
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
f-Block hydride complexes – synthesis, structure and reactivity
Richard
Drummond Turnbull
 and
Nicola L.
Bell
*
and
Nicola L.
Bell
*
School of Chemistry, University of Glasgow, Glasgow, UK G12 8QQ. E-mail: Nicola.Bell@Glasgow.ac.uk
First published on 26th June 2024
Abstract
Complexes formed between the heaviest and lightest elements in the periodic table yield the f-block hydrides, a unique class of compounds with wide-ranging utility and interest, from catalysis to light-responsive materials and nuclear waste storage. Recent developments in syntheses and analytics, such as exploiting low-oxidation state metal ions and improvements in X-ray diffraction tools, have transformed our ability to understand, access and manipulate these important species. This perspective brings together insights from binary metal hydrides, with molecular solution phase studies on heteroleptic complexes and gas phase investigations. It aims to provide an overview of how the f-element influences hydride formation, structure and reactivity including the sometimes-surprising power of co-ligands to tune their behaviour towards a variety of applications.
 Richard Drummond Turnbull | Richard Drummond Turnbull hails from Durham in the North-East of England and graduated from Newcastle University with an MChem (Hons) in Chemistry in 2018. He obtained his PhD in Inorganic Chemistry from the University of Manchester in 2023, conducted under the supervision of Prof. Stephen T. Liddle, before pursuing postdoctoral research in the Bell Group at the University of Glasgow. His principal research interests are in the synthesis and stabilisation of highly-reactive compounds, as well as the development of novel tools and methodologies to aid in this pursuit. |
 Nicola L. Bell | Dr Nicola Bell was awarded her PhD in 2013 from the University of Edinburgh before undertaking an EPSRC Doctoral Prize Fellowship with Prof. Polly Arnold and later Prof. Jason Love studying actinide imido and oxo complexes. A subsequent period at the University of St Andrews saw Nicola investigate copper catalysis before joining the University of Glasgow in 2019 working on Digital Chemistry. In 2023 Nicola began her independent career with an EPSRC Open Fellowship in Digital Inorganic Chemistry at the University of Glasgow. Dr Bell's research interests encompass f-block coordination chemistry, nuclear applications of chemistry and organometallic catalysis. |
Introduction
Although the reactivity of lanthanide and actinide metals towards hydrogen gas had been long observed, it was the Manhattan Project in the 1940s which first brought to light the utility and unique subtleties of hydride complexes of the f-elements.1 Distinct from their substantially more numerous main group and d-block counterparts, f-block hydrides are characterised by their predominantly ionic nature, high coordination numbers (which favour bridging hydride ligation) and relatively inaccessible redox activity. These properties, coupled with the inherent photophysical, magnetic and, for some, radioactive nature of the metal ions in these complexes, means that f-block hydrides still represent an important and accelerating field of inorganic chemistry.Solubilised heteroleptic lanthanide hydrides are found to be the key catalytically active species in a range of hydroelementation transformations2,3 while binary lanthanide hydrides have also been investigated for applications in methanol synthesis,4 dinitrogen fixation5 and neodymium magnet recycling.6 Useful optical properties exhibited by lanthanide oxyhydrides allow them to be widely utilised in light-responsive materials, such as those now-frequently applied to corrective lenses.7 At the same time, actinide hydrides represent important potential by-products of radiolysis processes occurring during nuclear waste storage and remediation, which must be understood to facilitate safe nuclear waste handling.8–10 Superhydrides of both the lanthanides and actinides have demonstrated promise as potential room temperature superconductors and as hydrogen storage materials.11–13 Actinides have also been demonstrated to activate more challenging substrates, such as converting dinitrogen to ammonia;14 their reactivity is often unique within the periodic table, making them interesting potential reagents and catalysts in their own right.
Despite their importance, f-block hydrides are often overlooked. In catalysis, their presence is implicated in a wide range of processes; however, the utility of lanthanide alkyl precatalyst handling, for example, means the true nature of the hydride involved in these processes is opaque—with consequences for the development of these reactions.
In actinide chemistry, much work to date has focussed on the hydrogenation of actinide metals in the solid state to gain insights into nuclear waste remediation processes; however, bridging the gap between these studies and the well-defined academic curiosities, isolated using complex supporting co-ligands over the past few decades, has been challenging. Despite this, these heteroleptic actinide hydrides provided valuable contributions towards our understanding of the role of f-orbitals in actinide bonding more generally.15
In this perspective, we seek to put into context the chemistry of f-block hydrides, including the synthesis, structure and reactivity of both lanthanide and actinide hydrides. We give an overview of the behaviour of binary hydrides (MHx) of each period such that the structure and synthesis of heteroleptic molecular hydrides finds context. Within each section, we discuss synthetic strategies, solid-state structure and analytical data on the resulting hydrides and their further reactivity. This work follows on from an extensive early review of the field by Ephritikhine in 1997,16 drawing upon improvements in analytical data (notably X-ray diffraction), novel synthetic strategies and newly discovered reactivity profiles in the ensuing three decades to provide an overview of the now mature field.
Homoleptic lanthanide hydrides
Solid phase binary hydrides – structural insights
In the first step of hydrogen's reaction with bulk metallic lanthanides a solid solution is formed, whereby dihydrogen molecules occupy tetrahedral interstitial sites within the Ln(0) crystal lattice. Specifically, one half of the sites (the second nearest neighbour sites), are occupied by dihydrogen (Fig. 1).17 This shifts to occupancy of all sites by a single hydride upon formation of LnH2 (which has the CaF2 structure; La–H = 2.45 Å). Recent work has demonstrated that Ln dihydrides (Ln = La, Ce) exist as electride materials with a [LnIII(Ht)2(e−)] formulation.18 Further hydrogens occupy octahedral sites but defects mean octahedral sites (Ho) can start filling before tetrahedral sites (Ht) are full. Transformation from LnH2 to LnH3 is accompanied by a transition from metal-like to insulating behaviour as the valence electrons are occupied in bonding, with the structure adopting a cubic BiF3 or trigonal HoH3 type lattice depending upon cation size (La–Ht = 2.43 Å; La–Ho = 2.81 Å). At the same time, a transition in optical properties from darkly coloured LnH2 to metallic LnH3 occurs.19,20 However, it should be noted that even at a >2.9 H![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ln ratio, conductivity can be high, with small defects in the lattice occupancy yielding superionic conductivity.21
:Ln ratio, conductivity can be high, with small defects in the lattice occupancy yielding superionic conductivity.21
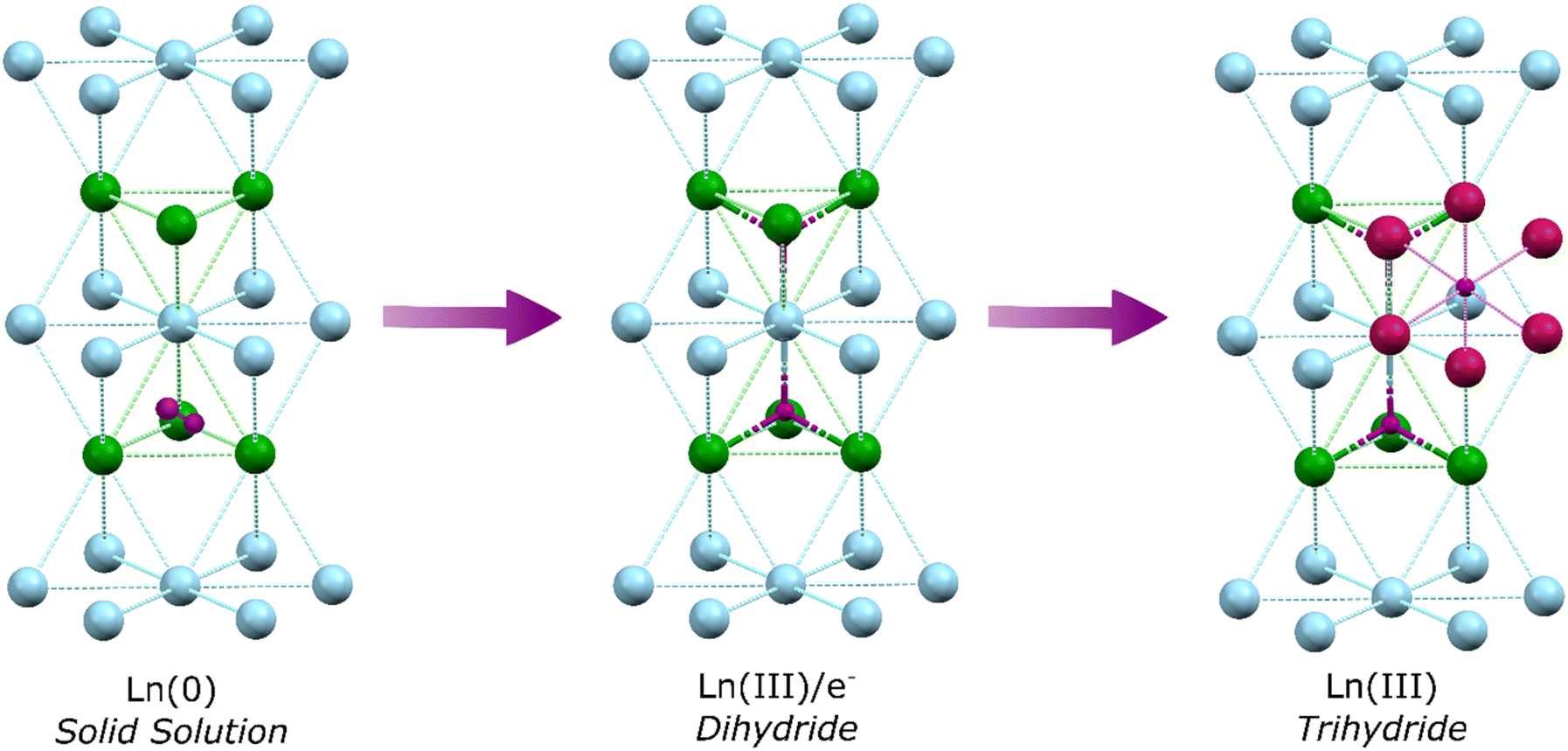 | ||
| Fig. 1 The formation of binary lanthanide hydrides from Ln(0) proceeds via a solid solution, with dihydrogen occupying nearest neighbour sites before oxidation first to lanthanide dihydride, with occupancy of tetrahedral vacancies, then Ln(III), through occupancy of octahedral sites with hydride. Ln ions: Blue (general), Green (tetrahedral site) or Pink (octahedral site) spheres. Hydrogen/hydride atoms: Purple spheres. | ||
Gas phase/molecular hydrides
Molecular binary lanthanide hydrides have been accessed using laser ablation of lanthanide metal atoms under H2 within noble gas matrices. Andrews used this method to investigate the vibrational spectra across a range of LnHx stoichiometries in the gas phase (i.e., molecular examples), thereby finding evidence for a non-linear structure for LaH2 in the gas phase, as well as the formation of the H2 adduct (H2)CeH2.22,23 This was further investigated across the series, finding that stable adducts of dihydrogen may form with La, Ce, Pr and Nd.24 Potential stability is more favourable with softer divalent lanthanides which bind molecular H2 more strongly than harder trivalent ions, with hydrogen adduct formation occurring via transfer of a 6s electron to the H2 molecule, and therefore representing the covalent extreme of lanthanide–hydrogen bonding. The trihydrides were also observed; later calculations determined that LnH3 complexes exhibit a pyramidal geometry, due to 5d orbital contributions to the bonding.15 Thus, whilst bonding in these f-block hydrides is mostly ionic, subtle influences, such as interaction with 5f orbitals for the early actinides or with accessible 4f and 5d orbitals for the lanthanides, can have dramatic effects on structure and reactivity.Lanthanide superhydrides
Higher stoichiometries, Ln4+x, have been shown to be stable under elevated pressure and temperature regimes and are of interest as next-generation superconductors. This is because in these species hydrogen may exist as metallic H(0).11 The best-studied of these is LnH10, which involves a clathrate-type arrangement of H atoms (32 H per Ln) encapsulating the lanthanide ion. These species can be synthesised above 106 bar pressure in a diamond anvil cell at high temperatures (1000 K). Raman spectroscopy shows no H–H bands, corresponding to bound diatomic hydrogen, despite the fact that LaH10 contains the shortest H–H distances other than molecular hydrogen.12Heteroleptic lanthanide hydrides
Synthetic strategies
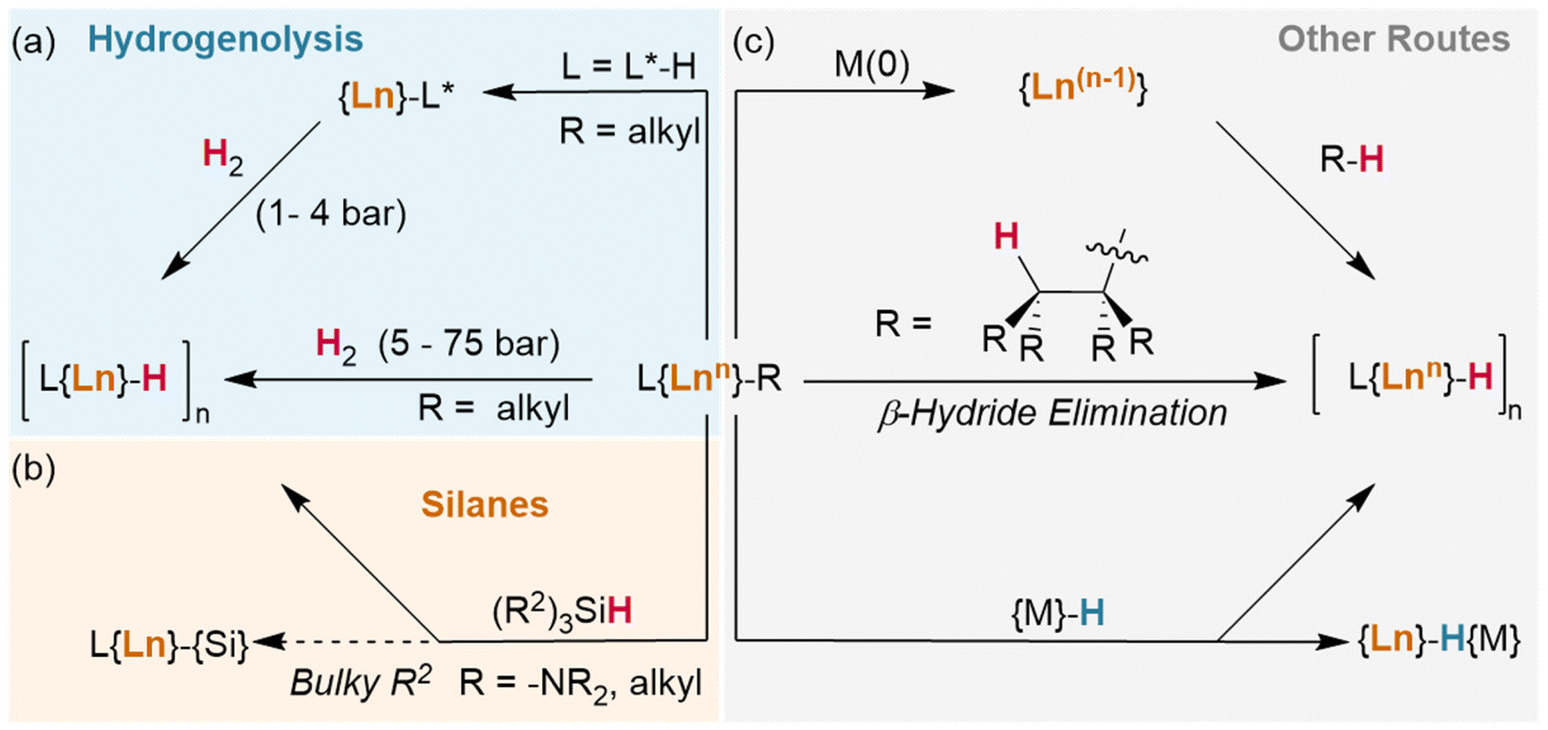 | ||
| Fig. 2 Synthetic routes to lanthanide hydrides from L{Ln}–R complexes (a) via hydrogenolysis. L* = metalated ligand, accessed via C–H activation/deprotonation. (b) Using silanes. (c) Other routes include reduction with alkali metals, β-hydride elimination and reactions with metal hydrides. | ||
Okuda et al. report the synthesis of the lutetium complex [(Me4TACD)Lu(R)2]+, which reacts with H2 at 1 bar pressure in thf (Fig. 3). The dialkyl precursor yields a mixture of a ligand-metalated trihydrido dimer, [(L*)Lu(μ2-H)3LuL]2+ or [{(κ5-N,N,N,N,C-CH2-Me3TACD)Lu}(μ2-H)3{Lu(κ4-N,N,N,N-Me4TACD)}]2+, and a hydrogenated tetrahydrido dimer, [{(L)Lu}2(μ2-H)4]2+ or [{(κ4-N,N,N,N-Me4TACD)Lu}2(μ2-H)4]2+.26 Based on this (as well as complementary reactions with silanes), the authors conclude that the metalate is an intermediate in the hydrogenation process. Many of the hydrides reported to date either arise directly from metalated complexes (e.g., with substituted metallocene co-ligands) or utilise ligands which are able to undergo metalation.27–40 Importantly, a survey of the literature demonstrates that these examples allow for hydrogenation at low pressures of H2 (1–4 bar). Lanthanide alkyl complexes of ligands which are not known to metalate (e.g., TpR) have only been reported under significantly higher pressures for hydrogenolysis (5–75 bar), often with longer reaction times.41–47 This suggests that the ability of the ligand to metalate provides a significantly lower energy pathway for hydrogenolysis reactions. Kinetic isotope experiments and DFT calculations with these metalated TACD-lutetium hydrides indeed appear to suggest that ligand metalation favours the association of H2 before cleaving the M–C bond.
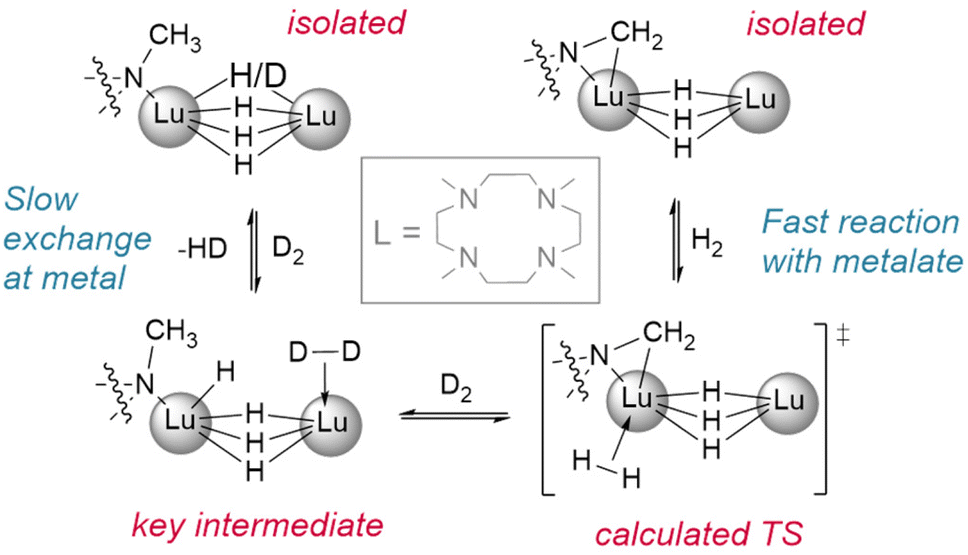 | ||
| Fig. 3 Demonstration of the reactivity of metalated ligands in the activation of H2. Deuteration studies show metal based H/D exchange is slow with the tetrahydrido dimer while the metalated trihydrido complex reacts with H2/D2 quickly. L = Me4-TACD.† | ||
The basicity of the alkyl is also shown to be a key factor for hydrogenolysis. Hou et al. demonstrate that hydrogenolysis of the tetrameric methanediide {(Cp′)Lu}4(μ2-CH2)4 (Cp′ = C5H4SiMe3)† yields tetrahedral cluster {(Cp′)Lu}4(μ2-Me)4(μ2-H)2(μ3-H)(μ4-H) through H2 addition across the Lu–CH2 bond.48 Further hydrogenolysis of the remaining four methyl ligands does not occur in this example. Similarly, treatment of the trimeric cluster {(Cp′)Lu}3(μ2-Me)6 under the same conditions (1 bar H2, 70 °C) yielded the corresponding tetramer, {(Cp′)Lu}4(μ2-Me)6(μ3-Me)(μ4-H), where only one of the eight possible methyl groups is protonated off (Fig. 4). This suggests a balance of donor properties, with strongly basic methane ligands competing with hydrides to stabilise the lanthanide complex. At the same time, this work suggests hydride structure persists in solution, at least to a degree sufficient to affect reactivity. These limits are important to understand when considering which lanthanide precatalysts to use in hydrogenation and hydroelementation reactions.
