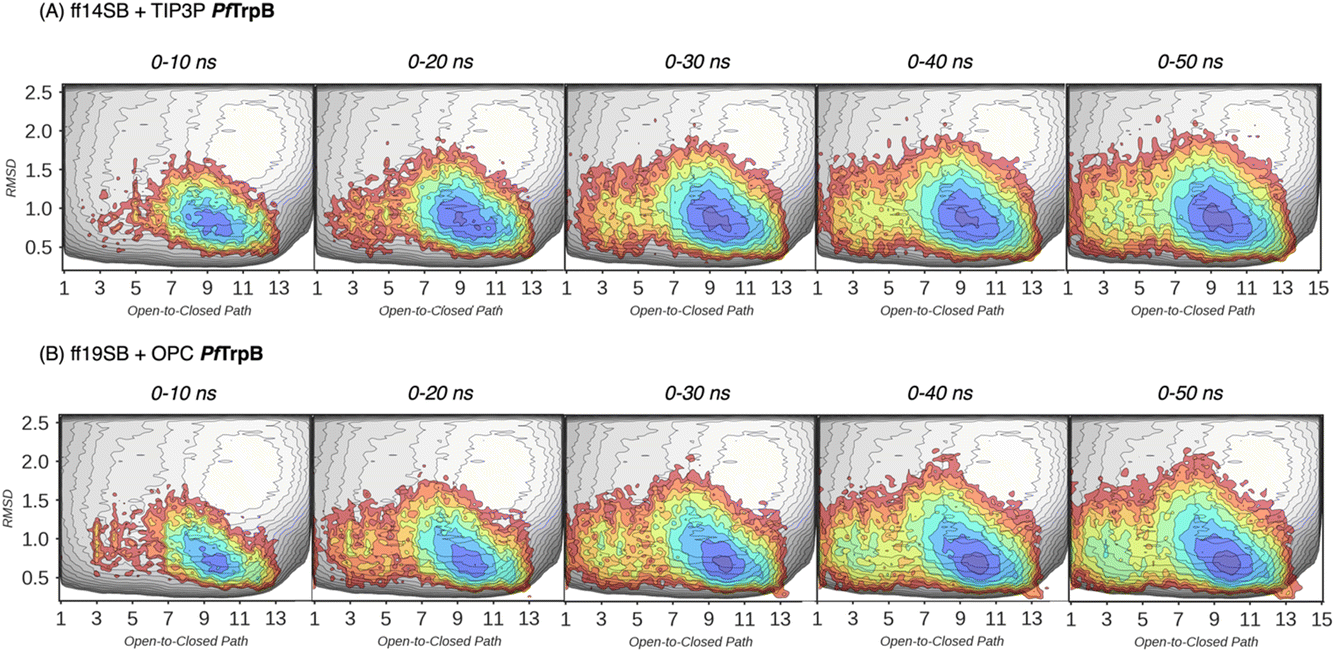
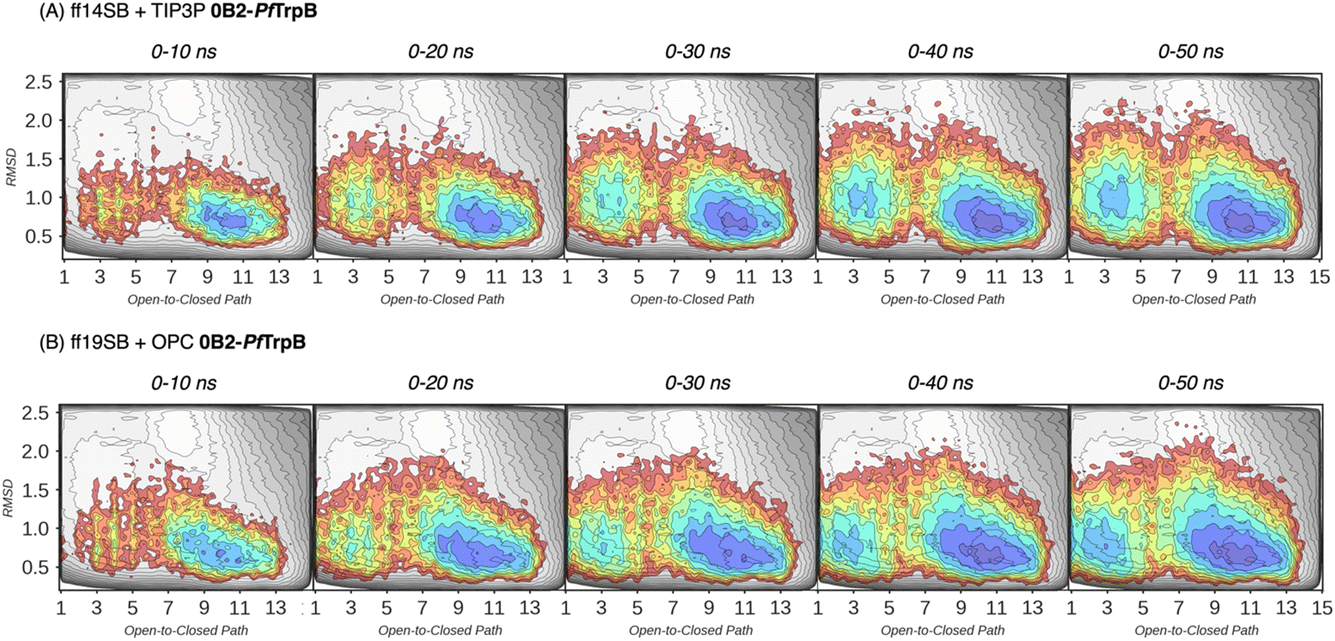
In our previous publication we found that the conformational heterogeneity of related TrpB variants exhibit different levels of stand-alone activity that could be estimated by performing 10 ns MD simulations, starting from the ensemble of structures generated by our template-based AF2 (tAF2) approach.33,34 This approach generates multiple structures by running AF2 with different starting templates and multiple sequence alignments (MSAs) of different depths, followed by short nanosecond timescale MD simulations. As we found in our previous study, the conformational landscapes reconstructed from these multiple replica 10 ns MD simulations starting at different tAF2 predictions, were qualitatively in line with the previously reconstructed computationally expensive FELs obtained from well-tempered multiple-walker metadynamics simulations.15 The computational cost associated with both approaches is dramatically different: estimations can be obtained in a matter of hours for the former, whereas multiple weeks are needed for the latter. Still, there were some major differences observed in the conformational landscapes, which we want to evaluate further in this study. In this section, we aimed to extend the simulation time of the MD simulations to test whether a better exploration of the conformational landscape could be obtained. To that end, we extended the 10 ns MD simulations to up to 50 ns and reconstructed and compared the FELs (see Fig. 3 and 4, and the Materials and methods section for a detailed description). We also tested the effect of using the recommended AMBER ff19SB force field and the improved OPC water model (as opposed to the previously used ff14SB and TIP3P). OPC was found to accurately reproduce the electrostatic properties of water, which is particularly important for balancing the interactions between the protein residues and water molecules, especially for disordered regions, as most water models tend to favour too compact structures.39,40
 | ||
| Fig. 3 Estimated FEL of PfTrpB. The estimated FEL from the accumulated multiple replica nanosecond timescale MD simulations performed starting at the ensemble of template-based AF2 predictions, is shown in colour on top of the previously reconstructed FEL of the PfTrpB variant (shown in grey scale).19 FELs are displayed every 10 ns of MD simulation time and using two different combinations of forcefield and water model: (A) ff14SB and TIP3P water, and (B) ff19SB and OPC water. The x axis denotes the ensemble of structures generated from X-ray data for the open-to-closed transition of the COMM domain, which ranges from 1–5 (open structures, O), 6–10 (partially closed, PC), to 11–15 (closed, C), the y axis is the mean square deviation (MSD, in Å2) from the path of O-to-C structures generated. Most stable conformations are shown in blue, whereas higher energy regions are shown in red. | ||
