
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-site catalysis of high-entropy hydroxides for sustainable electrooxidation of glucose to glucaric acid†
Xianhong
Wu‡
ag,
Zhi-Jian
Zhao‡
 b,
Xiangcheng
Shi‡
bh,
Liqun
Kang
c,
Pratteek
Das
a,
Sen
Wang
a,
Shengqi
Chu
d,
Hua
Wang
e,
Kenneth
Davey
f,
Bo
Zhang
*e,
Shi-Zhang
Qiao
*f,
Jinlong
Gong
*b and
Zhong-Shuai
Wu
*ai
b,
Xiangcheng
Shi‡
bh,
Liqun
Kang
c,
Pratteek
Das
a,
Sen
Wang
a,
Shengqi
Chu
d,
Hua
Wang
e,
Kenneth
Davey
f,
Bo
Zhang
*e,
Shi-Zhang
Qiao
*f,
Jinlong
Gong
*b and
Zhong-Shuai
Wu
*ai
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
bKey Laboratory for Green Chemical Technology of Ministry of Education, School of Chemical Engineering & Technology, Tianjin University, Tianjin 30072, P. R. China. E-mail: jlgong@tju.edu.cn
cMax Planck Institute for Chemical Energy Conversion, Stifstrasse 34-36, D-45470 Mülheim an der Ruhr, Germany
dInstitute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
eCAS Key Laboratory of Science and Technology on Applied Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: bo.zhang@dicp.ac.cn
fSchool of Chemical Engineering, The University of Adelaide, Adelaide, South Australia, Australia. E-mail: s.qiao@adelaide.edu.au
gCollege of Light Industry and Chemical Engineering, Dalian Polytechnic University, Dalian 116034, China
hJoint School of National University of Singapore and Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou 350207, Fujian, China
iDalian National Laboratory for Clean Energy, Chinese Academy of Sciences, Dalian 116023, China. E-mail: wuzs@dicp.ac.cn
First published on 26th March 2024
Abstract
The glucose electrooxidation reaction (GOR) is an environmentally benign way to generate high value-add glucaric acid. However, a lack of suitable catalysts for the GOR limits development. Here we report for the first time a practically robust, multi-site, synergistic catalyst of defect-rich high-entropy FeCoNiCu layered double-hydroxide nanosheets grown on nickel foam. We demonstrate a highly significant activity and stability for the GOR leading to a low potential of 1.22 V vs. RHE at a current density of 100 mA cm−2, together with an excellent glucose conversion of ∼100% and glucaric acid yield of >90%. We evidence that the Cu–Co bridge promotes dehydrogenation of the hydroxyl group, and that the Cu–Cu bridge boosts dehydrogenation on carbon to form an aldehyde group. We establish that the Cu–Ni bridge boosts the oxidation of the aldehyde group to carboxyl to exhibit an important advantage of multi-site synergistic catalysis of high entropy hydroxides. We confirm an energy-saving hybrid flow electrolytic cell, prototype coupled GOR with a nitrate reduction reaction (NO3−RR), that requires an applied voltage of just 1.07 and 1.32 V for an electrolytic current density of, respectively, 10 and 100 mA cm−2 for GOR‖NO3−RR, together with concurrent low-potential production of glucaric acid and NH3. We conclude that defect-rich high-entropy FeCoNiCu catalysis of high-entropy hydroxides can be used for the practical design of sustainable and environmentally benign electrooxidation of glucose to glucaric acid. Our findings will be of benefit to researchers and manufacturers in the electrocatalytic conversion of renewable biomass for high value-add chemicals.
Broader contextElectrocatalytic conversion of renewable biomass to produce high value-add chemicals not only alleviates the dependence on fossil energy but can also reduce carbon emissions. The glucose electrooxidation reaction (GOR) provides an environmentally friendly strategy for efficient generation of high value-added glucaric acid. However, a lack of efficient catalysts for the GOR with multiple electron reactions has limited its rapid development. Herein, a multi-site synergistic catalyst of high entropy hydroxide D-FeCoNiCu-LDH/NF with tandem active sites has been designed for the GOR. As a result, the D-FeCoNiCu-LDH/NF electrocatalyst displays excellent electrocatalyst activity for the GOR, which only needs a low potential of 1.22 V vs. RHE to achieve a current density of 100 mA cm−2, with an excellent glucose conversion of ∼100% and glucaric acid yield of 90%. The outstanding performance of high-entropy materials with multiple active sites for the GOR provides inspiration for the design of complex biomass electrooxidation catalysts. In addition, a coupled system of GOR and nitrate electro-reduction reaction (NO3−RR) has been assembled successfully, which achieved the co-production of glucaric acid and ammonia with low energy consumption. The coupled system improves energy utilization efficiency, which is of great significance to the development of sustainable energy technology. |
Introduction
Electrocatalytic conversion of renewable biomass to produce high value-added chemicals obviates dependence on fossil energy and carbon emissions.1–5 Glucose is an abundant biomass on earth. Amongst its oxidation products, glucaric acid is a ‘top valuable bio-refined product’ because it is used widely in medicine and industry including, treating cancer, lowering cholesterol and producing nylon-66.6–8 Major production methods for glucaric acid include chemical oxidation and microbial fermentation. However, chemical oxidation is limited under harsh reaction conditions using (precious) metal catalysts e.g. Au, Pt, Pd or Ru, with a low glucaric acid selectivity of <60%,9,10 and microbial fermentation suffers from an extensive reaction time for >2 days, together with problematic separation, resulting in a low glucaric acid selectivity of <20%.11 Electrochemical oxidation however uses mild reaction conditions, has greater sustainability and does not require additional toxic oxidant(s). These advantages make it highly competitive for the scalable production of glucaric acid.12 However, the reported electrocatalysts do not exhibit adequate activity, stability or selectivity, because of a high reaction energy barrier caused by multi-electron processes.12–14 A multi-site synergistic catalyst with high activity and stability is therefore advantageous for complex multi-electron biomass reactions.15,16High entropy materials (HEMs) reportedly exhibit outstanding performance in electrocatalysis17–22 because of abundant active sites, adjustable electron structure23 and an entropy stabilization effect.20,24 In particular, high entropy materials (HEMs) containing four (4) or more elements homogenously distributed in a single phase exhibit optional active sites that are a class of multi-site synergistic catalyst, for complex biomass electrooxidations. The multi-sites of HEMs ‘cooperatively’ catalyze different reaction steps of the GOR, respectively, including an aldehyde group and hydroxyl group oxidation. However, because of the structural uncertainty and diversity of multiple active sites, the catalytic mechanism(s) for HEMs remains inadequately understood. Reported findings, generally, have simplified these into a single catalytic site to hypothesize the catalytic mechanisms for HEMs,17,19 therefore masking the role of multiple elements in multi-electron biomass reaction. An improved understanding of the actual active center and fundamental mechanisms for different-element multi-site synergistic catalysts, could therefore be used for the rational design for sustainable biomass electrocatalytic reactions.
Here we report the synthesis of a ‘new’ multi-site synergistic catalyst of high-entropy defect-rich FeCoNiCu-layered double hydroxides (LDH) on nickel foam (D-FeCoNiCu-LDH/NF). We demonstrate this as a highly active and durable tandem catalyst for the glucose electrooxidation reaction (GOR) and show that just 1.22 V (vs. RHE) is required for a current density of 100 mA cm−2, together with near complete conversion of glucose and a high glucaric acid yield of >90%. We evidence using a judiciously combined experiment and theoretical computation that glucose is oxidized to gluconic acid, and then to glucaric acid in which the dehydrogenation of the hydroxyl group is rate-limiting. We establish for HEM catalysts that introducing Cu as the core and the synergistic effect with Ni, Co and Fe generate Cu–Ni, Cu–Co, and Cu–Cu bridge sites that act as tandem active sites that boost the oxidation rate and yield of glucaric acid. With HEMs as bifunctional catalysts, we confirm that the GOR‖NO3−RR coupled system assembled by the GOR anode with a nitrate reduction reaction (NO3−RR) cathode, co-produces glucaric acid and NH3 concurrently. Significantly, we show this system requires an applied voltage of only 1.07 and 1.32 V for an electrolytic current density of, respectively, 10 and 100 mA cm−2 and that it maintains long-term electrolysis stability for >50 h. We conclude therefore that the defect-rich high-entropy FeCoNiCu catalysis of high-entropy hydroxides is practical for sustainable and environmentally benign electrooxidation of glucose to glucaric acid. Our findings will be of benefit in electrocatalytic conversion of renewable biomass for high value-add chemicals.
