
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUtilizing three-terminal, interdigitated back contact Si solar cells as a platform to study the durability of photoelectrodes for solar fuel production†
Darci K.
Collins‡
 *ab,
Zebulon G.
Schichtl‡
a,
Nathan T.
Nesbitt
a,
Ann L.
Greenaway
a,
Valentin D.
Mihailetchi
c,
Daniel
Tune
c and
Emily L.
Warren
*a
*ab,
Zebulon G.
Schichtl‡
a,
Nathan T.
Nesbitt
a,
Ann L.
Greenaway
a,
Valentin D.
Mihailetchi
c,
Daniel
Tune
c and
Emily L.
Warren
*a
aMaterials, Chemical, and Computational Science Center (MCCS), National Renewable Energy Laboratory, Golden, Colorado 80401, USA. E-mail: emily.warren@nrel.gov; darci.collins@nrel.gov
bColorado School of Mines, Advanced Energy Systems Graduate Program, Golden, CO 80401, USA
cInternational Solar Energy Research Center (ISC) Konstanz, Rudolf–Diesel–Straße 15, 78467, Konstanz, Germany
First published on 2nd May 2024
Abstract
Unassisted photoelectrochemical (PEC) reactions, such as H2 generation and CO2 reduction, are limited by the durability of the immersed photoelectrode. Small band gap semiconductors, like Si, are efficient at utilizing a large portion of the solar spectrum but are not stable in aqueous environments without protection. While great strides have been made to improve stability under constant illumination, dark stability remains relatively unexamined and presents great challenges for durable PEC systems. Cathodic protection is an established electrochemical method for preventing metal electrode degradation in harsh conditions. Similar protection strategies cannot be applied to traditional two-terminal (2T) semiconductor photoelectrodes because of their inability to pass reverse bias current in the dark. New, three-terminal (3T) photovoltaic (PV) architectures introduce additional degrees of freedom in traditional 2T PEC operations by adding an extra electrical contact for an alternative low resistance path to protect the photoelectrode and drive electrochemical reactions, even in the dark. Here, we investigate bare 3T Si PV devices operating as photocathodes in aqueous methyl viologen electrolyte. The 3T architecture provides additional capabilities to PEC systems such as cathodic protection, the ability to drive reactions with or without illumination, and in situ switching between different operational modes. We show that 3T-based Si photocathodes maintain PEC activity after several hours of light/dark cycling. This work helps advance PEC use in real-world conditions where variable illumination must be considered.
Broader contextStoring solar energy in chemical bonds using electrochemistry could provide long-term, inexpensive energy storage. Solar-driven carbon dioxide reduction can generate hydrocarbon fuels using photoelectrochemical (PEC) systems that combine light absorption and electrochemical fuel production into a single device. Real-world PEC systems will need to be stable across the day and night, but few studies have investgated dark stability. In the dark, the photoelectrode sits at the solution potential, which allows deleterious reactions that cannot occur under illumination. Industrial scale construction projects improve the long-term durability of metal components by applying a negative (cathodic) voltage to them, preventing degradation. However, most PEC photoelectrodes are two-terminal (2T) devices and applying a cathodic potential in the dark will damage them. New advances in photovoltaic device construction have produced three-terminal (3T) devices that allow for the application of cathodic voltages in the dark. Here, we demonstrate protection of 3T Si solar cells used as photoelectrodes in a methyl viologen PEC system. We discuss light and dark working modes and demonstrate continued operation in cycled illumination conditions, utilizing cathodic protection in the dark to maintain activity. The 3T architecture is a critical step towards a working PEC system to enable energy storage in chemical bonds. |
1. Introduction
Photovoltaic (PV) device performance has seen significant advances over the past decade, driven by dramatic decreases in semiconductor photoabsorber costs, making electricity production from PV devices cost competitive with fossil fuels.1–3 Photoelectrochemical (PEC) systems integrate semiconductor photoabsorbers and electrocatalysis into a single device, using solar energy directly to drive fuel-forming reactions that store energy in chemical bonds.4,5 PEC systems for the reduction of H2O to H2 or CO2 to hydrocarbon fuels are an attractive way to store solar energy as the only inputs are H2O, CO2, and sunlight. PEC systems are primed to take advantage of the dramatic cost reductions and performance improvements in PV technologies to provide a route towards critical, long-term renewable energy storage. A major challenge limiting PEC systems is maintaining long term performance and efficiency, particularly under real-world operating conditions, where devices undergo day/night illumination cycles.6–8Many PEC prototypes have demonstrated high performance for fuel-forming reactions,5,9–12 but few studies have examined how these systems need to be operated under all illumination conditions.13 Preventing unfavorable reverse reactions and degradation of the semiconductor in solution, especially in the dark, remains a challenge. Photoelectrode durability studies tend to focus on demonstrating performance under continuous illumination14–17 or preventing degradation in the light.10,18–20 The introduction of protective layers (e.g. TiO2, MoS2, sacrificial ions, and polymer films)5,11,21–27 has greatly extended the illuminated operation of photoelectrodes. However, under real outdoor operating conditions, a semiconductor photoabsorber operating as a photoelectrode needs to be stable under day/night (diurnal) lighting conditions.8,28–32 Small band gap semiconductors (e.g. Si) utilize more of the solar spectrum than large band gap semiconductors (e.g. metal oxides) but suffer from corrosion or passivation when in aqueous electrolyte. Si readily oxidizes in room temperature air or water.33 Under illumination, photocathode stability is less of an issue since the excess of electrons at the surface inhibits oxidative corrosion processes, but in the absence of electrons, dark degradation mechanisms can proceed. There is thus a great need to understand and improve the stability of photoelectrodes operating overnight.
It would be convenient to have an excess of electrons at the surface in the dark to inhibit oxidative corrosion, however, applying negative voltage in the dark passes reverse bias current on two-terminal (2T) devices and could damage the semiconductor or require prohibitively large voltages, as it does in PV devices. Passing protective cathodic current in the dark is a well-known electrochemical phenomenon for metals. In typical electrochemical systems utilizing cathodic protection, metal surfaces exposed to corrosive environments are subject to a cathodic electrical bias to prevent or slow the rate of degradation.34 Cathodic protection is used in metals to provide a supply of electrons at the interface which can be oxidized instead of the material itself.35 The additional electrons from a protective cathodic voltage in the dark (or photogenerated electrons) can prevent thermodynamically favorable self-oxidation reaction.36 Cathodic protection is widely used in industrial applications,37–39 including marine corrosion protection,40 where it extends the lifetime of the structures and reduces repair costs.39 Utilizing a different PV architecture as a photoelectrode that could leverage cathodic protection in the dark would be of great interest to realizing dark stability in PEC systems.