 | ||
| Fig. 4 Estimated FEL of 0B2-PfTrpB. The estimated FEL from the accumulated multiple replica nanosecond timescale MD simulations performed starting at the ensemble of template-based AF2 predictions, is shown in colour on top of the previously reconstructed FEL of the PfTrpB variant (shown in grey scale).19 FELs are displayed every 10 ns of MD simulation time and using two different combinations of forcefield and water model: (A) ff14SB and TIP3P water, and (B) ff19SB and OPC water. The x axis denotes the ensemble of structures generated from X-ray data for the open-to-closed transition of the COMM domain, which ranges from 1–5 (open, O), 6–10 (partially closed, PC), to 11–15 (closed, C), the y axis is the mean square deviation (MSD, in Å2) from the path of O-to-C structures generated. Most stable conformations are shown in blue, whereas higher energy regions are shown in red. | ||
In Fig. 3 and 4, the reconstructed FELs from the multiple replica 10 ns MD simulations from our previous study are compared to the new FELs generated from 50 ns MD. The FEL obtained from well-tempered multiple-walker metadynamics is also shown in grey for comparison.15 Although the 10 ns MD simulations already suggested some differences in the conformational heterogeneity between systems, the extension of the simulations up to 50 ns confirmed the estimations. Still, the analysis of the conformational space sampled every 10 ns of MD simulation show that, especially in the 20–30 ns timeframe, the obtained FEL is very similar to the one explored after 50 ns (see Fig. 3, 4, S1 and S2†). This is especially relevant for the open conformation of the COMM domain in the evolved 0B2-PfTrpB, which after 20–30 ns is substantially more sampled and thus stabilised as expected from the metadynamics FEL. This stabilisation is even more evident in the FELs from the ff19SB-OPC simulations. Thanks to the improved water model and forcefield, and as found for disordered proteins, ff19SB-OPC stabilises a much more open conformation of the COMM (Fig. S2 and S3†). For wild-type PfTrpB the open conformation is more sampled in the 50 ns FEL, but it is not stabilised as in the case of 0B2-PfTrpB in line with its inferior conformational heterogeneity. Altogether, the extension of the multiple replica MD simulations up to 20–30 ns show a better agreement with the computationally intensive FELs reconstructed from the well-tempered metadynamics simulations.19 This analysis suggests that the TrpB conformational landscape can be properly sampled and estimated from the developed tAF2 when coupled with 20–30 ns MD starting from 60, and 59 structures from tAF2 for PfTrpB, and 0B2-PfTrpB, respectively. As shown in Fig. S4–S6,† ff19SB/OPC provides a better description of the secondary structure of TrpB as compared to X-ray data, thus this combination is more appropriate for evaluating TrpB conformational dynamics.