Results and discussion
Structural characterization of electrocatalysts
To synthesize the multiple active site HEMs, FeCoNiCuCr-LDH was initially synthesized on Ni-foam, denoted as FeCoNiCuCr-LDH/NF, via electrodeposition followed by the removal of Cr to re-construct defect-rich FeCoNiCu-LDH/NF (D-FeCoNiCu-LDH/NF) using cyclic voltammetry (CV) (see the Methods section). It was observed that ultrathin FeCoNiCuCr-LDH nanosheets are vertically electrodeposited on Ni foam with an open 3D structure. A scanning electron microscope (SEM) image evidenced the growth of a ‘dense’ network of FeCoNiCuCr-LDH nanosheets on the surface of Ni foam (Fig. S1a and b, ESI†). The transmission electron microscopy (TEM) image confirmed the nanosheet structure of FeCoNiCuCr-LDH with a diameter of several hundred nm (Fig. S1c, ESI†). High resolution TEM (HRTEM) analysis of FeCoNiCuCr-LDH evidenced lattice fringes of ca. 0.25 nm that were identified as the spacing corresponding to the (012) facet of LDHs (Fig. S1d, ESI†). Elemental mapping analysis (Fig. S2, ESI†) established a homogenous distribution of Fe, Co, Ni, Cu, Cr and O elements in FeCoNiCuCr-LDH as a result of high disorder of high entropy.19,21 X-ray diffraction (XRD) pattern confirmed the crystallographic structure for FeCoNiCuCr-LDH/NF (Fig. S3, ESI†). The peaks at 13.1, 26.1, 36.8, 42.6 and 61.6° corresponding to (003), (006), (012), (018) and (113) crystal lattice facets, were found to agree well with the characteristic phases of LDHs,25 without any impurity phase.D-FeCoNiCu-LDH/NF was accomplished via electrochemical activation of FeCoNiCuCr-LDH/NF to etch the Cr element (Fig. 1a). SEM and TEM images of D-FeCoNiCu-LDH evidenced that the 2D structure was preserved following electrochemical activation (Fig. 1b and c). Atomic force microscopy (AFM) confirmed a uniform thickness of 2 to 3 nm across the nanosheet (Fig. S4, ESI†). A spherical aberration-corrected TEM image revealed the highly crystalline nature of D-FeCoNiCu-LDH (Fig. 1d). The corresponding Fourier transform pattern (Fig. 1e) confirmed the hexagonal phase of D-FeCoNiCu-LDH corresponding to the (012) and (110) planes of LDH. The disappearance of the LDH signal in the XRD pattern (Fig. S3, ESI†) is, in all probability, attributable to the existence of defects and lattice distortion. The elemental mapping analysis (Fig. 1f) established a homogenous distribution of Fe, Co, Ni, Cu and O elements in D-FeCoNiCu-LDH, demonstrating structural stability during electrochemical activation. Significantly, no Cr element was observed from D-FeCoNiCu-LDH (Fig. S5, ESI†), evidencing a ‘complete’ etching of Cr by electrochemical activation. It is concluded therefore that these findings confirm the ordered crystal structure and single-phase feature of D-FeCoNiCu-LDH HEMs. The molar ratio of Fe, Co, Ni and Cu in D-FeCoNiCu-LDH is optimal at 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:9:8, as evidenced via inductively coupled plasma optical emission spectroscopy (ICP-OES).
:3:9:8, as evidenced via inductively coupled plasma optical emission spectroscopy (ICP-OES).
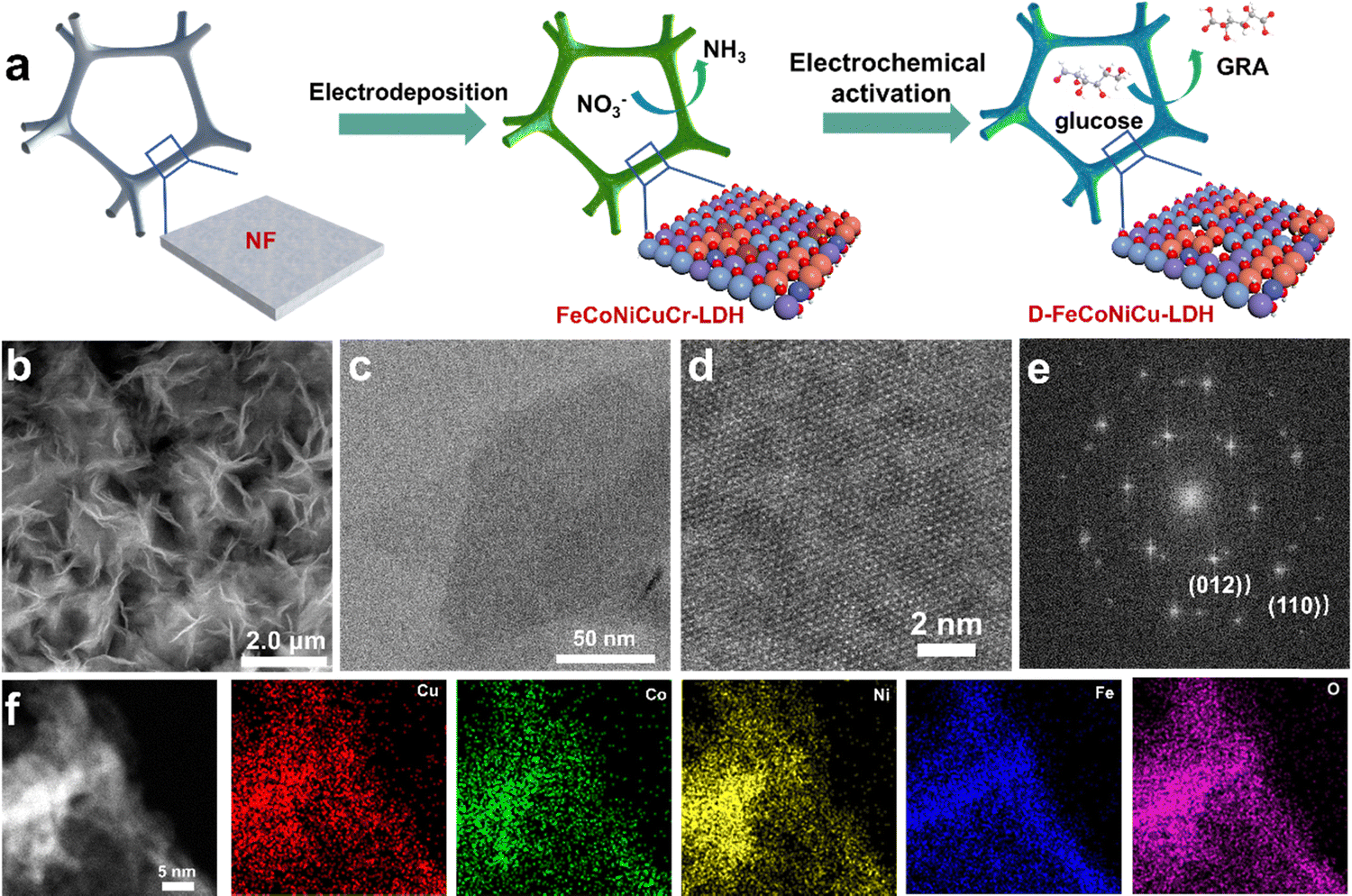 | ||
| Fig. 1 Morphology of the high-entropy hydroxide electrocatalyst D-FeCoNiCu-LDH/NF. (a) Schematic for the synthesis of D-FeCoNiCu-LDH/NF. (b) SEM image of D-FeCoNiCu-LDH/NF. (c) TEM image of D-FeCoNiCu-LDH peeled from NF. (d) High-angle annular dark-field scanning transmission electron microscopy image and (e) corresponding fast Fourier transform pattern for (d), showing an ordered crystal structure. (f) EDS mapping evidencing the presence and homogenous distribution of Fe, Co, Ni, Cu and O elements in D-FeCoNiCu-LDH. | ||
