
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into lithium inventory quantification of LiNi0.5Mn1.5O4–graphite full cells†
Wurigumula
Bao
 *a,
Weiliang
Yao
b,
Yixuan
Li
c,
Baharak
Sayahpour
c,
Bing
Han
c,
Ganesh
Raghavendran
c,
Ryosuke
Shimizu
c,
Ashley
Cronk
b,
Minghao
Zhang
c,
Weikang
Li
c and
Ying Shirley
Meng
*ac
*a,
Weiliang
Yao
b,
Yixuan
Li
c,
Baharak
Sayahpour
c,
Bing
Han
c,
Ganesh
Raghavendran
c,
Ryosuke
Shimizu
c,
Ashley
Cronk
b,
Minghao
Zhang
c,
Weikang
Li
c and
Ying Shirley
Meng
*ac
aPritzker School of Molecular Engineering, University of Chicago, Chicago, IL 60637, USA. E-mail: shirleymeng@uchicago.edu; wubao@uchicago.edu
bMaterials Science and Engineering, University of California San Diego, La Jolla, CA 92093, USA
cDepartment of NanoEngineering, University of California San Diego, La Jolla, CA 92093, USA
First published on 16th May 2024
Abstract
High voltage spinel cathode LiNi0.5Mn1.5O4 (LNMO) offers higher energy density and competitive cost compared to traditional cathodes in lithium-ion batteries, making it a promising option for high-performance battery applications. However, the fast capacity decay in full cells hinders further commercialization. The Li inventory evolution upon cycling in the LNMO–graphite pouch cell is systematically studied by developing lithium quantification methods on the cathode, anode, and electrolyte. The findings reveal that active Li loss is a primary factor contributing to capacity decay, stemming from an unstable anode interphase caused by crosstalk. This crosstalk primarily originates from electrolyte degradation on the cathode under high-voltage operation, leading to increased moisture and acidity, subsequently corroding the anode interphase. In response, two approaches including an aluminum oxide (Al2O3) surface coating layer on the cathode and lithium difluoro(oxalato)borate (LiDFOB) electrolyte additives are evaluated systematically, resulting in cycling stability enhancement. This study offers a quantitative approach to understanding the Li inventory loss in the LNMO–Gr system, providing unique insights and guidance into identifying critical bottlenecks for developing high voltage (>4.4 V) lithium battery technology.
Broader contextLithium-ion batteries experience capacity fading due to complex interplays between lithium inventory loss and active material loss. While advanced characterization techniques shed light on these mechanisms, quantifying lithium inventory changes across all cell components has remained elusive. In this work, we present innovative methods to quantify lithium inventory in the cathode, anode, and electrolyte of LNMO–Gr pouch cells, a high-voltage system prone to degradation. This represents the first study to quantitatively understand the full cell degradation mechanism. Our results reveal that lithium inventory loss, primarily resulting in solid electrolyte interphase (SEI) formation at the anode, is the major culprit behind capacity fading. These insights provide a deeper understanding of LNMO–Gr degradation and demonstrate the versatility of the methods for studying lithium inventory in diverse Li-ion battery configurations under demanding conditions. |
Introduction
The transition to renewable energy systems around the world has increased the interest in energy storage technologies, especially in lithium-ion batteries (LIBs). The high energy density and long cycle life of LIBs make them attractive for various applications. The high-voltage spinel LNMO (LiNi0.5Mn1.5O4) has attracted wide attention due to the high mass-specific energy density and high operating voltage (4.7 V).1 More importantly, it does not contain expensive cobalt or an excessive amount of lithium, making LNMO cost-effective and suitable for applications in power batteries and large-scale energy storage.2 However, LNMO has not been fully commercialized due to its severe degradation over time. Various degradation mechanisms have been proposed by different research groups and can be classified into two modes: lithium inventory loss and active material loss.3Fig. 1 shows a summary of the degradation mechanisms in the LNMO cathode and graphite anode cell. In this system, the cathode is the only source of active lithium. The loss of active lithium leads to capacity loss, and it is straightforward to track the degradation using Coulombic efficiency.4 In detail, three major degradation mechanisms are proposed on the cathode: (1) structure change; due to the J–T effect in the Mn–O framework, the distortion likely causes structure degradation.5 (2) Excessive cathode electrolyte interphase (CEI) formation. Upon electrolyte–cathode contact, irreversible reactions form a lithium-contained CEI layer, resulting in active Li loss.6 The stability of the CEI layer is closely tied to cycling conditions, such as the upper cut-off voltage and electrolyte chemistry.7,8 (3) Active material loss. The decomposition of electrolyte from various reactions typically triggers an acidity increase, causing corrosion of cathode active materials and the current collector, eventually resulting in active material loss.9,10 As for graphite (Gr) anodes, two primary mechanisms take precedence. (1) Excessive solid electrolyte interphase (SEI) formation. SEI stability can be influenced by the electrolyte and cathode.11 An additional SEI can be formed with the consumption of active Li, resulting in capacity loss.12 (2) Unwanted Li plating and inactive lithiated graphite (LixC6) trapping. Li plating on graphite anodes as a challenge for fast-charging Li-ion batteries occurs due to kinetic limitations, causing active lithium loss.13 This process often coincides with inactive LixC6, likely a result of plated Li volume expansion during charging, further contributing to active lithium loss.14,15 The electrolyte is another factor to take into consideration, as it is involved in the charge–discharge process. In a practical full cell, limited electrolyte is required to achieve high energy density. Therefore, electrolyte depletion may also induce capacity decay of LNMO–graphite full cells. The complex interplay among the LNMO cathode, graphite anode, and electrolyte makes these mechanism studies challenging and the dominant degradation mechanism may vary under different testing conditions.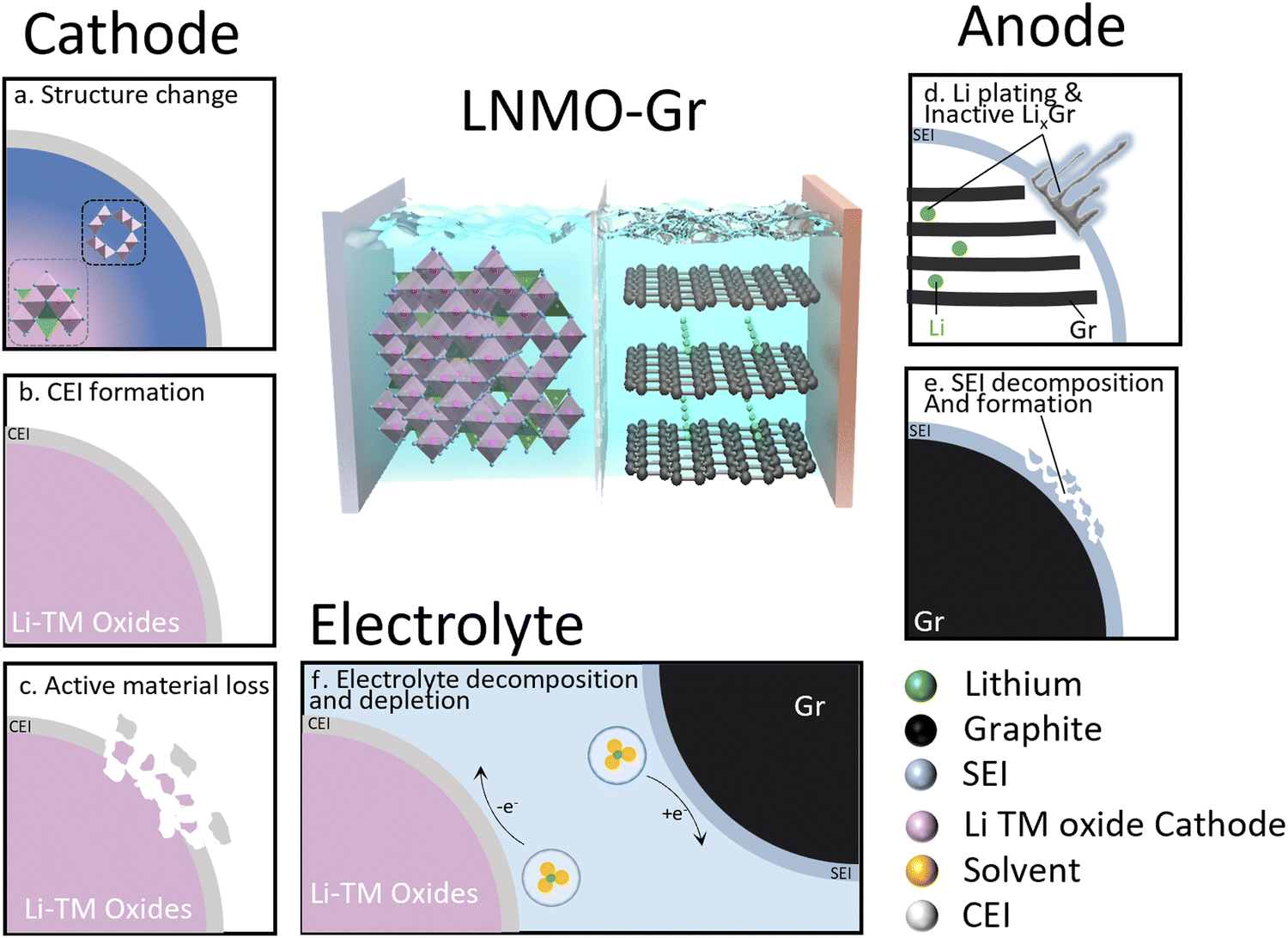 | ||