Applying the correlation-based SPM tool for identifying key conformationally-relevant positions
After the comparison of the FEL reconstructed from tAF2 and ns timescale MD simulations with those obtained with metadynamics simulations, we decided to evaluate the effect of the two strategies on the computed SPMs. As mentioned in the introduction, SPM is computed from the distance and correlation matrix obtained using the MD data, and therefore identifies nearby residues that are conformationally relevant. Two different thresholds are applied for SPM calculation and visualization: only the set of residues whose carbon alpha is on average less than 6 Å along the MD runs will be considered (this is the distance threshold), and only those edges that have a higher contribution (i.e., are more correlated) will be displayed (this is the significance threshold, which is often set to 0.3).41 In the SPM graphs, one can notice that different sphere sizes and edges are represented (Fig. 5–7). All spheres and edges displayed are weighted according to their relative importance: those edges connecting pairs of residues more correlated will be more often selected as the shortest paths, and thus will be represented with a thicker line and a bigger sphere. However, the size and thickness of spheres and edges is only qualitative, as the SPM outcome is dependent on the conformational changes explored along the MD runs.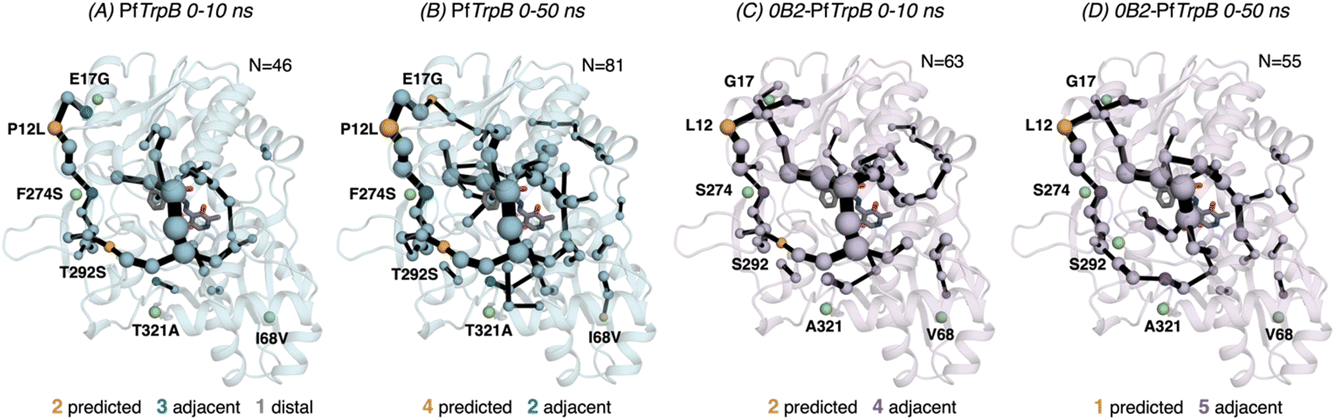 | ||
| Fig. 5 Evolution of the SPMs along the MD time. The number of identified conformationally relevant positions (N) is increased from 46 to 81 in the case of (A and B) PfTrpB, and slightly decreased from 63 to 55 in (C and D) 0B2-PfTrpB. The size of spheres and the thickness of edges in the SPM plots are weighted according to their importance for the conformational dynamics of the enzyme: those pairs of residues that have a higher contribution to the conformational dynamics are represented with a thicker edge and larger sphere. All SPM graphs have been generated using the default settings: a threshold of 6.0 Å for the distance matrix, and a 0.3 for the significance threshold. | ||
We generated the SPM graphs every 10 ns of the multiple replica MD, started from a different tAF2 output structure, and compared them with respect to the SPM generated from the metadynamics simulations.19 We also assessed whether the SPM graphs contained DE mutation sites, as we did in our previous publication.19 All SPM graphs generated at this point make use of the default parameters of distance and significance thresholds (set to 6 Å and 0.3, respectively, see methods). Similarly to what is found in the previous section regarding FEL convergence, smaller differences between the computed SPM are found after 20–30 ns of MD. It is interesting to observe that the SPM computed in the 0–10 ns timeframe contains a substantially reduced number of residues, and this number is expanded when the MD simulation time is increased (Fig. 5). This is particularly evident in PfTrpB, as the first SPM contains 46 residues and is further increased to up to 81. In the case of 0B2-PfTrpB the number of included residues differs only from 63 to 55 (see Fig. 5). Altogether this comparison suggests a higher interlinked communication in the most evolved variant, which can be successfully captured after short 20–30 ns MD simulations from the multiple tAF2 output structures. The comparison of the SPM with the one obtained for PfTrpB with the multiple-walker well-tempered metadynamics simulations19 reveals similar SPM pathways (Fig. 2 and 6). However, it should be mentioned that in terms of identifying DE mutations, the new SPM coming from the tAF2 and 20–30 ns MD simulations captures two additional mutations (T292S and I68V), which in the previously published SPM based on metadynamics, were not predicted. The same distance and significance thresholds were used for SPM construction, thus the conformational space sampled was the most important difference between both strategies (tAF2+MD versus metadynamics).