The in situ Raman spectroscopy confirmed the phase transformation of D-FeCoNiCu-LDH/NF in electrochemical activation (Fig. 2a). With increasing anodic potential, the hydroxide transforms to hydroxyl oxide (M–OOH).17,26,27 To assess the chemical states and local coordination structure of FeCoNiCuCr-LDH and D-FeCoNiCu-LDH, X-ray absorption fine structure (XAFS) characterizations were determined at Fe K-edge, Co K-edge, Ni K-edge and Cu K-edge. The X-ray absorption near edge structure (XANES) spectra in Fig. 2b and d of Fe and Ni evidenced that the valences for Fe and Ni species were, respectively, +3 and +2, in both FeCoNiCuCr-LDH and D-FeCoNiCu-LDH. The ‘weak’ pre-edge features, 7114.2 eV at the Fe K-edge and 8334.0 eV at the Ni K-edge, together with a ‘featureless’ rising edge evidenced that the Fe/Ni species had center-symmetric 6-coordinated octahedral geometry, a finding in good agreement with the LDH structure.28,29 However, the Co K-edge XANES (Fig. 2f) exhibited that the dominant Co species in D-FeCoNiCu-LDH were oxidized to Co3+ from Co2+ in FeCoNiCuCr-LDH. Similarly, the Co species in FeCoNiCuCr-LDH and D-FeCoNiCu-LDH remained configured in the octahedral coordination structure inferred from the non-pronounced pre-edge and main absorption edge. A pre-edge shoulder peak at ca. 8983.0 eV ‘disappeared’ from D-FeCoNiCu-LDH in Cu K-edge XANES (Fig. 2h) evidencing that the Cu+ component of FeCoNiCuCr-LDH was oxidized to Cu2+ during activation.30
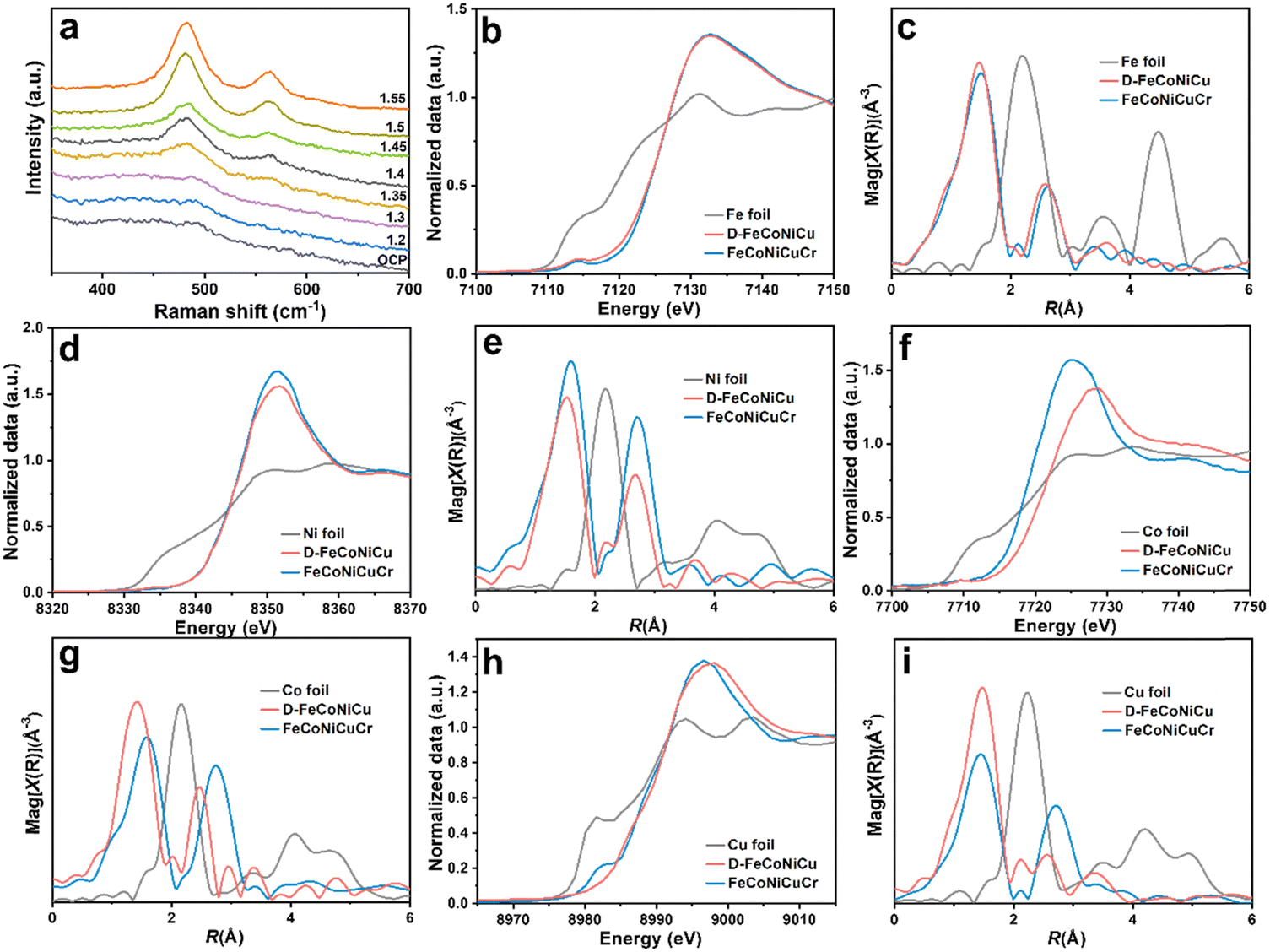 | ||
| Fig. 2 Structural characterization of the high-entropy hydroxide electrocatalyst D-FeCoNiCu-LDH/NF. (a) In situ Raman spectra for the FeCoNiCuCr-LDH/NF electrode and corresponding structural conversion under changed potentials in 1.0 M KOH. (b) Fe K-edge normalized XANES spectra and (c) Fe K-edge k2-weighted R-space EXAFS spectra for Fe foil, D-FeCoNiCu-LDH and FeCoNiCuCr-LDH. (d) Ni K-edge normalized XANES spectra and (e) Ni K-edge k2-weighted R-space EXAFS spectra for Ni foil, D-FeCoNiCu-LDH and FeCoNiCuCr-LDH. (f) Co K-edge normalized XANES spectra and (g) Co K-edge k2-weighted R-space EXAFS spectra for Co foil, D-FeCoNiCu-LDH and FeCoNiCuCr-LDH. (h) Cu K-edge normalized XANES spectra and (i) Cu K-edge k2-weighted R-space EXAFS spectra for Cu foil, D-FeCoNiCu-LDH and FeCoNiCuCr-LDH. | ||
To establish which metal species are more sensitive to the valence states, extended X-ray absorption fine structure (EXAFS) spectra were collected and quantitatively fitted to resolve the coordination structure for each metal site (Fig. 2, Fig. S6–S9 and Table S1, ESI†). The EXAFS spectra for FeCoNiCuCr-LDH and D-FeCoNiCu-LDH were fitted using two (2) scattering paths; the (1) first-shell coordinated by oxygen atoms (denoted as O) and (2) second-shell coordinated by the transition metal atoms (denoted as M). For Fe, Co and Ni, the first shells present a similar coordination number (C.N.) of approximately six (6), corresponding to the six (6) oxygen-coordinated octahedral structure, a finding in good agreement with XANES findings (Fig. 2c, e and g). Importantly, the bond length for M–O was consistent with the LDH structure.31,32 It is (slightly) different for Cu because the C.N. for the Cu–O scattering path was 3.0 and 3.4 for, respectively, FeCoNiCuCr-LDH and D-FeCoNiCu-LDH (Fig. 2i). This is because the Jahn–Teller effect for Cu2+ with a 3d9 configuration resulted in an elongated octahedral structure. For the second coordination shell (M–M), it was apparent that the C.N. and interatomic distances for Fe–M remained (nearly) identical for FeCoNiCuCr-LDH and D-FeCoNiCu-LDH. It was concluded therefore that the coordination structure for Fe in FeCoNiCuCr-LDH was preserved in D-FeCoNiCu-LDH following electrochemical activation, confirming that Fe3+ species is a structural stabilizer in HEMs. However, the coordination structures for Co, Ni and Cu in FeCoNiCuCr-LDH and D-FeCoNiCu-LDH were significantly different. The C.N. for Co–M, Ni–M and Cu–M reduced significantly from, respectively, 6.0 to 2.9, 9.2 to 7.7 and 6.6 to 2.5, accompanied by a decrease in interatomic distance from, respectively, 3.13 to 2.84 Å, 3.13 to 3.06 Å and 3.12 to 3.00 Å. It is strongly evidenced that the coordination structures of the Co, Ni and Cu species were highly significantly modified from FeCoNiCuCr-LDH to D-FeCoNiCu-LDH following removal of Cr from the structure via etching. Given that the C.N. for M–M scattering was strongly correlated with σ2 and the σ2 values that increased following activation, D-FeCoNiCu-LDH apparently possessed significantly more defects and disordered structure compared with FeCoNiCuCr-LDH to provide more active sites for the GOR.