New PV fabrication techniques have led to the development of three-terminal (3T) Si PV devices that have previously primarily been investigated for tandem PV applications.41 Interdigitated back contact (IBC) devices can be modified to add a conductive front surface field to create a 3T device (based on a modified ZEBRA™ process).42 A 3T Si can operate as a standard IBC PV device under illumination, a photocathode under illumination, or dark cathode via the additional contact providing an alternate low resistance pathway bypassing the p–n junction in the device that enables the use of cathodic current to pass in the dark with minimal applied voltage. There have been many previous studies on 3T PV devices,43–48 but to our knowledge only one study has applied 3T devices to PEC fuel-forming systems.49
In this work, we use bare 3T Si PV devices based on n-type IBC cells with degenerately doped n-type front surface fields (designated as nuIBC under the recently proposed taxonomy of 3T solar cells)41 as a platform to study the durability of photoelectrodes in the dark. We hypothesize that the alternative current pathway could be used for cathodic protection of the photoelectrode surface in dark conditions. The introduction of cathodic protection to Si photoelectrodes could improve stability of PEC systems in variable illumination conditions and allow for production of fuel under illumination or at night with an external energy source. We use PEC techniques to determine the operational lifetime of the bare 3T Si under dark conditions with a regenerative PEC system using the methyl viologen (MV2+/+) redox couple, which experimentally decouples the electrical characteristics of the photocathode from fuel-forming catalysis.50,51 By using methyl viologen we can characterize the stability of bare Si in an aqueous environment, without having to decouple the semiconductor stability from catalyst stability. This provides a foundation for future studies that can address integrated photocatalytic systems, since the system stability will be dependent on the specific combination of materials and device architecture.52 Here, we demonstrate cathodic protection as an effective protection strategy for Si under variable illumination conditions, and this work can be extended to fuel-forming PEC systems utilizing Si, or more generally, to PEC systems operating in diurnal lighting conditions.
2. Results and discussion
The 3T ZEBRA™ solar cells were fabricated out of crystalline n-type Czochralski-grown (CZ) Si wafers with degenerately doped p-type (p+) and n-type (n+) Si regions formed by laser ablation and low temperature diffusion.42 The p+-type and n+-type back contacts are referred to as R and Z, respectively (Fig. 1).41 The ZEBRA™ metallization scheme uses isolation paste in select areas between fingers and busbars, allowing flexible positioning of the busbars with respect to screen-printed Ag fingers. In the Gen 1 ZEBRA™ cells used in this work, the front of the cells had a diffused n+-Si front surface field (Fig. 1a). The front, electrochemical contact is denoted as F. A simplified equivalent circuit of the 3T device is shown in Fig. 1b.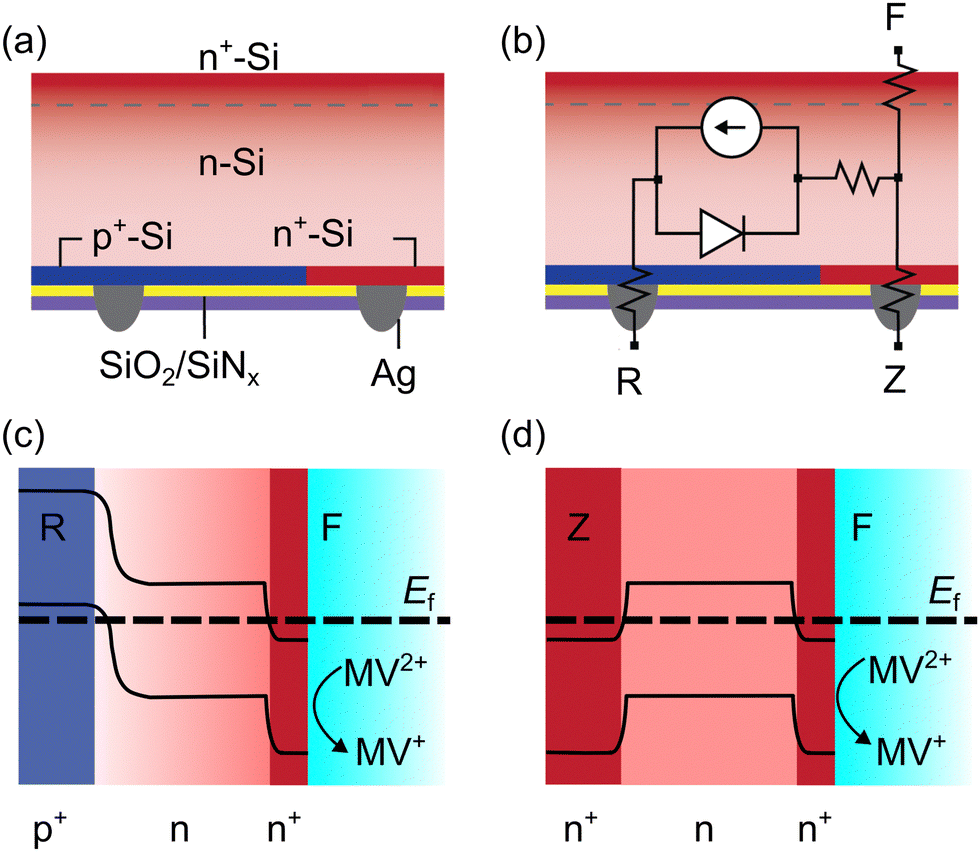 | ||
| Fig. 1 Cross sectional schematics of 3T Si devices: (a) device schematic with doping, passivation layers, and contacts; (b) equivalent electrical circuit, showing how FR and RZ pass current through a diode, while FZ measurements only have resistive losses; (c) schematic of one-dimensional band bending at equilibrium between the F (n+ front) and R (p+ back) contacts; (d) schematic of one-dimensional band bending at equilibrium between the F (n+ front) and Z (n+ back) contacts. | ||
Before the 3T Si can be used as a photoelectrode, the native oxide layer on the electrochemical (front n+ contact) surface must be removed via HF etching. Then, the 3T Si can be operated in three modes in solution, depending on which contacts are connected: FR, FZ, or RZ.41,43 When the device is operated as a photoelectrode, both FR and FZ modes have a wet, electrochemical contact. In RZ mode, the device operates like a traditional IBC solar cell and does not transfer charge carriers with solution. When configured for FR mode (operating under illumination), qualitative band bending diagrams suggest that after equilibrium is established electrons are directed towards the semiconductor-solution interface and holes are collected at the p+ back contact (Fig. 1c). Band bending diagrams for FZ mode (operating in the dark) indicate no large energy barrier for electrons at the solution interface or n+ back contact (Fig. 1d).
Unlike a traditional 2T PV device, where there is only one independent electrical variable (current, J or voltage, V), a 3T device has two independent electrical variables.41 For a 3T device, the full electrical performance cannot be described by a simple J–V curve unless one of the contacts is not connected (Fig. 2).43 A 3T PV device with a metallized front grid (F contact) was measured in the dark to show the different performance that is possible in FR and FZ modes. Fig. 2a shows a contour plot of the FR current (JFR) as a function of FR voltage (VFR) and FZ voltage (VFZ), while Fig. 2b shows the JFRvs. VFR when VFZ = 0 V, (indicated by the dashed line in Fig. 2a). These figures show that just like a 2T device, in FR mode, a 3T device does not pass any cathodic current in the dark under forward bias, regardless of the FZ voltage applied to the cell. Fig. 2c shows a contour plot of the FZ current (JFZ) as a function of VFR and VFZ in the dark, while Fig. 2d shows the JFZvs. VFZ when VFR = 0 V (indicated by the dashed line in Fig. 2c). As expected from the band diagram in Fig. 1, the data is linear, showing typical resistive behavior between the two n+ contacts. This means in FZ mode, a 3T device can pass cathodic current in the dark, limited only by resistive losses in the semiconductor.