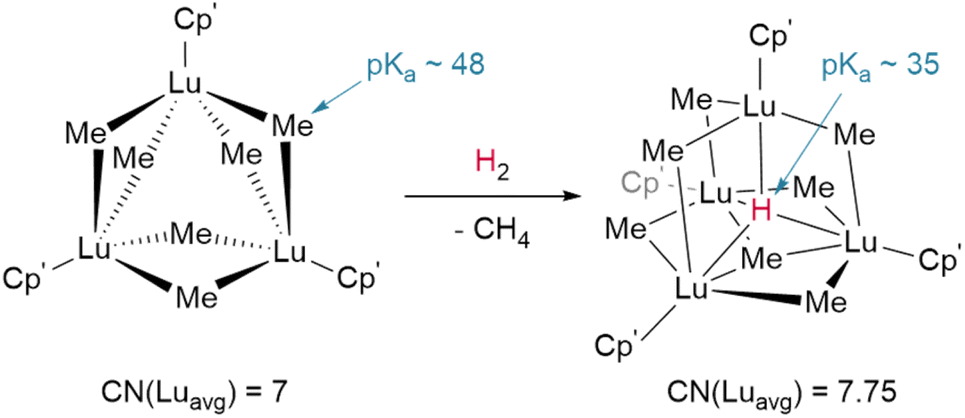 | ||
| Fig. 4 Despite the higher pKa of the methyl anion hydrogenolysis occurs with Hou's trimer due to the coordinative stabilisation provided by the μ4-hydride in the tetrameric product. CN = coordination number; Cp′ = (C5H4SiMe3).† | ||
In an advance on hydrogenolysis reactions, silanes have also been shown to add across Ln–{N(SiMe3)2} bonds in addition to lanthanide alkyls,63,64 and have even been shown to supplant Cp* (Cp* = C5Me5)† as a ligand.65 However, in rare circumstances, silanes have also been found to participate in other side reactions, including exchange with trimethylsilyl (TMS, Si′)† groups on Ln–{CH2SiMe3} ligands (Fig. 5a/b),66 although this may be mitigated with ligand steric bulk.67 In addition, the bulky silane H2Si(Si′)2 was shown to undergo reversed σ-bond metathesis with (Cp*)2Sm-CH(Si′)2 yielding the Ln–Si product rather than the hydride (Fig. 5c), likely due to steric hindrance preventing the formation of the expected four-centred transition state.68,69
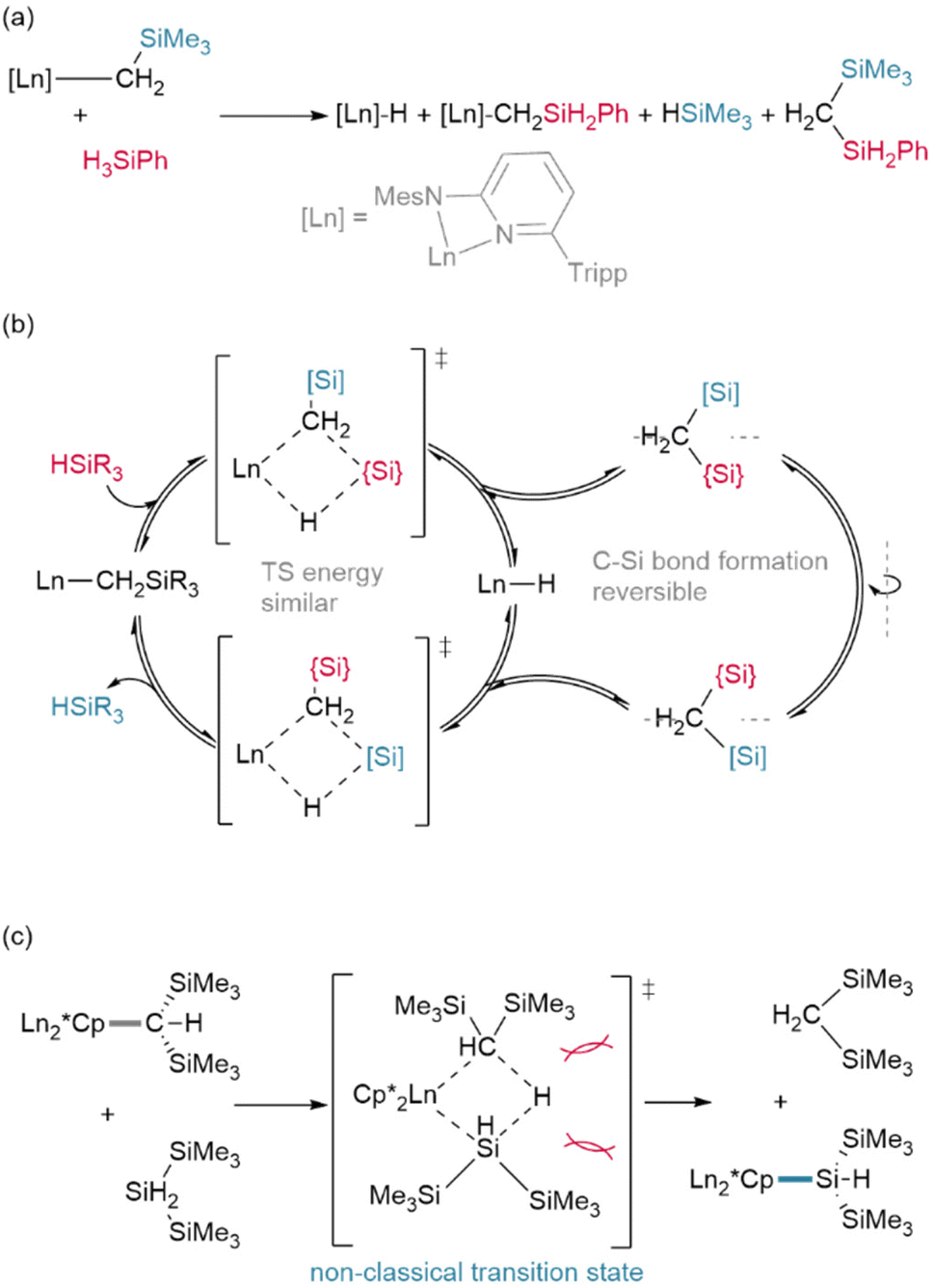 | ||
| Fig. 5 Alternate non-classical reaction pathways for silanes with lanthanide organometallics: (a) Trifonov et al. found a mixture of silyl products from reaction of their Ln–CH2SiMe3 complexes with PhSiH3 for Ln = Y, Yb (but not Lu) when their ligand was not sufficiently bulky for classical reactivity to occur (Mes = 1,3,5-trimethylphenyl; Tripp = 1,3,5-triisopropylphenyl). (b) A mechanistic explanation for this different behaviour is based upon the reversibility of σ-bond metathesis in these complexes and the similar electronic properties of the –[SiMe3] (blue) and –{SiH2Ph} (red) groups. Re-attack of the disilylmethane by-product of the reaction in a different orientation can explain all of the products observed. (c) Tilley et al. found that bulky silanes reacting with bulky Ln–CH(SiMe3)2 (Ln = Sm) organometallics formed Ln–Si bonds likely due to sterics preventing the formation of a classical 4-membered transition state. Instead the hydride likely occupies the position opposite the lanthanide in the TS for this transformation resulting in C–Si bond formation. | ||
Investigating reactions with silanes has also allowed researchers to uncover differences across the lanthanide series, with early lanthanide (Me3TACD)Ln(allyl)2 complexes reacting to give octahydrido tetramer {(Me3TACD)Ln}4(μ2-H)8 while the later lanthanides react with only one equivalent of silane to yield {(Me3TACD)Ln(allyl)}2(μ2-H)8.70 This may be due to kinetic rather than thermodynamic factors since the reaction of {(Cp*)2Ln}2(μ2-Me)2 with PhMeSiH2 was found to follow a stepwise mechanism, with a fast initial transformation to {(Cp*)2Ln}2(μ2-Me)(μ2-H) followed by slower hydrogenolysis of the final methyl ligand. The rate of both of these processes decreased across the lanthanide series.71
Finally, even the siloxane reagent HSi(OEt)3 has been shown to hydrogenate lanthanide complexes (Fig. 6); however, this only occurs from the phenylacetylide bridged dimer [{η5:κ2-N,N-(2,5-{C(Ph)2pz}-{N-(Me)pz})}Yb(μ2-CCPh)]2 (pz = pyrazole). Reaction with the classical Ln–CH2SiMe3 precursor instead yields only the bridged ethoxide analogue [{η5:κ2-N,N-(2,5-{C(Ph)2pz}-{N-(Me)pz})}Yb(μ2-OEt)]2. The different bond dissociation energies of the Ln–Cspvs. Ln–Csp3 ligands and the ability of the dimer to act cooperatively likely promotes an alternate reaction pathway in the former case.72
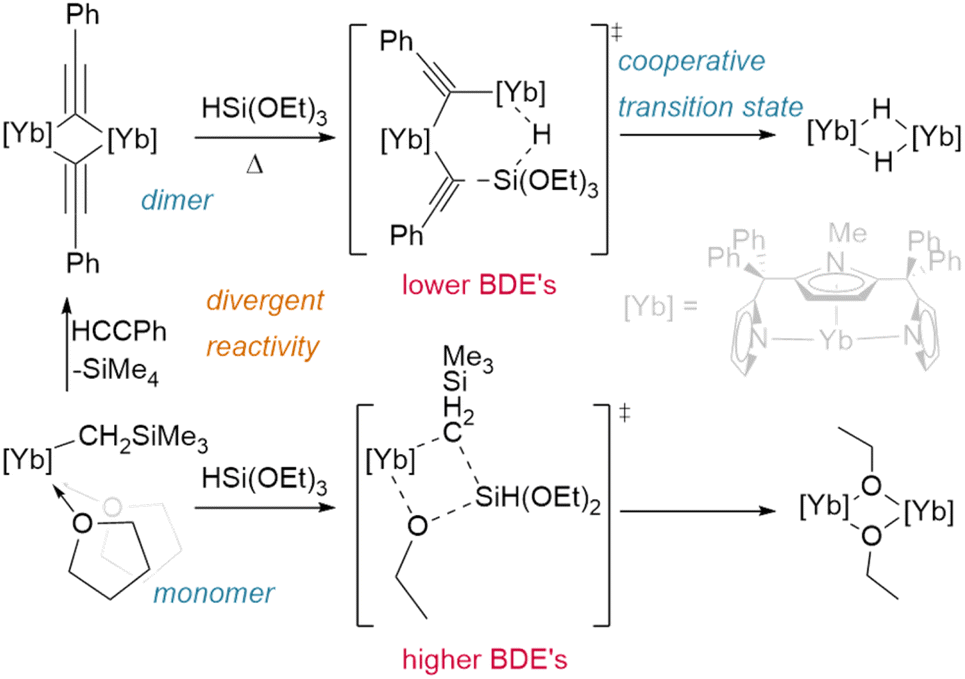 | ||
| Fig. 6 Divergent reactivity was shown between the monomeric ytterbium alkyl and dimeric ytterbium phenylacetylide upon reaction with HSi(OEt)3, with the dimer proposed to react cooperatively to yield the hydride product. | ||
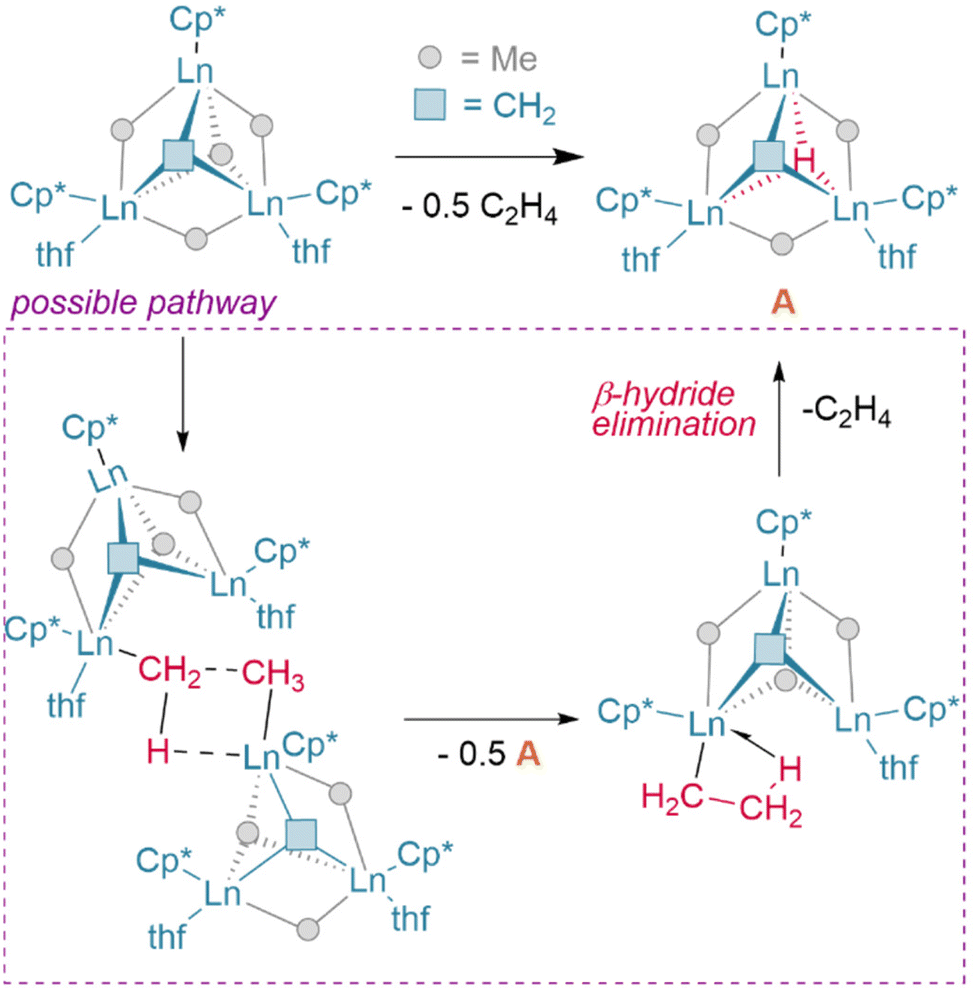 | ||
Fig. 7 Spontaneous C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond formation from a lanthanide alkyl/alkylidene complex may proceed via β-hydride elimination to yield the μ3-hydride product (A). C bond formation from a lanthanide alkyl/alkylidene complex may proceed via β-hydride elimination to yield the μ3-hydride product (A). | ||
:1 molar ratio of the corresponding lutetium(III) hydride and naphthalenide {(NNfc)Lu(thf)}2(C10H7) (Fig. 8b).82 Differential C–H activation behaviour between the lanthanides was also observed with the reduction of lanthanides bound within Meyer's 1,3,5-tris(aryloxo)mesitylene ligand, [(Ad,MeArO)3mes].† In this case, reduction with KC8 in the presence of crypt in thf/C6H6 generated increasing relative yields of a terminal hydride product upon moving from larger to smaller lanthanides, Gd (35%) to Er (45%)—while no hydride was observed under similar conditions for neodymium (Fig. 8c).83 Hydride formation was suggested to arise from activation at the benzylic position of the reduced central arene (vide infra). These data together suggest a key role for reduced arene functions in the formation of Ln–H bonds from Ln(II) complexes, rather than simply via abstraction from solvent.