X-ray photoelectron spectroscopy (XPS) can be interpreted to determine the surface chemical state of D-FeCoNiCu-LDH/NF (Fig. S10, ESI†). Consistent with XANES findings is that the Fe and Ni species are found to be the +3 and +2 valence state of Fe 3p and Ni 2p for, respectively, FeCoNiCuCr-LDH/NF and D-FeCoNiCu-LDH/NF (Fig. S10b and c, ESI†).33,34 The ‘minor’ amount of Cu+ of FeCoNiCuCr-LDH/NF is oxidized to Cu2+ of D-FeCoNiCu-LDH/NF (Fig. S10d, ESI†).19,34,35 Additionally, the O 1s XPS spectrum evidences that there are more M–O bonds for D-FeCoNiCu-LDH/NF compared with FeCoNiCuCr-LDH/NF (Fig. S10e, ESI†), whilst the M–OH bond was reduced because of conversion of transition metal hydroxide into oxide during activation.36 The signal for Co 2p of D-FeCoNiCu-LDH/NF was boosted following activation (Fig. S10f, ESI†), because of an outward diffusion of Co ions, leading to increased Co species exposed on the surface. Significantly, no Cr 2p signal for D-FeCoNiCu-LDH/NF (Fig. S10g, ESI†) was detected, because chromium oxide is dissolved in 1.0 M KOH electrolyte during electrochemical activation. It is concluded that in this way, the generation of a significant number of defects was promoted in the electrocatalyst.28
GOR performance and product analyses
The catalytic activity of D-FeCoNiCu-LDH/NF for the GOR was established in a mixed solution of 1.0 M KOH and 100 mM glucose. The D-FeCoNiCu-LDH/NF exhibited the lowest potential of 1.22 V vs. RHE for a current density of 100 mA cm−2 (ηj=100), a value that is 40 mV less than that for FeCoNiCuCr-LDH/NF. To exhibit a high current density of 500 mA cm−2 D-FeCoNiCu-LDH/NF requires a potential of just 1.3 V vs. RHE for the GOR (Fig. 3a and b). To evidence the highly significant GOR activity of D-FeCoNiCu-LDH/NF, for comparison, FeNiCoCu-LDH/NF, D-FeNiCo-LDH/NF, D-FeCoCu-LDH/NF and D-FeNiCu-LDH/NF electrocatalysts were parallelly prepared in the same manner as FeCoNiCuCr-LDH/NF and D-FeCoNiCu-LDH/NF. FeCoNiCu-LDH/NF exhibited a higher potential (ηj=100 = 1.32 V) than that for D-FeCoNiCu-LDH/NF, evidencing that defect sites boost catalytic reaction. Without Ni and Co elements, the catalytic activity for the GOR of D-FeNiCu-LDH/NF (ηj=100 = 1.28 V) and D-FeCoCu-LDH/NF (ηj=100 = 1.30 V) decreased compared with that for D-FeCoNiCu-LDH/NF. The activity for D-FeCoNi-LDH/NF (ηj=100 = 1.38 V) is significantly reduced because of the lack of Cu elements. It is concluded that these findings confirm that the synergistic effect of Cu, Ni and Co elements catalyzes the electrooxidation of glucose, and Cu-related active sites promote rapid GOR. In addition, the electrochemical active surface area (ECSA)-normalized GOR activity of D-FeCoNiCu-LDH/NF demonstrates the superiority over D-FeCoNi-LDH/NF, D-FeNiCu-LDH/NF and D-FeCoCu-LDH/NF, manifesting the increased intrinsic catalytic activity in D-FeCoNiCu-LDH/NF (Fig. S11 and S12, ESI†). Then, the open-circuit potential (OCP) that reflects variation of adsorbates in the Helmholtz layer was applied to study the glucose adsorption behavior on the electrocatalysts (Fig. S13, ESI†).37 The OCP dropped significantly when 0.1 M glucose was introduced into the reaction system. The voltage on D-FeCoNiCu-LDH/NF reduced by 0.164 V before and after introducing 100 mM glucose, which was 0.086 V higher than that on D-FeCoNi-LDH/NF, indicating stronger adsorption of glucose on D-FeCoNiCu-LDH/NF than on D-FeCoNi-LDH/NF. It is indicated that the Cu element contributed to the adsorption of glucose, benefiting the subsequent GOR.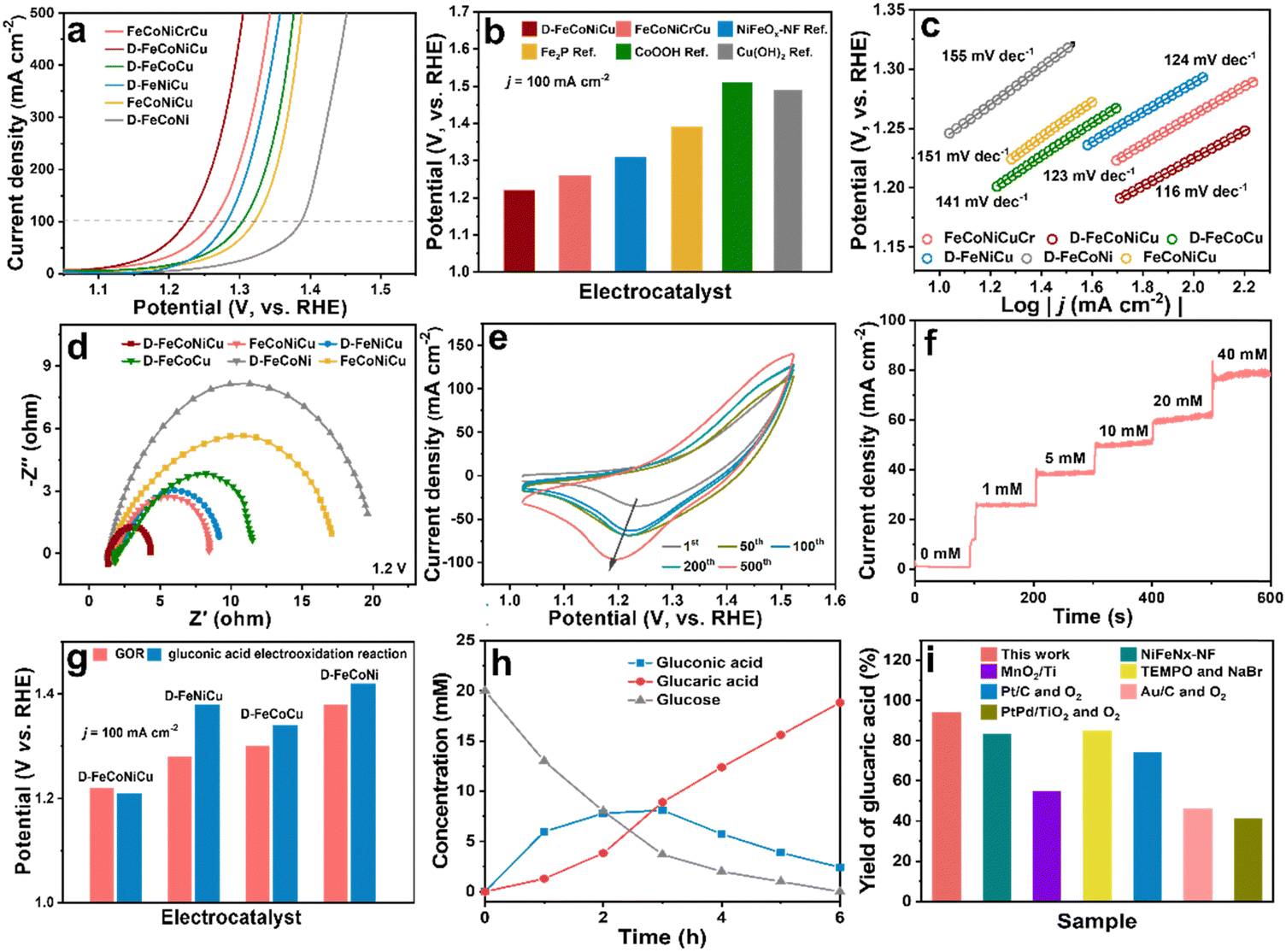 | ||
| Fig. 3 Electrocatalytic performance of high-entropy hydroxide electrocatalyst D-FeCoNiCu-LDH/NF for the GOR. (a) LSVs for D-FeCoNiCu-LDH/NF, FeCoNiCuCr-LDH/NF, FeCoNiCu-LDH/NF, D-FeNiCu-LDH/NF D-FeCoCu-LDH/NF and D-FoCoNi-LDH/NF for glucose electrooxidation in 1.0 M KOH with 0.1 M glucose. (b) Comparison between D-FeCoNiCu-LDH/NF, FeCoNiCuCr-LDH/NF and reported electrocatalysts for the GOR in potential required for ηj=100. (c) Tafel slopes and (d) EIS spectra at 1.2 V vs. RHE for D-FeCoNiCu-LDH/NF and controlled catalysts. (e) CV curves at differing cycles for FeCoNiCuCr-LDH/NF electrocatalyst in 1.0 M KOH solutions. (f) Time-dependent current density curves for D-FeCoNiCu-LDH/NF at 1.25 V vs. RHE in 1.0 M KOH with differing glucose concentration. (g) Comparison between D-FeCoNiCu-LDH/NF and controlled catalysts for the GOR and gluconic acid electrooxidation in the potential required for ηj=100. (h) Concentration of glucose and oxidation products as a function of time for chronoamperometric tests at 1.25 V vs. RHE. (i) Comparison between the yield of glucaric acid and selected, reported values. | ||
Compared with reported GOR catalysts, the D-FeCoNiCu-LDH/NF exhibits superior activity than Fe, Ni, Co and Cu-based electrocatalysts (Fig. 3b and Table S2, ESI†).12–14,38,39 D-FeCoNiCu-LDH/NF exhibited a reduced Tafel slope of 116 mV dec−1 compared with FeCoNiCuCr-LDH/NF, 123 mV dec−1, FeNiCoCu-LDH/NF, 151 mV dec−1, D-FeCoNi-LDH/NF, 155 mV dec−1, D-FeCoCu-LDH/NF, 141 mV dec−1 and D-FeNiCu-LDH/NF 124 mV dec−1, confirming faster kinetics for the GOR (Fig. 3c). From the electrochemical impedance spectroscopy (EIS) findings it was apparent that the D-FeCoNiCu-LDH/NF electrocatalyst exhibited lower charge-transfer impedance (Rct = 1.9 Ω), showing a faster charge transfer rate (Fig. 3d and Fig. S14, ESI†). Significantly, the active area of D-FeCoNiCu-LDH/NF increases with each subsequent CV sweep during electrochemical activation, contributing to exposure of additional defect sites for D-FeCoNiCu-LDH/NF together with Cr element etching, and promotes glucose conversion (Fig. 3e).40
During electrooxidation, the oxygen evolution reaction (OER) is a competitive reaction. In the new system the sluggish OER needs ηj=100 = 1.53 V in 1.0 M KOH – 310 mV more than that for GOR – to ensure a high Faraday efficiency (FE) for the GOR (Fig. S15, ESI†). The linear sweep voltammetry (LSV) for D-FeCoNiCu-LDH/NF for the GOR in 1.0 M KOH with differing glucose concentration (Fig. S16, ESI†) evidenced that current density increases with increasing glucose concentration, associated with the lower diffusion resistance of the electrocatalytic reaction from the solution to the surface of the electrode, thereby boosting the occurrence of electrocatalytic reaction(s). Importantly, the current density did not meaningfully change when the concentration was >100 mM, and the current density was near to zero with an applied voltage of 1.25 V vs. RHE in 1.0 M KOH, whilst it increased significantly when a low concentration glucose (1 mM) solution was injected into the electrolyte. Therefore, in consequence, it is concluded that the GOR occurs at a low concentration of glucose, ensuring a high conversion rate (Fig. 3f).