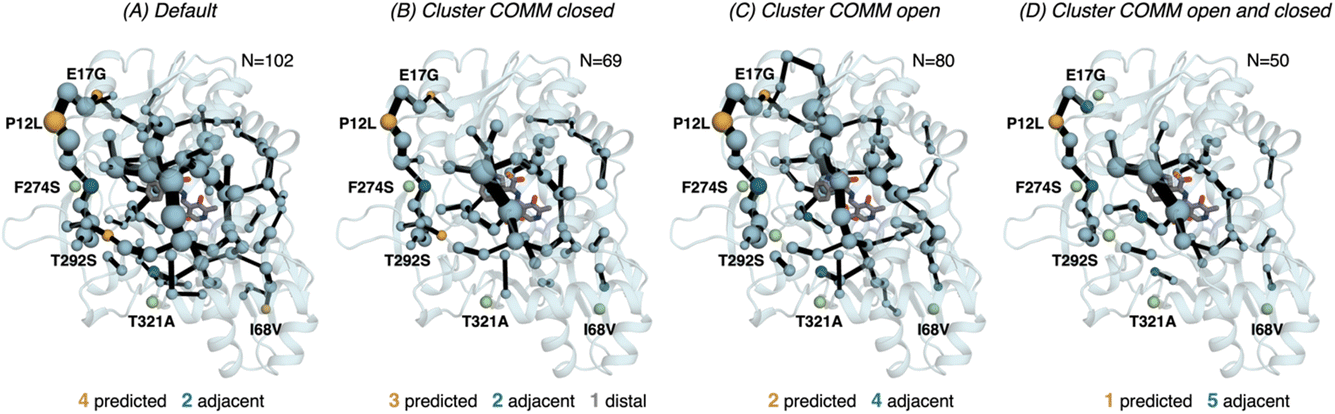 | ||
| Fig. 6 Computed SPM of PfTrpB. Representation of the generated SPM graphs using the last 20 ns of the MD trajectories with: (A) default settings (i.e., considering a threshold of 6.0 Å for the distance matrix computed from the MD data without any filtering of structures), (B) only the distance information from the structures that present a closed conformation of the COMM domain, (C) only the distance information from the open structures of the COMM, and (D) combining the distance information of (B) and (C). The number of contained residues in the SPMs is shown (N). Directed evolution (DE) mutations37 are labelled and highlighted in yellow if contained in the SPM, or green if not contained but making non-covalent interactions with SPM positions. The size of spheres and the thickness of edges in the SPM plots are weighted according to their importance for the conformational dynamics of the enzyme: those pairs of residues that have a higher contribution to the conformational dynamics are represented with a thicker edge and larger sphere. In all cases, a threshold for the significance of 0.3 is used. | ||
Finally, as the SPM outcome is dependent on the mean distances between all combinations of residues that compose the protein, we decided to evaluate how the SPM graphs differ when: (1) considering only open or closed states of the COMM domain, and (2) generating a new distance matrix combining the information from the individual open and closed distance matrices (see methods for a full description, and Fig. S7†). For PfTrpB, the generated SPM considering either closed or open states contain a reduced number of identified positions if compared with the SPM generated with the standard protocol (i.e., considering the full trajectory for computing the distance matrix). It is interesting to note that the SPM for the open state contains a larger number of residues in comparison with the one generated with closed states (80 versus 69), thus suggesting a reduced communication in the closed state in PfTrpB. This observation is in line with the inability of PfTrpB to adopt productively closed conformations, consistent with its lower catalytic activity as stand alone.19 The connection between the COMM domain and the active site is also different in both SPMs, as the one for the open state is more similar to the default SPM (see Fig. 6). In terms of identifying the DE mutations, a reduced number of positions is identified in comparison with the default parameters. This suggests that considering the whole ensemble of MD trajectories is a more appropriate strategy for SPM construction for capturing DE hotspots. Another interesting observation is that the SPM computed considering a distance matrix containing the information from the closed and open matrices (Fig. 6), contains a substantially lower number of positions (50 versus 102 for the default).
In 0B2-PfTrpB, the SPM for the closed state contains a much larger number of positions (110), in comparison with the one for the open state (55) and the default SPM (47, see Fig. 7). This comparison suggests that the distal mutations introduced in 0B2-PfTrpB stabilise the closed state, thus enhancing the communication between the COMM domain, the active site, and the interface region between the beta subunits. This fact is also in line with the reconstructed FELs reported in the previous section, and the well-tempered metadynamics published in the previous study that show the stabilisation of the catalytically competent closed state.19 The SPM for the closed state actually contains a higher number of DE mutations (3 are contained, 3 are adjacent), especially if compared with the default SPM (1 mutation is contained in the path, 4 are adjacent, 1 is distal). The SPM obtained from the distance matrix generated from the individual contributions of the closed and open state contains a reduced number of positions (37), and actually is very similar to the default SPM (47 residues identified).