| Fig. 1 Schematic of active Li inventory loss in Li transition metal (TM) oxide cathode/Graphite full cells. (a) Cathode structure change; (b) CEI formation; (c) active material loss; (d) Li plating and trapped LixC6; (e) SEI formation; (f) electrolyte decomposition. | ||
Various characterizations have been employed to understand capacity decay in LIBs quantitively, as listed in Table S1 (ESI†). Strehle et al.16 applied ex situ X-ray diffraction (XRD) to quantify the Li content of a LiNi0.8Mn0.1Co0.1O2 (NMC 811) cathode in NMC 811/Gr full cells, revealing that the active Li loss due to NCM-811 degradation at 45 °C is more than doubled compared to at 22 °C. McShane et al.17 utilized mass spectrometry titration techniques to quantify the inactive Li0/LixC6 and SEI in a fast charging graphite anode and demonstrated that plated Li causes extra SEI formation. Differential voltage analysis has been applied to quantify the active Li in the cathode and anode in an aged NMC 811/Gr full cell. The results demonstrate that active Li loss at the anode SEI dominates capacity loss.18 Electrolyte depletion has been quantitively investigated by Thompson et al.19 using inductive coupled plasma mass spectrometry (ICP-MS). They demonstrated that Li+ in the electrolyte had not been consumed or has been consumed at the same rate as the solvent in aged LiNi0.5Mn0.3Co0.2O2 (NMC 532)/Gr full cells. Many previous studies have primarily focused on lithium loss in one or two components of the battery. However, a comprehensive understanding of the reaction mechanisms in LIBs necessitates the evaluation of lithium inventory across the entire battery system, including the cathode, anode, and electrolyte.
In this study, we set up methodologies to quantify the Li inventory in cathode, anode, and electrolyte to understand the degradation mechanism in the LNMO–Gr cell. XRD was applied on the cathode to quantify the active Li. Titration gas chromatography (TGC) was adopted to quantify the inactive LixC6 in the graphite anode since only the LixC6 can react with the protic solvent to generate H2 while SEI cannot. ICP-MS was applied to evaluate the Li+ concentration of the electrolyte upon cycling. The results show that the SEI corrosion/formation due to the cathode crosstalk is the primary cause of capacity decay. Two strategies were proposed to prevent crosstalk: (1) cathode coating and (2) electrolyte additives. The results show that electrolyte additives achieve more improvement than a cathode coating. Combining both strategies results in increased capacity retention. These findings provide a comprehensive understanding of the degradation behavior of the high voltage LNMO–Gr system. The developed workflow can extend to the Li inventory study for various Li-ion battery systems under extreme operation conditions (such as high voltage and high temperature storage).
Results and discussion
Electrochemical performance evaluation
Single-layer pouch cells with a 2 mA h cm−2 level were prepared using LNMO as the cathode, graphite as the anode, and Gen2 (1 M LiPF6 in EC: EMC = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 wt%) as the electrolyte. The cycling performance is shown in Fig. 2a. The cell started from C/10 for 2 formation cycles and C/3 for long-term cycling with a voltage range of 3.5–4.85 V. The cell delivered 82% capacity retention after 140 cycles, which is not comparable to the LNMO half-cell using a lithium metal chip as the counter electrode, where no obvious degradation was observed up to 150 cycles, as shown in Fig. S1 (ESI†). The performance discrepancy between the half cell and full cell may result from the following reasons: (1) electrolyte depletion, and (2) Li inventory loss. To verify the above hypothesis, first, the anode and cathode from the cycled pouch cell were collected and reassembled into a coin full cell with fresh electrolyte, and the corresponding cycling performances are shown in Fig. 2b. The even lower capacity indicated that the electrolyte depletion was not the primary reason for the original capacity decay. The half-cell performance of the harvested LNMO and Gr electrode with fresh electrolyte was further determined to check the electrode degradation, and the related data are shown in Fig. 2d and f, respectively. Compared to the fresh LNMO (Fig. 2c), the cycled LNMO exhibited a higher OCV of 4.6 V (pristine LNMO half cell: ∼2.8 V) and delivered a lower charge capacity of 108.6 mA h g−1 but a similar discharge capacity of 135.4 mA h g−1 in the 1st cycle. These results suggest that Li inventory loss occurred on the cathode. With the compensation of active Li from a fresh Li metal chip, the cycled cathode can be recovered, as evidenced by the similar charge–discharge profile observed in the 2nd (Fig. 2d) compared to the fresh LNMO (Fig. 2c). On the anode side, the cycled Gr, in comparison to the fresh Gr (Fig. 2e), exhibits an OCV of 1 V but similar charge–discharge profiles. Half-cell reassembly tests exclude electrolyte depletion and electrode structure degradation as the predominant reasons for capacity decay in the LNMO–Gr full cell; rather, it is the loss of active Li. Therefore, understanding how the lithium inventory changes quantitatively in each component is essential to further improve the performance of the LNMO–Gr cell.