Because the oxidation energy barrier for the hydroxyl group is greater than that for the aldehyde group, particular electrocatalysts can only catalyze glucose to gluconic acid.39 To establish that D-FeCoNiCu-LDH/NF significantly catalyzes glucose to glucaric acid, the catalytic activity for gluconic acid electrooxidation was tested in 1.0 KOH and 100 mM gluconic acid mixed solution. The catalyst activity for D-FeCoNiCu-LDH/NF for the gluconic acid electrooxidation reaction (ηj=100 = 1.21 V) was found to be similar to that for the GOR, confirming that D-FeCoNiCu-LDH/NF exhibits high catalytic activity during the entire glucose oxidation (Fig. 3g and Fig. S17, ESI†). Whilst the D-FeCoCu-LDH/NF (ηj=100 = 1.34 V), D-FeNiCu-LDH/NF (ηj=100 = 1.38 V) and D-FeCoNi-LDH/NF (ηj=100 = 1.42 V) exhibit significantly greater voltage for gluconic acid electrooxidation due to a lack of, respectively, Ni, Co or Cu elements, it evidences that the synergistic effect(s) of Cu, Ni and Co related active sites catalyze the oxidation of hydroxyl groups together.
Time-course conversion of glucose into gluconic acid and glucaric acid was monitored by high performance liquid chromatography (HPLC, Fig. 3h), and the chronoamperometric measurements of the GOR carried out at a constant potential of 1.25 V vs. RHE. The concentration of glucose decreased to a value not distinguishable from zero, giving a conversion close to 100%. In parallel, the concentration of gluconic acid initially increases and then decreases, and the concentration of glucaric acid increases to 18.8 mM with a yield of 94%, confirming that gluconic acid is the initial oxidation product from glucose that is oxidized to glucaric acid. Significantly, the faradaic efficiency for the GOR is ca. 95%, evidencing high power utilization.
In addition to activity, the stability and durability of D-FeCoNiCu-LDH/NF for the GOR is an important performance metric. To establish this, four (4) successive cycles of chronoamperometric measurements were determined (Fig. S18a, ESI†). It was found that the conversion of glucose remained unchanged and the yield of glucaric acid was 87 to 94%. Though the glucose concentration increased to 50 and 100 mM, the conversion of glucose remained ca. 100% with a >90% yield of glucaric acid (Fig. S18b, ESI†). It is concluded therefore that these findings confirm the ‘excellent’ stability of the D-FeCoNiCu-LDH/NF catalyst. The D-FeCoNiCu-LDH/NF electrocatalyst following the GOR was characterized via SEM, TEM and HRTEM to assess stability. It was found that the sheet-structure for D-FeCoNiCu-LDH/NF remained following the GOR (Fig. S19a–c, ESI†). Elemental mapping analysis evidenced a uniform distribution of Fe, Co, Cu, Ni and O elements of D-FeCoNiCu-LDH/NF following the GOR (Fig. S19, ESI†). Additionally, the Fe 2p, Cu 2p, Ni 2p, Co 2p and O 1s XPS spectra for D-FeCoNiCu-LDH/NF remained all but unchanged in peak intensity and valence states following the GOR compared with the fresh structure (Fig. S20, ESI†). Therefore it was concluded that D-FeCoNiCu-LDH/NF is highly-significantly stable in the reaction, and it retains its original structure following GOR. Significantly, the new D-FeCoNiCu-LDH/NF electrocatalyst exhibits a high yield of glucaric acid of 90% compared with reported catalysts for the GOR, even with additional 2,2,6,6-tereamethyl-1-piperidinyloxy (TEMPO) as the oxidant agent (Fig. 3i).12,41 Significantly, conventional thermal catalytic methods depend on precious metals, Pt, Pd and Au, as catalysts under potentially dangerous high-pressure oxygen using a low concentration of glucose, leading to by-products including, tartronic acid, oxalic acid and 2-keto-D-gluconic acid with a low yield of glucaric acid.9,10,42 Importantly, this new electrooxidation of glucose catalyzed by D-FeCoNiCu-LDH/NF with a high concentration of glucose under mild conditions will be practically more reliable and low-cost for the mass production of high-yield glucaric acid.
Bifunctional HEMs for the GOR‖NO3−RR coupled system
In addition to the GOR, D-FeCoNiCu-LDH/NF with diverse active sites also exhibits a high catalytic activity in the electrooxidation of other biomasses including, urea (ηj=100 = 1.35 V), glycerol (ηj=100 = 1.30 V), glycolaldehyde (ηj=100 = 1.19 V) and 5-hydroxymethyfurfural (HMF) (ηj=100 = 1.40 V) confirming a versatile practical applicability for HEMs (Fig. 4b and c). It was found additionally via liquid chromatograph-mass spectrometer analyses, that glycolic acid and oxalic acid were apparent in glycolaldehyde electrooxidation products (Fig. S21, ESI†), confirming that the aldehyde group is initially oxidized to the carboxyl group, and the hydroxyl group oxidized to the aldehyde group and, subsequently, to the carboxyl group – an important finding consistent with that for the GOR.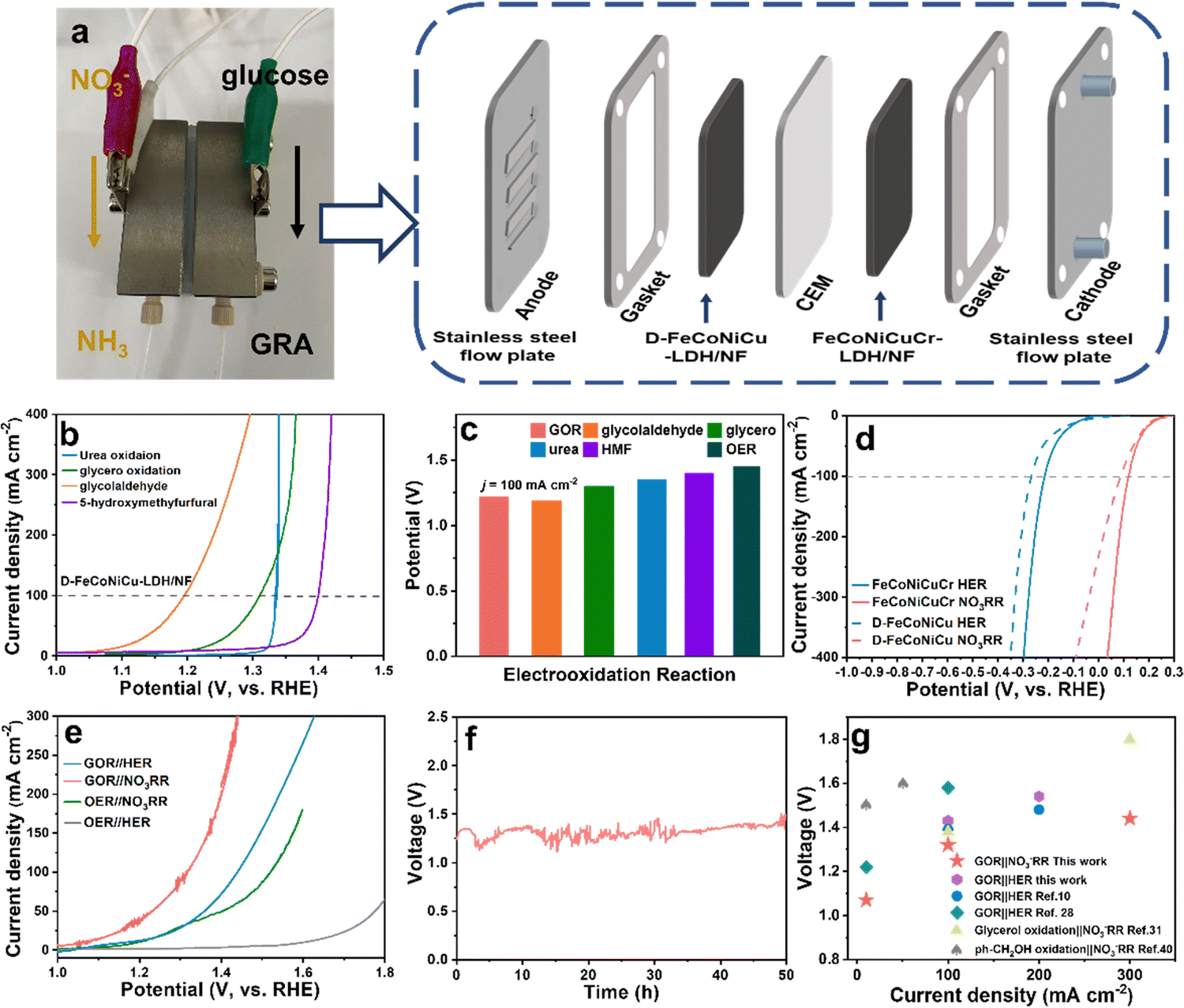 | ||