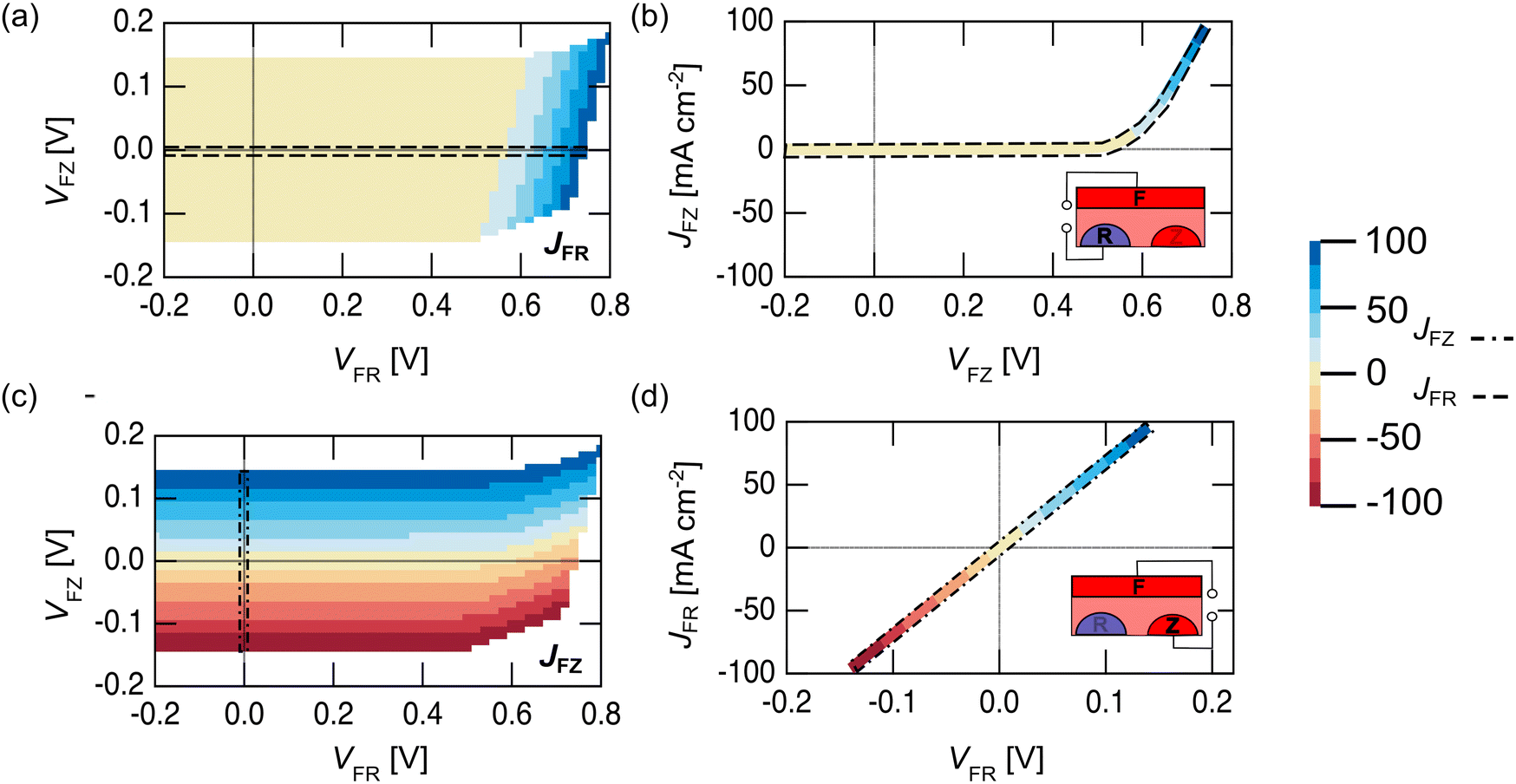 | ||
| Fig. 2 Electrical characterization of a nuIBC 3T Si solar cell in the dark. The front of the device (F) was contacted with an evaporated Ag grid as described in the experimental section. (a) Contour plot of JFR as a function of VFZ and VFR; (b)JFR–VFR data corresponding to VFZ = 0 (black dashed line in the JFR contour plot); (c) contour plot of JFZ as a function of VFZ and VFR; (d) JFZ–VFZ data corresponding to VFR = 0 (black dashed line in the JFZ contour plot). Inset cartoons in (c) and (d) indicate the connected contacts of the device. | ||
Schematics of the electrochemical characterization configurations for this work are shown in Fig. 3 (a photograph of the actual cell is shown in Fig. S1, ESI†). We used a MV2+/+ redox couple in an aqueous solution (50 mM MV2+, 0.1 M potassium hydrogen phthalate, 0.5 M K2SO4, pH 3.5) to assess the PEC performance of bare Si. Si is a rather poor electrocatalyst for H2 generation or CO2 reduction, so it either must have a catalyst or be characterized with a fast, outer-sphere redox couple. The MV2+/+ redox couple is used to demonstrate cathodic protection on the bare semiconductor architecture. To build up the concentration of MV+, pre-electrolysis was performed in situ to reduce MV2+ to MV+ immediately prior to the characterization of the device (Fig. 3a). This is necessary to fix the solution potential (Esoln) for extracting stable PEC measurements since both species must be present in solution for the Nernstian potential to be well-defined.53 Pre-electrolysis was terminated once Esoln reached approximately −0.5 V vs. Ag/AgCl with a concentration of less than 1 mM of MV+. This potential provided enough MV+ to conduct experiments while limiting the light loss through the solution, as MV+ is dark blue and absorbs light that could reach the photoelectrode.54 Because oxygen can convert the MV+ back into MV2+ and change the solution potential during operation, the PEC cell was constantly purged with wet N2. During pre-electrolysis the Pt counter electrode was also behind a ground glass frit to minimize the oxygen present in solution. While the MV2+/+ redox system has some complexities, it provides a valuable way to electrochemically characterize the 3T Si without the addition of a catalyst, allowing us to completely decouple the light absorption and electrocatalytic components of the PEC system. Obviously, for real-world applications a catalyst would be added to Si to facilitate electrocatalysis. However, durability measurements would then characterize the Si and the catalyst. It is important to first establish the bare 3T Si photocathode durability before adding additional components.
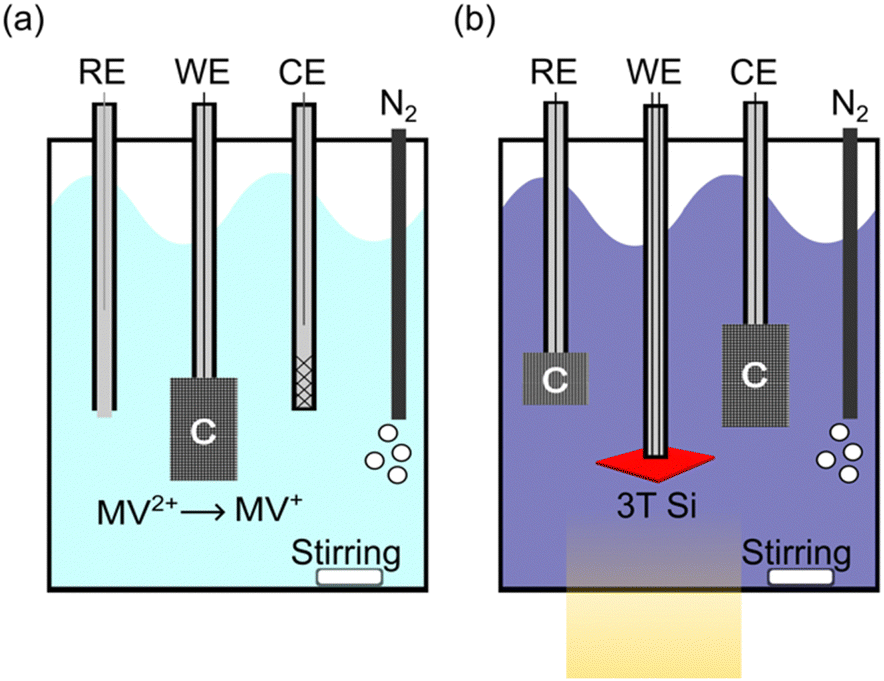 | ||
| Fig. 3 Schematic of the experimental set up for (a) the in situ pre-electrolysis of MV2+ to MV+ and (b) for photoelectrochemical/electrochemical characterization of 3T Si with respect to the MV2+/MV+ solution potential using carbon cloth electrode (C). | ||
After pre-electrolysis, the cell was reconfigured for PEC measurements of the 3T Si working electrode (Fig. 3b). During PEC characterization and durability studies, the working electrode was the 3T device and both the counter and reference electrodes were carbon cloth. A carbon-cloth reference was used as a pseudo reference, making the reference electrode potential dependent on the concentrations of MV2+ and MV+. If too much of the MV+ species is produced, the solution will strongly absorb visible light and Esoln will be more negative. Small fluctuations from Esoln = −0.5 V vs. Ag/AgCl can shift the current density of the 3T device by up to 1.5 mA cm−2 (Fig. S2, ESI†).