:7 wt%) as the electrolyte. The cycling performance is shown in Fig. 2a. The cell started from C/10 for 2 formation cycles and C/3 for long-term cycling with a voltage range of 3.5–4.85 V. The cell delivered 82% capacity retention after 140 cycles, which is not comparable to the LNMO half-cell using a lithium metal chip as the counter electrode, where no obvious degradation was observed up to 150 cycles, as shown in Fig. S1 (ESI†). The performance discrepancy between the half cell and full cell may result from the following reasons: (1) electrolyte depletion, and (2) Li inventory loss. To verify the above hypothesis, first, the anode and cathode from the cycled pouch cell were collected and reassembled into a coin full cell with fresh electrolyte, and the corresponding cycling performances are shown in Fig. 2b. The even lower capacity indicated that the electrolyte depletion was not the primary reason for the original capacity decay. The half-cell performance of the harvested LNMO and Gr electrode with fresh electrolyte was further determined to check the electrode degradation, and the related data are shown in Fig. 2d and f, respectively. Compared to the fresh LNMO (Fig. 2c), the cycled LNMO exhibited a higher OCV of 4.6 V (pristine LNMO half cell: ∼2.8 V) and delivered a lower charge capacity of 108.6 mA h g−1 but a similar discharge capacity of 135.4 mA h g−1 in the 1st cycle. These results suggest that Li inventory loss occurred on the cathode. With the compensation of active Li from a fresh Li metal chip, the cycled cathode can be recovered, as evidenced by the similar charge–discharge profile observed in the 2nd (Fig. 2d) compared to the fresh LNMO (Fig. 2c). On the anode side, the cycled Gr, in comparison to the fresh Gr (Fig. 2e), exhibits an OCV of 1 V but similar charge–discharge profiles. Half-cell reassembly tests exclude electrolyte depletion and electrode structure degradation as the predominant reasons for capacity decay in the LNMO–Gr full cell; rather, it is the loss of active Li. Therefore, understanding how the lithium inventory changes quantitatively in each component is essential to further improve the performance of the LNMO–Gr cell.
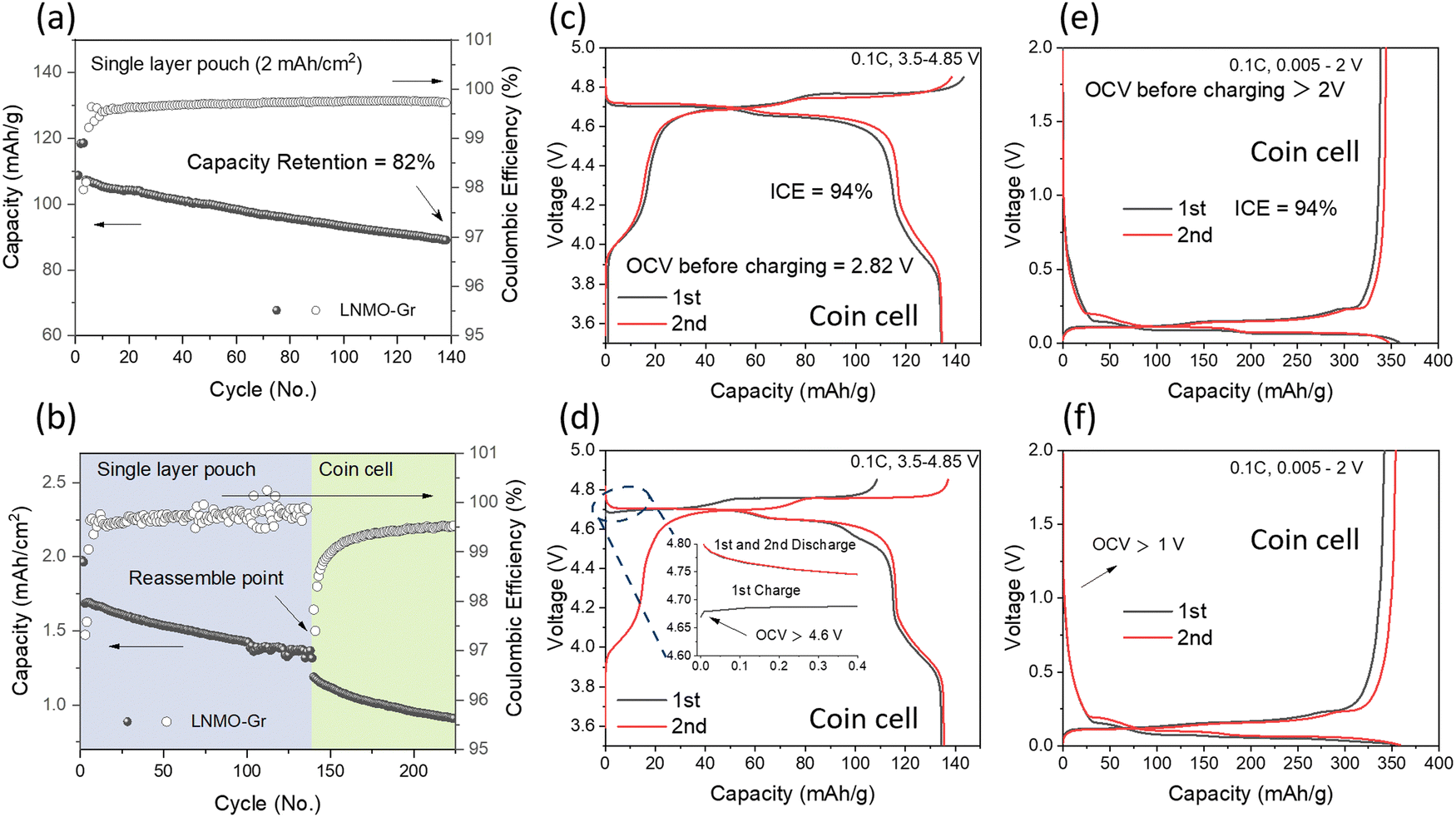 | ||
| Fig. 2 LNMO–Gr single layer pouch cell (a) cycling performance and (b) reassembly test in a coin cell; Half-cell charge–discharge profiles of (c) fresh LNMO, (d) harvested LNMO from the pouch cell, (e) fresh graphite, and (f) harvested graphite from the pouch cell. | ||
Methodology setup for Li inventory quantification
Li is present in the cathode (Licathode) and electrolyte (Lielectrolyte) when the cell is assembled. Various chemical/electrochemical reactions occur in the full cell after cycling, leading to Li redistribution as follows:| Licathode + Lielectrolyte → Licathodebulk + LiCEI + Lielectrolyte + LiSEI + LiinactiveLixC6 | (1) |
LixC6 are considered as inactive Li. To precisely measure the lithium content in each component of the battery, we utilized specific analytical techniques tailored to each part. XRD was used to analyze the cathode, TGC was applied to the anode, and ICP-MS was employed to assess the electrolyte.
Active Li quantification on the LNMO cathode via XRD
LNMO crystallizes in a spinel structure with Li+ ions located on the tetrahedral sites and the transition metal ions on the octahedral sites in an oxygen cubic close packed network,20 as shown in Fig. S2a (ESI†). The charging of cathodes is tied to the decrease of lithium ions as the lithium ions leave the structure, similar to the discharging process. From a structural viewpoint, changes in lithium concentration lead to a change in ion occupancy that could change the lattice constant.21 The relative lattice parameter evolution of the supercell with different Li amounts is plotted in Fig. 3a based on the density functional theory (DFT) calculation. Both the lattice parameter and lattice volume show a linear relationship along with Li content which provides an avenue to quantifying the Li inventory of LNMO cathodes via XRD.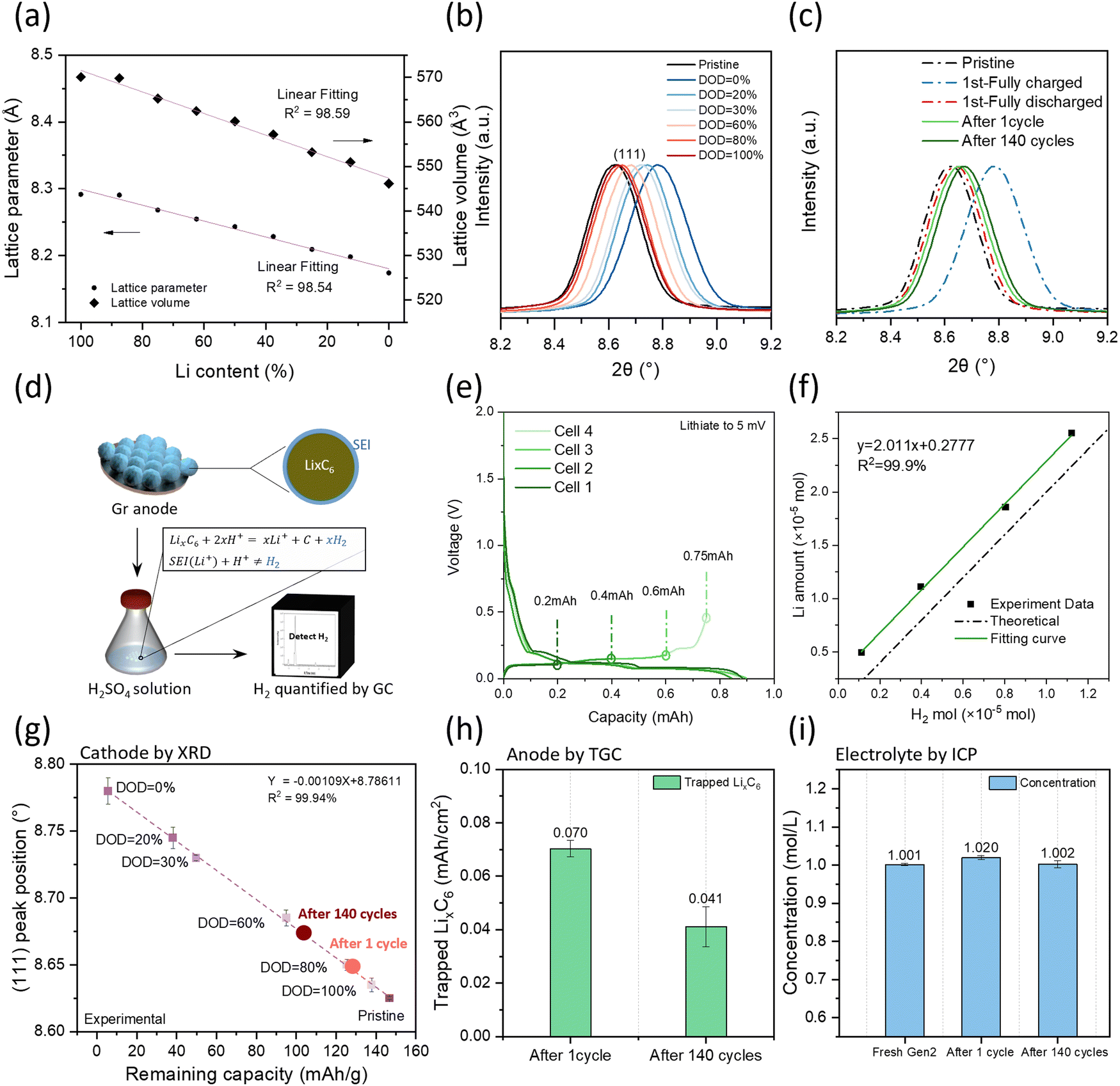 | ||
| Fig. 3 (a) Linear fitting of the LNMO structure change from computational results; (b) experimental XRD results of LNMO with different DOD states; (c) XRD results of cycled LNMO, (d) TGC methodology for quantifying LixC6 in the graphite anode; (e) charge and discharge curve of the Gr anode with the designed charge capacity (all the cells are lithiated to 5 mV and delithiated to 0.2 mA h, 0.4 mA h. 0.6 mA h and 0.75 mA h); (f) TGC verification curve for LixC6 and H2; (g) linear fitting of the remaining capacity and (111) peak position on LNMO with different DOD states; (h) TGC results of the graphite anode, and (i) Li concentrations in electrolyte after different cycles. | ||
To build a relationship between lattice parameters and the Li inventory, several half cells were performed with identical cell configurations to establish the methodology, stopping them at different depths of discharge (DOD) controlled by discharge capacity. The charge–discharge profile is shown in Fig. S2b (ESI†). The LNMO materials were collected from each cell for XRD analysis. As shown in Fig. S2c (ESI†), all the LNMO samples exhibit similar X-ray diffraction patterns with minimal impurity phases. Notably, the peak position shifted towards the right side with smaller DOD values, as detailed in Fig. 3b. The 2θ angle position of the (111) peak is quantified by the first derivative method for each sample. The remaining capacity in the cathode is then calculated using the equation as shown below.
| Remaining capacity = Theoretical capacity (147 mA h g−1) − (charge capacity − discharge capacity) | (2) |
Fig. 3g illustrates the relationship between the (111) peak position and the remaining capacity of each sample. A linear regression model was applied to the dataset and demonstrated a strong fit, with an R2 value of 99.94%, indicating excellent model fitting. The fitted equation is provided in the figure for subsequent Li quantification.
Fig. 3c shows the XRD data of LNMO collected from pouch cells with different cycling numbers, together with the pristine, fully charged, and fully discharged LNMO materials. The peak position was extracted, and the remaining capacity was calculated based on a fitted calibration curve and is shown in Fig. 3g; the results are listed in Table S2 (ESI†). The delivered capacities were obtained from the cycler, while the remaining capacities were calculated from the calibration curve. The difference between delivered capacity and remaining capacity is due to the kinetics limitation, which depends on the voltage range, C-rate, and temperature. LNMO initially exhibits a kinetically trapped capacity of 10 mA h g−1, which increases to 16 mA h g−1 after long-term cycling, possibly due to impedance growth. The increased amount of kinetically trapped lithium after 140 cycles at the cathode side is around 6 mA h g−1. Considering that the total delivered capacity loss is 24 mA h g−1, nearly one-fourth of the capacity lost is due to the cathode kinetically trapped lithium.
Li quantification on a graphite anode and in electrolyte
The inactive Li in the cycled Gr anode exists in the form of Li-containing SEI compounds (SEI Li+) and inactive LixC6. Quantifying the amount of LixC6 is achievable using the TGC method. The TGC method's principle relies on the fact that LixC6 can react with a protic solution to generate hydrogen gas (H2). The total amount of H2 gas is quantified by the peak area in the GC test, as illustrated in Fig. 3d. Previous work has demonstrated that the accuracy of the measurement is influenced by the choice of protic solution.22 Therefore, to develop the TGC method for LixC6, a LixC6 standard sample is prepared by using a Li||Gr half-cell cycle at current C/20, as shown in Fig. S3a (ESI†). The Gr anode first lithiated to 5 mV and then delithiated to 0.2 mA h. The delithiation state is chosen because the capacity change during the delithiation process is only related to the Li amount in LixC6.22 Since the cells are cycled at a low current rate, the major Li loss in the 1st cycle is attributed to the SEI formation. Therefore, the amount of LixC6 can be estimated using the following equation:| CapacityLixC6 = CapacityLithiation × ICE − Capacitydelithiation | (3) |
Five solutions H2O, 0.5 M H2SO4, 2 M H2SO4, 3 M H2SO4 and 4 M H2SO4 are selected to determine the optimal titration solvent, and the results are presented in Table S3 (ESI†). Unlike metallic Li, which can be quantified using pure water, H2O only titrates 83.0% of the LixC6. In contrast, 0.5 M H2SO4 titrates 87.2%, 2 M H2SO4 titrates 95.3% and 3 M H2SO4 titrates 98.2%. However, increasing the H2SO4 concentration to 4 M leads to additional side reactions between the Cu current collector and H2SO4 solution, impacting the titration accuracy. Fig. S3b (ESI†) displays the TGC results of Cu foil in 2 M, 3 M and 4 M H2SO4 solutions. Gases are detected when the Cu foil is immersed in the 4 M H2SO4. The results indicate that the H+ concentration influences the titration accuracy, emphasizing the necessity of acid to titrate the remaining LixC6 in the graphite anode, while the concentration should be kept below 4 M.
After identifying the optimal solvent, the linear relationship between the remaining LixC6 and the generated H2 amount is verified. Additional cells are made, and the delithiation capacity is well controlled as shown in Fig. 3e. Electrodes with varying amounts of LixC6 are taken from coin cells and stored in sealed vials. Subsequently, 1 mL of 3 M H2SO4 solution is injected into the vial, reacting with LixC6 and generating H2. The TGC results are presented in Fig. 3f. The detected H2 amount from the TGC method aligned well with the Li amount calculated by the capacity from eqn (3). It exhibits a linear relationship with 2 as the slope, indicating that as long as there is LixC6, the 3 M H2SO4 solution can convert it to H2 and be quantified by the GC.
The amount of inactive Li in the form of LixC6 in the cycled graphite anode was investigated by TGC and the results are depicted in Fig. 3h. It is noteworthy that the amount of inactive LixC6 decreases after long-term cycling, suggesting the instability of the anode interphase. The lithium content in the electrolyte was monitored via ICP-MS as shown in Fig. 3i. It was observed that the Li concentration did not exhibit significant changes before and after long-term cycling (Li concentration of Gen2:1 mol L−1), indicating simultaneous decomposition of salt and solvent.
The TEM and XPS data further underpin the TGC results. Fig. 4a–d show the morphology and structure of the cathode and anode interphase. After the 1st cycle at C/3, as shown in Fig. 4a, a thin and uniform CEI layer with a thickness of 1.5 nm is observed on the cathode surface. After 140 cycles the thickness of CEI is 3.5 nm from the TEM results as shown in Fig. 4c. No significant changes in CEI layer are observed after cycling, indicating that the consumed Li inventory on the cathode interphase in the form of CEI is negligible. Assuming that the entirety of the CEI consists of LiF, the consumption of Li in the CEI is still less than 1% after 140 cycles, as indicated in Fig. S4 and Table S4 (ESI†). However, a notable difference is observed in the anode interphase. A thicker and uneven SEI layer with a thickness of 40–200 nm is observed after the 1st cycle at C/3 compared to the CEI layer, as shown in Fig. 4b and d. After 140 cycles, the thickness of the SEI layer increased to 300–900 nm, indicating a large amount of active Li consumption on the anode interphase. The SEAD patterns detected LiF in the SEI and a stronger F signal is detected in EDX after 140 cycles compared to the 1st cycle sample. XPS F1s spectra further verified that the dominant inorganic SEI component is LiF, as shown in Fig. 4e. Besides, XPS depth profiling was further applied to understand the distribution of elements in SEI portraits, as shown in Fig. 4f and Fig. S5 (ESI†). The presence of C 1s and O 1s signals indicates solvent decomposition along with cycling, and P 2p and F 1s indicate salt decomposition. Compared to the 1st cycle sample, the SEI components after long-term cycling vary significantly. The C atomic ratio decreases as etching time increases, implying that organic components dominate the top of SEI. A weak Mn signal is detected after long-term cycling, revealing that the transition metal deposition is not the major SEI component. A significant increase in the Li, P, and F atomic ratio on the anode surface (at 120 s and 240 s etching) compared to the 1st cycle sample is observed, which indicates salt decomposition. This decomposition is triggered by by-products of solvent decomposition. Extensive studies have shown that the EC solvent can decompose, producing H2O either electrochemically23 or through a chemical reaction with O2 released24 by the cathode under high voltage. While layered cathodes such as LCO and NMC indeed release oxygen under high voltage, leading to solvent decomposition, LNMO, which has a spinel structure, does not typically release oxygen, as supported by most evidence.25 Thus, in systems using LNMO, EC is prone to electrochemical decomposition. The H2O will further react with LiPF6 salts and generate strong acid F2PO2H/HF, which will corrode the cathode and anode SEI. Considering only salts contained the F, and no significant change was observed in Li concentration before and after long-term cycling, the results further verified that electrolyte depletion happened mostly on the anode surface.
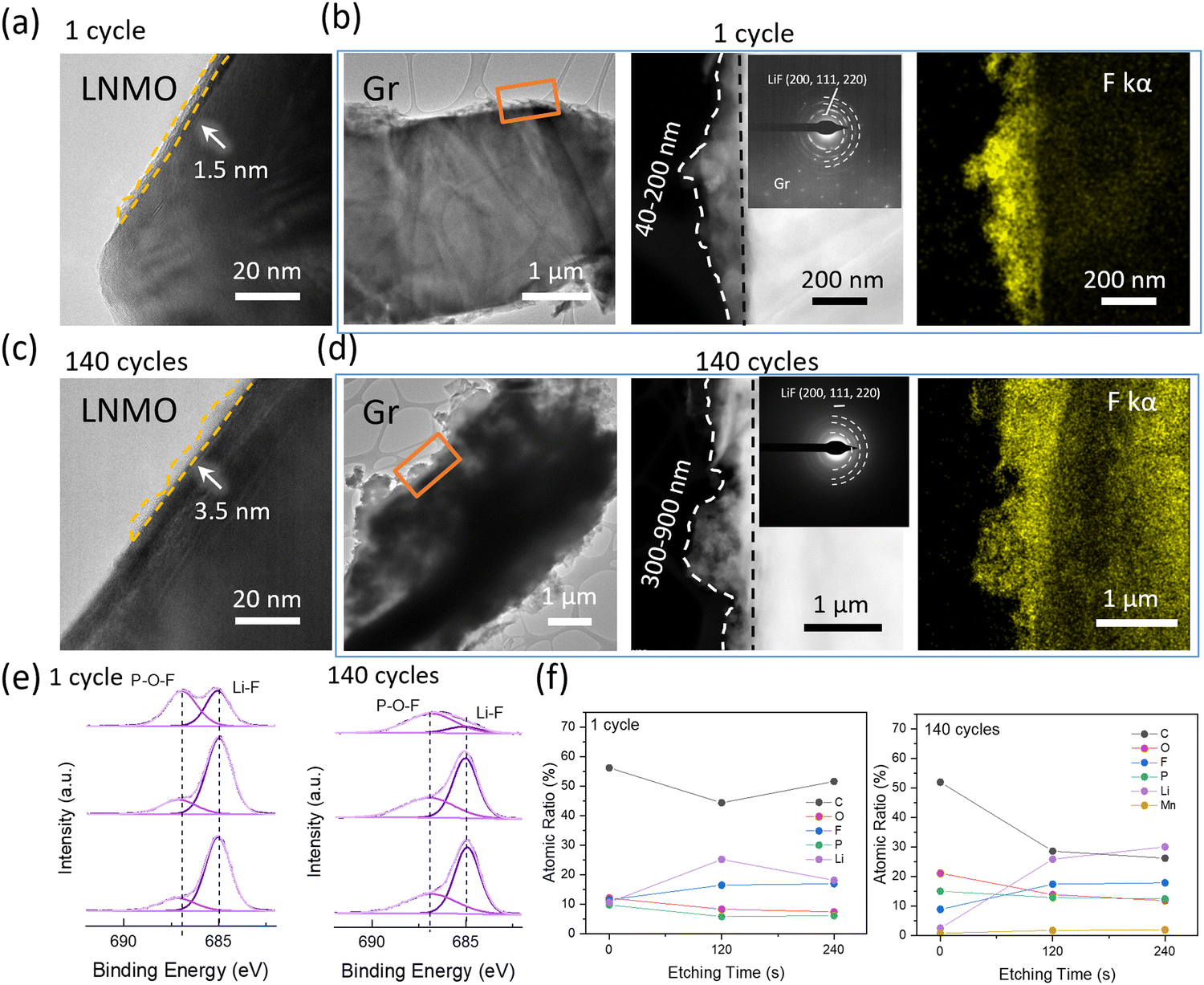 | ||
| Fig. 4 HRTEM images of the LNMO interphase at (a) the 1st cycle after the formation and (c) 140 cycles. HRTEM images of the Gr interphase at (b) the 1st cycle after formation and (d) 140 cycles, and the corresponding SEAD pattern and EDS mapping results of F. (e) XPS spectra of F 1s on the Gr interphase after different cycles. (f) Atomic ratio of C, O, F, P and Li on the Gr interphase from the XPS depth profile. | ||
All the pertinent Li inventory data are summarized in Fig. 5 and Table S5 (ESI†). After the 1st cycle at C/3, 7% of Li was kinetically trapped in the cathode, 12.5% of Li formed SEI and 4% were trapped as LixC6. After 140 cycles, there was no significant change in kinetically trapped Li in the cathode and LixC6, indicating the intact of the cathode and anode. The results align well with the reassembling testing in Fig. 2d and f, where the capacity of the cathode and anode recovered once compensated with active Li. Most of the Li inventory loss is due to the SEI formation (>27%), leading to active Li consumption and electrolyte depletion. This finding is consistent with our previous study on electrolyte decomposition, where increased acidity results from the electrolyte decomposition on the cathode side.26 Fig. S6 (ESI†) displays the optical image of the disassembled pouch cells. The LNMO material delaminated from the Al foil after one cycle at C/3, and the electrolyte changed to a dark brown color. The separator color remained consistent with the electrolyte color. These issues are exacerbated after long-term cycling in the LNMO cell, indicating severe electrolyte degradation.