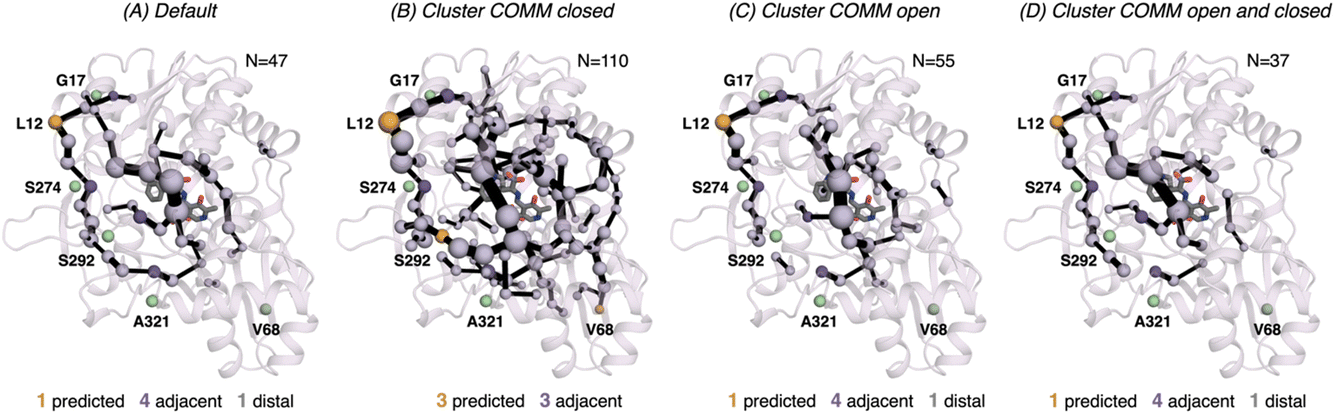 | ||
| Fig. 7 Computed SPM of 0B2-PfTrpB. Representation of the generated SPM graphs using the last 20 ns of the MD trajectories with: (A) default settings (i.e., considering a threshold of 6.0 Å for the distance matrix computed from the MD data without any filtering of structures), (B) only the distance information from the structures that present a closed conformation of the COMM domain, (C) only the distance information from the open structures of the COMM, and (D) combining the distance information of (B) and (C). The number of contained residues in the SPMs is shown (N). Directed evolution (DE) mutations37 are labelled and highlighted in yellow if contained in the SPM, or green if not contained but making non-covalent interactions with SPM positions. The size of spheres and the thickness of edges in the SPM plots are weighted according to their importance for the conformational dynamics of the enzyme: those pairs of residues that have a higher contribution to the conformational dynamics are represented with a thicker edge and larger sphere. In all cases, a threshold for the significance of 0.3 is used. | ||
Practical use of SPM for computational enzyme redesign
In the previous section, we evaluated the potential of our SPM methodology for identifying the conformationally-relevant positions, which we have seen in previous studies coincide with DE hotspots.11 We also found that the two matrices that are needed for SPM construction can be generated using the developed tAF2 approach when coupled to 30–50 ns of MD simulations. In fact, the SPM graphs generated using this strategy were found to be more successful in identifying DE hotspots found in the case of PfTrpB.37 Based on these observations, we recommend the following approach for using SPM for enzyme redesign: (1) generation of multiple conformations of the same protein by providing to AF2, MSA of different depths as we did in our previous publication;33 (2) running of 30–50 ns MD simulations from the ensemble of structures generated in (1); (3) computation of the correlation and distance matrices using the last 20 ns of the multiple replica MD simulations; (4) generation of the SPM graph considering a distance threshold of 6 Å and a significance threshold of 0.3.41 The generated SPM paths provide relevant information on the conformational dynamics of the starting enzyme scaffold. This reduces the number of positions for mutagenesis, but still too many residues are identified. Multiple strategies can be applied to reduce the number of hotspots further, but as we found in previous studies a successful strategy is to combine SPM with ASR,15 and/or incorporate evolutionary information from the generated MSA. One possible strategy to identify the key positions for mutagenesis and the specific amino-acid change is to evaluate the conformational dynamics of a reference enzyme to generate a reference SPM. By comparing both SPMs, i.e., the one for the starting scaffold and the one of the reference enzyme, we can search for non-conserved SPM positions connected to key structural elements or catalytic residues. Although this is not explored in this study, we have applied this methodology for enhancing the stand-alone activity of tryptophan synthases,15 and the esterase activity of hydroxynitrile lyases.14 SPM graphs can also be of interest for selecting the best starting scaffold for an enzyme redesign task. By evaluating the conformational dynamics and thus the SPM graphs of multiple potential initial scaffolds, one could use SPM to decide which might be the most appropriate scaffold exhibiting closer conformational dynamics to the reference enzyme.Discussion and conclusions
Tryptophan synthase (TrpS) is an enzyme composed of two different subunits, which exhibits a complex mechanism, and possesses a finely tuned set of structural variations that require optimisation to enhance its function.15,19,37,38,42–45 Our previous investigations into the structural dynamics of this enzyme complex and individual TrpB enzymes have revealed that by modifying the relative stability of the different conformational states of the catalytic COMM domain (open, partially-closed, and closed conformations), we can optimise the various steps involved in its catalytic process. Such structural changes are crucial for optimising the active site of the enzyme for efficient catalysis, substrate binding, and product release. When we computationally analysed different TrpB variants with distinct catalytic activities, we discovered that enhanced stand-alone activity depends on the enzyme’s ability to adopt closed conformations of the COMM domain independently of its binding partner, and its high structural flexibility to facilitate substrate binding and product release.19 We also found that our SPM methodology was able to capture conformationally relevant positions involved in the open-to-closed conformational change of the COMM domain, and some of these were mutation points in a previous DE study for the generation of the stand-alone 0B2-PfTrpB variant.19 This finding allowed us to design new stand-alone TrpB variants based on the SPM methodology in combination with ASR.15 However, reconstructing the FEL associated with these structural changes is computationally demanding, limiting its application to only a few selected enzyme systems. In addition to that, multiple residues are usually identified in the constructed SPMs. Both constraints pose a significant challenge when attempting to computationally design new stand-alone enzyme variants in a routine and fast manner.In this study, we aimed to evaluate the potential of the SPM tool, especially if combined with our previously developed template based AF2 (tAF2) approach coupled to MD simulations,33 for quickly estimating the conformational heterogeneity and identifying key conformationally relevant positions. We first compared the reconstructed FEL obtained from the tAF2 method in conjunction with short nanosecond timescale MD simulations using two different forcefields and water models. The conclusions derived from these new FELs match those of the computationally much more demanding well-tempered multiple walker metadynamics simulations. The most evolved 0B2-PfTrpB exhibits a much more stabilised closed conformation of the COMM domain, as well as a higher conformational heterogeneity, as described by the previously reported metadynamics simulations.19 Our analysis indicates that the TrpB conformational landscape can be properly sampled and estimated from the developed tAF2 approach especially when coupled with 20–30 ns MD simulations. Still, as reported in our earlier study,33 10 ns MD simulations starting from the multiple outputs from tAF2 can provide some hints about the conformational heterogeneity of the systems. The comparison between ff14SB/TIP3P and ff19SB/OPC indicates that the latter seems to provide a better description of the open and closed states of TrpB.