To understand the behavior of 3T nuIBC Si photocathodes, we first used cyclic voltammetry (CV) to look at the energy conversion behavior of each 3T mode (Fig. 4). The cathodic sweep of CVs in FR mode under illumination and FZ mode in the dark are shown in Fig. 4a. The functionalities of the 3T Si modes are comparable to the familiar 2T photocathode architectures (Fig. S3, ESI†). In FR mode, the photoelectrochemical behavior shows a positive voltage onset and a light-limited cathodic current (Fig. 4a). This is similar behavior to what is observed for a standard 2T photocathode device (e.g. p-Si). In FZ mode the device does not form a rectifying junction with the electrolyte and can pass cathodic current freely (similar to a heavily doped, e.g. n+-Si dark cathode) (Fig. 4a). However, these modes should not be considered as simple 2T analogues. Rather, relating the behavior of each operational mode of the 3T to their respective 2T helps predict performance and compare the 3T with previous measurements of 2T devices. While in FR mode, the illuminated open circuit potential (VOC,FR) was 0.55 V vs. Esoln, and the short circuit current density (JSC,FR) of the 3T Si in FR mode was –13.7 mA cm−2. When operating FZ mode in the dark, the reduction of MV2+ begins at 0 V vs. Esoln and at approximately –0.2 V vs. Esoln the current passed was equivalent to JSC,FR under illumination. As expected from dark, dry measurements in FZ mode at small applied potentials, the 3T Si photocathode can pass the same (or more) current density as in FR mode under illumination, enabling the same reaction to be driven in the dark and in the light.
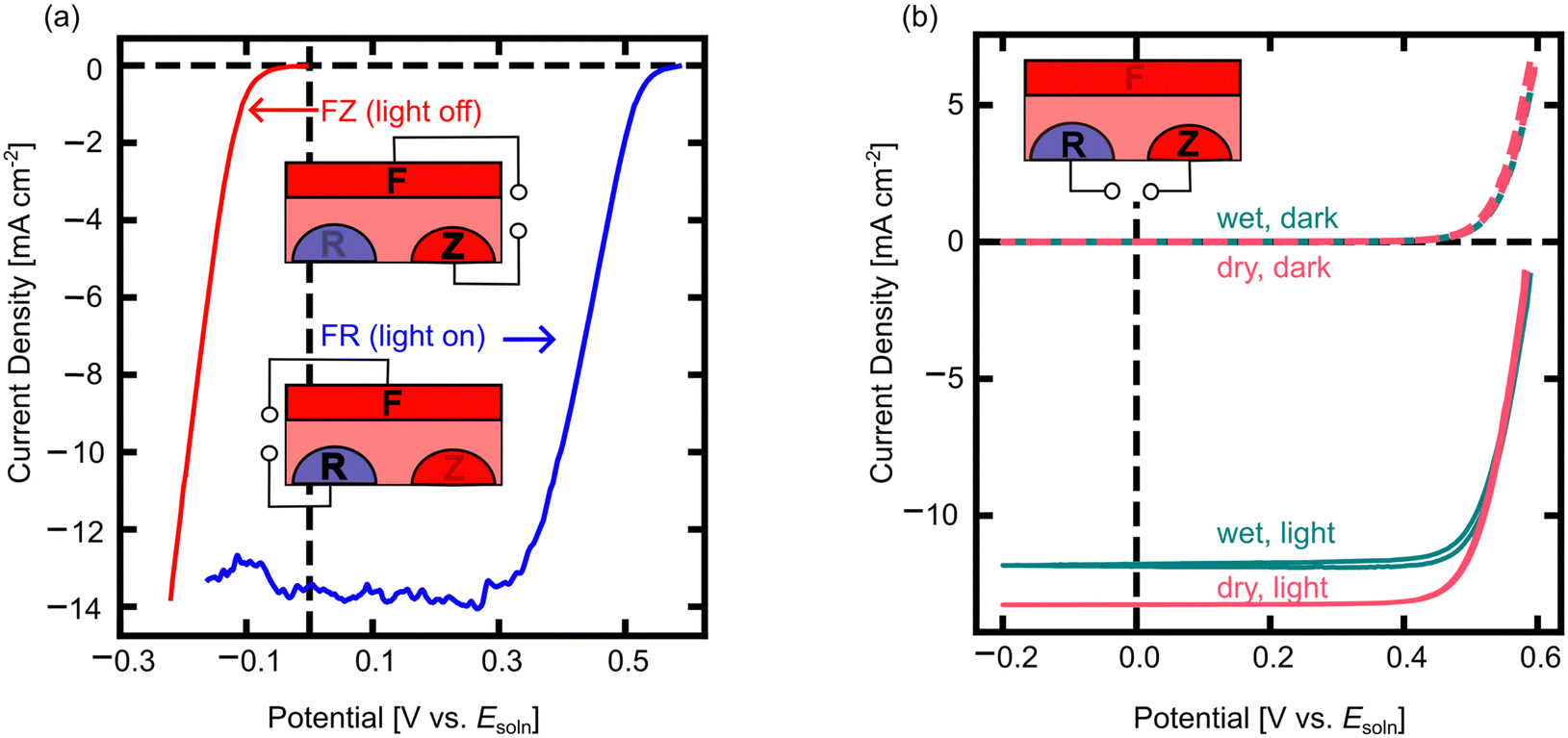 | ||
| Fig. 4 Photoelectrochemical, electrochemical, and electrical characterization of the three-operating modes of 3T nuIBC Si in both the light and in the dark. (a) Illuminated FR mode CV and dark FZ mode CV vs. Esoln immersed in MV2+/+ solution. (b) Illuminated and dark RZ mode CVs for an electrode measured in air (dry) and in MV2+/+ electrolyte (wet, Esoln = –0.5 V vs. Ag/AgCl). All illuminated data measured under simulated AM1.5G spectra. Insets show schematics of the contacts used for FR, FZ and RZ measurements. | ||
It is important to monitor the illumination intensity in the MV2+/+ solution, since the reduced MV+ absorbs some of the incident light. Usually this requires monitoring with a carefully calibrated photodiode closely aligned with the working electrode,50,51 but the 3T photoelectrode construction demonstrated here can act as an in situ photodiode in RZ mode, as no current is passed through the electrochemical cell, similar to the operation of a normal IBC cell (Fig. 4b). The J–V performance of dry RZ measurements reveals the maximum photocurrent of the device under investigation. The wet RZ measurements were performed after the pre-electrolysis of the MV2+/+ solution. The maximum photocurrent obtained in RZ mode with the reduced solution was ∼1.5 mA cm−2 less than the dry measurement (Fig. 4b). The dry cell setup and the MV2+ (not reduced) solution do not absorb significantly in the visible range. However, at Esoln = –0.5 V vs. Ag/AgCl, a broad absorption peak centered around 600 nm appears with a maximum absorbance near 0.5.54 In RZ mode, no charges pass to solution, therefore the reduction in current density is from the absorption of light by the MV+ species in solution.