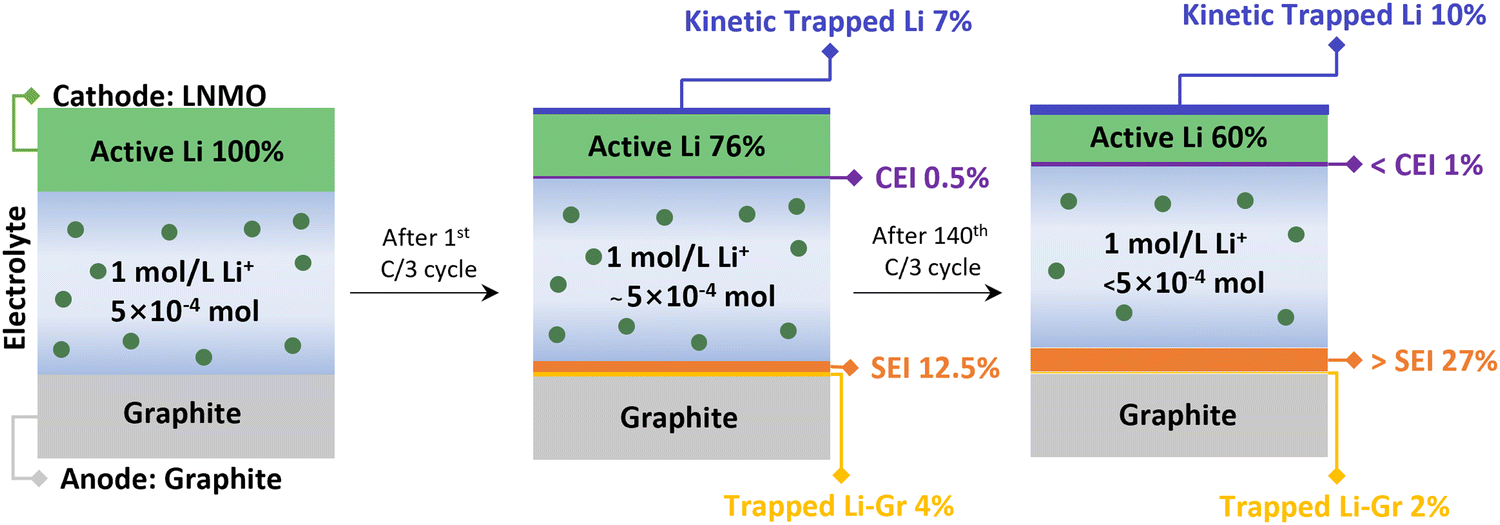 | ||
| Fig. 5 Schematic of Li inventory evolution in the LNMO–graphite pouch cell system with Gen2 electrolyte. | ||
Further electrolyte evaluation was conducted via Karl Fisher titration to monitor the moisture level, and the results are shown in Fig. S7a (ESI†). Compared with the fresh Gen2 electrolyte, an increased moisture level was observed in the electrolyte from the cycled LNMO–Gr cell, which indicates solvent decomposition. The moisture induces the decomposition of LiPF6 salts, resulting in acid formation. A quick pH value check on the electrolyte is also performed, as shown in Fig. S8 (ESI†), the much darker red color implied the high acidity, which can potentially corrode the cathode and anode interphase. The ICP-MS results in Fig. S7b (ESI†) show that after long-term cycling, Mn and Ni concentration in the LNMO–Gr pouch cell electrolyte increased, implying the obvious crosstalk between the cathode and anode through the electrolyte media.
Preventing crosstalk by cathode coating and stable interphase formation by electrolyte additives
The quantitative and qualitative results confirm that the by-products from electrolyte decomposition under high voltage operation corrode the anode interphase, resulting in active Li loss. Hence stabilizing the cathode under high voltage is crucial for preventing crosstalk. Both cathode surface coating27,28 and electrolyte additive29–31 have proven to be effective in passivating the cathode interphase. To prevent the cross talk in the LNMO–Gr system, two strategies were systematically investigated. One involves Al2O3 surface coating on the LNMO cathode surface using an atomic layer deposition (ALD) technique.32 The other one introduces boron-contained electrolyte additives LiDFOB. Fig. S9a (ESI†) shows the cycling performance of the LNMO–Gr full cell with Al2O3 coated/uncoated LNMO in Gen2 electrolyte with/without LiDFOB electrolyte additives. Compared with the pristine LNMO–Gr (black dots), which shows 77.85% capacity retention after 150 cycles with 99.48% average coulombic efficiency, improvements are observed in both strategies. A slight enhancement is observed in the Al2O3 coated LNMO–Gr cell (red dots), which shows 82.44% capacity retention after 150 cycles with the average coulombic efficiency of 99.71%. A significant improvement is obtained with LiDFOB electrolyte additives (blue dots), showing 87.32% capacity retention after 150 cycles with an average coulombic efficiency of 98.99%. The lower average coulombic efficiency in the cell with LiDFOB additives is likely due to the scavenger effect of LiDFOB with the HF generated in the electrolyte, which is one of the benefits of Boron-based electrolyte additives.33 A similar trend is observed in the high temperature storage evaluation in Fig. S9b (ESI†). The cells are stored at 100% SOC at 55 °C for 24 hours, and room temperature cycling is performed to check the capacity retention. Without coating and electrolyte improvement, fast capacity decay is observed. After repeating the storage test 4 times, only 55.5% capacity retention is obtained. With Al2O3 coating, a slight improvement of 60% capacity retention is observed, lower than the improvement that is achieved with the LiDFOB electrolyte additives, which shows 70% capacity retention after storage. The results indicate that both strategies can prevent crosstalk from the cathode, with electrolyte additives showing significant improvement in cycling performance compared with the Al2O3 coating. A better cycling performance with 86.47% capacity retention after 150 cycles and a significant improvement of 82% capacity retention after a high temperature storage test were achieved (green dots) by combining both an ALD cathode coating and LiDFOB electrolyte additives. The results provide insights into the need for comprehensive cell engineering for LNMO commercialization.Conclusions
In summary, Li inventory quantification methodologies were developed to understand the primary cause of capacity decay in the high-voltage LNMO–Gr system. XRD, TGC, and ICP-MS were applied to the LNMO cathode, Gr anode, and electrolyte for Li inventory investigation, respectively. It was revealed that the excessive SEI formation consumes the most active Li, resulting in capacity loss of the LNMO–Gr cell. This conclusion was further supported by TEM and XPS results. Moisture and pH tests revealed electrolytes degraded during high voltage operation. Charging to 4.85 V leads to by-products from parasitic reactions on the cathode surface migrating to the anode, corroding the SEI that consumes more active Li. Both Al2O3 coating and LiDFOB electrolyte additives were applied to prevent crosstalk from the cathode. Combining the two strategies resulted in even better cycling performance, highlighting the importance of novel electrolyte design and interphase protection for high voltage cycling stability. We believe that this study provides a universal method to study the Li inventory change for lithium-ion batteries, Similar methodologies can be adapted to a broader range of chemistries to achieve a more comprehensive understanding for future secondary battery development.Experimental methods
Electrochemical test
The LNMO powder and Gr electrode is from Haldor Topsoe (Denmark) and Ningbo Institute of Materials Technology & Engineering (NIMTE). For the half-cell testing, the LNMO electrode and Gr electrode were assembled into a 2032 type coin cell in an Ar-filled glove box. Li metal (1 mm thick, 0.5 inches in diameter) was employed as the counter electrode. The electrolyte was Gen2 (EC:EMC 1/4 3:7 wt% with 1 mol L−1 LiPF6) from Gotion company (USA), and the electrolyte amount was fixed as 50 μL per cell. The Gr half-cell was lithiated to 5 mV and delithiated to different states for TGC samples during lithiation by limiting either the cut-off voltage or the cut-off capacity. For the full cell testing, the Gr electrode was paired with a 2 mA h cm−2 LNMO cathode and assembled in a 2032 type coin cell with a negative to positive ratio (N/P) of 1.05. The full cell was cycled between 3.5 V and 4.85 V at room temperature at a current density (1C = 147 mA h g−1, based on LNMO) of C/20 for the first two cycles and C/3 for the subsequent cycles.
The pouch cells were first assembled in the atmosphere without electrolyte, and then the dry pouch was dried at 80 overnight under vacuum before the electrolyte injection. The cathode size is 44 mm × 30 mm, and anode size is 45 mm × 32 mm. After electrolyte injection, the pouch cell was vacuum sealed in the glove box. The electrochemical performances of all the electrodes were tested at room temperature either using a Neware Battery Test System, Arbin BT2000 instruments (Arbin instrument, USA), and the electrolyte injection amount is 500 μL.
Characterizations
Author contributions
W. B., W. L., and Y. S. M. conceived the ideas. W. B. and W. L. prepared electrodes and cycled the cells. W. B. fabricated the half-cell, conducted electrochemical cycling, and carried out TGC measurements. W. B. wrote the manuscript. B. H. and M. Z. performed TEM experiments and data analysis. W.B., W.L. and R.S. designed and conducted the XPS experiments and ICP experiments. Y. L. prepared the electrolyte additives, G. R. and W. Y. prepared and tested the pouch cell. G. R. prepared the XRD sample, B. S. conducted the XRD measurements and W. L. performed XRD refinement. A. C. performed the moisture test. All authors discussed the results and commented on the manuscript.Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the Office of Vehicle Technologies of the US Department of Energy and U.S. Department of the Army's Tank & Automotive Research Development and Engineering Command (TARDEC) through the Battery R&D program under contract DE-EE0008442. TEM was performed at the San Diego Nanotechnology Infrastructure (SDNI) of UCSD, a member of the National Nanotechnology Coordinated Infrastructure supported by the National Science Foundation (Grant ECCS-1542148). The authors acknowledge the use of facilities and instrumentation at the UC Irvine Materials Research Institute (IMRI), which is supported in part by the National Science Foundation through the UC Irvine Materials Research Science and Engineering Center (DMR-2011967). Specifically, the XPS work was performed using instrumentation funded in part by the National Science Foundation Major Research Instrumentation Program under grant no. CHE-1338173. The DFT calculation used Stampede 2 at the Texas Advanced Computing Center (TACC) through allocation DMR110008 from the Extreme Science and Engineering Discovery Environment (XSEDE), which was supported by the National Science Foundation (Grant 1548562). The authors also acknowledge the use of facilities and instrumentation supported by NSF through the UC San Diego Materials Research Science and Engineering Center (UCSD MRSEC), DMR-2011924. The ICP-MS in this work was conducted at the Environmental and Complex Analysis Laboratory (ECAL) in the Chemistry and Biochemistry department at UC San Diego. The authors acknowledge Neware Instruments for the Neware battery test system and MTI for cell assembling tool donation.References
- S. Park, S.-W. Oh, S. H. Kang, I. Belharouak, K. Amine and Y.-K. Sun, Comparative study of different crystallographic structure of LiNi0. 5Mn1. 5O4− δ cathodes with wide operation voltage (2.0–5.0 V), Electrochim. Acta, 2007, 52(25), 7226–7230 CrossRef CAS.
- W. Li, Y.-G. Cho, W. Yao, Y. Li, A. Cronk, R. Shimizu, M. A. Schroeder, Y. Fu, F. Zou and V. Battaglia, Enabling high areal capacity for Co-free high voltage spinel materials in next-generation Li-ion batteries, J. Power Sources, 2020, 473, 228579 CrossRef CAS.
- C. R. Birkl, M. R. Roberts, E. McTurk, P. G. Bruce and D. A. Howey, Degradation diagnostics for lithium ion cells, J. Power Sources, 2017, 341, 373–386 CrossRef CAS.
- J. Xiao, Q. Li, Y. Bi, M. Cai, B. Dunn, T. Glossmann, J. Liu, T. Osaka, R. Sugiura and B. Wu, Understanding and applying coulombic efficiency in lithium metal batteries, Nat. Energy, 2020, 5(8), 561–568 CrossRef CAS.
- S. Liu, B. Wang, X. Zhang, S. Zhao, Z. Zhang and H. Yu, Reviving the lithium-manganese-based layered oxide cathodes for lithium-ion batteries, Matter, 2021, 4(5), 1511–1527 CrossRef CAS.
- S. P. Kühn, K. Edström, M. Winter and I. Cekic-Laskovic, Face to face at the cathode electrolyte interphase: From interface features to interphase formation and dynamics, Adv. Mater. Interfaces, 2022, 9(8), 2102078 CrossRef.
- H. Jia, Y. Xu, S. D. Burton, P. Gao, X. Zhang, B. E. Matthews, M. H. Engelhard, L. Zhong, M. E. Bowden and B. Xiao, Enabling ether-based electrolytes for long cycle life of lithium-ion batteries at high charge voltage, ACS Appl. Mater. Interfaces, 2020, 12(49), 54893–54903 CrossRef CAS PubMed.
- H. Jia, X. Zhang, Y. Xu, L. Zou, J.-M. Kim, P. Gao, M. H. Engelhard, Q. Li, C. Niu and B. E. Matthews, Toward the practical use of cobalt-free lithium-ion batteries by an advanced ether-based electrolyte, ACS Appl. Mater. Interfaces, 2021, 13(37), 44339–44347 CrossRef CAS PubMed.
- M. Metzger, P. Walke, S. Solchenbach, G. Salitra, D. Aurbach and H. A. Gasteiger, Evaluating the high-voltage stability of conductive carbon and ethylene carbonate with various lithium salts, J. Electrochem. Soc., 2020, 167(16), 160522 CrossRef CAS.
- J. G. Han, K. Kim, Y. Lee and N. S. Choi, Scavenging materials to stabilize LiPF6-containing carbonate-based electrolytes for Li-ion batteries, Adv. Mater., 2019, 31(20), 1804822 CrossRef PubMed.