We also assessed the differences in the SPM graphs obtained via the tAF2 approach coupled to nanosecond timescale MD simulations. SPM requires the correlation and distance matrix, therefore we decided to evaluate in more detail the effect of the distance matrix on the generated output graphs. We compared the SPM computed using the last 20 ns of the 50 ns MD trajectory, with the SPMs derived from the distance matrix containing only either closed or open states of the COMM domain. We also evaluated the effect on the SPM when a distance matrix combining the information from the individual open and closed distance matrices was used. The SPM of PfTrpB using 20 ns of the 50 ns MD data is comparable to the one obtained with the multiple replica well-tempered metadynamics simulations.19 However, the SPM generated via the tAF2 approach coupled to short MD simulations predicts two additional DE mutations (positions T292S and I68V). This suggests that the tAF2-MD approach, at least in TrpB, is a suitable strategy for identifying key positions targeted with DE. The SPMs obtained considering only open or closed states include a reduced number of DE mutations. In the case of PfTrpB, the SPM generated for both closed and open states shows fewer identified positions, as compared to the one produced considering the last 20 ns of the MD trajectory. Notably, the SPM for the open state has more residues than the one for the closed state, indicating a limited communication in the closed state of PfTrpB. This aligns with the inability of PfTrpB to adopt productive closed conformations,19 consistent with its lower stand-alone catalytic activity. These SPMs also identify a reduced number of DE mutations. Interestingly, the SPM for the closed state of the stand-alone 0B2-PfTrpB variant features a larger number of positions compared to the open state SPM and the default. This suggests that the introduced distal mutations stabilise the closed state and enhance the communication between the COMM domain, the active site, and the interface region between the beta subunits. This higher communication is in line with the stabilisation of the catalytically competent closed state, as we observed in our previous publications.19,33 The SPM for the closed state also contains more distal DE mutations, especially when compared to the default SPM. This also highlights the potential of tAF2-MD-SPM for rationalising the effect of DE mutations and design.
In this study we show that 20 ns MD simulations using ff19SB with OPC water model and starting from the multiple output structures provided by the developed tAF2 protocol,33 allow the estimation of the conformational heterogeneity of systems differing in only a few mutations (98.4% of sequence identity). Most importantly, these simulations allow the construction of SPM graphs that are comparable to those obtained after performing multiple-walker well-tempered metadynamics simulations. SPM can also be used to study the conformationally relevant positions at the different open and closed states of the protein, which identify fewer DE mutations but are useful to study how the introduced mutation altered the inter-residue communication. This study demonstrates the potential application of the developed tAF2-MD-SPM for the fast computational evaluation, redesign and ranking of new enzyme variants. This is exciting for achieving the ultimate goal of computationally redesigning new enzymes with nature-like catalytic efficiencies.
Materials and methods
MD simulations
The starting structures for the two systems (PfTrpB and 0B2-PfTrpB) were previously obtained with the predictions of the X-ray template-based AF2 approach.33 Using AMBER 20,46 two different force field and water model conditions were tested: ff14SB/TIP3P and ff19SB/OPC. Two replicas of 50 ns MD simulation at Q2 intermediate were calculated, starting with 60 and 59 AF2 structures for PfTrpB and 0B2-PfTrpB systems, respectively. The MD calculations using ff14SB/TIP3P were elongated, starting from the 10 ns simulation previously studied.33 We followed exactly the same protocol for the MD equilibration and production run as described in ref. 33.SPM calculations
For the default shortest path map (SPM) analysis,11,20,41 the MD simulations of each system (i.e., PfTrpB and 0B2-PfTrpB) using ff19SB/OPC were used. The process involves calculating the inter-residue mean distance and dynamic cross-correlation (DCC) matrices from the MD simulation data. Based on these matrices, a graph is constructed, where residue pairs with a mean distance of less than 6 Å during the simulation are connected with a line. The weight of these edges is determined by the Pearson correlation coefficient (dij = −log|Cij|), where shorter lines indicate higher correlation between the motions of the residue pairs. This generates a first complex graph in which the edge lengths between residues are the key features and is the basis for SPM construction. This complex graph is further processed to elucidate the shortest path lengths, emphasizing those connections that are most influential in the enzymes’ conformational dynamics.11 We compute the number of times each edge connecting a given pair of residues is included in the shortest path to go through all residues of the protein. This process ranks each edge according to their frequency of use: those edges that have been included a higher number of times in the shortest paths will have a higher contribution and therefore will be represented using a thicker line in the SPM graph. Similarly, the size of the two spheres connected by the edge will be weighted according to the edge thickness. It should be therefore noted that whereas in the first complex graph edge lengths identified the most important positions for the conformational dynamics, in the SPM only the set of edges and spheres having a higher contribution are represented in the 3D structure with different sphere sizes and edge lengths. This reduction in the number of displayed positions is the so-called significance threshold than is often set to 0.3, but it can be tuned by the user as we described in a recent publication.41 The final SPM graph is overlaid with the 3D structure of the respective enzyme. SPM calculations can be done using the recently released SPMweb server, in which the distance and significance thresholds can be manually changed by the user.41For the evaluation of the SPM graph considering only open or closed states of the COMM domain, two clusters were obtained fitting the trajectories in two Gaussian distributions described with the open-to-closed path, using Gaussian Mixture Model implemented in the scikit-learn package.47 For each open and closed state, a distance and a DCC matrix were obtained. From here, the SPM procedure is the same as followed in the default part, after obtaining the matrices. The SPM graph was finally assessed generating a new distance matrix that combines the individual open and closed distance matrices. By taking the information of the clusters previously computed, a single distance matrix was created. However, the DCC matrix was constructed with all data without filtering by the open/closed clusters. The SPM analysis method was then equal to the default procedure as described above. As discussed in the Results section, the SPM graphs were constructed using different trajectory lengths.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Generalitat de Catalunya for the consolidated group TCBioSys (SGR 2021 00487), Spanish MICIN for grant projects PID2021-129034NB-I00 and PDC2022-133950-I00. S. O. is grateful to the funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (ERC-2015-StG-679001, ERC-2022-POC-101112805, and ERC-2022-CoG-101088032), and the Human Frontier Science Program (HFSP) for project grant RGP0054/2020. C. D. was supported by the Spanish MINECO for a PhD fellowship (PRE2019-089147), and G. C. by a research grant from ERC-StG (ERC-2015-StG-679001) and ERC-POC (ERC-2022-POC-101112805).Notes and references
- N. Tokuriki and D. S. Tawfik, Science, 2009, 324, 203–207 CrossRef CAS PubMed.
- E. Campbell, M. Kaltenbach, G. J. Correy, P. D. Carr, B. T. Porebski, E. K. Livingstone, L. Afriat-Jurnou, A. M. Buckle, M. Weik, F. Hollfelder, N. Tokuriki and C. J. Jackson, Nat. Chem. Biol., 2016, 12, 944–950 CrossRef CAS PubMed.
- R. M. Crean, J. M. Gardner and S. C. L. Kamerlin, J. Am. Chem. Soc., 2020, 142, 11324–11342 CrossRef CAS PubMed.
- M. A. Maria-Solano, E. Serrano-Hervás, A. Romero-Rivera, J. Iglesias-Fernández and S. Osuna, Chem. Commun., 2018, 54, 6622–6634 RSC.
- D. Petrović, V. A. Risso, S. C. L. Kamerlin and J. M. Sanchez-Ruiz, J. R. Soc. Interface, 2018, 15, 20180330 CrossRef PubMed.
- O. Khersonsky and D. S. Tawfik, Annu. Rev. Biochem., 2010, 79, 471–505 CrossRef CAS PubMed.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed.
- S. Marti, M. Roca, J. Andres, V. Moliner, E. Silla, I. Tunon and J. Bertran, Chem. Soc. Rev., 2004, 33, 98–107 RSC.
- D. D. Boehr, R. Nussinov and P. E. Wright, Nat. Chem. Biol., 2009, 5, 789–796 CrossRef CAS PubMed.
- G. G. Hammes, S. J. Benkovic and S. Hammes-Schiffer, Biochemistry, 2011, 50, 10422–10430 CrossRef CAS PubMed.
- S. Osuna, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, e1502 CAS.
- C. Calvó-Tusell, M. A. Maria-Solano, S. Osuna and F. Feixas, J. Am. Chem. Soc., 2022, 144, 7146–7159 CrossRef PubMed.
- C. Curado-Carballada, F. Feixas, J. Iglesias-Fernández and S. Osuna, Angew. Chem., Int. Ed., 2019, 58, 3097–3101 CrossRef CAS PubMed.
- G. Casadevall, C. Pierce, B. Guan, J. Iglesias-Fernandez, H.-Y. Lim, L. R. Greenberg, M. E. Walsh, K. Shi, W. Gordon, H. Aihara, R. L. E. III, R. Kazlauskas and S. Osuna, bioRxiv, 2023, 2023.2008.2023.554512 Search PubMed.
- M. A. Maria-Solano, T. Kinateder, J. Iglesias-Fernández, R. Sterner and S. Osuna, ACS Catal., 2021, 11, 13733–13743 CrossRef CAS PubMed.
- E. C. Campbell, G. J. Correy, P. D. Mabbitt, A. M. Buckle, N. Tokuriki and C. J. Jackson, Curr. Opin. Struct. Biol., 2018, 50, 49–57 CrossRef CAS PubMed.
- A. Broom, R. V. Rakotoharisoa, M. C. Thompson, N. Zarifi, E. Nguyen, N. Mukhametzhanov, L. Liu, J. S. Fraser and R. A. Chica, Nat. Commun., 2020, 11, 4808 CrossRef CAS PubMed.
- R. Otten, R. A. P. Pádua, H. A. Bunzel, V. Nguyen, W. Pitsawong, M. Patterson, S. Sui, S. L. Perry, A. E. Cohen, D. Hilvert and D. Kern, Science, 2020, 370, 1442–1446 CrossRef CAS PubMed.
- M. A. Maria-Solano, J. Iglesias-Fernández and S. Osuna, J. Am. Chem. Soc., 2019, 141, 13049–13056 CrossRef CAS PubMed.
- A. Romero-Rivera, M. Garcia-Borràs and S. Osuna, ACS Catal., 2017, 7, 8524–8532 CrossRef CAS PubMed.