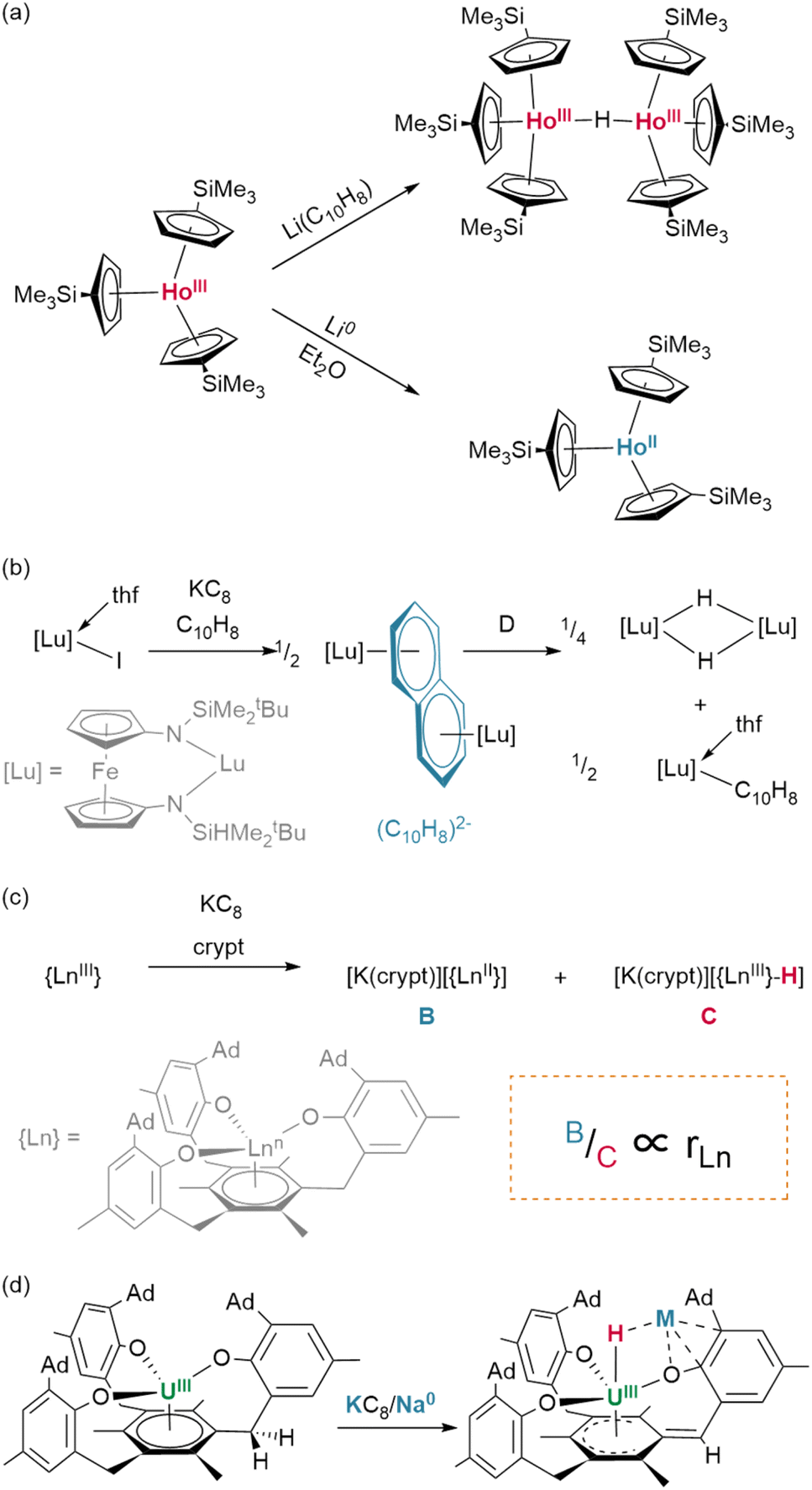 | ||
| Fig. 8 Role of arenes in hydride formation under reducing conditions. (a) Hydride formation is observed via reduction with lithium naphthalenide but not lithium metal. (b) Diaconsecu's rationally prepared lutetium naphthalenide yields the hydride and a lanthanide aryl cleanly upon heating. (c) Meyer demonstrated the importance of ion size with the ratio of Ln(II) product to lanthanide hydride found to be proportional to metal ionic radius. (d) Lanthanide behaviour can be compared to the uranium reactivity in which the ligand activated species was able to be trapped, demonstrating benzylic C–H activation within this scaffold. crypt = 2,2,2-cryptand. | ||
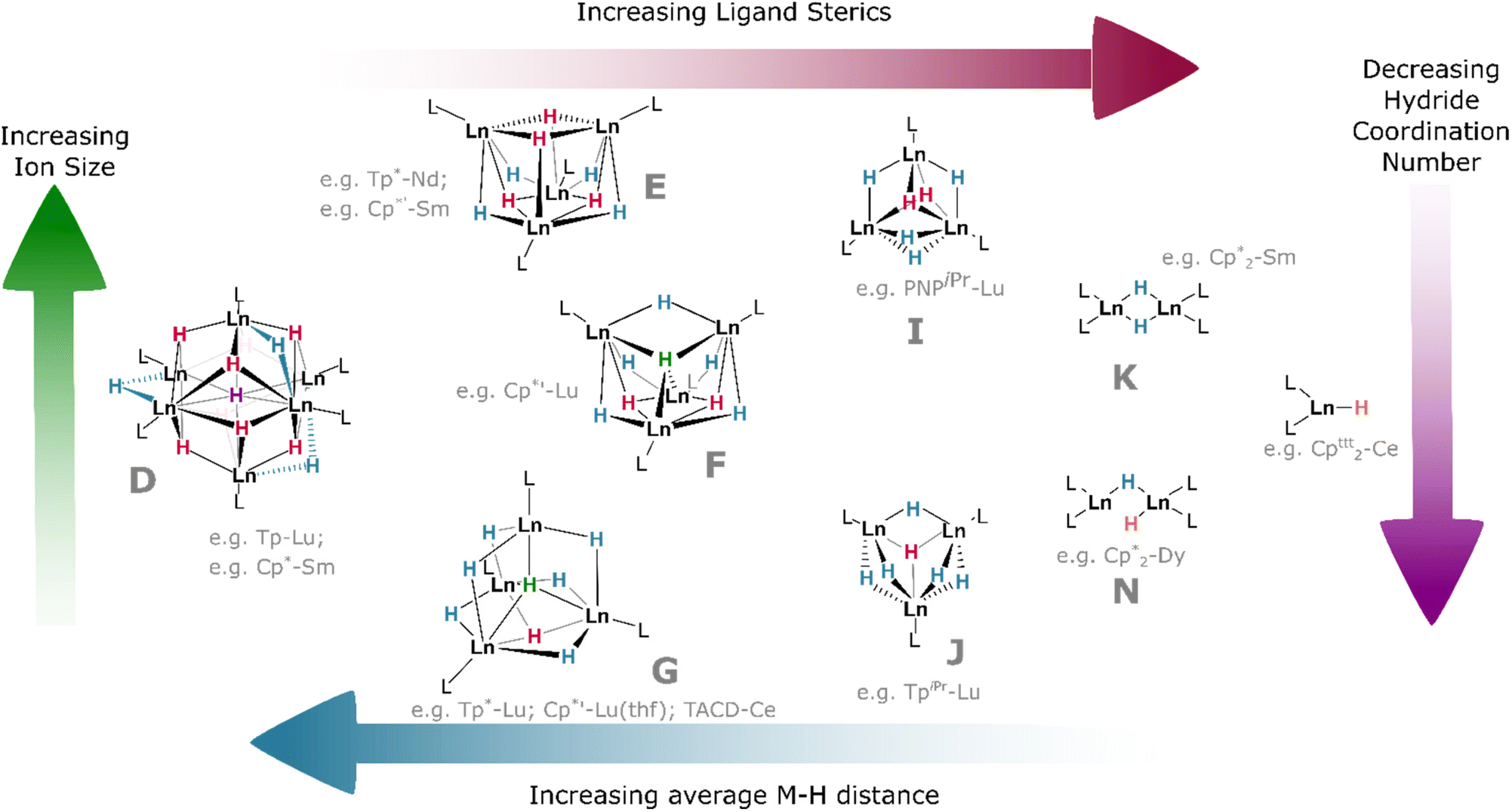 | ||
| Fig. 9 Trends in structures of Ln hydride clusters with examples of L and Ln for each set. Ligand abbreviations: Cp* = C5(CH3)5; Cp′* = C5(CH3)4(SiMe3); Cpttt = C5H2(tBu)3; Tp* = tris(3,5-dimethylpyrazolyl)borane; TpiPr = tris(3-isopropyl-5-methyl)pyrazolyl)borane; PNPiPr = N(o-C6H4P(iPr)2)2; TACD = κ4-N,N,N,N-Me4-1,4,7,10-tetraazadodecane. | ||
With dicationic {XM}2+ fragments, small co-ligands such as X = Cp* and Tp (Tp = tris(pyrazolyl)borane)† can give rise to large hexamers. Few examples exist; however, treatment of Ln(III) complexes (Tp)Yb(CH2SiMe3)2 or (Tp)Lu(CH2SiMe3)2 with H2 yields an octahedron of lanthanide ions as a {Ln6H12} cluster with three μ2 hydrides, eight face-bridging μ3 hydrides, and a single, central μ6 ligand—the highest hydride coordination number observed outside of lithium complexes.86 In contrast, treatment of Sm(II) precursor K[(Cp*)2SmII(CH2SiMe3)] with phenylsilane yields a trigonal prism of oxidised Sm(III) ions, containing a similar central bridging μ6 hydride, which is tricapped with potassium ions, yielding a {Sm6H15} core cluster (Fig. 9D).65
The slightly larger Cp′* ligand (Cp′* = C5Me4(SiMe3))† yields tetramers38 with μ3 or μ4 coordination modes which had been predicted by theory.87 It should be noted that the μ6 and μ4 bridging modes of the hydride ligands within lanthanide clusters mirror the ionic binding of hydride in octahedral (Ho) and tetrahedral (Ht) sites, respectively, in the binary lanthanide hydrides (Fig. 1), with indistinguishable bond lengths in the latter case reinforcing the ionic nature of these bonds. In the tetramers, the lanthanides occupy the vertices of a tetrahedron, and the average coordination number of the eight hydrides increases concomitantly with lanthanide ionic radius; lutetium complexes, for example, have a greater tendency to include μ4 hydrides than the larger lanthanide analogues, with the small {(Cp′*)Lu}4(H)8 complex comprising one μ4, two μ3, and five μ2 hydrides in an overall C2v symmetric central cluster (Fig. 9F).58 Complexes of the larger metals yield clusters which have been crystallised as thf solvates: {(Cp′*)Nd}4(thf)2(H)8 (and earlier lanthanides) are found with four μ3 (2.18–2.34 Å) and four μ2 (2.12–2.39 Å) hydrides, with overall C2 cluster symmetry (Fig. 9E).38 This trend is also reflected with different co-ligands: {(Tp*)Lu}4(H)8 (Tp* = tris(3,5-dimethylpyrazolyl)borane)† incorporates one μ4, one μ3, and six μ2 hydrides, giving a C3v-symmetric cluster (Fig. 9G); while the neodymium example {(Tp*)Nd}4(H)8 contains one μ4 and seven μ2 hydrides in a cluster of C2v symmetry (cf.Fig. 9F).44 That smaller ions tend to construct higher-order bridging interactions is likely due to the contraction of the M–H bonds along the lanthanide series, which declines from 2.40(3) Å/2.26(5) Å (μ3/μ2) in {(Cp′*)Pr}4(thf)2(H)8 (Fig. 9E) to 2.13 Å/2.06 Å/2.16 Å (μ4/μ3/μ2) in {(Cp′*)Lu}4(thf)(H)8 (Fig. 9G); indirectly, this is also observed in the concomitant increase in M–H stretching frequency (1307 to 1323 cm−1) as the series is progressed. Additionally, the average M⋯M distance in these clusters spans the range 3.86–3.45 Å (La–Lu). The larger metal ions (La–Sm) retain two solvent molecules per cluster, falling to one at gadolinium, whilst the lutetium example has been characterised as existing without solvent incorporation.58
As co-ligand size increases, lower-order clusters become more prevalent. Illustratively, with the TpiPr ligand (TpiPr = tris(3-isopropyl-5-methyl)pyrazolyl)borane)† on Lu(III), a trimer is formed with one μ3 hydride and five μ2 hydrides. Bulkier TptBu,Me (TptBu,Me = tris(3-tert-butyl-5-methylpyrazolyl)borane)† imposes a dimeric structure to its lower-valent Yb(II) monohydride complex,45 while the even larger TpAd,Me ligand (TpAd,Me = tris(3-adamantyl-5-methylpyrazolyl)borane)† enforces a monomeric structure.41 The high symmetry of some of these clusters can lead to interesting magnetic properties.88
Within the same ligand class, there are identifiable trends in M–H bonding due to clustering. With hexameric [(Tp)Lu(H)2]6, the average coordination number of the hydrides is 2.8, resulting in a longer M–H bond length of 2.25 Å (2.08–2.48 Å). For the tetrameric analogue with Tp*, the average hydride coordination number falls to 2.4, resulting in a mean M–H length of 2.11 Å (1.87–2.38 Å). Finally, for the trimer resulting from bulky TpiPr, the average hydride coordination number declines further to 2.2, which gives bond lengths of ca. 2.09 Å (1.99–2.25 Å).
In bis(cyclopentadienyl) ‘metallocene’ complexes, lower topology clusters are also favoured, in line with the lower M:H ratio (vide supra). Unsubstituted {(Cp)2La}3(H)3 exists as a distorted trimer with two μ2 ligands and a single μ3 hydride as a result of the large ionic radius of this ion. Whereas Cp′ complexes of Dy and Tb exist as trimers with three μ2 hydrides, Cp*-ligated complexes of the same metals exist as dimers; [(Cp*)2Dy(μ2-H)]2 exhibits one bridging and one terminal hydride, whilst its Tb and Gd analogues exist as C2-symmetric dimers with two μ2 hydrides. With the very large Cp′* ligand, monomeric complexes become isolable across the series: solvated Ce, Dy and Lu examples exhibit bond lengths ranging from 2.27–2.14 Å (Ce–Lu).25,37
In (μ2-H) species, the lanthanide contraction results in a general decrease in the average M–H bond length across the series (2.38(11) to 2.08(11) Å, La–Lu; see Table 1), and, as the bridging M–H bond length increases, so too does cluster order. Conversely, M–H distance increases upon moving from μ2 to μ3 bridging modes, then decreases in μ4 cases—in the latter case, because the tetrahedral ligand draws the cluster tighter.89
| Subset | μ2-M–H (Å) | μ3-M–H (Å) | μ4-M–H (Å) |
|---|---|---|---|
| Dimer | 2.17(15) | — | — |
| Trimer | 2.20(20) | 2.19(13) | — |
| Tetramer | 2.22(18) | 2.36(18) | 2.20(12) |
| Lanthanum | 2.38(11) | 2.56(14) | 2.43(4) |
| Lutetium | 2.08(11) | 2.20(14) | 2.11(5) |
| All Lanthanides | 2.20(17) | 2.29(18) | 2.20(12) |
| All Actinides | 2.25(14) | 2.35(15) | — |
| Compound | IR Band (cm−1) | Assignment | Ref. |
|---|---|---|---|
| Ln–H | 1100–1300 | M–H | 16 |
| [Cp*2LaH]2 | 1120 | M–H (bridging) | 93 |
| [Cp*2LuH]2 | 1345 | M–H (bridging) | 93 |
| [Cp2YH(thf)]2 | 1315 | M–H (bridging) | 94 |
| [CpMe2YH(thf)]2 | 1240 | M–H (bridging) | 94 |
| Cp*2YH(thf) | 1295 | M–H (terminal) | 95 |
| [Cp*YH]2 | 1272 | M–H (bridging) | 96 |
| {Cp′*Lu}4(H)8 | 1304 | M–H (bridging) | 58 |
| {Cp′*Lu}4(H)8(thf) | 1288 | M–H (bridging) | 58 |
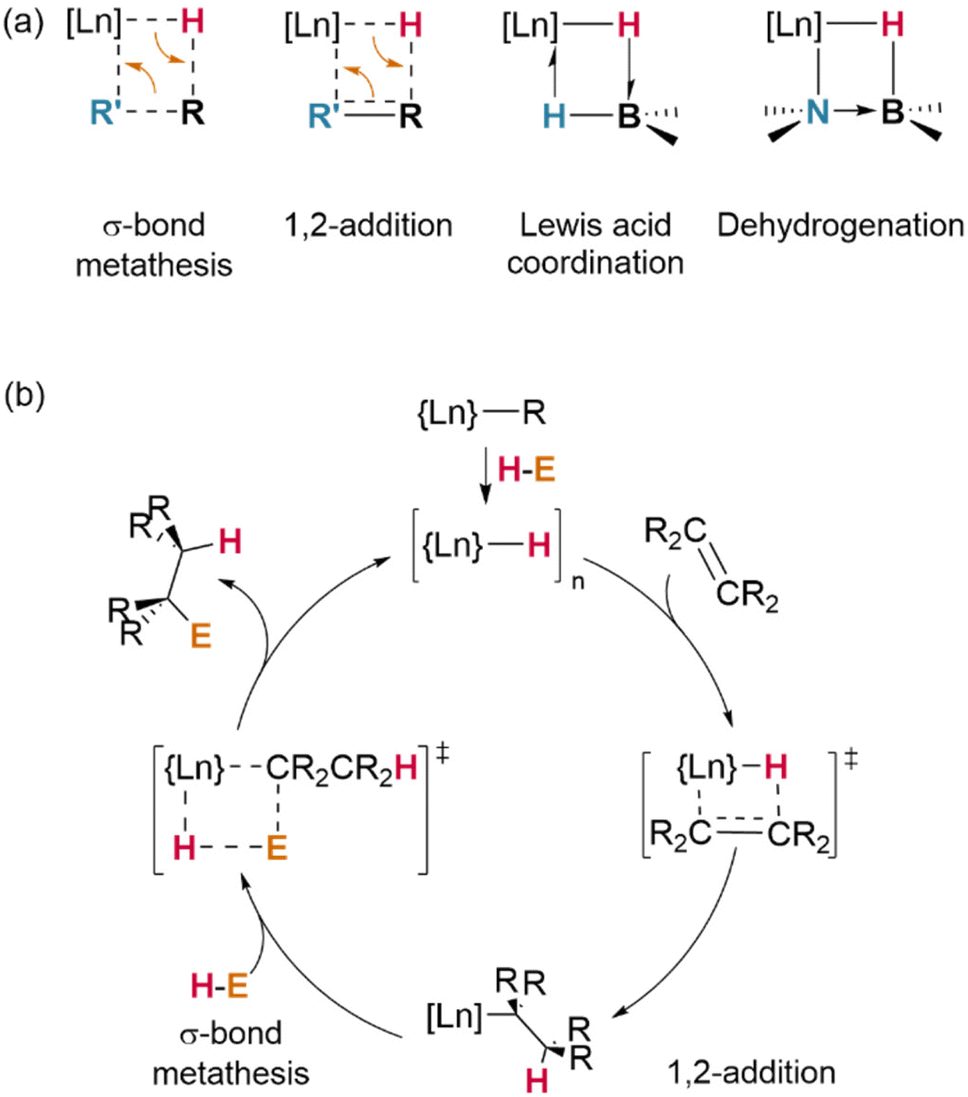 | ||
| Fig. 10 Overview of primary mechanisms of reactivity of lanthanide hydrides. (a) Key transition states for primary reactions types. (b) General catalytic scheme for Lanthanide hydride catalysed hydroelementation processes. | ||
Amines45 (E = NR2) readily react in this way—and so, too, does ammonia itself (E = NH2). However, importantly, the latter was shown to react selectively with the tetrameric cluster {Cp*Ln}4(H)8, yielding a heptaammine product which retains the same cluster topology, then undergoes a slower reaction (ca. 2 days) with the final, central, tetrahedral μ4-hydride; this seems to demonstrate that hydride structure—which may differ from that of a precatalyst—persists in solution, and that its impact on reactivity can be significant.99 Ammonium salts (E = NR3) have also been used similarly to generate cationic lanthanide fragments.42
In another demonstrative case, σ-bond metathesis is mechanistically implicated in catalytic H/D exchange processes which occur between Ln–H centres and certain hydrocarbon substrates (preferentially, those possessed of hydrogens on sp2-hybridised carbons, or those with low steric hindrance).100 Where alternative reaction pathways (insertion/addition; vide infra) are also possible, the strong driving force of H2 loss is a significant factor in favouring σ-bond metathesis; terminal alkynes (E = –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR) most often undergo H–C activation with loss of H2 to yield Ln–CCR complexes, whereas less acidic alkenes undergo insertion across the Ln–H bond.54 However, steric effects are also important in determining the reaction outcome: at a sterically-encumbered Yb centre, the disubstituted allene H2CCCMe2 preferentially reacts by σ-bond metathesis, whilst less bulky H2CCC(H)Cy undergoes 1,2-insertion.41
CR) most often undergo H–C activation with loss of H2 to yield Ln–CCR complexes, whereas less acidic alkenes undergo insertion across the Ln–H bond.54 However, steric effects are also important in determining the reaction outcome: at a sterically-encumbered Yb centre, the disubstituted allene H2CCCMe2 preferentially reacts by σ-bond metathesis, whilst less bulky H2CCC(H)Cy undergoes 1,2-insertion.41
Metathesis of σ-bonds is also the primary process responsible for many ligand activation reactions at lanthanides. As demonstrated by Evans and Furche, in [(Cp*)2Ln(H)]2, the activation of Cp* methyl substituents occurs preferentially in complexes of the early (larger) lanthanides—although, in their experiments, a mixture of species was observed in all cases.28
Whilst dehalogenation of alkanes by lanthanide hydrides has long been known, Andersen and Maron have more recently demonstrated C–F bond metathesis using a bulky Ce(IV) hydride (Fig. 11).29 Likewise, dehydrogenative coupling of terminal alkynes with silanes, mediated by a catalytically-active hydride species, is proposed to occur through σ-bond metathesis.72
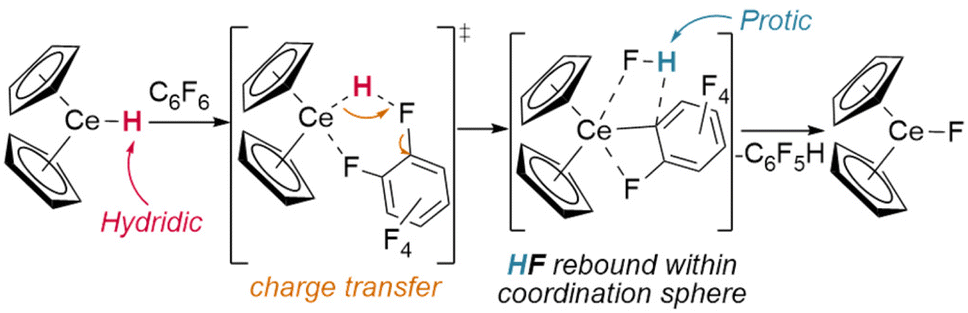 | ||
| Fig. 11 Calculated mechanism of Cpttt2CeH reactions with C6F6 (shown as calculated with Cp (i.e. C5H5) for computational simplicity). | ||
Lacking the driving force of H2 elimination, the likelihood of alkynes undergoing insertion reactivity over σ-bond metathesis is subject to a fine balance of effects. It has been shown that bis(trimethylsilyl)acetylene undergoes Si–C bond metathesis with [(TptBu,Me)YbIIH]2, yielding the dimer [(TptBu,Me)YbII(μ2-η1:η2-CCSiMe3)]2 (Fig. 12)—but bis(trimethylsilyl)but-1,3-diyne instead undergoes an addition reaction to yield a monomeric, η3-bound product, (TptBu,Me)YbII(η3-Me3SiCC-CC(H)SiMe3).45
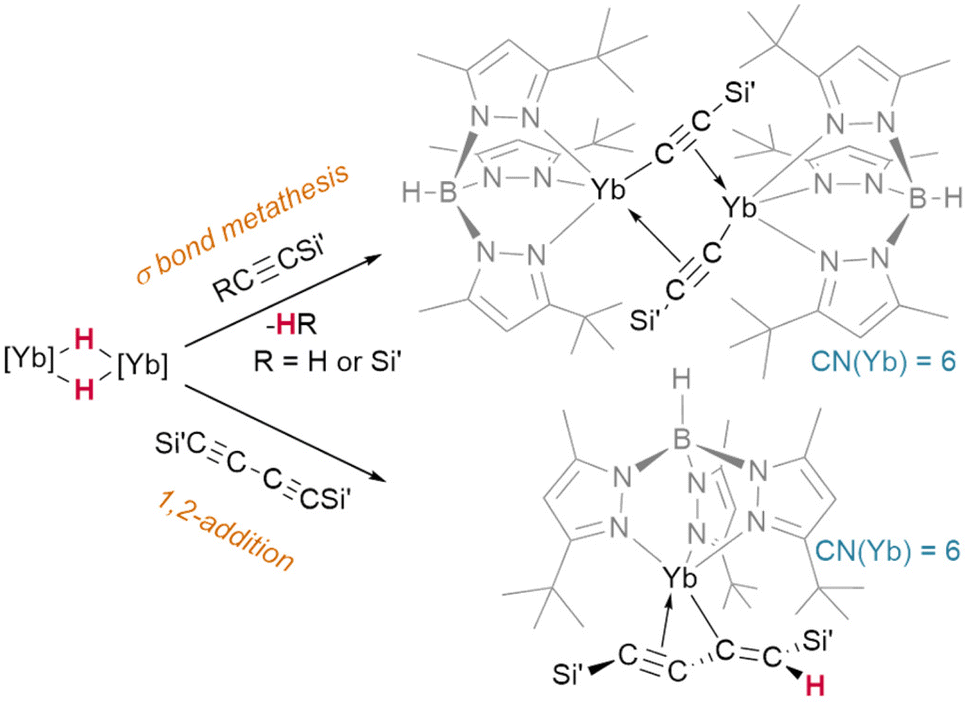 | ||
| Fig. 12 Divergent pathways for reactions of ytterbium hydrides with silyl acetylenes ([Yb] = TptBu,MeYbII) may be driven by maintaining a high coordination number at the metal. | ||
Hill and Anker demonstrated that, after olefin insertion into the hydride of [(NacNaciPr)YbIIH]2 (NacNaciPr = N,N′-bis(diisopropylphenyl)-β-pentanediimide), it is activation of the benzene-d6 solvent which subsequently regenerates the hydride (deuteride) and affords the resultant hydroarylation product (Fig. 13).63 This reactivity contrasts with classical Ln(III) alkyl activation of benzene, which results in the formation of lanthanide aryl species—and may proceed through a cooperative transition state similar to that proposed by Zhu.72
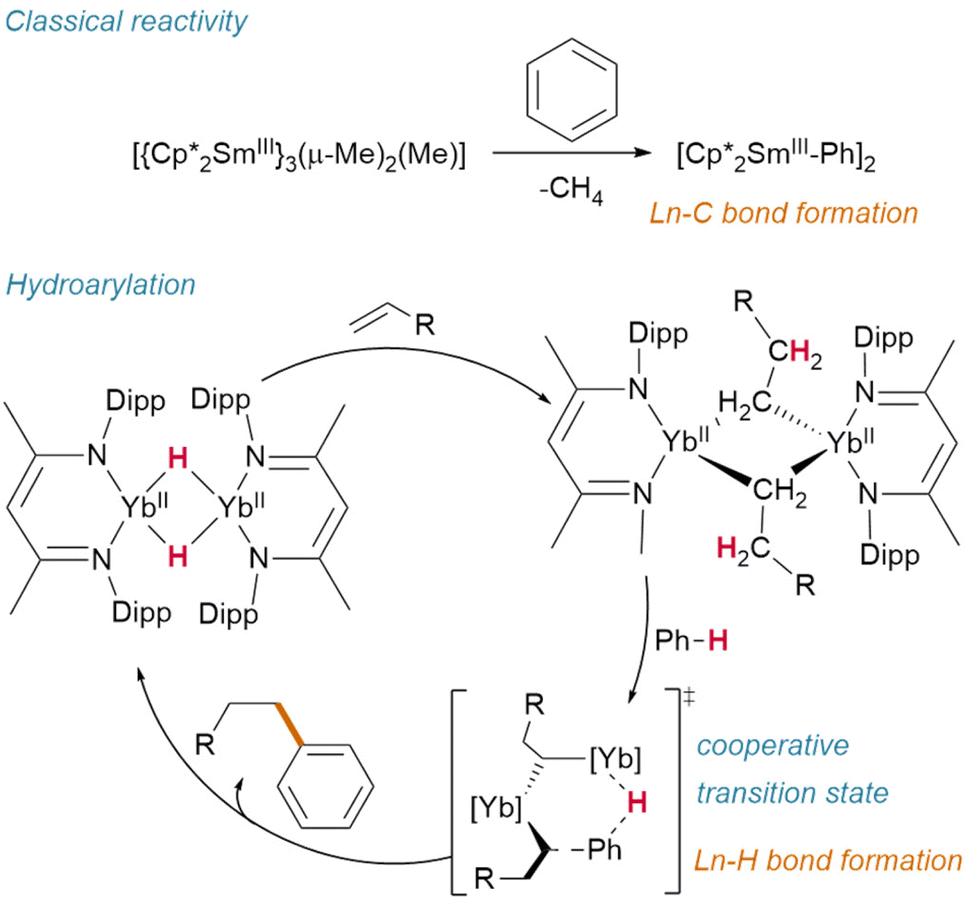 | ||
| Fig. 13 Hydroarylation catalysed by an ytterbium(II) hydride complex contrasts with classical sigma bond metathesis reactivity observed for Sm(III). | ||
Reduction of the ansa-cyclopentadienyl ligated dimer {(1η5,1η5-CpAn)DyIII(thf)}2(μ2-H)2 (CpAn = (Cp′)2SiMe2) resulted in an unusual ligand rearrangement: in its new, desolvated form, [K(18-crown-6)][{1η5:2η5-CpAn}2DyIII/II2(μ2-H)2], the supporting ligands bridge between the two metal centres—although it retains the pre-existing {Dy2H2} core (Fig. 14a).106
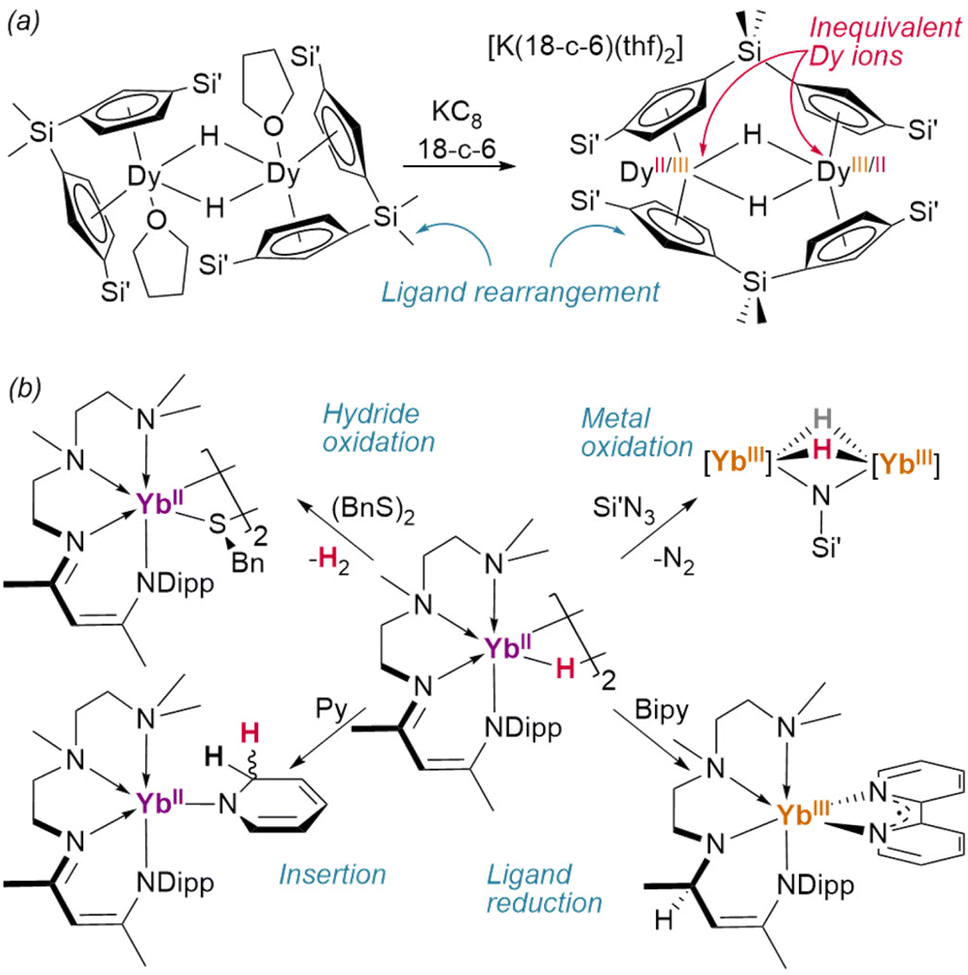 | ||
| Fig. 14 Redox reactions of lanthanide hydrides. (a) Single electron reduction of the ansa-metallocene ligated dysprosium hydride yields a ligand rearrangement product with unsymmetrical Dy charge density and a calculated Dy⋯Dy ground state interaction. Si′ = SiMe3; 18-c-6 = 18-crown-6. (b) Different redox reactivity of a YbII hydride complex. [Yb] = {κ4-TMEDA-NacNacDipp}Yb; Dipp = 2,6-diisopropylphenyl; Bipy = 2,2′-bipyridine; Py = pyridine. | ||
Intriguingly, low-valent Yb(II) dihydride [{κ4-TMEDA-NacNacDipp}YbII(μ2-H)]2 has been shown to reduce azidotrimethylsilane without perturbing the hydrides, affording a dihydrido–imido Yb(III) complex (Fig. 14b).107 In this case, further reaction with Se(0) dehydrogenated the complex entirely—while with (BnS)2 direct dehydrogentation of the hydride was observed to yield the Yb(III) product.55 This complex was also shown to reduce 2,2-bipyridine, during which reaction hydride transfer to the NacNac backbone occurs. A TpAd,iPr complex of the same ion was also shown to reduce cyclooctatetraene to its dianion, yielding the inverse sandwich product {(TpAd,iPr)YbIII}2(COT) (COT = cyclooctatetraene).
Lewis acid coordinated lanthanide hydrides
In addition to homometallic clusters of lanthanide hydrides, a wealth of complexes of the form Ln⋯H–E or Ln⋯H–M have been reported, ranging from silane complexes containing β-agostic interactions109,110 (and potentially α-agostic interaction111) to complexes of the BH4 anion112 and those of heavier main group and transition metals (Fig. 15).113,114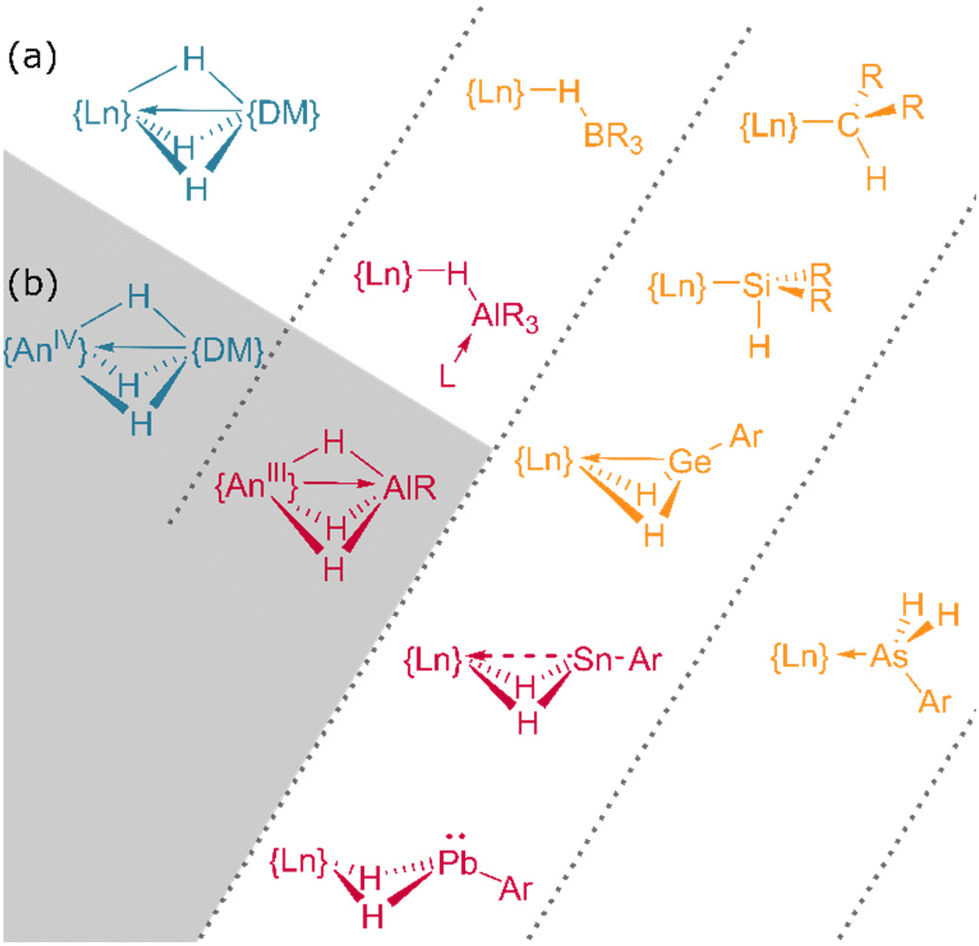 | ||
| Fig. 15 The bonding interactions of heterobimetallic f-block hydrides. (a) Lanthanide complexes. (b) Actinide complexes. DM = d-block metal. Further examples can be found in ref. 109 and 123. | ||
Borohydrides108
Complexes of lanthanides with tetrahydroborate ligands—both Ln(II) and Ln(III)—are myriad in the literature,112 owing in part to the utility of the [BH4]− anion to act as a pseudohalide in salt metathesis transformations.115 This borohydride ligand can also adopt multiple coordination modes (terminal, κ2, and κ3), flexibly enabling the synthesis of complexes spanning the lanthanide series. Unlike conventional halides, borohydrides conveniently contain two NMR-active nuclei, discernible IR bands, and are also able to react through protonolysis routes—making these ligands both synthetically and analytically valuable. Borohydride derivatives such as [HBR3],108 [HB(C6F6)3],116 and tris(pyrazolyl)borane117 are also able to engage in M⋯H–B bridging interactions, stabilising their complexes. The bonding in these complexes is non-intuitive and has been studied in depth concluding that for early transition metals bonding is mostly ionic with some electronic effects modulating the orientation of bridging hydrogens.118,119Main group hydrides
In contrast to tetrahydroborato ligands ([BH4]−), tetrahydroaluminato ligands ([AlH4]−) demonstrate a prevalence to bind Lewis bases at the Al(III), generating a 5-coordinate centre upon coordination to a lanthanide and highlighting the ionic nature of the hydride bonding (Fig. 16).120 In 2013, Anwander et al. sought to access homometallic hydrides by treatment of bulky TptBu,MeLuMe2 with two equivalents of HAlMe2.113 Instead, they isolated the ‘masked monomeric dihydride’ insertion product TptBu,MeLu(HAlMe2)2, with a bridging Lu⋯H–Al interaction (Lu–H = 2.04(3), 2.09(2) Å) well within the range for homobimetallic TpLn⋯H–Ln interactions 1.87–2.09 Å.113 Similarly, Farnaby and Weetman recently demonstrated the insertion of trihydroaluminium across a {Tp2Ln-N′′} bond, yielding a bridged hydride, Tp2Ln(μ2-H)2Al(H)N′′, with rapidly-exchanging bridging and terminal hydrides.121 The Anwander group have gone on to expand the scope of Ln⋯H–E complexes, providing examples with E = Ge, Sn and Pb.114 In NBO calculations based on their data, they found increasing importance of a direct Ln–E interaction upon moving from Pb to Ge, while the Ln–H bond concurrently lengthens. The bonding, therefore, may be intermediate between that of group 13 and that of group 15 complex [Cp3Dy-As(H)2Mes], which contains only a Dy–As bond (Fig. 15).109,122,123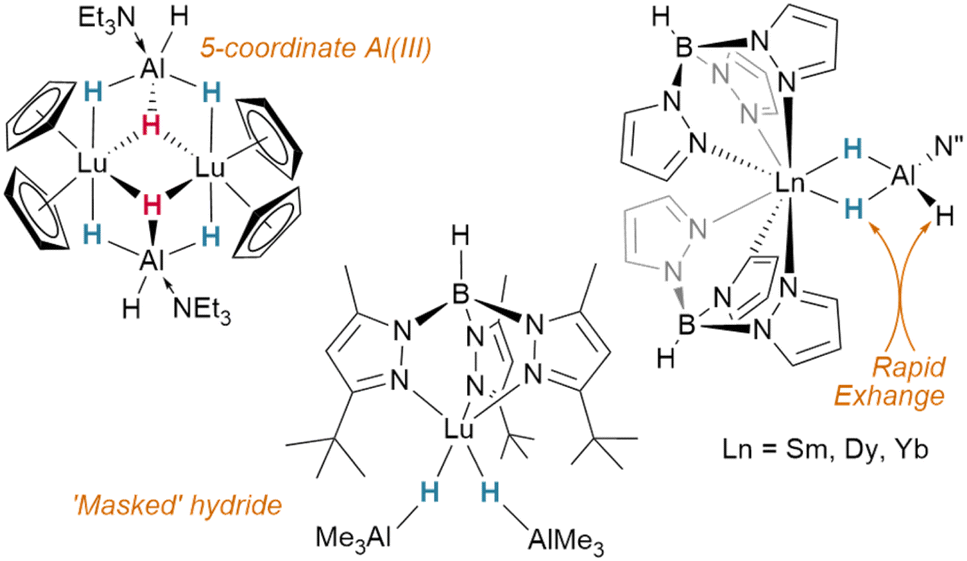 | ||
| Fig. 16 Examples of aluminohydride complexes of lanthanides. | ||
Transition metal hydrides
Heterobimetallic complexes with transition metal hydrides have also attracted interest due to the excellent hydrogen storage properties of materials such as LaNi5Hx. Complexes of lanthanide hydrides with Mo, W, Re, Ru, Os, and Ir are all known and these have been synthesised by a range of routes including alkane elimination,124 salt elimination,125 CH activation126 and H2 elimination.37 Transition metals can lend the lanthanide clusters redox activity which would otherwise be inaccessible, enabling facile C–H bond activation in Cp, PPh3 and PR3 co-ligands.126–130 These oxidative addition-like C–H activation steps provide both bridging organometallic and hydride ligands, which buttress direct Ln–TM bonds.128 In Ln/Mo clusters, H2 addition was even observed by single-crystal X-ray diffraction.131Homoleptic actinide hydrides
The use of the actinides in synthetic chemistry is distinguished from that of the lanthanides in two key respects. First and foremost, the radioactivity of many actinides—particularly the transuranic elements—makes necessary numerous additional precautions and, in the latter case, their handling remains unfeasible in all but the most specialist of laboratory environments; the natural consequence is that markedly less work has been conducted in this area. It is the second respect which makes the effort worthwhile: although their bonding interactions are predominantly electrostatic, the early actinides—especially uranium—are softer, more polarizable metals, for which the valence 5f and 6d orbitals possess sufficient radial extension to impart some degree of covalent character to An–E bonds. As a result, the chemistry of these elements diverges conspicuously from that of the lanthanides—moreover, exhibiting a unique combination of behaviours to which no other region of the periodic table can entirely attest.132Observations from gas phase and matrix isolation experiments
Early actinide hydrides, AnHx (An = Th, U; x = 1–4), have, like their lanthanide analogues, been observed by pulsed laser ablation studies in argon. Under these conditions, where x = 1–3, IR spectroscopic measurements indicate that Th–H bonds are somewhat stronger than their uranium analogues—although those of UH4 complexes are stronger still.133,134 Dimeric, hydride-bridged {An2H2} species are also known for uranium, whereas isostructural thorium analogues have not been observed; calculations imply that a dithorium-bonded {HThThH} formulation may be more favourable in this case.135 Thorium(IV) dihydrogen complexes are also stabilised due to the highly ionic nature of the bonding in ThH4 generating polarized H2 complexes which exhibit Crabtree's “dihydrogen bond”-type interactions. This stands in contrast to the uranium example, which has more 5f orbital involvement in bonding. Dihydrogen complexes have also been predicted by these methods and observed in the gas phase and recently in solution (vide infra).136,137
Synthesis, isolation and reactivity
Although straightforwardly preparable as a black powder by high-temperature exposure of metallic uranium turnings to H2 atmosphere (1 bar, up to 250 °C), uranium trihydride (UH3) remains a relatively uncommon starting material for the synthesis of molecular uranium complexes.138–140 The metal initially reacts to generate the metastable α-UH3 (orthorhombic) form, which is then transformed into stable β-UH3 (cubic) at higher temperatures. Somewhat curiously, UH3 formation is reversible at higher temperatures—a feature which has led actinide materials to be considered for the production and storage of ultrapure hydrogen.141Based on its purportedly superior reactivity, some early reports describe the preferential use of UH3 over metallic uranium in synthesis;140 it is presumably its characterisation as intensely pyrophoric which has rather limited the frequency of its applications in the intervening time.138 That UH3 can exhibit this particular behaviour is not in doubt; however, the mechanism of its pyrophoricity is not straightforward. By exposure of UH3 to dry air at elevated temperatures, it has been demonstrated that ignition occurs initially in the gas phase above the metal hydride, consuming H2 produced in the ensuing reaction and generating sufficient heat to ignite the solid material.142 Exposure of UH3 to anoxic water vapour likewise induces oxidation to uranium oxides; however, even at 100 °C, the rate of conversion slows considerably after ca. 80% consumption of UH3, and temperatures of up to 500 °C are required to achieve complete conversion to U3O8.10 A particularly curious complication arises from the work of Hayton and colleagues, whose rationally-synthesised UH3 powder exhibits essentially no reactivity towards atmospheric gases, and reacts even with aqueous 1 M H2SO4 on the timescale of hours.143 Such sluggishness has also been apparent in our own investigations of similarly-prepared samples of UH3; we are aware of no other precedent for this potent display of lethargy from so reportedly aggressive a reagent. It is plausible that its behaviour depends strongly on its purity and crystalline phase in the solid state; although passivating oxide films have previously been implicated, the preparation of UH3 as a fine, black powder would necessitate the oxidation of a very considerable surface area, with corresponding impacts on subsequent reaction products and yields, to completely arrest its reactivity. The isolated binary hydride does, of course, undergo reactions with mild oxidants, and under mild reaction conditions yields an array of valuable uranium synthons—including UBr3(dme)2, UX4(dme)2 (X = Cl, Br, OTf; dme = 1,2-dimethoxyethane), and UI4(OEt2)2.143,144
Whilst PaH3 is known, with forms isostructural to α- and β-UH3, the formulations of other actinide hydrides are broadly different. Two key forms of thorium hydride are known: ThH2 and Th4H15 (with an H:Th ratio of ca. 3.75:1). The former is obtained by hydrogenation at 200–350 °C, whilst increased temperatures and H2 pressures instead yield the higher hydride. Crystalline ThH2 occurs in the I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) space group, wherein the thorium centres are found in an 8-coordinate, square antiprismatic arrangement (Fig. 17). Th4H15 occurs in space group I3d, in which hydrides adopt a 12-coordinate, cuboctahedral arrangement about each thorium centre. The typical hydrides of the transuranic actinides are AnH2 formulations, adopting the same fluorite (CaF2) structure as their lanthanide congeners, or hexagonal AnH3 structures.141
space group, wherein the thorium centres are found in an 8-coordinate, square antiprismatic arrangement (Fig. 17). Th4H15 occurs in space group I3d, in which hydrides adopt a 12-coordinate, cuboctahedral arrangement about each thorium centre. The typical hydrides of the transuranic actinides are AnH2 formulations, adopting the same fluorite (CaF2) structure as their lanthanide congeners, or hexagonal AnH3 structures.141
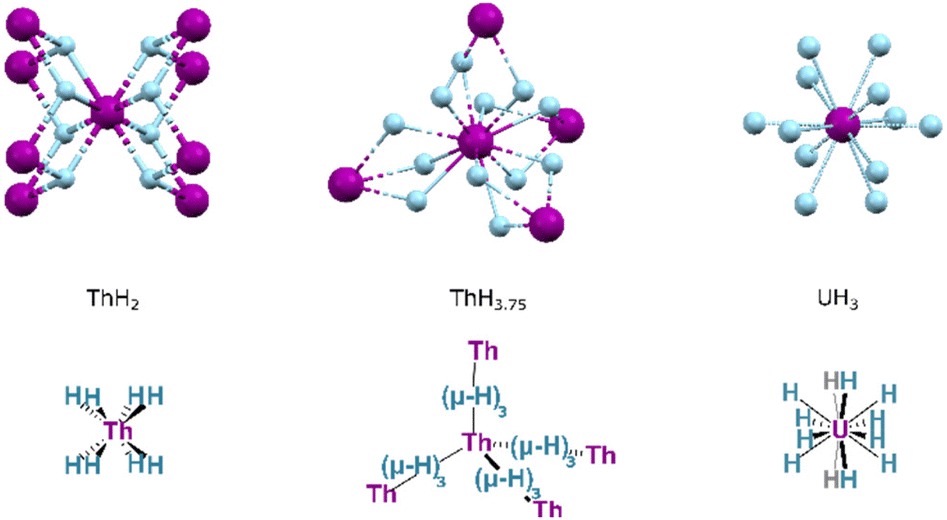 | ||
| Fig. 17 The local structure of key binary actinide hydrides ThH2, Th4H15 and UH3 illustrating the coordination geometry of the metal in each. Purple Spheres: An ions; Blue Spheres, Hydrides. | ||
Heteroleptic actinide hydrides
A significant portion of the work with heteroleptic actinide hydrides was undertaken as early as the 1980s, and it is conspicuous even amongst other areas of f-block chemistry that the field has developed slowly in the intervening years: only 42 such crystallographically-characterised examples are present in the CSD. Some interest in this area arises from the relatively unusual electronic structures of actinide complexes; in particular, bonding in U, Np and Pu complexes is understood to have the potential to involve some degree of covalency, albeit predominated by ionic character.145 This raises questions about the nature of An–H bonding, and offers opportunities to consider complexes of these elements in comparison to other actinide elements, as well as to the lanthanides.145Synthetic routes
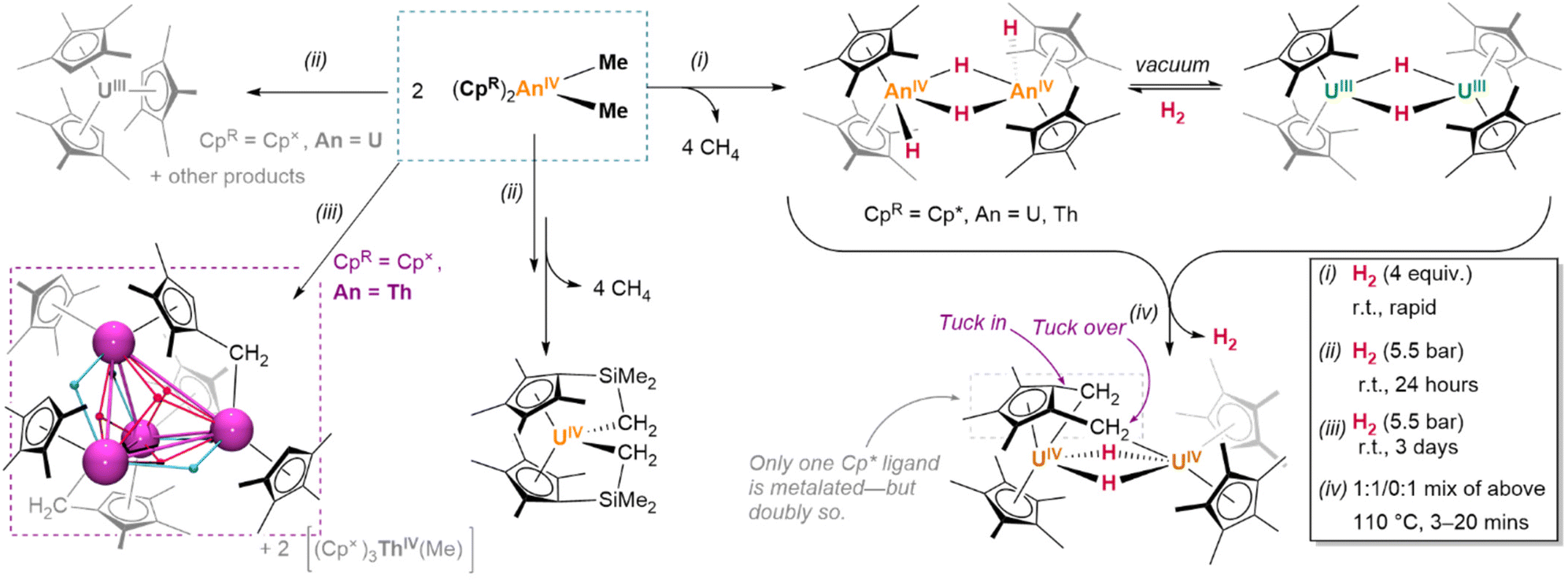 | ||
| Fig. 18 Hydrogenolysis of actinide alkyl complexes: thorium complexes tend to form stable hydrides while uranium complexes are able to undergo redox processes which complicate their reactivity. Ligand metalation of the Cp′* ligands could arise via similar hydride intermediates observed for the Cp* analogue. Actinide ions: Purple spheres; Bridging hydrides: Blue and red spheres. | ||
Despite these considerations (or perhaps because of them), uranium hydrides are frequently accessible under milder hydrogenation conditions than lanthanides (ca. 1 bar H2, −40 °C to ambient temperature). For example, diuranium(IV) siloxide complex K2[{(SiO†O)3U}2(μ-O)(μ-H)2] (SiO†O = OSi(OtBu)3), based on a ligand which is not known to metalate, is formed from an oxo-bridged diuranium(III) precursor upon treatment with H2 (1 bar, r.t.).150
With less accessible redox activity, preparations of thorium hydrides often more closely resemble those of lanthanide congeners; syntheses are somewhat more affected by steric and electronic differences in the coordination sphere of the alkyl precursor complex than the feasibility of ligand cyclometalation. The preparation of a range of thorium hydride clusters, [(CpR)Th(μ2-H)3]n (n = 5–13, vide infra), from thorium monocyclopentadienyl trialkyl precursors, necessitates higher pressures of H2 (10–60 bar), elevated temperatures (up to 60 °C), and extended reaction times (12–96 h).151 Likewise, thorium trialkyl complexes bearing a single, extremely hindered pentaarylcyclopentadienyl ligand undergo a combined protonolysis and hydrogenolysis upon treatment with HCp* or HCp× (Cp× = C5Me4H) and H2, affording the corresponding (CpAr)(CpR)Th(H)2(thf) (CpAr = C5(3,5-tBu2C6H3)5, CpR = Cp* or Cp×) species—although reactions are reported only at relatively high pressures of H2 (10–20 bar).152 Only some preformed thorium bis(cyclopentadienyl) complexes are known to react under much lower H2 pressures; conspicuously, however, this occurs only with supporting ligand systems susceptible to metalation. Representatively, the thorium Cp× complex (Cp×)2Th(Me)2 yields a stable tetranuclear octahydride upon exposure to hydrogen gas, in which two of the metallocene methyl substituents are metalated.148 Considering that greater steric occlusion of the actinide centre in uranium bis(cyclopentadienyl) complexes appears to favour metalation, facilitating facile hydrogenation, the same effect is likely also to be operative here. Conversely, increasing the number of alkyl ligands appears to impede conversion to the hydride; as observed in lanthanide cases, this may be due to the formation of stable, highly-bridged hydride/alkyl intermediates.
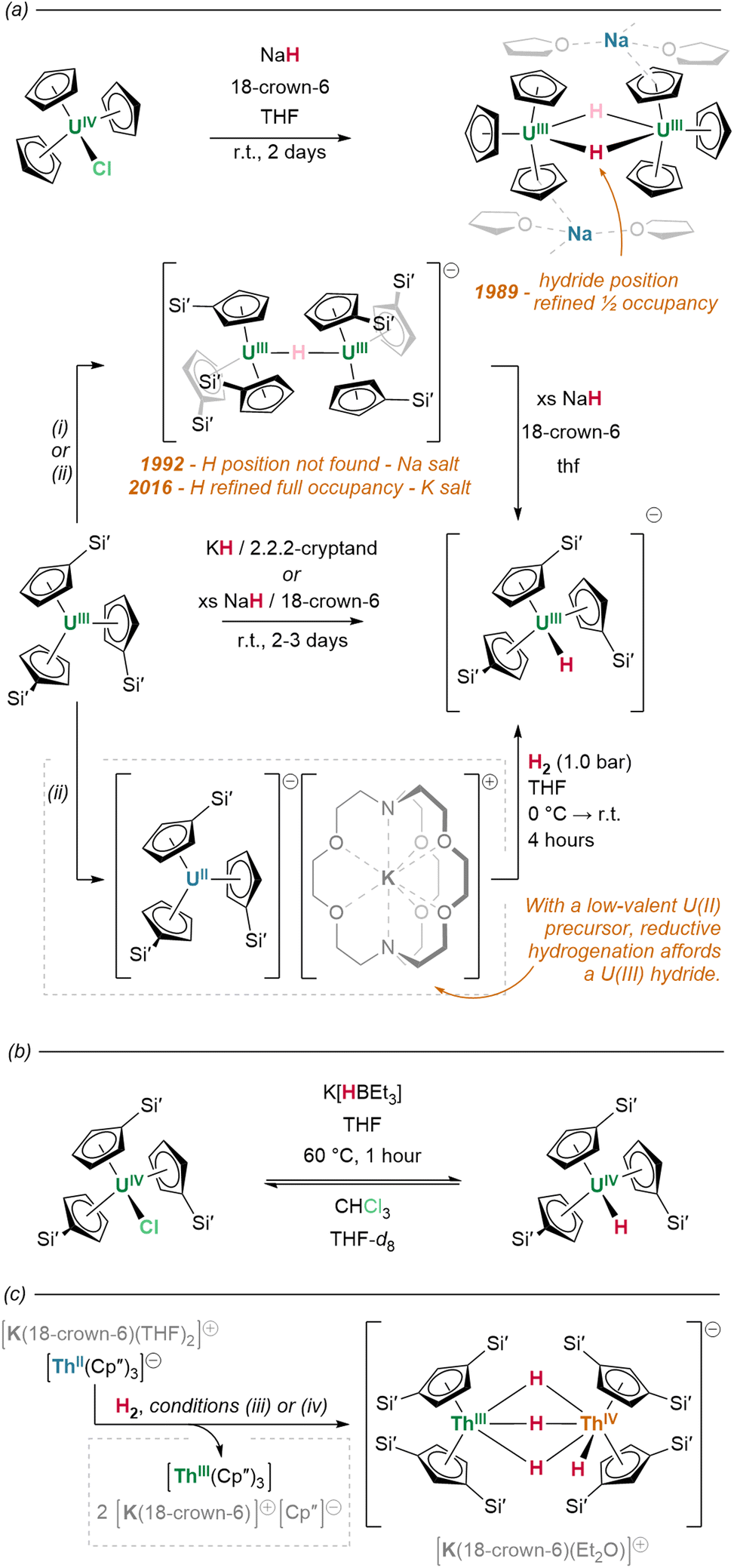 | ||
| Fig. 19 Synthesis of hydrides via reactions with hydride transfer agents or via low-valent actinide species show similarities in their behaviour. (a) In 1989, salt metathesis of Cp3UCl with NaH yielded a mono-hydride bridged dimer which was originally assigned as a crystallographically disordered species with bent hydride bridges exhibiting M–H–M angles around 160°. In 1992, treatment of UIIICp′3 with NaH yielded a dimer with a proposed linear M–H–M angle; however, no hydride was found. In 2016, Evans et al. reported the K analogue and were able to refine the H position in the linear bound site. (i) NaH, 18-crown-6, thf, 2 days; (ii) KC8 18-crown-6, Et2O, −35 °C (iii) H2 (4.1 bar), 18 h, solvent free; (iv) H2 (1 bar), 20 min, thf, 0 °C – r.t. | ||
Alternatively, but in similar fashion to lanthanide examples, the exchange of An–C for An–H linkages is also achievable by actinide complexes through the σ-bond metathesis of silanes. Complementarily to the original, hydrogenative preparations of [(Cp*)2An(H)2]2 (An = U, Th), Kiplinger and colleagues report that treatment of (Cp*)2An(Me)2 with PhSiH3 (toluene, 50 °C, 30 minutes) effects the near-quantitative conversion of these alkyl complexes to the respective hydrides.162 Reactions with K(HBEt3) salts may also generate hydrides with concomitant loss of K(RBEt3): treatment of tris(cyclopentadienyl) halide precursor (Cp′)3UIVCl with K(HBEt3) effects the abstraction and exchange of chloride for hydride, yielding (Cp′)3UIV(H) (Fig. 19b).157
What solid state structural data does exist suggests a marked change in behaviour compared to the lighter elements of the f-block. In Marks’ 1979 report of the first crystallographically-characterised example of a molecular actinide hydride, neutron diffraction identified [(Cp*)2Th(μ2-H)(H)]2 as a bimetallic tetrahydride complex, wherein the metal centres each possess one terminal hydride ligand, and are bridged between themselves by the two remaining μ2 hydrides. The Th–H contacts in the complex differ significantly between coordination modes, with terminal linkages the shorter of the two configurations (terminal: 2.03(1); bridging: 2.29(3) Å).165 In solution, however, these hydrides exchange readily between modes at room temperature, as evidenced by the indistinguishability of their 1H resonances (19.25 ppm) on the NMR timescale.146 Although this resonance occurs significantly downfield of those associated with typical, diamagnetic late transition metal hydrides, deshielding in complexes of large d0 ions is known to produce a similar effect.166
Similar, dinuclear structures, [(L)2An(H)2]2, occur in a significant proportion of the thorium-containing examples reported to date,146,147,163,165,167–169 although mononuclear complexes are more common,152,161,163,169–172 and clusters containing three or more hydride-bridged thorium centres are also represented to varying but lesser degrees.151,173,174 Amongst uranium examples, dinuclear species predominate,146,147,149,150,153–155,158,159,168 but a sizeable number of mononuclear complexes are also known.159,160,172,175–178 Notably, until this year, no higher-order uranium hydride clusters had been reported: Zhu, Maron and colleagues’ tetranuclear, ansa-bis(cyclopentadienyl)-supported octahydride cluster {(CpCMe2CMe2Cp)UIV}4(μ2-H)4(μ3-H)4 represents only the first such example. Significantly, X-ray diffraction reveals a coplanar arrangement of its uranium centres and conspicuously short U–U distances (3.65–3.70 Å) which approach the sum of single-bond covalent radii for a U–U interaction; the intriguing possibility that this indicates electron delocalisation between the uranium centres themselves is somewhat supported by NBO analyses and the calculation of a U–U Wiberg bond index of 0.39. NBO analyses further characterise U–H bonding modes as delocalised, 3c–2e or 4c–2e bonds which are strongly polarised towards H.179
For thorium, complex morphologies depend strongly upon the steric influence of supporting ligand systems—although the differences observed result no less from stoichiometry than the identity of the chosen ligand. With one additional Cp* ligand, the mononuclear, tris(cyclopentadienyl) analogue of [(Cp*)2Th(H)2]2, (Cp*)3Th(H), possesses a single, conspicuously unreactive, terminal hydride; its Th–H contact (2.33(13) Å) compares well with that of its prior congener, although the 1H resonance of its singular hydride (15.4 ppm) appears somewhat further upfield.169 With one fewer ligand per thorium centre, [(Cp*)Th(H)3]7 exists as a heptanuclear thorium(IV) cluster with all eight hydrides coalescing into a single 1H NMR shift at 16.44 ppm.151
Uranium examples exhibit marginally less steric strain, differing from those above by oxidation state and cluster order. The direct analogues comprise dinuclear species [(Cp*)2UIII(μ2-H)]2 and [(Cp*)2UIV(H)2]2;147 of further note is the “tuck-in tuck-over” bimetallic complex {(Cp*)2U}(μ2-H)2{U(Cp*)(C5Me3{CH2}{μ2-CH2})}, in which two of the methyl substituents of a single pentamethylcyclopentadienyl ligand are metalated and in contact with different uranium centres.149
The hydride positions in these latter examples have never been determined crystallographically, and without neutron diffraction data, further structural analysis is only intermittently possible. In a related uranium(III) diphosphine complex, (Cp*)2UIII(H)(dmpe) (dmpe = 1,2-(dimethylphosphino)ethane), its hydride position was deduced from geometric considerations;177 a reported thorium(IV) congener, [(Cp*)4ThIV2(H)2(dmpe)][BPh4]2, is proposed to adopt a bimetallic configuration but has not been crystallographically characterised.169 Despite the crystallographic identification of (N′′)3UIV(H), the position of its hydride ligand could not be refined; its presence can only be inferred from the tetravalent oxidation state of the metal.172
Considered modifications made to the steric profiles of ligands may represent a route to more subtly influence complex structures (Fig. 20). With only the absence of one ligand methyl substituent, mononuclear (Cp×)3ThIV(H) remains a close comparator of (Cp*)3ThIV(H);163 in the monoligated mode, however, the corresponding cluster is octanuclear [(Cp×)ThIV(μ2-H)3]8. Conversely, exchanging a methyl for a trimethylsilyl substituent affords hexanuclear [(Cp′*)ThIV(μ2-H)3]6. Stepping beyond minor alterations, two even more sterically substantial congeners, [(CpR)ThIV(μ2-H)3]5 (CpR = Cpttt, Cp′′′; Cpttt = 1,2,4-(tBu)3C5H2; Cp′′′ = 1,2,4-(SiMe3)3C5H2), crystallise instead as a pentamer. These thorium clusters differ from comparable lanthanide examples in that only μ2- and μ3-bridging hydrides have yet been identified in crystallographically-authenticated structures; however, it remains the case that average hydride coordination number increases with cluster order. Between thorium examples, morphologies differ dramatically: while [(Cp×)Th(H)3]8 adopts a distorted square-antiprismatic arrangement of its eight thorium centres, [(Cp*)ThIV(H)3]7 is a distorted pentagonal bipyramid, [(Cp′′′)ThIV(H)3]6 approximates an octahedron, and [(Cpttt)ThIV(μ2-H)3]5 a square-based pyramid. In [(Cp′)12Th13H40], although hydride positions could not be refined, a tetrahedron of {(Cp′)Th}3 units encapsulates a single thorium centre.151
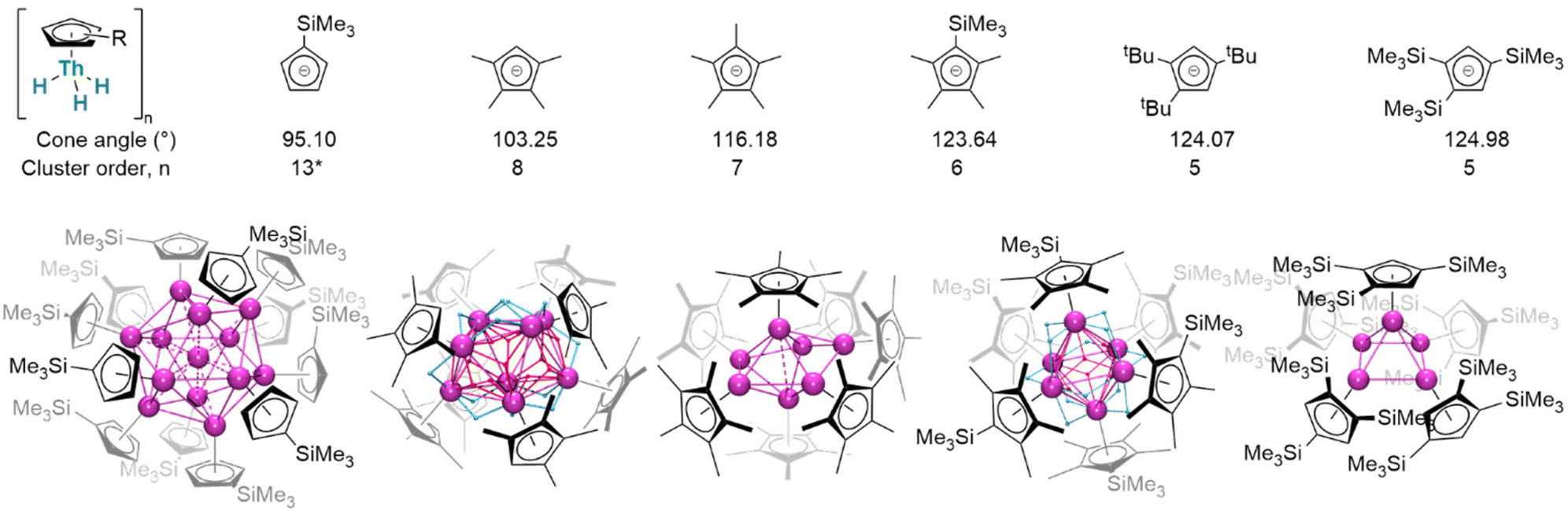 | ||
| Fig. 20 Cluster order decreases with increasing with cyclopentadienyl ligand cone angle, measured from solid state structures. * The Cp′ ligand results in a complex cluster of the form {(Cp′Th)3(H)10}4Th in which the position of the hydrides in unknown. Despite its non-standard nature it is clear than this ligand can, and does, facilitate for formation of higher-order clusters than its smaller analogues. This cluster is also reminiscent of the structure of binary hydride Th4H15. | ||
IR spectroscopy can provide complementary insights where other techniques are limited. In general, An–H vibrations appear at slightly higher frequencies (1200 to 1500 cm−1) than those of their lanthanide congeners, and there are also discernible differences in vibrational energies between the hydrides of isostructural thorium and uranium complexes, such as (N′′)3An(H) (An = Th, νTh–H = 1480 cm−1; An = U, νU–H = 1430 cm−1). This discernibility is also retained upon deuteration, whereupon bands are instead observed at 1060 and 1020 cm−1 respectively.181,182 Unfortunately, the availability and assignment of IR spectral data in the literature has been somewhat limited to date; its inclusion in future publications is likely to offer new insights.
In other respects, their chemistry can differ dramatically. Whilst the potentiality of these f-block species has not yet been fully-established by comprehensive studies, in individual cases, detailed investigations have begun to illustrate the divergence and potential utility of their reactivity. A case worth considering in this context is that of Meyer and colleagues’ trivalent scaffold complex, {(Ad,MeArO)3mes}UIII, which undergoes an isomerization process upon reduction with KC8 or Na0 wherein H˙, abstracted from the benzylic methylene linker of one arm of the supporting ligand, migrates to the metal centre (Fig. 8d); here, it bridges between the uranium centre and an alkali metal centre, which is itself η4-coordinated to the most proximate aryloxide moiety of the ligand. However, abstraction of the respective alkali metal centres by treatment with an appropriate encapsulation agent (18-crown-6, 15-crown-5) also induces the insertion of hydride into the central arene ring, disrupting its aromaticity.175 Such an actinide-mediated rearrangement is at least suggestive of a novel dearomatisation strategy. Of course, as is true in this case, not all such reactivity is welcome; the aforementioned decomposition of suitably-ligated hydride species, eliminating H2 to yield cyclometalated ‘tuck-in’ complexes, is indicative of the precarious balance of factors which keep these complexes stable. When reactions are reversible—as in the case of (COTTIPS2)(Cp*)U(H), which exists in an equilibrium balanced by its ‘tuck-in’ analogue and H2—this can somewhat complicate the interpretation of complex reactivity.176
Of some further interest is the potential for hydrides to exhibit redox activity, even in the absence of a typically redox-active metal (e.g. thorium, for which the tetravalent oxidation state overwhelmingly predominates): [(Cp*)2ThIV(μ2-H)(H)]2, generated in situ, reacts with S8 to form (Cp*)2ThIV(S5) (Fig. 21); its uranium(IV) analogue reacts with 2,2′-bipyridine to yield (Cp*)2UIV(bipy), wherein the reduction of the 2,2′-bipyridyl ligand is achieved without any ultimate change in the metal oxidation state.162
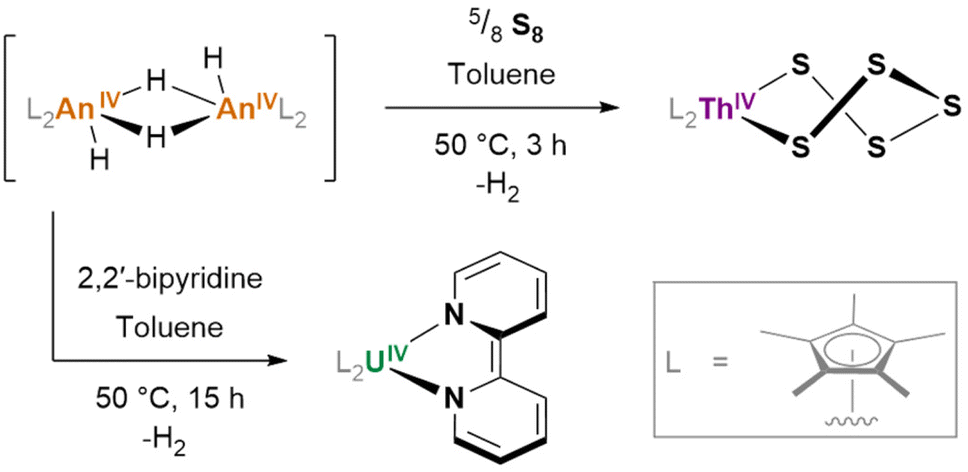 | ||
| Fig. 21 Actinide hydride reduction behaviour towards substrates. {Cp*Th}2(H)4 generated in situ reacts with elemental sulfur to yield form a S52− coordinated product. The analogous uranium hydride, {Cp*U}2(H)4, reacts with 2,2′-bipyridine to coordinate the reduced bipyridyl dianion (bipy2−). | ||
Alternatively, reactions of actinide hydrides with small molecules demonstrate the surprising potential for actinide hydride-mediated functionalization of organic fragments. Treatment of [(Cp*)2U(μ2-H)]2 with MeCN (6 equiv.) induces the condensation of three MeCN units to (C6N3H7)2−, which binds in a κ2-N,N′ fashion, bridging the two uranium(IV) centres (Fig. 22, top left). Of further significance is that dithorium(IV) tetrahydride complex [(Cp*)2Th(μ2-H)(H)]2 may be considered to possess similar reactivity to U(III) containing [(Cp*)2U(H)]2. It, likewise, undergoes reaction with MeCN to generate (C6N3H7)2− (Fig. 22, top right).168 Previously-mentioned Cs[{(SiO†O)3U}2(μ2-H)(μ2-NH)] also undergoes reaction with MeCN to give Cs[{(SiO†O)3U}2(μ3-NH)(μ2-NCHCH3)] (Fig. 22, middle), in which the μ3-imido ligand also coordinates Cs+. In the presence of CO2 (2 equiv.), insertion into both the U–H and U–NH linkages of Cs[{(SiO†O)3U}2(μ2-H)(μ2-NH)] yields Cs[{(SiO†O)3U}2(μ2-HCOO)(μ2-NHCOO)].153 In a further expansion on this concept, sequential reactions of related K2[{(SiO†O)3U}2(μ2-O)(μ2-H)2] with a range of small molecules demonstrate the efficacy of transfer hydrogenation at actinide centres, enabling the assembly of entire organic fragments (Fig. 22, bottom). By a two-electron process, the complex reductively couples MeCN to form (NC(CH3)NCH2CH2)2−, a dianionic fragment which may be isolated in a μ2:κ2 bridging mode between its two uranium centres alongside its pre-existing μ2-oxo. Alternatively, treatment with CO results in the formation of an (OCH2)2− fragment, whereafter further treatment with H2 (1 bar, r.t.) affords (OCH3)− and reintroduces a hydride ligand. From either the (OCH2)2− bridged complex, or after further reaction of the (OCH3)− bridged complex with CO2, separable organic compounds CH2DOD—or, in the latter case, a mixture of formate, bicarbonate, and methanol—may be identified by NMR spectroscopy after treatment of the complex with D2O.150
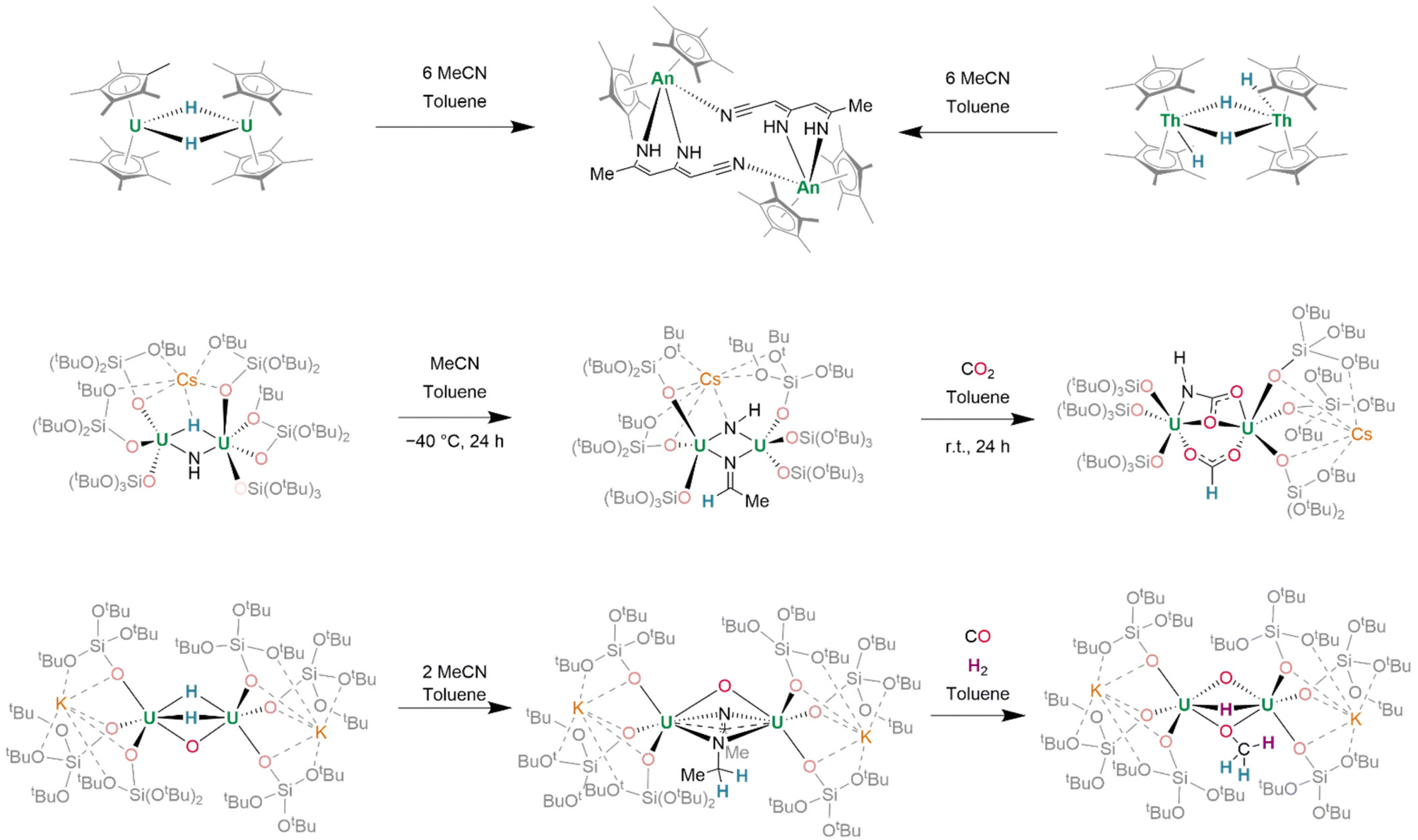 | ||
| Fig. 22 Reactivity of actinide hydrides towards unsaturated small molecules. (a) Actinide metallocene hydrides facilitate β-coupling of nitriles. (b) and (c) U(IV) mono and dihydrides can facilitate hydride transfer to nitriles with C–N coupling in the latter case. In both cases further transfer of the hydrides to other unsaturated small molecules is possible. | ||
Bridging and Lewis acid coordinated actinide hydrides
Such divergent reactivity suggests a structural cause. Coordination of group XIII metal hydrides to actinide centres characteristically involves An⋯H–M bridging interactions, wherein the hydrides span the metal centres in a 3c–2e bonding configuration. Sharing either two or three hydrides between actinide and group XIII metal centres, both μ2- and μ3-bridging modes are observed crystallographically in the isomorphous, homoleptic, tetravalent borohydrides, [U(BH4)4] and [Th(BH4)4].193,194 That there is a degree of covalency in these linkages is supported by their volatility: [U(BH4)4] sublimes at temperatures as low as 30 °C, and the volatility of its transuranic analogues is reportedly even greater; these materials exist as pyrophoric liquids which decompose readily at room temperature.195,196 Conspicuously, sublimation of its thorium congener requires temperatures at least 100 degrees higher.196 Furthermore, in an intriguingly straightforward procedure, it has been demonstrated by Daly and colleagues that U(BH4)4 is reducible to uranium(III) merely by heating the tetravalent borohydride to 100 °C under argon, offering facile access to this low oxidation state.197
Evidently, the capacity for variable bridging modes is important: alongside their relative lability, these factors likely offer complexes a degree of flexibility in their coordination spheres which cannot be achieved using comparable halides. The magnitude of U⋯H interactions have been quantified spectroscopically in hydrobis(mercaptoimidazolyl)borate (timMe) complex U{B(H)(R)(timMe)2}2(thf)2 (R = H, Ph), wherein a trivalent uranium centre is stabilised by tridentate ligands capable of enforcing a single U⋯H–B interaction per ligand (Fig. 23a). Straightforward IR spectroscopic observation of a shift in B–H stretching frequencies between the free (2446 cm−1) and coordinated (2376 cm−1) ligand is suggestive of a κ3-H,S,S-binding mode—although U–H distances are significantly elongated (R = H, 2.31(9) Å; R = Ph, 2.54(9) Å) relative to direct actinide hydrides.198 In a further illustrative case, Bart and colleagues have observed a sterically-induced change in the coordination mode of a tris(pyrazolyl)borate ligand, which occurs upon introduction of imide ligands bearing substituents of differing steric size. Whereas the supporting ligands of (Tp*)2UIV(NR) (R = mes, Ph) both adopt typical κ3-N,N,N coordination modes to the uranium(IV) centre, the more substantial adamantyl substituent in (Tp*)2UIV(NAd) enforces a change to an unsymmetrical configuration, wherein one ligand instead binds in a κ3-N,N,H fashion with one of the pyrazolyl arms of the ligand in the apical position Fig. 23b.199
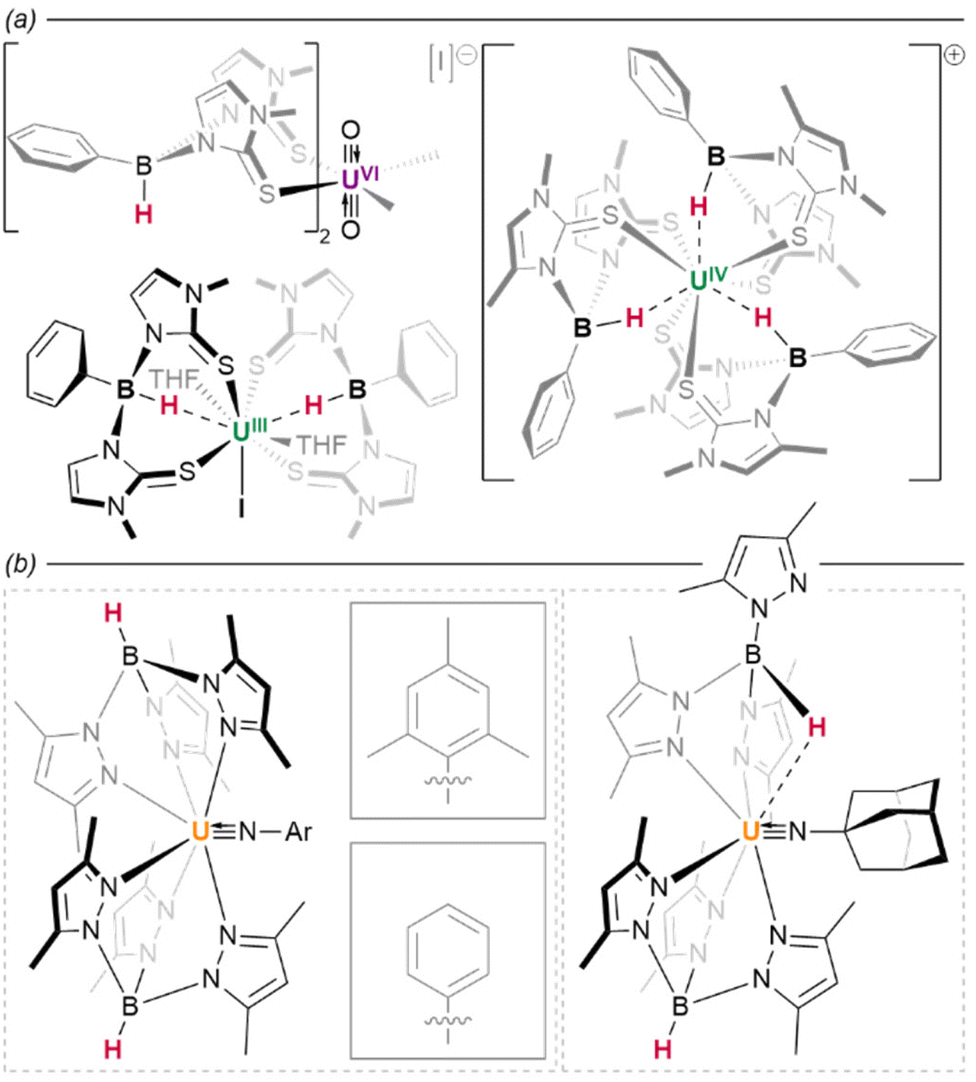 | ||
| Fig. 23 Borohydride complexes of actinides. (a) With bis(pyrazolyl)borane and bis(methimazolyl)boranes coordination of the B–H bond to the actinide centre is facile. (b) With tris(pyrazolyl)boranes the flexibility in coordination mode may allow for formation of complexes with more sterically encumbered co-ligands. | ||
There are few examples of heterobimetallic actinide complexes incorporating transition metal hydrides; however, recent work by the Arnold and Camp groups has also characterised hydride-supported An → M interactions with iridium. The isolated complexes, {(Cp*)Ir(H)3}4UIV and {(Cp*)Ir(H)3}4ThIV, wherein the respective actinide centres are twelve- and ten-coordinate, are calculated to possess notably high Wiberg An–M bond indices (U → Ir = 0.97; Th → Ir = 0.65).201
Conclusions
In this perspective, we have illustrated the key synthetic pathways for the synthesis of lanthanide and actinide hydrides, with hydrogenolysis and σ-bond metathesis reactions from metal alkyls being the most widely utilised. Our analysis suggests that sequential hydrogenolysis of metal alkyls is slower as the number of alkyl groups increases on a metal due to the formation of highly bridged hydride intermediates. In addition, hydrogenolysis appears to be facilitated greatly by metalated ligand intermediates which contain ring-strained metallacycles. Only a few examples of hydrides formed from, or with, low-oxidation state f-block metals are known; however, what has been reported to date suggests a key role for reduced arene ligands in their formation by this pathway.In terms of structure, the order of clusters is shown to grow as co-ligand stoichiometry and sterics decrease, in order to retain high coordination numbers on the metals. The structure of these clusters also changes with metal ion size with smaller lanthanides, in particular, able to invoke μ4 and even μ6 coordination modes for the hydride, mirroring the ionic bonding in binary hydrides. At the opposite end of the covalency spectrum, evidence has now been presented for the formation of both lanthanide and actinide dihydrogen complexes, though current evidence suggests bonding is very different between the two periods.