To determine the effect of cathodic protection on the nuIBC 3T photoelectrodes, we performed durability experiments (Fig. 5). First, we examined changes to the current/voltage behavior of the 3T photoelectrode without cathodic protection in the dark. The 3T Si electrode was left in the dark in the previously described MV2+/+ electrolyte at open circuit potential for hour-long increments. After each hour, a CV in FR mode was measured under illumination; this was repeated for 6 hours. The cathodic sweeps of the FR CVs are shown in Fig. 5a. The initial CV had a JSC,FR of –9.0 mA cm−2. The JSC,FR decreased each hour until hour 5, where it plateaued at approximately –6.0 mA cm−2. After the final CV, the 3T electrode was re-etched in hydrofluoric acid (Fig. S4, ESI†). The subsequent CV regained the original shape, although the JSC,FR remained reduced at around –6.0 mA cm−2. This indicates that some, but not all, of the degradation after 6 hours was from an oxide layer forming on the surface. To further understand the change in performance of the electrodes without protection, galvanic electrochemical impedance spectroscopy (GEIS) was performed before and after 3 hours in steady state dark operation without protective voltages (Fig. S5, ESI†). Subsequent CVs were taken in FR mode after the EIS experiment (Fig. S5, ESI†). The EIS data was taken in dark FZ mode. The increase in the real and imaginary components of impedance, as seen by the large increase in the resistive elements of the fitted circuit, suggests that over time, the charge transfer resistance of the photoelectrode is increasing, likely from SiOx formation.
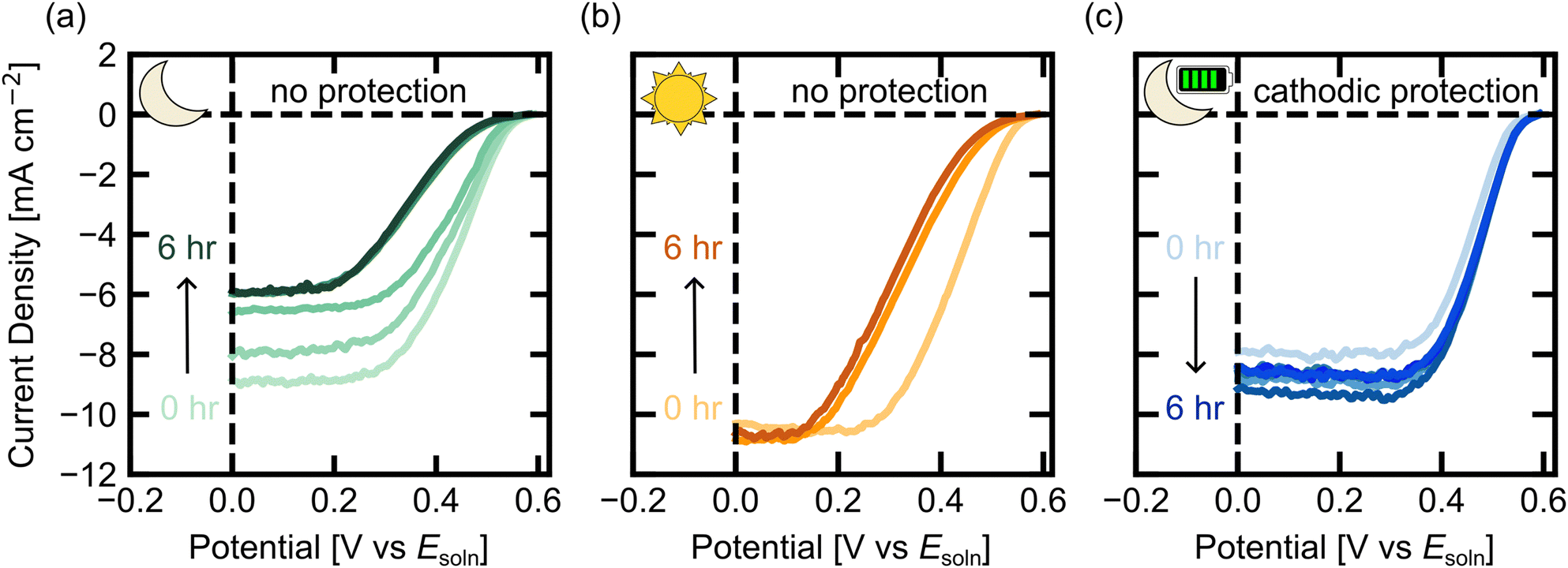 | ||
| Fig. 5 Durability studies of the 3T Si electrodes in dark conditions in MV2+/+ electrolyte. (a)–(c) FR mode light CVs under simulated AM1.5G illumination (a) were taken each hour in between steady state dark operation holding an electrode in FZ mode open circuit (JFZ = 0), (b) were taken every three hours in between continuous illumination in between open circuit (JFZ = 0) illuminated operation (c) were taken each hour in between steady state dark operation holding an electrode in FZ mode at –0.16 V vs. Esoln. | ||
The durability experiment without cathodic protection was repeated for a p-Si photocathode, where the VOC decreased by more than 100 mV in 24 hours (Fig. S6, ESI†) with the tabulated VOC values in Table S1 (ESI†). Clearly, unprotected 2T and 3T Si photocathodes have losses in activity over prolonged exposure to aqueous solution in the dark.
Next, we performed a durability experiment under illumination to characterize the performance of 3T Si in solution without any cathodic protection. The 3T Si electrode was left in the light in the MV2+/+ electrolyte at open circuit potential. After 3 hours and 6 hours, illuminated CVs were measured (Fig. 5b). The initial CV had a JSC,FR of –10.2 mA cm−2. The JSC,FR remained nearly constant throughout the 6 hour experiment. However, resistive losses in the shape of the CV are apparent after both 3 hours and 6 hours, indicating loss in performance, but not as drastic as the 3T Si photocathode in the dark for 6 h.
Then, we examined cathodic protection applied through the extra electrical contact (Z) to the 3T Si photoelectrode in the dark. In this experiment, rather than letting the photoelectrode sit at open circuit in the dark, the electrode was held at a constant potential in FZ mode (–0.16 V vs. Esoln) for 6 one-hour intervals. This FZ voltage was chosen because it produced the same current in the dark as the maximum power point current under illumination in FR mode. At the end of each hour in the dark, FR and RZ mode CVs were recorded under illumination and the cathodic FR sweeps of the CVs are shown in Fig. 5c (Fig. S7, ESI† shows the full forward and reverse sweeps). Initially, the FR CV had slightly lower activity compared to the previous dark no cathodic protection experiment, but the activity improved in the subsequent, hourly CVs. However, unlike the unprotected dark experiment, every subsequent CV had similar JSC,FR, with an average of −8.5 ± 0.5 mA cm−2 (Fig. 5c) and no apparent resistive losses in the CV shape. Changes in light intensity through the solution caused the JSC,FR in each CV to vary by ∼±1 mA cm−2 as observed in the RZ measurements (Fig. S8, ESI†). As the experiment progressed, more of the light absorbing MV+ species was produced, which made Esoln more negative, increasing the light absorbed by the solution, and decreasing JFR. Thus, the change in JSC,FR in the CV hour-to-hour is mostly due to the small variations in solution potential.