| Fig. 4 Assembly and performance of the GOR‖NO3−RR flow electrolyzer. (a) Photograph and schematic for the flow electrolyzer for the GOR at the anode and NO3−RR at the cathode. (b) LSVs for D-FeCoNiCu-LDH/NF for urea-, glycerol-, glycolic aldehyde- and HMF-oxidation in 1.0 M KOH with, respectively, 0.33 M urea, 0.1 M glycerol, 0.1 M glycolic aldehyde, or 10 mM HMF. (c) Potential vs. RHE required for a current density of 100 mA cm−2 with D-FeCoNiCu-LDH/NF as the electrocatalyst for the GOR, urea-, glycerol-, glycolic aldehyde- and HMF-oxidation reaction, and OER. (d) LSVs of FeCoNiCuCr-LDH/NF and D-FeCoNiCu-LDH/NF for the HER and NO3−RR in 1.0 M KOH, respectively, with or without 0.1 M NO3−. (e) LSV curves for the GOR‖NO3−RR, GOR‖HER, OER‖NO3−RR and OER‖HER hybrid electrolytic cells. (f) Long-term stability for the GOR‖NO3−RR hybrid electrolytic cell with D-FeCoNiCu-LDH/NF as an electrocatalyst in the anode and FeCoNiCuCr-LDH/NF as an electrocatalyst in the cathode at a current density of 30 mA cm−2 using a flow electrolytic cell. (g) Comparison of electrolytic voltage between the GOR‖NO3−RR, GOR‖HER hybrid electrolytic cell and reported hybrid electrolytic cells of organics electrooxidation coupled with the NO3−RR or HER. | ||
The hydrogen evolution reaction (HER) and NO3−RR produce high value-added chemicals, respectively, H2 and NH3. However the matched anodic OER has a high electrodynamic potential of 1.23 V vs. RHE, increased energy consumption and low value-added O2.43 In contrast, coupling the GOR with the HER or NO3−RR produces glucaric acid at the anode and H2 or NH3 at the cathode with a lower potential. Cathodic HER in 1.0 M KOH and NO3−RR in 1.0 M KOH and 0.1 M KNO3 solution was established using D-FeCoNiCu-LDH/NF and FeCoNiCuCr-LDH/NF as electrocatalysts. The FeCoNiCuCr-LDH/NF electrocatalyst required a lower potential of −0.22 and 0.12 V vs. RHE than D-FeCoNiCu-LDH/NF to exhibit a current density of 100 mA cm−2 for, respectively, the HER and NO3−RR (Fig. 4d). Because of the significantly lower potential for the GOR in comparison with the OER (Fig. 2h), the GOR‖NO3−RR couple required an applied voltage of just 1.07 and 1.32 V to exhibit an electrolytic current density of 10 and 100 mA cm−2 (Fig. 4a) values significantly less than for the OER‖NO3−RR couple with η10 = 1.20 V and η100 = 1.52 V (Fig. 4e). Similarly, the GOR‖HER, η10 = 1.17 V and η100 = 1.43 V couple exhibited a lower potential than for OER‖HER, η10 = 1.60 V. The GOR‖NO3−RR couple worked stably for 50 h (Fig. 4f) strongly evidencing long-term durability. The ultraviolet-visible (UV-Vis) spectrophotometer findings confirm that the product in the NO3−RR is NO2−, and NH3 in NO3−RR (NO3− → NO2− → NH3) and the ammonia can be produced continuously in the GOR‖NO3−RR flow cell (Fig. S22 and S23, ESI†).44,45 Importantly, the applied voltage for the GOR‖NO3−RR couple is ‘superior’ to the state-of-the-art hybrid electrolytic cell of organics electrooxidation coupled with the NO3−RR or HER (Fig. 4g and Table S3, ESI†).12,13,35,46
It is concluded therefore, that this new coupling of NO3−RR and GOR is a practically facile, energy-attractive and environmentally benign means for generating high value-added chemicals at both the anode and cathode.
Theoretical computation
To determine the role(s) of multiple active sites and establish a mechanism for multi-site synergistic catalysis of HEMs for the GOR, theoretical computation was used. To model the structure of HEMs possible configurations have a compositional space that will increase exponentially with the number of elements N.47 An accurate structural searching method is therefore required to reliably model D-FeCoNiCu-LDH. The globally optimized surface of D-FeCoNiCu-LDH was established via a machine learning-accelerated evolutionary algorithm-based structure search.48,49 The initial structure was a NiOOH one-layer nanosheet consisting of Fe, Co, Ni and Cu, based on in situ Raman spectra (Fig. S24, ESI†) and AFM imaging. Metals in LDH can present a 6-coordinated octahedral geometry, consistent with XANES and EXAFS findings. The energy searching curves as a function of number of local evaluations are presented in Fig. 5a, and the structure is determined as in Fig. 5b.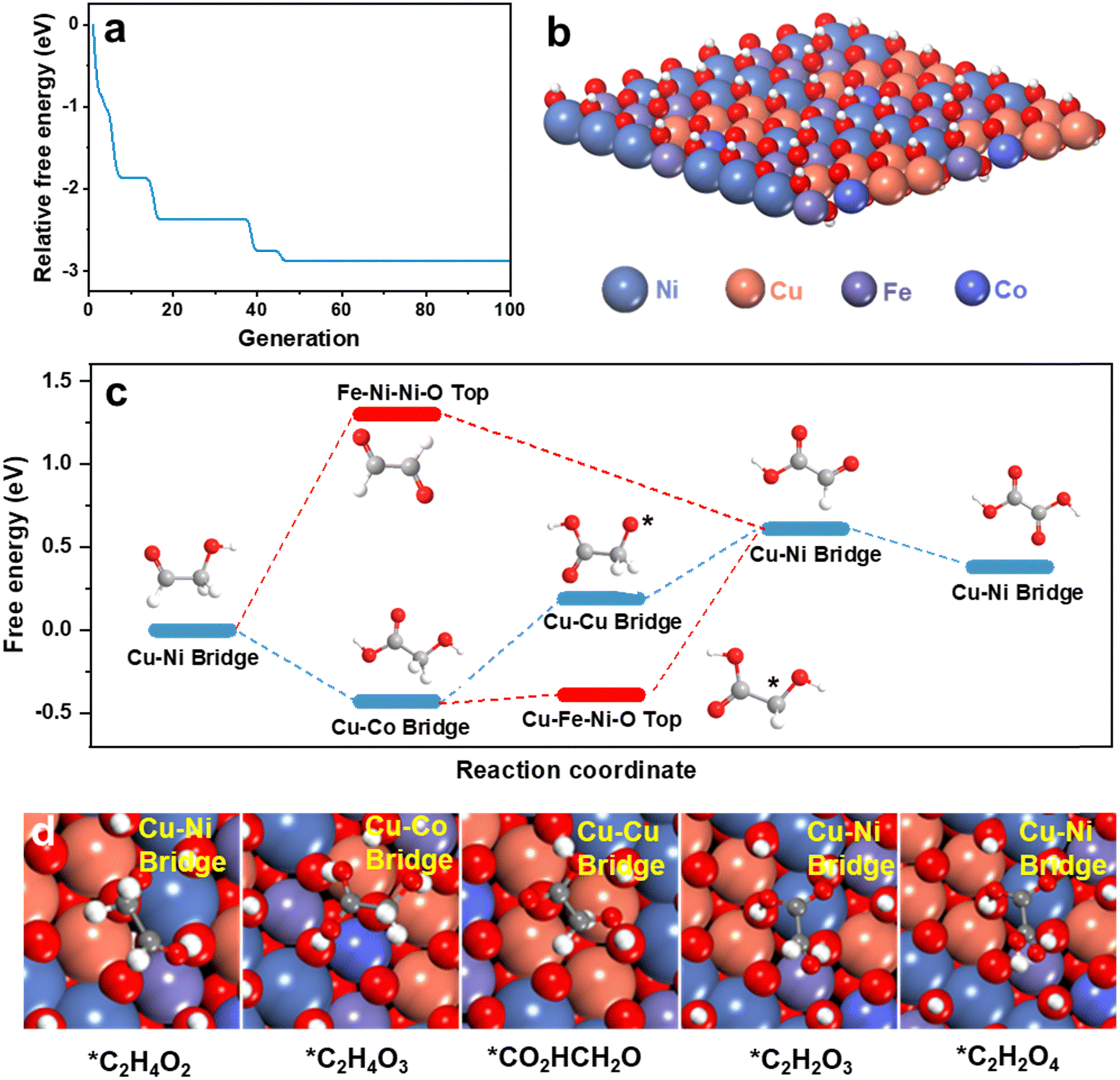 | ||
| Fig. 5 Theoretical computations. (a) Energy-search curves for the D-FeCoNiCu-LDH structure as a function of the number of generations, using an evolutionary algorithm-based structure-search. The searches shown are the mean value of three (3) replicates. (b) Stable structure determined for D-FeCoNiCu-LDH. (c) Reaction pathway for glycolaldehyde (*C2H4O2) to oxalic acid (*C2H2O4). (d) Schematic for active adsorption sites for differing reactants. | ||
To determine the multi-site synergistic catalysis mechanism for the GOR on D-FeCoNiCu-LDH/NF, the oxidation pathway was computed (Fig. 5c). Because of the size and complexity of glucose, glycolaldehyde, the ‘small’ molecule involving both an aldehyde and hydroxyl group, together with the same C/O ratio as glucose, was selected as a functional group surrogate for the reaction pathway for glucose. Reportedly there are strong similarities between the adsorption and reaction of these two (2) molecules on multiple heterogeneous catalysts.50–55 The new experimental findings reported here, confirm D-FeCoNiCu-LDH/NF exhibits high electrocatalytic activity for both glucose and glycolaldehyde oxidation, and that the aldehyde group is preferentially oxidized, evidencing that these two reactions have similar reaction pathways (Fig. 4b).