- S. Klein, P. Bärmann, L. Stolz, K. Borzutzki, J.-P. Schmiegel, M. Börner, M. Winter, T. Placke and J. Kasnatscheew, Demonstrating apparently inconspicuous but sensitive impacts on the rollover failure of lithium-ion batteries at a high voltage, ACS Appl. Mater. Interfaces, 2021, 13(48), 57241–57251 CrossRef CAS PubMed.
- H. Jia, B. Billmann, H. Onishi, J. Smiatek, S. Roeser, S. Wiemers-Meyer, R. Wagner, M. Winter and I. Cekic-Laskovic, LiPF6 stabilizer and transition-metal cation scavenger: a bifunctional bipyridine-based ligand for lithium-ion battery application, Chem. Mater., 2019, 31(11), 4025–4033 CrossRef CAS.
- W. Cai, Y.-X. Yao, G.-L. Zhu, C. Yan, L.-L. Jiang, C. He, J.-Q. Huang and Q. Zhang, A review on energy chemistry of fast-charging anodes, Chem. Soc. Rev., 2020, 49(12), 3806–3833 RSC.
- H. Charalambous, O. J. Borkiewicz, A. M. Colclasure, Z. Yang, A. R. Dunlop, S. E. Trask, A. N. Jansen, I. D. Bloom, U. Ruett and K. M. Wiaderek, Comprehensive Insights into Nucleation, Autocatalytic Growth, and Stripping Efficiency for Lithium Plating in Full Cells, ACS Energy Lett., 2021, 6(10), 3725–3733 CrossRef CAS.
- D. P. Finegan, A. Quinn, D. S. Wragg, A. M. Colclasure, X. Lu, C. Tan, T. M. Heenan, R. Jervis, D. J. Brett and S. Das, Spatial dynamics of lithiation and lithium plating during high-rate operation of graphite electrodes, Energy Environ. Sci., 2020, 13(8), 2570–2584 RSC.
- B. Strehle, F. Friedrich and H. A. Gasteiger, A Comparative Study of Structural Changes during Long-Term Cycling of NCM-811 at Ambient and Elevated Temperatures, J. Electrochem. Soc., 2021, 168(5), 050512 CrossRef CAS.
- E. J. McShane, A. M. Colclasure, D. E. Brown, Z. M. Konz, K. Smith and B. D. McCloskey, Quantification of inactive lithium and solid–electrolyte interphase species on graphite electrodes after fast charging, ACS Energy Lett., 2020, 5(6), 2045–2051 CrossRef CAS.
- W. M. Dose, C. Xu, C. P. Grey and M. F. De Volder, Effect of anode slippage on cathode cutoff potential and degradation mechanisms in Ni-rich Li-ion batteries, Cell Rep. Phys. Sci., 2020, 1(11), 100253 CrossRef.
- L. Thompson, W. Stone, A. Eldesoky, N. Smith, C. M. McFarlane, J. Kim, M. Johnson, R. Petibon and J. Dahn, Quantifying changes to the electrolyte and negative electrode in aged NMC532/graphite lithium-ion cells, J. Electrochem. Soc., 2018, 165(11), A2732 CrossRef CAS.
- A. Bhatia, C. Leviel, M. Hallot, J. P. Pereira-Ramos, C. Lethien, P. Roussel and R. Baddour-Hadjean, Probing the Electrochemical Li Insertion–Extraction Mechanism in Sputtered LiNi0. 5Mn1. 5O4 Thin Film Cathode for Li-Ion Microbattery, Adv. Mater. Interfaces, 2022, 9(25), 2200733 CrossRef CAS.
- L. Chladil, D. Kunický, T. Kazda, P. Vanýsek, O. Čech and P. Bača, In-situ XRD study of a Chromium doped LiNi0. 5Mn1. 5O4 cathode for Li-ion battery, J. Energy Storage, 2021, 41, 102907 CrossRef.
- W. Bao, C. Fang, D. Cheng, Y. Zhang, B. Lu, D. H. Tan, R. Shimizu, B. Sreenarayanan, S. Bai and W. Li, Quantifying lithium loss in amorphous silicon thin-film anodes via titration-gas chromatography, Cell Rep. Phys. Sci., 2021, 2(10), 100597 CrossRef CAS.
- B. L. Rinkel, D. S. Hall, I. Temprano and C. P. Grey, Electrolyte oxidation pathways in lithium-ion batteries, J. Am. Chem. Soc., 2020, 142(35), 15058–15074 CrossRef CAS PubMed.
- C. Jayawardana, N. Rodrigo, B. Parimalam and B. L. Lucht, Role of electrolyte oxidation and difluorophosphoric acid generation in crossover and capacity fade in lithium ion batteries, ACS Energy Lett., 2021, 6(11), 3788–3792 CrossRef CAS.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, Chemical versus electrochemical electrolyte oxidation on NMC111, NMC622, NMC811, LNMO, and conductive carbon, J. Phys. Chem. Lett., 2017, 8(19), 4820–4825 CrossRef CAS PubMed.
- Y. Li, W. Li, R. Shimizu, D. Cheng, H. Nguyen, J. Paulsen, S. Kumakura, M. Zhang and Y. S. Meng, Elucidating the Effect of Borate Additive in High-Voltage Electrolyte for Li-Rich Layered Oxide Materials, Adv. Energy Mater., 2022, 12(11), 2103033 CrossRef CAS.
- H.-M. Cho, M. V. Chen, A. C. MacRae and Y. S. Meng, Effect of surface modification on nano-structured LiNi0. 5Mn1. 5O4 spinel materials, ACS Appl. Mater. Interfaces, 2015, 7(30), 16231–16239 CrossRef CAS PubMed.
- M. Hallot, B. Caja-Munoz, C. Leviel, O. I. Lebedev, R. Retoux, J. Avila, P. Roussel, M. C. Asensio and C. Lethien, Atomic layer deposition of a nanometer-thick Li3PO4 protective layer on LiNi0. 5Mn1. 5O4 films: dream or reality for long-term cycling?, ACS Appl. Mater. Interfaces, 2021, 13(13), 15761–15773 CrossRef CAS PubMed.
- J. Alvarado, M. A. Schroeder, M. Zhang, O. Borodin, E. Gobrogge, M. Olguin, M. S. Ding, M. Gobet, S. Greenbaum and Y. S. Meng, A carbonate-free, sulfone-based electrolyte for high-voltage Li-ion batteries, Mater. Today, 2018, 21(4), 341–353 CrossRef CAS.
- A. Hofmann, A. Höweling, N. Bohn, M. Müller, J. R. Binder and T. Hanemann, Additives for Cycle Life Improvement of High-Voltage LNMO-Based Li-Ion Cells, ChemElectroChem, 2019, 6(20), 5255–5263 CrossRef CAS.
- T. Yang, H. Zeng, W. Wang, X. Zhao, W. Fan, C. Wang, X. Zuo, R. Zeng and J. Nan, Lithium bisoxalatodifluorophosphate (LiBODFP) as a multifunctional electrolyte additive for 5 V LiNi 0.5 Mn 1.5 O 4-based lithium-ion batteries with enhanced electrochemical performance, J. Mater. Chem. A, 2019, 7(14), 8292–8301 RSC.
- W. Li, D. Cheng, R. Shimizu, Y. Li, W. Yao, G. Raghavendran, M. Zhang and Y. S. Meng, Artificial cathode electrolyte interphase for improving high voltage cycling stability of thick electrode with Co-free 5 V spinel oxides, Energy Storage Mater., 2022, 49, 77–84 CrossRef.
- B. Zhang, L. Wang, X. Wang, S. Zhou, A. Fu, Y. Yan, Q. Wang, Q. Xie, D. Peng and Y. Qiao, Sustained releasing superoxo scavenger for tailoring the electrode-electrolyte interface on Li-rich cathode, Energy Storage Mater., 2022, 53, 492–504 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee00842a |
| This journal is © The Royal Society of Chemistry 2024 |