The reactivity of lanthanide hydrides comprises primarily σ-bond metathesis and 1,2-addition reactions due to their limited access to redox reactivity; however, these two powerful mechanisms have been exploited to great effect in catalytic polymerisation, hydrogenation and hydroelementation.
In this work, we have outlined how cluster structure and nuclearity can influence reactivity, with highly bridged hydrides reacting more slowly with some substrates. This should inform choice of co-ligand for lanthanide pre-catalysts which invoke hydrides in the catalytic cycle. For actinides, different pathways may operate, allowing for coupling of nitrile ligands for example, rather than simple 1,2-addition; transfer hydrogenation of small molecules; and redox-like reactivity for thorium(IV) hydrides.
Bridging hydrides are also shown to stabilise a wide range of complexes of the form Ln⋯H–E/Ln⋯H–M with varying Ln–E/Ln–M interactions from the extreme of Ln–E bonds with agostic interactions to An → M species where a low-valent actinide is able to act as a Lewis base toward an electron-deficient metal centre.
Together, this survey presents an emerging picture of the field of f-block hydrides which is beginning to understand how to employ fine electronic and structural control to exploit the distinctive characteristics of complexes which span the full length and breadth of the periodic table.
Author contributions
This manuscript was conceived by Dr Bell and written together by both Dr Bell and Dr Drummond Turnbull, including preparation of all figures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the EPSRC (Open Fellowship EP/W02702X/1) and the University of Glasgow (LKAS Fellowship) for funding this work.Notes and references
- J. Hutchinson, J. Alwin, A. McSpaden, W. Myers, M. Rising and R. Sanchez, Nucl. Technol., 2021, 207, S62–S80 CrossRef.
- H. Liu and M. S. Eisen, Synthesis, 2020, 52, 629–644 CrossRef CAS.
- W. Chen, J. Li and C. Cui, Synlett, 2020, 32, 962–970 Search PubMed.
- H. Sugiyama, T. Nakao, M. Miyazaki, H. Abe, Y. Niwa, M. Kitano and H. Hosono, ACS Catal., 2022, 12, 12572–12581 CrossRef CAS.
- H. Yan, W. Gao, J. Cui, W. Zhang, Q. Pei, Q. Wang, Y. Guan, S. Feng, H. Wu, H. Cao, J. Guo and P. Chen, J. Energy Chem., 2022, 72, 1–7 CrossRef CAS.
- A. Habibzadeh, M. A. Kucuker and M. Gökelma, ACS Omega, 2023, 8, 17431–17445 CrossRef CAS PubMed.
- F. Nafezarefi, H. Schreuders, B. Dam and S. Cornelius, Appl. Phys. Lett., 2017, 111, 103903 CrossRef.
- J. M. Haschke and T. H. Allen, J. Alloys Compd., 2001, 320, 58–71 CrossRef CAS.
- A. Banos, N. J. Harker and T. B. Scott, Corros. Sci., 2018, 136, 129–147 CrossRef CAS.
- A. Banos and T. B. Scott, Sci. Rep., 2020, 10, 9479 CrossRef CAS PubMed.
- F. Peng, Y. Sun, C. J. Pickard, R. J. Needs, Q. Wu and Y. Ma, Phys. Rev. Lett., 2017, 119, 107001 CrossRef PubMed.
- Z. M. Geballe, H. Liu, A. K. Mishra, M. Ahart, M. Somayazulu, Y. Meng, M. Baldini and R. J. Hemley, Angew. Chem., Int. Ed., 2018, 57, 688–692 CrossRef CAS PubMed.
- D. V. Semenok, A. G. Kvashnin, A. G. Ivanova, V. Svitlyk, V. Y. Fominski, A. V. Sadakov, O. A. Sobolevskiy, V. M. Pudalov, I. A. Troyan and A. R. Oganov, Mater. Today, 2020, 33, 36–44 CrossRef CAS.
- M. Falcone, L. Chatelain, R. Scopelliti, I. Živković and M. Mazzanti, Nature, 2017, 547, 332–335 CrossRef CAS PubMed.
- L. Perrin, L. Maron and O. Eisenstein, Faraday Discuss., 2003, 124, 25–39 RSC.
- M. Ephritikhine, Chem. Rev., 1997, 97, 2193–2242 CrossRef CAS PubMed.
- P. Vajda, Solid State Ionics, 2004, 168, 271–279 CrossRef CAS.
- H. Mizoguchi, M. Okunaka, M. Kitano, S. Matsuishi, T. Yokoyama and H. Hosono, Inorg. Chem., 2016, 55, 8833–8838 CrossRef CAS PubMed.
- R. Griessen, J. N. Huiberts, M. Kremers, A. T. M. van Gogh, N. J. Koeman, J. P. Dekker and P. H. L. Notten, J. Alloys Compd., 1997, 253–254, 44–50 CrossRef CAS.
- F. J. A. den Broeder, S. J. van der Molen, M. Kremers, J. N. Huiberts, D. G. Nagengast, A. T. M. van Gogh, W. H. Huisman, N. J. Koeman, B. Dam, J. H. Rector, S. Plota, M. Haaksma, R. M. N. Hanzen, R. M. Jungblut, P. A. Duine and R. Griessen, Nature, 1998, 394, 656–658 CrossRef CAS.
- W. Zhang, J. Cui, S. Wang, H. Cao, A. Wu, Y. Xia, Q. Jiang, J. Guo, T. He and P. Chen, Nature, 2023, 616, 73–76 CrossRef CAS PubMed.
- S. P. Willson and L. Andrews, J. Phys. Chem. A, 2000, 104, 1640–1647 CrossRef CAS.
- X. Wang, L. Andrews, I. Infante and L. Gagliardi, J. Phys. Chem. A, 2009, 113, 12566–12572 CrossRef CAS PubMed.
- I. Infante, L. Gagliardi, X. Wang and L. Andrews, J. Phys. Chem. A, 2009, 113, 2446–2455 CrossRef CAS PubMed.
- O. T. Summerscales, E. R. Batista, B. L. Scott, M. P. Wilkerson and A. D. Sutton, Eur. J. Inorg. Chem., 2016, 4551 CrossRef.
- W. Fegler, A. Venugopal, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2013, 52, 7976 CrossRef CAS PubMed.
- S.-S. Liu, S. Gao, J. W. Ziller and W. J. Evans, Dalton Trans., 2014, 43, 15526 RSC.
- M. E. Fieser, T. J. Mueller, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, Organometallics, 2014, 33, 3882 CrossRef CAS.
- L. Maron, E. L. Werkema, L. Perrin, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2005, 127, 279 CrossRef CAS PubMed.
- H. Kulinna, T. P. Spaniol and J. Okuda, J. Organomet. Chem., 2013, 744, 49 CrossRef CAS.
- A. Venugopal, F. Tuna, T. P. Spaniol, L. Ungur, L. F. Chibotaru, J. Okuda and R. A. Layfield, Chem. Commun., 2013, 49, 901 RSC.
- A. Venugopal, W. Fegler, T. P. Spaniol, L. Maron and J. Okuda, J. Am. Chem. Soc., 2011, 133, 17574 CrossRef CAS PubMed.
- M. T. Dumas, G. P. Chen, J. Y. Hu, M. A. Nascimento, J. M. Rawson, J. W. Ziller, F. Furche and W. J. Evans, J. Organomet. Chem., 2017, 849–850, 38 CrossRef CAS.
- E. Abinet, D. Martin, S. Standfuss, H. Kulinna, T. P. Spaniol and J. Okuda, Chem. – Eur. J., 2011, 17, 15014 CrossRef CAS PubMed.
- S. Arndt, P. Voth, T. P. Spaniol and J. Okuda, Organometallics, 2000, 19, 4690 CrossRef CAS.
- K. R. D. Johnson, B. L. Kamenz and P. G. Hayes, Organometallics, 2014, 33, 3005 CrossRef CAS.
- Y. Takenaka and Z. Hou, Organometallics, 2009, 28, 5196 CrossRef CAS.
- M. Nishiura, J. Baldamus, T. Shima, K. Mori and Z. Hou, Chem. – Eur. J., 2011, 17, 5033 CrossRef CAS PubMed.
- G. Desurmont, Y. Li, H. Yasuda, T. Maruo, N. Kanehisa and Y. Kai, Organometallics, 2000, 19, 1811 CrossRef CAS.
- P. W. Roesky, U. Denninger, C. L. Stern and T. J. Marks, Organometallics, 1997, 16, 4486 CrossRef CAS.
- X. Shi, P. Deng, T. Rajeshkumar, L. Zhao, L. Maron and J. Cheng, Chem. Commun., 2021, 57, 10047 RSC.
- J. Cheng, T. Shima and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 1857 CrossRef CAS PubMed.
- J. Cheng, M. J. Ferguson and J. Takats, J. Am. Chem. Soc., 2010, 132, 2 CrossRef CAS PubMed.
- J. Cheng, K. Saliu, M. J. Ferguson, R. McDonald and J. Takats, J. Organomet. Chem., 2010, 695, 2696 CrossRef CAS.
- G. M. Ferrence, R. McDonald and J. Takats, Angew. Chem., Int. Ed., 1999, 38, 2233 CrossRef CAS PubMed.
- Y. Wang, I. D. Rosal, G. Qin, L. Zhao, L. Maron, X. Shi and J. Cheng, Chem. Commun., 2021, 57, 7766 RSC.
- J. Cheng and Z. Hou, Chem. Commun., 2012, 48, 814 RSC.
- T. Li, M. Nishiura, J. Cheng, W. Zhang, Y. Li and Z. Hou, Organometallics, 2013, 32, 4142 CrossRef CAS.
- M. Konkol, T. P. Spaniol, M. Kondracka and J. Okuda, Dalton Trans., 2007, 4095 RSC.
- W. Chen, H. Song, J. Li and C. Cui, Angew. Chem., Int. Ed., 2020, 59, 2365 CrossRef CAS PubMed.
- J. Liu, W. Chen, J. Li and C. Cui, ACS Catal., 2018, 8, 2230 CrossRef CAS.
- T. Dube, S. Gambarotta and G. P. A. Yap, Organometallics, 2000, 19, 817 CrossRef CAS.
- T. Dube, S. Gambarotta and G. Yap, Organometallics, 2000, 19, 121 CrossRef CAS.
- C. Wang, L. Xiang, X. Leng and Y. Chen, Dalton Trans., 2017, 46, 1218 RSC.
- Q. Wen, B. Feng, L. Xiang, X. Leng and Y. Chen, Inorg. Chem., 2021, 60, 13913 CrossRef CAS PubMed.
- O. Tardif, M. Nishiura and Z. Hou, Tetrahedron, 2003, 59, 10525 CrossRef CAS.
- D. Cui, M. Nishiura, O. Tardif and Z. Hou, Organometallics, 2008, 27, 2428 CrossRef CAS.
- O. Tardif, M. Nishiura and Z. Hou, Organometallics, 2003, 22, 1171 CrossRef CAS.
- D. M. Lyubov, A. M. Bubnov, G. K. Fukin, F. M. Dolgushin, M. Yu. Antipin, O. Pelce, M. Schappacher, S. M. Guillaume and A. A. Trifonov, Eur. J. Inorg. Chem., 2008, 2090 CrossRef CAS.
- W. Rong, D. He, M. Wang, Z. Mou, J. Cheng, C. Yao, S. Li, A. A. Trifonov, D. M. Lyubov and D. Cui, Chem. Commun., 2015, 51, 5063 RSC.
- A. A. Trifonov, E. A. Fedorova, G. K. Fukin and M. N. Bochkarev, Eur. J. Inorg. Chem., 2004, 4396 CrossRef CAS.
- A. A. Trifonov, G. G. Skvortsov, D. M. Lyubov, N. A. Skorodumova, G. K. Fukin, E. V. Baranov and V. N. Glushakova, Chem. – Eur. J., 2006, 12, 5320 CrossRef CAS PubMed.
- G. M. Richardson, I. Douair, S. A. Cameron, J. Bracegirdle, R. A. Keyzers, M. S. Hill, L. Maron and M. D. Anker, Nat. Commun., 2021, 12, 3147 CrossRef CAS PubMed.
- C. Ruspic, J. Spielmann and S. Harder, Inorg. Chem., 2007, 46, 5320 CrossRef CAS PubMed.
- Z. Hou, Y. Zhang, O. Tardif and Y. Wakatsuki, J. Am. Chem. Soc., 2001, 123, 9216 CrossRef CAS PubMed.
- D. M. Lyubov, A. V. Cherkasov, G. K. Fukin, S. Yu. Ketkov, A. S. Shavyrin and A. A. Trifonov, Dalton Trans., 2014, 43, 14450 RSC.
- D. M. Lyubov, C. Doring, S. Yu. Ketkov, R. Kempe and A. A. Trifonov, Chem. – Eur. J., 2011, 17, 3824 CrossRef CAS PubMed.
- N. S. Radu and T. D. Tilley, J. Am. Chem. Soc., 1995, 117, 5863–5864 CrossRef CAS.
- I. Castillo and T. D. Tilley, Organometallics, 2001, 20, 5598–5605 CrossRef CAS.
- D. Martin, J. Kleemann, E. Abinet, T. P. Spaniol, L. Maron and J. Okuda, Eur. J. Inorg. Chem., 2013, 3987 CrossRef CAS.
- A. Z. Voskoboynikov, I. N. Parshina, A. K. Shestakova, K. P. Butin, I. P. Beletskaya, L. G. Kuz'mina and J. A. K. Howard, Organometallics, 1997, 16, 4041–4055 CrossRef CAS.
- X. Zhu, D. Guo, Z. Huang, T. Sheng, S. Wang, M. Pan, L. Zha and S. Zhou, Inorg. Chem., 2020, 59, 14152 CrossRef CAS PubMed.
- W. J. Evans, J. H. Meadows, A. L. Wayda, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1982, 104, 2015–2017 CrossRef CAS.
- D. A. Buschmann, L. Schumacher and R. Anwander, Chem. Commun., 2022, 58, 9132 RSC.
- E. C. Moinet, O. Tardif, C. Maichle-Mössmer and R. Anwander, Chem. Commun., 2023, 59, 5261–5264 RSC.
- K. Lv and D. Cui, Organometallics, 2010, 29, 2987–2993 CrossRef CAS.
- S. Schäfer, S. Kaufmann, E. S. Rösch and P. W. Roesky, Chem. Soc. Rev., 2023, 52, 4006–4045 RSC.
- D. Schuhknecht, K.-N. Truong, T. P. Spaniol, L. Maron and J. Okuda, Chem. Commun., 2018, 54, 11280 RSC.
- G. M. Richardson, M. J. Evans, T. Rajeshkumar, J. A. J. McCone, S. A. Cameron, L. Maron, C. Jones and M. D. Anker, Chem. – Eur. J., 2024, 30(27), e202400681 CrossRef CAS PubMed.
- Y. K. Gun'ko, P. B. Hitchcock and M. F. Lappert, Organometallics, 2000, 19, 2832 CrossRef.
- D. N. Huh, L. E. Darago, J. W. Ziller and W. J. Evans, Inorg. Chem., 2018, 57, 2096 CrossRef CAS PubMed.
- W. Huang, F. Dulong, S. I. Khan, T. Cantat and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 17410–17413 CrossRef CAS PubMed.
- M. E. Fieser, C. T. Palumbo, H. S. L. Pierre, D. P. Halter, V. K. Voora, J. W. Ziller, F. Furche, K. Meyer and W. J. Evans, Chem. Sci., 2017, 8, 7424 RSC.
- S. P. Nolan and T. J. Marks, J. Am. Chem. Soc., 1989, 111, 8538–8540 CrossRef CAS.
- L. Perrin, L. Maron, O. Eisenstein, D. J. Schwartz, C. J. Burns and R. A. Andersen, Organometallics, 2003, 22, 5447–5453 CrossRef CAS.
- J. Cheng, K. Saliu, G. Y. Kiel, M. J. Ferguson, R. McDonald and J. Takats, Angew. Chem., Int. Ed., 2008, 47, 4910 CrossRef CAS PubMed.
- Y. Luo and Z. Hou, J. Phys. Chem. C, 2008, 112, 635–638 CrossRef CAS.
- K. R. McClain, H. Kwon, K. Chakarawet, R. Nabi, J. G. C. Kragskow, N. F. Chilton, R. D. Britt, J. R. Long and B. G. Harvey, J. Am. Chem. Soc., 2023, 145, 8996–9002 CrossRef CAS PubMed.
- T. Stewart, M. Nishiura, Y. Konno, Z. Hou, G. J. McIntyre and R. Bau, Inorg. Chim. Acta, 2010, 363, 562 CrossRef CAS.
- P. L. Watson and G. W. Parshall, Acc. Chem. Res., 1985, 18, 51–56 CrossRef CAS.
- D. J. Mindiola, Organometallics, 2020, 39, 1135–1138 CrossRef CAS.