The applied cathodic potential was recorded with chronoamperometry (CA) to monitor the current densities during each 1-h period in the dark (Fig. S9, ESI†). The initial current density was −10.5 mA cm−2, which remained steady over the first hour in the dark. The second hour started at a lower current density of −9.5 mA cm−2 and increased slightly over the hour to −10.0 mA cm−2. The steady state current densities achieved in the third hour was again −10.5 mA cm−2 and over the next few hours there was a current loss of ∼1.5 mA cm−2 likely due to the changing solution potential over the hour.
Scanning electron microscopy (SEM) micrographs and energy-dispersive X-ray spectroscopy (EDS) data was taken on a cathodically protected 3T nuIBC Si electrode after the durability testing. Salt crystals were observed on the electrode surface (Fig. S10, ESI†). EDS showed C, O, Cl, Si, S, and K all present on the sample. An oxide layer had likely grown as the sample was transferred to the SEM chamber. The Cl, K, S, and C are concentrated on the crystalline salt feature. The additional losses in performance in both cases could have been to the salt formation on the surface lowering active area and therefore lowering the current density.
To understand the difference between cathodic protection in FR mode and FZ mode, a CV was taken, and a durability experiment was performed. First, a cathodic FR sweep of the 3T nuIBC Si photoelectrode in the dark was recorded from 0.6 V vs. Esoln to −4.0 V vs. Esoln (Fig. S11, ESI†). In the dark in FR mode, it took an applied voltage of approximately −3.0 V vs. Esoln to pass 1 mA cm−2 of cathodic current. To pass current densities higher than −1 mA cm−2 in FR mode in the dark, the photoelectrode would need to operate at significantly higher voltages. To protect the 3T Si electrode in the dark in FR mode the electrode was held at the current passed at the maximum power point from the light CV (∼−8.2 mA cm−2). It required nearly −4.5 V vs. Esoln to maintain a constant current of −8.2 mA cm−2 (Fig. S12, ESI†). Although the large negative voltages were able to maintain CV performance in FR mode, as apparent in the initial CV and CV after FR cathodic protection (Fig. S13, ESI†), that large voltage required to protect the 3T Si photocathode in the dark in FR mode is impractical for any solar fuel system.
For real world PEC applications, catalysts will be used for fuel forming reactions, so it is important to understand if cathodic protection can still be used for a photoelectrode coupled to a catalyst. Pt is commonly used HER catalyst that has been paired with Si for many PEC demonstrations.8 To demonstrate a model system, we evaporated a Pt grid (5 nm Pt, 10 nm Ti adhesion layer) on the front of a 3T electrode and performed the same MV2+/+ measurements. Samples were not HF-dipped prior to measurement, meaning the entire Si surface was coated in Pt or a native oxide layer. First, we tested if cathodic protection could extend to the catalyst by applying a potential of −0.3 V vs. Esoln in FZ mode for 1 hour (Fig. S14, ESI†) and saw slightly larger JSC,FR after durability testing. We then studied the durability without cathodic protection, keeping the photoelectrode in the dark at open circuit for 6 hours after the initial CV (Fig. S14, ESI†). There was no significant change in the CVs between 0 and 6 hours, but a slight decrease in the open circuit voltage. This is not surprising, since the Si surface was coated in SiO2 or catalyst, which can act as a protection layer. It is known that adding a catalyst to Si can slow or prevent degradation of the photoelectrode.55,56
3. Conclusions
Three-terminal semiconductor devices provide more degrees of freedom in the operation of PEC systems, such as the ability to provide cathodic protection to the electrode surface or to drive reactions with or without illumination. Going forward, cathodic protection could be an important approach for improving the practicality of PEC systems by providing a mechanism to decrease or prevent semiconductor degradation in the dark. The 3T photocathode construction described here extends the PEC activity of bare Si photocathodes in aqueous solution to over 6 hours of cycled illumination.The initial J–V characteristics of the protected 3T Si were preserved for over 6 hours in the dark, whereas resistive losses in unprotected Si appeared after 1 hour and 3 hours for 3T Si in the light, and a 33% reduction in photocurrent was observed after 6 hours in the dark. The photocurrent losses seen in the protected 3T Si are from the small changes in solution potential affecting the amount of light absorbed by the MV+ species, which are seen in both RZ and FR measurements. The addition of a Pt catalyst to the photoelectrode surface also enhanced performance, and it did not impact the use of a cathodic protection scheme. The prolonged operation of the 3T Si under cycled illumination conditions and low-cost fabrication approach suggests the 3T Si architecture can be utilized in scalable PEC-based systems. Continued work on this system will include adding catalysts for specific fuel-forming reactions where cathodic protection could also extend to the catalytic components of PEC systems. This integrated photocathode, capable of being switched between operational modes to account for the intermittency of sunlight, is a critical milestone towards a deployable PEC system.
4. Experimental
4.1. Materials
3T IBC Si solar cells were fabricated by ISC Konstanz using a modified ZEBRA process.42 The methyl viologen electrolyte solution was prepared by dissolving methyl viologen dichloride (MV2+, 98%, Sigma Aldrich, 0.05 M), potassium hydrogen phthalate (KHP, 99.95%, Sigma Aldrich, 0.1 M) buffer solution, and potassium sulfate supporting electrolyte (99%, Sigma Aldrich 0.5 M) in DI water. The pH of the MV2+ solution was adjusted dropwise with concentrated hydrochloric acid (36.5–38%, J.T. Baker) until the pH was 3.5 as measured by a pH probe (SPER Scientific).4.2 Electrode fabrication
The 3T Si solar cells were assembled into photoelectrodes by soldering insulated copper wires (Jonard Tools) with Sn/Pb solder (Sn60/Pb40, Aim Solder) to each of the bus bars on the back of the solar cell (Fig. S15, ESI†). The wires were threaded through a glass tube with the solar cell mounted perpendicular to the glass tubing. The edges and back of the Si photoelectrode were covered with epoxy (EA 9460, Loctite). The counter and reference electrodes were made using carbon cloth (99%, fuel cell earth) connected directly to a Pt wire (99.9%, uGems), attached to a copper wire surrounded by glass, and epoxied in place to minimize any direct contact between the electrolyte and the Pt wire.The p-Si (1 Ω-cm, Virginia Semi) and n+-Si (0.001–0.003 Ω-cm, Siltronic Corp.) controls were prepared as photoelectrodes by cutting single side polished Si wafers into square pieces. The backside of the Si was lightly scratched with a diamond scribe and GaIn eutectic was applied to make ohmic contact. Tinned Cu wire was coiled then pulled through a 6 mm diameter glass tube, attached to the Si with a small amount of Ag paste (SPI), and epoxied in place. Electrode areas were measured using a scanner as reported in detail elsewhere.57 Electrode areas were 1.5–1.9 cm2.
For the platinized Si electrode, an e-beam evaporator Temescal FC2000 was used to evaporate Pt and Ti on the electrode. Prior to the evaporation, the Si photoelectrode was dipped in 10% hydrofluoric acid for 1 min to remove the native oxide layer then quickly placed in the vacuum chamber of the evaporator with a shadow mask. The films were deposited at 5 A s−1 for a 10 nm Ti adhesion layer and a 5 nm Pt layer.