- J. M. Gardner, M. Biler, V. A. Risso, J. M. Sanchez-Ruiz and S. C. L. Kamerlin, ACS Catal., 2020, 10, 4863–4870 CrossRef CAS.
- T. Devamani, A. M. Rauwerdink, M. Lunzer, B. J. Jones, J. L. Mooney, M. A. O. Tan, Z.-J. Zhang, J.-H. Xu, A. M. Dean and R. J. Kazlauskas, J. Am. Chem. Soc., 2016, 138, 1046–1056 CrossRef CAS PubMed.
- M. Schupfner, K. Straub, F. Busch, R. Merkl and R. Sterner, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 346–354 CrossRef CAS PubMed.
- G. Csárdi and T. Nepusz, InterJournal, 2006, 1695–1704 Search PubMed.
- H. A. Bunzel, J. L. R. Anderson, D. Hilvert, V. L. Arcus, M. W. van der Kamp and A. J. Mulholland, Nat. Chem., 2021, 13, 1017–1022 CrossRef CAS PubMed.
- A. W. Senior, R. Evans, J. Jumper, J. Kirkpatrick, L. Sifre, T. Green, C. Qin, A. Žídek, A. W. R. Nelson, A. Bridgland, H. Penedones, S. Petersen, K. Simonyan, S. Crossan, P. Kohli, D. T. Jones, D. Silver, K. Kavukcuoglu and D. Hassabis, Nature, 2020, 577, 706–710 CrossRef CAS PubMed.
- A. Ourmazd, K. Moffat and E. E. Lattman, Nat. Methods, 2022, 19, 24–26 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- E. Callaway, Nature, 2020, 588, 203–204 CrossRef CAS PubMed.
- D. del Alamo, D. Sala, H. S. McHaourab and J. Meiler, eLife, 2022, 11, e75751 CrossRef CAS PubMed.
- R. A. Stein and H. S. McHaourab, PLoS Comput. Biol., 2022, 18, e1010483 CrossRef CAS PubMed.
- H. K. Wayment-Steele, A. Ojoawo, R. Otten, J. M. Apitz, W. Pitsawong, M. Hömberger, S. Ovchinnikov, L. Colwell and D. Kern, Nature, 2024, 625, 832–839 CrossRef CAS PubMed.
- G. Casadevall, C. Duran, M. Estévez-Gay and S. Osuna, Protein Sci., 2022, 31, e4426 CrossRef CAS PubMed.
- G. Casadevall, C. Duran and S. Osuna, JACS Au, 2023, 3, 1554–1562 CrossRef CAS PubMed.
- Y. Hioki, K. Ogasahara, S. J. Lee, J. Ma, M. Ishida, Y. Yamagata, Y. Matsuura, M. Ota, M. Ikeguchi, S. Kuramitsu and K. Yutani, Eur. J. Biochem., 2004, 271, 2624–2635 CrossRef CAS PubMed.
- S. J. Lee, K. Ogasahara, J. C. Ma, K. Nishio, M. Ishida, Y. Yamagata, T. Tsukihara and K. Yutani, Biochemistry, 2005, 44, 11417–11427 CrossRef CAS PubMed.
- A. R. Buller, S. Brinkmann-Chen, D. K. Romney, M. Herger, J. Murciano-Calles and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14599–14604 CrossRef CAS PubMed.
- A. R. Buller, P. van Roye, J. K. B. Cahn, R. A. Scheele, M. Herger and F. H. Arnold, J. Am. Chem. Soc., 2018, 140, 7256–7266 CrossRef CAS PubMed.
- S. Piana, A. G. Donchev, P. Robustelli and D. E. Shaw, J. Phys. Chem. B, 2015, 119, 5113–5123 CrossRef CAS PubMed.
- P. S. Shabane, S. Izadi and A. V. Onufriev, J. Chem. Theory Comput., 2019, 15, 2620–2634 CrossRef CAS PubMed.
- G. Casadevall, J. Casadevall, C. Duran and S. Osuna, Protein Eng., Des. Sel., 2024, 37, gzae005 CrossRef PubMed.
- A. R. Buller, P. van Roye, J. Murciano-Calles and F. H. Arnold, Biochemistry, 2016, 55, 7043–7046 CrossRef CAS PubMed.
- M. Herger, P. van Roye, D. K. Romney, S. Brinkmann-Chen, A. R. Buller and F. H. Arnold, J. Am. Chem. Soc., 2016, 138, 8388–8391 CrossRef CAS PubMed.
- J. Murciano-Calles, D. K. Romney, S. Brinkmann-Chen, A. R. Buller and F. H. Arnold, Angew. Chem., Int. Ed., 2016, 55, 11577–11581 CrossRef CAS PubMed.
- D. K. Romney, J. Murciano-Calles, J. E. Wehrmuller and F. H. Arnold, J. Am. Chem. Soc., 2017, 139, 10769–10776 CrossRef CAS PubMed.
- K. B. D. A. Case I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, G. Giambasu, M. K. Gilson, H. Gohlke, A. W. Goetz, R. Harris, S. Izadi, S. A. Izmailov, K. Kasavajhala, A. Kovalenko, R. Krasny, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, V. Man, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, A. Onufriev, F. Pan, S. Pantano, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, L. Wilson, R. M. Wolf, X. Wu, Y. Xiong, Y. Xue, D. M. York and P. A. Kollman, AMBER 2020, 2020 Search PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and E. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3fd00156c |
| This journal is © The Royal Society of Chemistry 2024 |