- R. D. Dicken, A. Motta and T. J. Marks, ACS Catal., 2021, 11, 2715–2734 CrossRef CAS.
- G. Jeske, H. Lauke, H. Mauermann, P. N. Swepston, H. Schumann and T. J. Marks, J. Am. Chem. Soc., 1985, 107, 8091–8103 CrossRef CAS.
- W. J. Evans, J. H. Meadows, A. L. Wayda, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1982, 104, 2008–2014 CrossRef CAS.
- M. Booij, B. J. Deelman, R. Duchateau, D. S. Postma, A. Meetsma and J. H. Teuben, Organometallics, 1993, 12, 3531–3540 CrossRef CAS.
- K. H. Den Haan, Y. Wielstra and J. H. Teuben, Organometallics, 1987, 6, 2053–2060 CrossRef CAS.
- R. Waterman, Organometallics, 2013, 32, 7249–7263 CrossRef CAS.
- T. Ziegler, E. Folga and A. Berces, J. Am. Chem. Soc., 1993, 115, 636–646 CrossRef CAS.
- T. Shima and Z. Hou, Dalton Trans., 2010, 39, 6858 RSC.
- M. E. Thompson, S. M. Baxter, A. R. Bulls, B. J. Burger, M. C. Nolan, B. D. Santarsiero, W. P. Schaefer and J. E. Bercaw, J. Am. Chem. Soc., 1987, 109, 203–219 CrossRef CAS.
- X. Zhou and M. Zhu, J. Organomet. Chem., 2002, 647, 28–49 CrossRef CAS.
- A. Motta, I. L. Fragalà and T. J. Marks, Organometallics, 2006, 25, 5533–5539 CrossRef CAS.
- H. Yasuda, in Lanthanides: Chemistry and Use in Organic Synthesis, ed. S. Kobayashi, Springer Berlin Heidelberg, Berlin, Heidelberg, 1999, pp. 255–283, DOI:10.1007/3-540-69801-9_7.
- D. Cui, O. Tardif and Z. Hou, J. Am. Chem. Soc., 2004, 126, 1312 CrossRef CAS PubMed.
- P. Cui, T. P. Spaniol, L. Maron and J. Okuda, Chem. – Eur. J., 2013, 19, 13437 CrossRef CAS PubMed.
- J. C. Wedal, L. M. Anderson-Sanchez, M. T. Dumas, C. A. Gould, M. J. Beltrán-Leiva, C. Celis-Barros, D. Páez-Hernández, J. W. Ziller, J. R. Long and W. J. Evans, J. Am. Chem. Soc., 2023, 145, 10730–10742 CrossRef CAS PubMed.
- Q. Wen, T. Rajeshkumar, L. Maron, X. Leng and Y. Chen, Angew. Chem., Int. Ed., 2022, 61(25), e202200540 CrossRef CAS PubMed.
- W. J. Evans, J. M. Perotti and J. W. Ziller, Inorg. Chem., 2005, 44, 5820 CrossRef CAS PubMed.
- B. L. L. Réant, S. T. Liddle and D. P. Mills, Chem. Sci., 2020, 11, 10871–10886 RSC.
- A. Pindwal, K. Yan, S. Patnaik, B. M. Schmidt, A. Ellern, I. I. Slowing, C. Bae and A. D. Sadow, J. Am. Chem. Soc., 2017, 139, 16862–16874 CrossRef CAS PubMed.
- X. Pan, C. Wu, H. Fang and C. Yan, Inorg. Chem., 2022, 61, 14288–14296 CrossRef CAS PubMed.
- M. Visseaux and F. Bonnet, Coord. Chem. Rev., 2011, 255, 374–420 CrossRef CAS.
- C. Schadle, D. Schadle, K. Eichele and R. Anwander, Angew. Chem., Int. Ed., 2013, 52, 13238 CrossRef PubMed.
- M. Widemann, F. S. W. Aicher, M. Bonath, K. Eichele, C. Maichle-Mössmer, H. Schubert, P. Sirsch, R. Anwander and L. Wesemann, Chem. – Eur. J., 2022, 28, e202201032 CrossRef CAS PubMed.
- F. Ortu, Chem. Rev., 2022, 122, 6040–6116 CrossRef CAS PubMed.
- B. M. Schmidt, A. Pindwal, A. Venkatesh, A. Ellern, A. J. Rossini and A. D. Sadow, ACS Catal., 2019, 9, 827–838 CrossRef CAS.
- K. O. Saliu, J. Takats and M. J. Ferguson, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m643–m644 CrossRef CAS PubMed.
- Z. Xu and Z. Lin, Coord. Chem. Rev., 1996, 156, 139–162 CrossRef CAS.
- T. V. Fetrow, J. Zgrabik, R. Bhowmick, F. D. Eckstrom, G. Crull, B. Vlaisavljevich and S. R. Daly, Angew. Chem., Int. Ed., 2022, 61, e202211145 CrossRef CAS PubMed.
- S. Y. Knjazhanskij, B. M. Bulychev, O. K. Kireeva, V. K. Belsky and G. L. Soloveichik, J. Organomet. Chem., 1991, 414, 11–22 CrossRef.
- T. Chowdhury, F. Murphy, A. R. Kennedy, C. Wilson, J. H. Farnaby and C. E. Weetman, Inorg. Chem., 2024, 63(21), 9390–9394 CrossRef CAS PubMed.
- T. Pugh, V. Vieru, L. F. Chibotaru and R. A. Layfield, Chem. Sci., 2016, 7, 2128–2137 RSC.
- J. Du, P. J. Cobb, J. Ding, D. P. Mills and S. T. Liddle, Chem. Sci., 2024, 15, 13–45 RSC.
- T. Bauer, F. R. Wagner and R. Kempe, Chem. – Eur. J., 2013, 19, 8732–8735 CrossRef CAS PubMed.
- M. L. H. Green, A. K. Hughes, D. M. Michaelidou and P. Mountford, J. Chem. Soc., Chem. Commun., 1993, 591–593, 10.1039/c39930000591.
- T. Shima and Z. Hou, Chem. Lett., 2008, 37, 298 CrossRef CAS.
- A. P. Sobaczynski, T. Bauer and R. Kempe, Organometallics, 2013, 32, 1363 CrossRef CAS.
- T. Bauer, F. R. Wagner and R. Kempe, Chem. – Eur. J., 2013, 19, 8732 CrossRef CAS PubMed.
- T. Shima and Z. Hou, Organometallics, 2009, 28, 2244 CrossRef CAS.
- D. Kawai, T. Shima, M. Nishiura and Z. Hou, J. Organomet. Chem., 2017, 847, 74 CrossRef CAS.
- T. Shima, Y. Luo, T. Stewart, R. Bau, G. J. McIntyre, S. A. Mason and Z. Hou, Nat. Chem., 2011, 3, 814 CrossRef CAS PubMed.
- S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 8604–8641 CrossRef CAS PubMed.
- P. F. Souter, G. P. Kushto, L. Andrews and M. Neurock, J. Phys. Chem. A, 1997, 101, 1287–1291 CrossRef CAS.
- P. F. Souter, G. P. Kushto, L. Andrews and M. Neurock, J. Am. Chem. Soc., 1997, 119, 1682–1687 CrossRef CAS.
- M. Straka and P. Pyykkö, J. Am. Chem. Soc., 2005, 127, 13090–13091 CrossRef CAS PubMed.
- J. Raab, R. H. Lindh, X. Wang, L. Andrews and L. Gagliardi, J. Phys. Chem. A, 2007, 111, 6383–6387 CrossRef CAS PubMed.
- X. Wang, L. Andrews and L. Gagliardi, J. Phys. Chem. A, 2008, 112, 1754–1761 CrossRef CAS PubMed.
- J. E. Burke and C. S. Smith, J. Am. Chem. Soc., 1947, 69, 2500–2502 CrossRef CAS PubMed.
- L. Natrajan, M. Mazzanti, J.-P. Bezombes and J. Pécaut, Inorg. Chem., 2005, 44, 6115–6121 CrossRef CAS PubMed.
- T. Schleid, G. Meyer and L. R. Morss, J. Less-Common Met., 1987, 132, 69–77 CrossRef CAS.
- L. Havela, D. Legut and J. Kolorenč, Rep. Prog. Phys., 2023, 86, 056501 CrossRef PubMed.
- F. Le Guyadec, X. Génin, J. P. Bayle, O. Dugne, A. Duhart-Barone and C. Ablitzer, J. Nucl. Mater., 2010, 396, 294–302 CrossRef CAS.
- D. D. Schnaars, G. Wu and T. W. Hayton, Dalton Trans., 2008, 6121–6126, 10.1039/B809184F.
- J.-C. Berthet, M. Nierlich and M. Ephritikhine, C. R. Chim., 2002, 5, 81–87 CrossRef CAS.
- J. Su, E. R. Batista, K. S. Boland, S. E. Bone, J. A. Bradley, S. K. Cary, D. L. Clark, S. D. Conradson, A. S. Ditter, N. Kaltsoyannis, J. M. Keith, A. Kerridge, S. A. Kozimor, M. W. Löble, R. L. Martin, S. G. Minasian, V. Mocko, H. S. La Pierre, G. T. Seidler, D. K. Shuh, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, J. Am. Chem. Soc., 2018, 140, 17977–17984 CrossRef CAS PubMed.
- J. M. Manriquez, P. J. Fagan and T. J. Marks, J. Am. Chem. Soc., 1978, 100, 3939–3941 CrossRef CAS.
- W. J. Evans, K. A. Miller, S. A. Kozimor, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2007, 26, 3568–3576 CrossRef CAS.
- N. A. Siladke, C. L. Webster, J. R. Walensky, M. K. Takase, J. W. Ziller, D. J. Grant, L. Gagliardi and W. J. Evans, Organometallics, 2013, 32, 6522–6531 CrossRef CAS.
- W. J. Evans, K. A. Miller, A. G. DiPasquale, A. L. Rheingold, T. J. Stewart and R. Bau, Angew. Chem., Int. Ed., 2008, 47, 5075–5078 CrossRef CAS PubMed.
- M. Falcone, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2019, 141, 9570–9577 CrossRef CAS PubMed.
- R. Chen, G. Qin, S. Li, A. J. Edwards, R. O. Piltz, I. Del Rosal, L. Maron, D. Cui and J. Cheng, Angew. Chem., Int. Ed., 2020, 59, 11250–11255 CrossRef CAS PubMed.
- G. Qin, Y. Wang, X. Shi, I. Del Rosal, L. Maron and J. Cheng, Chem. Commun., 2019, 55, 8560–8563 RSC.
- M. Falcone, L. N. Poon, F. Fadaei Tirani and M. Mazzanti, Angew. Chem., Int. Ed., 2018, 57, 3697–3700 CrossRef CAS PubMed.
- C. T. Palumbo, R. Scopelliti, I. Zivkovic and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 3149–3157 CrossRef CAS PubMed.
- C. T. Palumbo, L. Barluzzi, R. Scopelliti, I. Zivkovic, A. Fabrizio, C. Corminboeuf and M. Mazzanti, Chem. Sci., 2019, 10, 8840–8849 RSC.
- D.-D. Zhai, H.-Z. Du, X.-Y. Zhang, Y.-F. Liu and B.-T. Guan, ACS Catal., 2019, 9, 8766–8771 CrossRef CAS.
- J.-C. Berthet, J.-F. Le Maréchal and M. Ephritikhine, J. Chem. Soc., Chem. Commun., 1991, 360–361, 10.1039/C39910000360.
- J.-F. Le Maréchal, C. Villiers, P. Charpin, M. Lance, M. Nierlich, J. Vigner and M. Ephritikhine, J. Chem. Soc., Chem. Commun., 1989, 308–310, 10.1039/C39890000308.
- J.-C. Berthet, C. Villiers, J.-F. Le Maréchal, B. Delavaux-Nicot, M. Lance, M. Nierlich, J. Vigner and M. Ephritikhine, J. Organomet. Chem., 1992, 440, 53–65 CrossRef CAS.
- M. R. MacDonald, M. E. Fieser, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 13310–13313 CrossRef CAS PubMed.
- W. Ren, N. Zhao, L. Chen, H. Song and G. Zi, Inorg. Chem. Commun., 2011, 14, 1838–1841 CrossRef CAS.
- J. K. Pagano, J. M. Dorhout, R. Waterman, K. R. Czerwinski and J. L. Kiplinger, Chem. Commun, 2015, 51, 17379–17381 RSC.
- R. R. Langeslay, M. E. Fieser, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2016, 138, 4036–4045 CrossRef CAS PubMed.
- C. J. Windorff, M. R. MacDonald, K. R. Meihaus, J. W. Ziller, J. R. Long and W. J. Evans, Chem. – Eur. J., 2016, 22, 772–782 CrossRef CAS PubMed.
- R. W. Broach, A. J. Schultz, J. M. Williams, G. M. Brown, J. M. Manriquez, P. J. Fagan and T. J. Marks, Science, 1979, 203, 172–174 CrossRef CAS PubMed.
- Z.-L. Xue, T. M. Cook and A. C. Lamb, J. Organomet. Chem., 2017, 852, 74–93 CrossRef.
- C. M. Fendrick, L. D. Schertz, V. W. Day and T. J. Marks, Organometallics, 1988, 7, 1828–1838 CrossRef CAS.
- W. J. Evans, K. A. Miller and J. W. Ziller, Angew. Chem., Int. Ed., 2008, 47, 589–592 CrossRef CAS PubMed.
- W. J. Evans, G. W. Nyce and J. W. Ziller, Organometallics, 2001, 20, 5489–5491 CrossRef CAS.
- E. Zhou, W. Ren, G. Hou, G. Zi, D.-C. Fang and M. D. Walter, Organometallics, 2015, 34, 3637–3647 CrossRef CAS.
- W. Ren, E. Zhou, B. Fang, G. Zi, D.-C. Fang and M. D. Walter, Chem. Sci., 2014, 5, 3165–3172 RSC.
- R. A. Andersen, A. Zalkin and D. H. Templeton, Inorg. Chem., 1981, 20, 622–623 CrossRef CAS.
- D. L. Clark, S. K. Grumbine, B. L. Scott and J. G. Watkin, J. Am. Chem. Soc., 1995, 117, 9089–9090 CrossRef CAS.
- D. L. Clark, S. K. Grumbine, B. L. Scott and J. G. Watkin, Organometallics, 1996, 15, 949–957 CrossRef CAS.
- H. S. La Pierre, H. Kameo, D. P. Halter, F. W. Heinemann and K. Meyer, Angew. Chem., Int. Ed., 2014, 53, 7154–7157 CrossRef CAS PubMed.
- J. A. Higgins, F. G. N. Cloke and S. M. Roe, Organometallics, 2013, 32, 5244–5252 CrossRef CAS.
- M. R. Duttera, P. J. Fagan, T. J. Marks and V. W. Day, J. Am. Chem. Soc., 1982, 104, 865–867 CrossRef CAS.
- J.-C. Berthet, J.-F. Le Maréchal, M. Lance, M. Nierlich, J. Vigner and M. Ephritikhine, J. Chem. Soc., Dalton Trans., 1992, 1573–1577, 10.1039/DT9920001573.
- K. Li, I. Del Rosal, Y. Zhao, L. Maron and C. Zhu, Angew. Chem., Int. Ed., 2024, e202405494, DOI:10.1002/anie.202405494.
- P. Hrobarik, V. Hrobarikova, A. H. Greif and M. Kaupp, Angew. Chem., Int. Ed., 2012, 51, 10884–10888 CrossRef CAS PubMed.
- H. W. Turner, S. J. Simpson and R. A. Andersen, J. Am. Chem. Soc., 1979, 101, 2782–2782 CrossRef CAS.
- S. J. Simpson, H. W. Turner and R. A. Andersen, Inorg. Chem., 1981, 20, 2991 CrossRef CAS.
- A. K. Dash, J. Q. Wang and M. S. Eisen, Organometallics, 1999, 18, 4724–4741 CrossRef CAS.
- N. Tsoureas, L. Maron, A. F. R. Kilpatrick, R. A. Layfield and F. G. N. Cloke, J. Am. Chem. Soc., 2020, 142, 89–92 CrossRef CAS PubMed.
- I. J. Brackbill, T. Rajeshkumar, L. Maron, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2024, 146(2), 1257–1261 CrossRef CAS PubMed.
- T. J. Marks and J. R. Kolb, Chem. Rev., 1977, 77, 263–293 CrossRef CAS.
- P. L. Arnold, J. H. Farnaby, M. G. Gardiner and J. B. Love, Organometallics, 2015, 34, 2114–2117 CrossRef CAS.
- P. L. Arnold, C. J. Stevens, J. H. Farnaby, M. G. Gardiner, G. S. Nichol and J. B. Love, J. Am. Chem. Soc., 2014, 136, 10218–10221 CrossRef CAS PubMed.
- N. Tsoureas, A. Mansikkamäki and R. A. Layfield, Chem. Commun., 2020, 56, 944–947 RSC.
- J. T. Boronski, L. R. Doyle, J. A. Seed, A. J. Wooles and S. T. Liddle, Angew. Chem., Int. Ed., 2020, 59, 295–299 CrossRef CAS PubMed.
- D. Baudry, E. Bulot, P. Charpin, M. Ephritikhine, M. Lance, M. Nierlich and J. Vigner, J. Organomet. Chem., 1989, 371, 155–162 CrossRef CAS.
- R. R. Ryan, K. V. Salazar, N. N. Sauer and J. M. Ritchey, Inorg. Chim. Acta, 1989, 162, 221–225 CrossRef CAS.
- D. Männig and H. Nöth, Z. Anorg. Allg. Chem., 1986, 543, 66–72 CrossRef.
- A. C. Dunbar, J. C. Wright, D. J. Grant and G. S. Girolami, Inorg. Chem., 2021, 60, 12489–12497 CrossRef CAS PubMed.
- R. H. Banks, N. M. Edelstein, R. R. Rietz, D. H. Templeton and A. Zalkin, J. Am. Chem. Soc., 1978, 100, 1957–1958 CrossRef CAS.
- H. R. Hoekstra and J. J. Katz, J. Am. Chem. Soc., 1949, 71, 2488–2492 CrossRef CAS.
- T. V. Fetrow, J. P. Grabow, J. Leddy and S. R. Daly, Inorg. Chem., 2021, 60, 7593–7601 CrossRef CAS PubMed.
- L. Maria, I. C. Santos and I. Santos, Dalton Trans., 2018, 47, 10601–10612 RSC.
- A. Dauth and J. A. Love, Dalton Trans., 2012, 41, 7782–7791 RSC.
- A. B. Altman, A. C. Brown, G. Rao, T. D. Lohrey, R. D. Britt, L. Maron, S. G. Minasian, D. K. Shuh and J. Arnold, Chem. Sci., 2018, 9, 4317–4324 RSC.
- C. Z. Ye, I. Del Rosal, M. A. Boreen, E. T. Ouellette, D. R. Russo, L. Maron, J. Arnold and C. Camp, Chem. Sci., 2023, 14, 861–868 RSC.
Footnote |
| † The following abbreviations will be used throughout this work: Si′ = –SiMe3; N′′ = N(SiMe3)2; Cp* = C5(CH3)5; Cp** = C5(Ph)5; Cp× = C5(CH3)4H; Cp′ = C5H4(SiMe3); Cp′′ = 1,3-(SiMe3)2C5H3; Cp′′′ = 1,2,4-(SiMe3)3C5H2; Cp′* = C5(CH3)4(SiMe3); Cp׆ = C5Me4(tBu); Cp† = C5H4tBu; Cp‡ = 1,3-(tBu)2C5H3; Cpttt = 1,2,4-(tBu)3C5H2; CpAn = {3-(SiMe3)C5H4}2SiMe2; Tp* = tris(3,5-dimethylpyrazolyl)borane; TpiPr = tris((3-isopropyl-5-methyl)pyrazolyl)borane; TptBu,Me = tris((3-tert-butyl-5-methylpyrazolyl)borane; TpAd,Me = tris((3-adamantyl-5-methylpyrazolyl)borane; OSiO† = –OSi(OtBu)3; (Ad,MeArO)3mes = 1,3,5-tris{methylene(3-adamantyl-5-methylphen-2-ol)}mesitylene; Me4TACD = 1,4,7,10-tetramethylazadodecane. |
| This journal is © The Royal Society of Chemistry 2024 |