4.3 Photoelectrochemical characterization
The electrodes were arranged in a cylindrical electrochemical cell with a quartz bottom and four ground-glass ports. An LED solar simulator (Pico, G2V Optics Inc.) was used as an illumination source. The spectral irradiance vs. wavelength can be seen in Fig. S16 (ESI†). The illumination on the electrochemical cell was adjusted using an in-house calibrated Si photodiode (FDS100, Thorlabs). As discussed in the main text, the MV+ species is highly absorbing but has some transmission at wavelengths above ∼800 nm.54Esoln of −0.50 V vs. Ag/AgCl was chosen to balance light absorption and stability of the solution potential. At this potential, 85% of the incident light still reaches the photoelectrode, and PEC measurements can be made.Prior to each photoelectrochemical experiment (except those with Pt), the Si photoelectrode was dipped in 10% hydrofluoric acid for 1 min to remove the native oxide layer then quickly placed in the MV2+/+ electrolyte. Each experiment used ∼30 mL of the buffered MV solution and was purged with N2 for 20 min prior to use. N2 was continuously purged through a water bubbler and into the electrochemical cell during experimentation. Pre-electrolysis of the MV was performed using a carbon cloth working electrode, a platinum counter electrode behind a glass frit, and an Ag/AgCl reference electrode (saturated KCl, BASi). For the pre-electrolysis, −0.54 V vs. Ag/AgCl was applied to the large carbon cloth working electrode to build up the concentration of the reduced MV+ species until the solution potential was −0.50 V vs. Ag/AgCl as measured between a small carbon cloth electrode and the Ag/AgCl reference. The solution potential was kept between −0.51 V and −0.49 V vs. Ag/AgCl during all experiments and was monitored with a multimeter checking the potential between carbon cloth and Ag/AgCl reference in the same solution. The solution was continuously stirred with a magnetic stir bar placed near the photoelectrode. Photoelectrochemical experimental data was taken using a Biologic SP-300 potentiostat. Each CV had a scan rate of 50 mV s−1. The CA data shown in Fig. S9 (ESI†) was held at a constant potential of −0.16 V vs. Esoln.
GEIS data was taken using a Biologic SP-300 potentiostat with a constant current set to 0 mA. The scan was taken from 200 kHz to 40 mHz with a perturbation amplitude of 100 μA and a wait period of 0.1 before each frequency. The data was fit using the Biologic Z-fit software (Fig. S5, ESI†).
4.4 Physical characterization
Scanning electron microscopy (SEM) micrographs were acquired using a secondary electron detector in a through-lens configuration on a FEI Nova NanoSEM 630 operating at 6 keV accelerating voltage and 3.9 nA beam current at a working distance of 5 mm. Energy-dispersive X-ray spectroscopy (EDS) was performed using an Oxford Instruments Ultim Max detector.Author contributions
D. K. C. and Z. G. S. contributed equally to this work in methodology, software, validation, investigation, and writing the original draft. N. N. contributed to conceptualization, initial experiments, and methodology. A. L. G. contributed to drafting. V. M. and D. T. contributed to resources. E. L. W. contributed to conceptualization, funding acquisition, methodology, project management, drafting, resources, and supervision. All authors contributed to manuscript edits and revision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank John Geisz for helping with acquisition of dark electrical 3T data, John Magnum for helping with acquisition of SEM/EDS data, Jeiwan Tan for helping with EIS analysis, and Anna Kundmann for help with evaporation. This work was performed in part at National Renewable Energy Laboratory, operated by Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under Contract no. DE-AC36-08GO28308. This material is based solely on work performed by the Liquid Sunlight Alliance, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Fuels from Sunlight Hub under Award DE-SC0021266. The views expressed in this article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.References
- H. Cui, G. Heath, T. Remo, D. Ravikumar, T. Silverman, M. Deceglie, M. Kempe and J. Engel-Cox, Sol. Energy Mater. Sol. Cells, 2022, 238, 111592 CrossRef CAS.
- A. Razzaq, T. G. Allen, W. Liu, Z. Liu and S. De Wolf, Joule, 2022, 6, 514–542 CrossRef CAS.
- H. Trivedi, A. Meshram and R. Gupta, J. Environ. Chem. Eng., 2023, 11, 109501 CrossRef CAS.
- Gurudayal, D. Perone, S. Malani, Y. Lum, S. Haussener and J. W. Ager, ACS Appl. Energy Mater., 2019, 2, 4551–4559 CrossRef CAS.
- W.-H. Cheng, M. H. Richter, I. Sullivan, D. M. Larson, C. Xiang, B. S. Brunschwig and H. A. Atwater, ACS Energy Lett., 2020, 5, 470–476 CrossRef CAS.
- D. Li, K. Yang, J. Lian, J. Yan and S. (Frank) Liu, Adv. Energy Mater., 2022, 12, 2201070 CrossRef CAS.
- L. K. Putri, B.-J. Ng, W.-J. Ong, S.-P. Chai and A. R. Mohamed, Adv. Energy Mater., 2022, 12, 2201093 CrossRef CAS.
- D. Bae, B. Seger, O. Hansen, P. C. K. Vesborg and I. Chorkendorff, ChemElectroChem, 2019, 6, 106–109 CrossRef CAS.
- Y. J. Jang, I. Jeong, J. Lee, J. Lee, M. J. Ko and J. S. Lee, ACS Nano, 2016, 10, 6980–6987 CrossRef CAS PubMed.
- M. Ben-Naim, R. J. Britto, C. W. Aldridge, R. Mow, M. A. Steiner, A. C. Nielander, L. A. King, D. J. Friedman, T. G. Deutsch, J. L. Young and T. F. Jaramillo, ACS Energy Lett., 2020, 5, 2631–2640 CrossRef CAS.
- R. Fan, J. Mao, Z. Yin, J. Jie, W. Dong, L. Fang, F. Zheng and M. Shen, ACS Appl. Mater. Interfaces, 2017, 9, 6123–6129 CrossRef CAS PubMed.
- V. Andrei, R. A. Jagt, M. Rahaman, L. Lari, V. K. Lazarov, J. L. MacManus-Driscoll, R. L. Z. Hoye and E. Reisner, Nat. Mater., 2022, 21, 864–868 CrossRef CAS PubMed.
- C. Pornrungroj, V. Andrei, M. Rahaman, C. Uswachoke, H. J. Joyce, D. S. Wright and E. Reisner, Adv. Funct. Mater., 2021, 31, 2008182 CrossRef CAS.
- T. H. Jeon, G. Moon, H. Park and W. Choi, Nano Energy, 2017, 39, 211–218 CrossRef CAS.
- K. Sun, R. Liu, Y. Chen, E. Verlage, N. S. Lewis and C. Xiang, Adv. Energy Mater., 2016, 6, 1600379 CrossRef.
- D. Bae, T. Pedersen, B. Seger, B. Iandolo, O. Hansen, P. C. K. Vesborg and I. Chorkendorff, Catal. Today, 2017, 290, 59–64 CrossRef CAS.
- M. Ben-Naim, C. W. Aldridge, M. A. Steiner, R. J. Britto, A. C. Nielander, L. A. King, T. G. Deutsch, J. L. Young and T. F. Jaramillo, ACS Appl. Mater. Interfaces, 2022, 14, 20385–20392 CrossRef CAS PubMed.
- S. Chen, D. Huang, P. Xu, W. Xue, L. Lei, M. Cheng, R. Wang, X. Liu and R. Deng, J. Mater. Chem. A, 2020, 8, 2286–2322 RSC.
- M. Liao, J. Feng, W. Luo, Z. Wang, J. Zhang, Z. Li, T. Yu and Z. Zou, Adv. Funct. Mater., 2012, 22, 3066–3074 CrossRef CAS.
- M. Liu, C.-Y. Nam, C. T. Black, J. Kamcev and L. Zhang, J. Phys. Chem. C, 2013, 117, 13396–13402 CrossRef CAS.
- L. A. King, T. R. Hellstern, J. Park, R. Sinclair and T. F. Jaramillo, ACS Appl. Mater. Interfaces, 2017, 9, 36792–36798 CrossRef CAS PubMed.