To establish the active site of D-FeCoNiCu-LDH/NF for the GOR, high-throughput screening amongst possible adsorption sites of the intermediates along the oxidation pathway of glycolaldehyde was undertaken (Fig. S25–S27 and Tables S4–S10, ESI†). The oxidation pathway, as presented as Fig. 5c, was determined via linking the most favorable adsorption of each intermediate. For glycolaldehyde (*C2H4O2) as the reactant, favorable adsorption is a precondition for the boosted oxidation kinetics for the reaction following, and higher site activity.56 It is concluded therefore that the Cu–Ni bridge site exhibits stronger adsorption (−1.64 eV) for glycolaldehyde than other sites of D-FeCoNiCu-LDH/NF, and other optimal adsorption sites for *C2H4O2 are the, (1) Cu–Cu–Cu–O top site, (2) Cu–Fe–Ni–O top site, (3) Cu–Ni–Ni hollow site, and (4) Cu–Cu–Ni hollow site. The introduction of Cu therefore boosts the adsorption of reactants, initially boosting rapid GOR, a finding and conclusion that is consistent with experiment results (Fig. 3a and Fig. S13, ESI†). For the following oxidation, there are two (2) possible pathways, namely, the (1) aldehyde group is preferentially oxidized to glycolic acid (*C2H4O3), and (2) the hydroxyl group is initially oxidized to form glyoxal (*C2H2O2). As presented in Fig. 5c, the oxidation of *C2H4O2 to *C2H2O2 requires overcoming a significant reaction energy barrier of 1.12 eV, whilst oxidation of *C2H4O2 to *C2H4O3 happens spontaneously. The oxidation of glycolaldehyde prefers therefore to take place in the aldehyde group to form *C2H4O3, rather than to form *C2H2O2. This finding is consistent with experiment, confirming that gluconic acid is the initial oxidation product from glucose, and that the Cu–Co bridge site is the optimal adsorption site for *C2H4O3.
Subsequently, *C2H4O3 is oxidized via preferentially losing H from its hydroxyl group, rather than its methylene group, forming glyoxylic acid (*C2H2O3), (Fig. 5c and d). The Cu–Cu bridge and Cu–Ni bridge sites are therefore optimal for dehydrogenation product and *C2H2O3. The dehydrogenation of the hydroxyl group is rate-limiting in the reaction. A synergistic effect of Cu, Co and Ni boosts the reaction and ensures a high yield of (final) product. Importantly, this is consistent with experiment showing that the lack of Cu, Ni or Co elements significantly decreases catalytic activity (Fig. 3g). The formation of the final product oxalic acid (*C2H2O4) from glyoxylic acid is thermodynamically favorable. Therefore, the intermediate product glucuronic acid is practically difficult to detect during the GOR. Apart from *C2H2O2 which is not favorable, the adsorption configurations with relatively low adsorption energy are more likely to be next to Cu-related active sites, such as the Cu–Ni bridge, Cu–Co bridge and Cu–Cu bridge, through the *C2H4O3 pathway. It is most probable that the preferred pathway does not occur on a single site, rather it consists of multiple different sites that contain Cu and other metals that evidence the synergetic effect of HEMs. It is concluded therefore that the Cu-relative site is the main active site for the GOR, and the addition of Cu mainly contributes to boosted GOR.
Conclusions
A robust, multi-site synergistic catalyst of high entropy hydroxide D-FeCoNiCu-LDH/NF with tandem active sites is practical for efficient, environmentally benign GOR. Cu–Co and Cu–Cu bridge sites boost oxidation of hydroxyl to aldehyde groups, and the Cu–Ni bridge promotes oxidation of aldehyde groups to carboxyl. This confirmed mechanism for multi-active HEMs can be applied to complex biomass electrooxidation reactions. Importantly, the (new) GOR‖NO3−RR hybrid electrolysis is cost-effective for co-production of high value-added NH3 and glucaric acid. Our findings will benefit researchers and manufacturers in electrocatalytic conversions of renewable biomass for high value-added chemicals.Author contributions
Z.-S. W., J. G., S. Q., and B. Z. conceived the experiments and supervised this project. X. W. performed materials syntheses, characterizations and electrochemical performance testing. Z.-J. Z. and X. S. carried out the theoretical computations. L. K. analyzed the EXFAS data. S. C. carried out XAS characterizations. B. Z. and H. W. carried out the HPLC analysis and S. W. participated in drawing. X. W., X. S., Z.-J. Z., P. D., B. Z., K. D. and Z.-S. W. wrote the manuscript. All the authors discussed the results and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant 22125903, 51872283, 22209174, 22078317, 22121004, U22A20409), the National Key R&D Program of China (Grant 2022YFA1504100, 2023YFB4005204), the Dalian National Laboratory For Clean Energy (DNL), CAS, DNL Cooperation Fund, CAS (DNL202016, DNL202019), DICP (DICP I2020032), the Joint Fund of the Yulin University, and the Dalian National Laboratory for Clean Energy (YLU-DNL Fund 2021002, YLU-DNL Fund 2021009). We gratefully acknowledge the Beijing Synchrotron Radiation Facility and Shanghai Synchrotron Radiation Facility for conducting the XANES/EXAFS and XPS experiments from the Vacuum Interconnected Nanotech Workstation (NANO-X).Notes and references
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- L. T. Mika, E. Csefalvay and A. Nemeth, Chem. Rev., 2018, 118, 505–613 CrossRef CAS PubMed.
- C. Liu, H. Wang, A. M. Karim, J. Sun and Y. Wang, Chem. Soc. Rev., 2014, 43, 7594–7623 RSC.
- T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484–2496 CAS.
- Z. Cheng, Q. Dong, G. Pu, J. Song, W. Zhong and J. Wang, Small, 2024 DOI:10.1002/smll.202400389.
- Z. Zhang and G. W. Huber, Chem. Soc. Rev., 2018, 47, 1351–1390 RSC.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- W. Deng, L. Yan, B. Wang, Q. Zhang, H. Song, S. Wang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 4712–4719 CrossRef CAS PubMed.
- J. Lee, B. Saha and D. G. Vlachos, Green Chem., 2016, 18, 3815–3822 RSC.
- S. Solmi, C. Morreale, F. Ospitali, S. Agnoli and F. Cavani, ChemCatChem, 2017, 9, 2797–2806 CrossRef CAS.
- Y. Jiang, K. Liu, H. Zhang, Y. Wang, Q. Yuan, N. Su, J. Bao and X. Fang, ACS Sustainable Chem. Eng., 2017, 5, 6116–6123 CrossRef CAS.
- W. J. Liu, Z. Xu, D. Zhao, X. Q. Pan, H. C. Li, X. Hu, Z. Y. Fan, W. K. Wang, G. H. Zhao, S. Jin, G. W. Huber and H. Q. Yu, Nat. Commun., 2020, 11, 265 CrossRef CAS PubMed.
- P. Du, J. Zhang, Y. Liu and M. Huang, Electrochem. Commun., 2017, 83, 11–15 CrossRef CAS.
- E. V. K. Matthijs, P. J. M. van der Ham, M. T. M. Koper, A. Asadi Tashvigh and J. H. Bitter, Angew. Chem., Int. Ed., 2023, 62, e202306701 CrossRef PubMed.
- S.-K. Geng, Y. Zheng, S.-Q. Li, H. Su, X. Zhao, J. Hu, H.-B. Shu, M. Jaroniec, P. Chen, Q.-H. Liu and S.-Z. Qiao, Nat. Energy, 2021, 6, 904–912 CrossRef CAS.
- W. He, J. Zhang, S. Dieckhofer, S. Varhade, A. C. Brix, A. Lielpetere, S. Seisel, J. R. C. Junqueira and W. Schuhmann, Nat. Commun., 2022, 13, 1129 CrossRef CAS PubMed.
- L. Fan, Y. Ji, G. Wang, J. Chen, K. Chen, X. Liu and Z. Wen, J. Am. Chem. Soc., 2022, 144, 7224–7235 CrossRef CAS PubMed.
- W. Chen, S. Luo, M. Sun, X. Wu, Y. Zhou, Y. Liao, M. Tang, X. Fan, B. Huang and Z. Quan, Adv. Mater., 2022, 34, 2206276 CrossRef CAS PubMed.
- L. Zhang, W. Cai and N. Bao, Adv. Mater., 2021, 33, 2100745 CrossRef CAS PubMed.
- C. Zhan, Y. Xu, L. Bu, H. Zhu, Y. Feng, T. Yang, Y. Zhang, Z. Yang, B. Huang, Q. Shao and X. Huang, Nat. Commun., 2021, 12, 6261 CrossRef CAS PubMed.
- S. Wang, K. Gu, D. Wang, C. Xie, T. Wang, G. Huang, Y. Liu, Y. Zou and L. I. Tao, Angew. Chem., Int. Ed., 2021, 60, 20253–20258 CrossRef PubMed.
- Y. Sun and S. Dai, Sci. Adv., 2021, 7, eabg1600 CrossRef CAS PubMed.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. Garcia-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. G. de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. De Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- J. Cavin, A. Ahmadiparidari, L. Majidi, A. S. Thind, S. N. Misal, A. Prajapati, Z. Hemmat, S. Rastegar, A. Beukelman, M. R. Singh, K. A. Unocic, A. Salehi-Khojin and R. Mishra, Adv. Mater., 2021, 33, 2100347 CrossRef CAS PubMed.
- M. Zhang, Y. Liu, B. Liu, Z. Chen, H. Xu and K. Yan, ACS Catal., 2020, 10, 5179–5189 CrossRef CAS.
- H. Huang, C. Yu, X. Han, H. Huang, Q. Wei, W. Guo, Z. Wang and J. Qiu, Energy Environ. Sci., 2020, 13, 4990–4999 RSC.
- B. S. Yeo and A. T. Bell, J. Phys. Chem. C, 2012, 116, 8394–8400 CrossRef CAS.
- P. Zhai, M. Xia, Y. Wu, G. Zhang, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, Z. Fan, C. Wang, X. Zhang, J. T. Miller, L. Sun and J. Hou, Nat. Commun., 2021, 12, 4587 CrossRef CAS PubMed.
- J. Chen, H. Li, S. Chen, J. Fei, C. Liu, Z. Yu, K. Shin, Z. Liu, L. Song, G. Henkelman, L. Wei and Y. Chen, Adv. Energy Mater., 2021, 11, 2003412 CrossRef CAS.
- L. J. A. Macedo, A. Hassan, G. C. Sedenho and F. N. Crespilho, Nat. Commun., 2020, 11, 316 CrossRef CAS PubMed.
- H. Yi, S. Liu, C. Lai, G. Zeng, M. Li, X. Liu, B. Li, X. Huo, L. Qin, L. Li, M. Zhang, Y. Fu, Z. An and L. Chen, Adv. Energy Mater., 2021, 11, 2002863 CrossRef CAS.
- R. Gao and D. Yan, Adv. Energy Mater., 2020, 10, 1900954 CrossRef CAS.
- J. Zhang, L. Yu, Y. Chen, X. F. Lu, S. Gao and X. W. Lou, Adv. Mater., 2020, 32, 1906432 CrossRef CAS PubMed.
- S. Zhang, Y. Zhao, R. Shi, C. Zhou, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Energy Mater., 2020, 10, 1901973 CrossRef CAS.
- S. Li, P. Ma, C. Gao, L. Liu, X. Wang, M. Shakouri, R. Chernikov, K. Wang, D. Liu, R. Ma and J. Wang, Energy Environ. Sci., 2022, 15, 3004–3014 RSC.
- X. Su, Y. Wang, J. Zhou, S. Gu, J. Li and S. Zhang, J. Am. Chem. Soc., 2018, 140, 11286–11292 CrossRef CAS PubMed.
- Y. Lu, T. Liu, C. L. Dong, Y. C. Huang, Y. Li, J. Chen, Y. Zou and S. Wang, Adv. Mater., 2021, 33, 2007056 CrossRef CAS PubMed.
- Y. Q. Zhu, H. Zhou, J. Dong, S. M. Xu, M. Xu, L. Zheng, Q. Xu, L. Ma, Z. Li, M. Shao and H. Duan, Angew. Chem., Int. Ed., 2023, 62, e202219048 CrossRef CAS PubMed.
- Y. Zhang, B. Zhou, Z. Wei, W. Zhou, D. Wang, J. Tian, T. Wang, S. Zhao, J. Liu, L. Tao and S. Wang, Adv. Mater., 2021, 33, 2104791 CrossRef CAS PubMed.
- X. Lu, K.-H. Wu, B. Zhang, J. Chen, F. Li, B.-J. Su, P. Yan, J.-M. Chen and W. Qi, Angew. Chem., Int. Ed., 2021, 60, 14528–14535 CrossRef CAS PubMed.
- D. Bin, H. Wang, J. Li, H. Wang, Z. Yin, J. Kang, B. He and Z. Li, Electrochim. Acta, 2014, 130, 170–178 CrossRef CAS.
- X. Jin, M. Zhao, M. Vora, J. Shen, C. Zeng, W. Yan, P. S. Thapa, B. Subramaniam and R. V. Chaudhari, Ind. Eng. Chem. Res., 2016, 55, 2932–2945 CrossRef CAS.
- Y. H. Feng, N. Ran, X. L. Wang, Q. A. Liu, J. C. Wang, L. J. Liu, K. Suenaga, W. W. Zhong, R. G. Ma and J. J. Liu, Adv. Energy Mater., 2023, 13, 2302452 CrossRef CAS.
- S. Han, H. Li, T. Li, F. Chen, R. Yang, Y. Yu and B. Zhang, Nat. Catal., 2023, 6, 402–414 CrossRef CAS.
- F. Y. Chen, Z. Y. Wu, S. Gupta, D. J. Rivera, S. V. Lambeets, S. Pecaut, J. Y. T. Kim, P. Zhu, Y. Z. Finfrock, D. M. Meira, G. King, G. Gao, W. Xu, D. A. Cullen, H. Zhou, Y. Han, D. E. Perea, C. L. Muhich and H. Wang, Nat. Nanotechnol., 2022, 17, 759–767 CrossRef CAS PubMed.
- S. Ye, Z. Chen, G. Zhang, W. Chen, C. Peng, X. Yang, L. Zheng, Y. Li, X. Ren, H. Cao, D. Xue, J. Qiu, Q. Zhang and J. Liu, Energy Environ. Sci., 2022, 15, 760–770 RSC.
- R. Singh, A. Sharma, P. Singh, G. Balasubramanian and D. D. Johnson, Nat. Comput. Sci., 2021, 1, 54–61 CrossRef PubMed.
- X. Shi, X. Lin, R. Luo, S. Wu, L. Li, Z.-J. Zhao and J. Gong, JACS Au, 2021, 1, 2100–2120 CrossRef CAS PubMed.
- X. Shi, D. Cheng, R. Zhao, G. Zhang, S. Wu, S. Zhen, Z.-J. Zhao and J. Gong, Chem. Sci., 2023, 14, 8777–8784 RSC.
- J. R. McManus, M. Salciccioli, W. Yu, D. G. Vlachos, J. G. Chen and J. M. Vohs, J. Phys. Chem. C, 2012, 116, 18891–18898 CrossRef CAS.
- J. R. McManus, E. Martono and J. M. Vohs, Catal. Today, 2014, 237, 157–165 CrossRef CAS.
- B. Caglar, M. O. Ozbek, J. W. Niemantsverdriet and C. J. Weststrate, J. Phys. Chem. C, 2015, 119, 22915–22923 CrossRef CAS.
- K. Xiong, W. Yu, D. G. Vlachos and J. G. Chen, ChemCatChem, 2015, 7, 1402–1421 CrossRef CAS.
- Q. T. Trinh, B. K. Chethana and S. H. Mushrif, J. Phys. Chem. C, 2015, 119, 17137–17145 CrossRef CAS.
- W. Yu, M. A. Barteau and J. G. Chen, J. Am. Chem. Soc., 2011, 133, 20528–20535 CrossRef CAS PubMed.
- Z. Lang, Z. Zhuang, S. Li, L. Xia, Y. Zhao, Y. Zhao, C. Han and L. Zhou, ACS Appl. Mater. Interfaces, 2019, 12, 2400–2406 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee00221k |
| ‡ These authors contributed equally: Xianhong Wu, Zhi-Jian Zhao, and Xiangcheng Shi. |
| This journal is © The Royal Society of Chemistry 2024 |