- D. Bae, B. Seger, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, Chem. Soc. Rev., 2017, 46, 1933–1954 RSC.
- S. Hu, M. R. Shaner, J. A. Beardslee, M. Lichterman, B. S. Brunschwig and N. S. Lewis, Science, 2014, 344, 1005–1009 CrossRef CAS PubMed.
- H. J. Fu, I. A. Moreno-Hernandez, P. Buabthong, K. M. Papadantonakis, B. S. Brunschwig and N. S. Lewis, Energy Environ. Sci., 2020, 13, 4132–4141 RSC.
- C. G. O. Bruziquesi, M. C. P. Stolzemburg, R. R. de Souza, M. Rodriguez, M. L. Rocco, P. E. A. Salomão, A. E. Nogueira, Z. E. López-Cabaña, M. C. Pereira and A. C. Silva, Int. J. Hydrogen Energy, 2023, 48, 3456–3465 CrossRef CAS.
- G. Liu, F. Zheng, J. Li, G. Zeng, Y. Ye, D. M. Larson, J. Yano, E. J. Crumlin, J. W. Ager, L. Wang and F. M. Toma, Nat. Energy, 2021, 6, 1124–1132 CrossRef CAS.
- G. A. Rome, F. Intia, T. R. Klein, Z. G. Schichtl, A. C. Tamboli, E. L. Warren and A. L. Greenaway, ChemElectroChem, 2023, 10, e202300209 CrossRef CAS.
- F. Nandjou and S. Haussener, J. Phys. D: Appl. Phys., 2017, 50, 124002 CrossRef.
- T. A. Kistler, G. Zeng, J. L. Young, L. Weng, C. Aldridge, K. Wyatt, M. A. Steiner, O. Solorzano, F. A. Houle, F. M. Toma, A. Z. Weber, T. G. Deutsch and N. Danilovic, Adv. Energy Mater., 2020, 10, 2002706 CrossRef CAS.
- T. A. Kistler, M. Y. Um and P. Agbo, J. Electrochem. Soc., 2020, 167, 066502 CrossRef CAS.
- K. M. K. Yap, S.-W. Lee, M. A. Steiner, J. E. Avilés Acosta, D. Kang, D. Kim, E. L. Warren, A. C. Nielander and T. F. Jaramillo, Chem Catal., 2023, 3, 100641 CrossRef CAS.
- Gurudayal, J. W. Beeman, J. Bullock, H. Wang, J. Eichhorn, C. Towle, A. Javey, F. M. Toma, N. Mathews and J. W. Ager, Energy Environ. Sci., 2019, 12, 1068–1077 RSC.
- W. Liao and S. Lee, J. Appl. Phys., 1996, 80, 1171–1176 CrossRef CAS.
- C. Christodoulou, G. Glass, J. Webb, S. Austin and C. Goodier, Corros. Sci., 2010, 52, 2671–2679 CrossRef CAS.
- Y. Bu and J.-P. Ao, Green Energy Environ., 2017, 2, 331–362 CrossRef.
- S. Chen and L.-W. Wang, Chem. Mater., 2012, 24, 3659–3666 CrossRef CAS.
- P. Pedeferri, Constr. Build. Mater., 1996, 10, 391–402 CrossRef.
- U. M. Angst, Corrosion, 2019, 75, 1420–1433 CrossRef CAS PubMed.
- N. Krishnan, D. K. Kamde, Z. Doosa Veedu, R. G. Pillai, D. Shah and R. Velayudham, J. Build. Eng., 2021, 42, 102467 CrossRef.
- Y. Yang, M. Sun, Z. Chen, H. Xu, X. Wang, J. Duan and B. Hou, Chem. Eng. J., 2023, 458, 141458 CrossRef CAS.
- E. L. Warren, W. E. McMahon, M. Rienäcker, K. T. VanSant, R. C. Whitehead, R. Peibst and A. C. Tamboli, ACS Energy Lett., 2020, 5, 1233–1242 CrossRef CAS PubMed.
- R. Kopecek, J. Libal, J. Lossen, V. D. Mihailetchi, H. Chu, C. Peter, F. Buchholz, E. Wefringhaus, A. Halm, J. Ma, L. Jianda, G. Yonggang, Q. Xiaoyong, W. Xiang and D. Peng, in 2020 47th IEEE Photovoltaic Specialists Conference (PVSC), IEEE, Calgary, AB, Canada, 2020, pp. 1008–1012 Search PubMed.
- J. F. Geisz, W. E. McMahon, J. Buencuerpo, M. S. Young, M. Rienäcker, A. C. Tamboli and E. L. Warren, Cell Rep. Phys. Sci., 2021, 2, 100677 CrossRef CAS.
- F. Gota, M. Langenhorst, R. Schmager, J. Lehr and U. W. Paetzold, Joule, 2020, 4, 2387–2403 CrossRef CAS.
- E. L. Warren, M. G. Deceglie, M. Rienäcker, R. Peibst, A. C. Tamboli and P. Stradins, Sustainable Energy Fuels, 2018, 2, 1141–1147 RSC.
- T. Nagashima, K. Okumura, K. Murata and Y. Kimura, Conference Record of the Twenty-Eighth IEEE Photovoltaic Specialists Conference - 2000 (Cat. No. 00CH37036), 2000, pp. 1193–1196 Search PubMed.
- K. T. VanSant, E. L. Warren, J. F. Geisz, T. R. Klein, S. Johnston, W. E. McMahon, H. Schulte-Huxel, M. Rienäcker, R. Peibst and A. C. Tamboli, iScience, 2022, 25, 104950 CrossRef CAS PubMed.
- M. Schnabel, H. Schulte-Huxel, M. Rienäcker, E. L. Warren, P. F. Ndione, B. Nemeth, T. R. Klein, M. F. A. M. van Hest, J. F. Geisz, R. Peibst, P. Stradins and A. C. Tamboli, Sustainable Energy Fuels, 2020, 4, 549–558 RSC.
- G. Segev, J. W. Beeman, J. B. Greenblatt and I. D. Sharp, Nat. Mater., 2018, 17, 1115–1121 CrossRef CAS PubMed.
- E. L. Warren, S. W. Boettcher, M. G. Walter, H. A. Atwater and N. S. Lewis, J. Phys. Chem. C, 2011, 115, 594–598 CrossRef CAS.
- S. W. Boettcher, J. M. Spurgeon, M. C. Putnam, E. L. Warren, D. B. Turner-Evans, M. D. Kelzenberg, J. R. Maiolo, H. A. Atwater and N. S. Lewis, Science, 2010, 327, 185–187 CrossRef CAS PubMed.
- F. Nandjou and S. Haussener, ACS Appl. Mater. Interfaces, 2022, 14, 43095–43108 CrossRef CAS PubMed.
- A. Bard, Standard potentials in aqueous solution, Routledge, 2017 Search PubMed.
- E. L. Warren, PhD thesis, California Institute of Technology, 2013 DOI:10.7907/8Q2S-DX81.
- Y. Wang, W. Tian, F. Cao, D. Fang, S. Chen and L. Li, Nanotechnology, 2018, 29, 425703 CrossRef PubMed.
- B. Guo, A. Batool, G. Xie, R. Boddula, L. Tian, S. U. Jan and J. R. Gong, Nano Lett., 2018, 18, 1516–1521 CrossRef CAS PubMed.
- O. J. Alley, K. Wyatt, M. A. Steiner, G. Liu, T. Kistler, G. Zeng, D. M. Larson, J. K. Cooper, J. L. Young, T. G. Deutsch and F. M. Toma, Front. Energy Res., 2022, 10, 884364 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee00349g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |