
Synthesis of triazole-fused tetracyclic spirooxindole derivatives via metal-free Huisgen cycloaddition†
Sandip
Maiti
,
Nabin
Parui
,
Joydev
Halder
and
Jyotirmayee
Dash
 *
*
School of Chemical Sciences, Indian Association for the Cultivation of Science Jadavpur, Kolkata-7000032, India. E-mail: ocjd@iacs.res.in
First published on 6th August 2024
Abstract
We report an efficient, metal free method for synthesizing tetracyclic spirooxindole derivatives from N-protected isatins and propargyl bromide via Huisgen cycloaddition. This simple and practicle method provides access to spirooxindoles containing five-, six-, or seven-membered rings fused to a triazole ring.
The spirooxindole core holds significance due to its prevalence in numerous natural products1 and biologically active compounds.1–4 The tricyclic skeleton as seen in horsfiline (Ia; Fig. 1), linked with a pyrrolidine, is known for its traditional use in herbal medicine for its antitumor properties.5 Similarly, coerulescine (Ib; Fig. 1) and elacomine (II, Fig. 1) exhibit antitumor activities.5 Compound III (Fig. 1) is a promising lead compound for inhibitors of β-secretase (BCAE-1) involved in Alzheimer's disease,6 while spirotryprostatin B (IV; Fig. 1) inhibits G2/M progression of mammalian tsFT210 cells.7 Spirooxindoles fused with piperidine systems are used to treat anaemia (HIF PHD 1–3, V).8 Compounds VI,9VII,10 and VIII11(Fig. 1) are known as selective anticancer agents. Given their widespread significance, several synthetic methods such as transition metal catalysis,4a–d organocatalysis,4e,f photocatalysis4g have been documented in the literature to access this class of compounds.
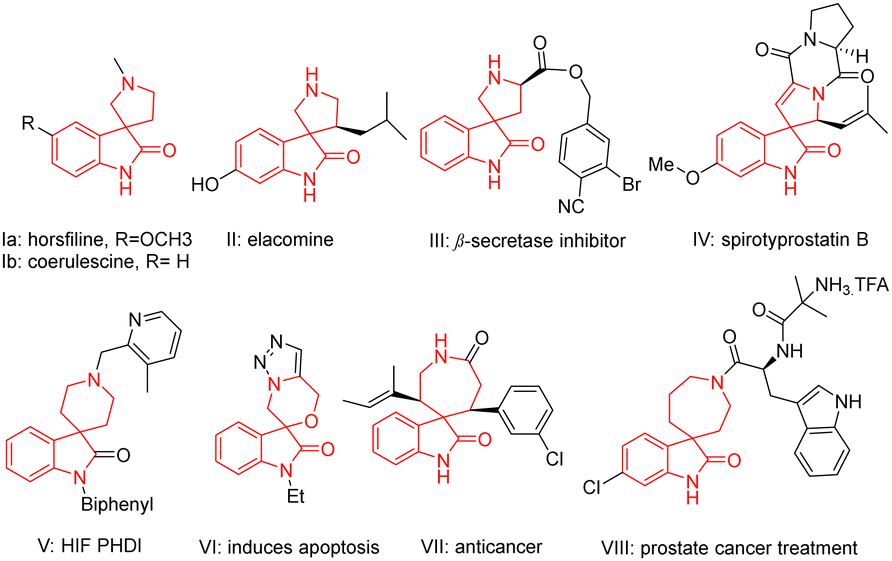 | ||
| Fig. 1 Some bioactive and naturally occurring spirooxindoles. | ||
The synthesis of 3-spirooxindoles has been achieved using various methods,12 including Ru-catalyzed ring-closing enyne metathesis and Co-mediated Pauson–Khand cyclization.13 Morpholine-fused spirooxindole derivatives with 1,2,3-triazole moieties, like compound VI show cytotoxic effects against various cancer cell lines, highlighting their therapeutic potential.9 Our work has focused on synthesizing hydantoin, thiazolidinedione, and oxindole-based spirocycles using ring-closing metathesis.14 Building upon our work on AAC15 and spirooxindoles,14c we aim to develop a unified method for synthesizing tetracyclic spirooxindoles with five-, six-, or seven-membered heterocycles fused to a triazole ring (Scheme 1).
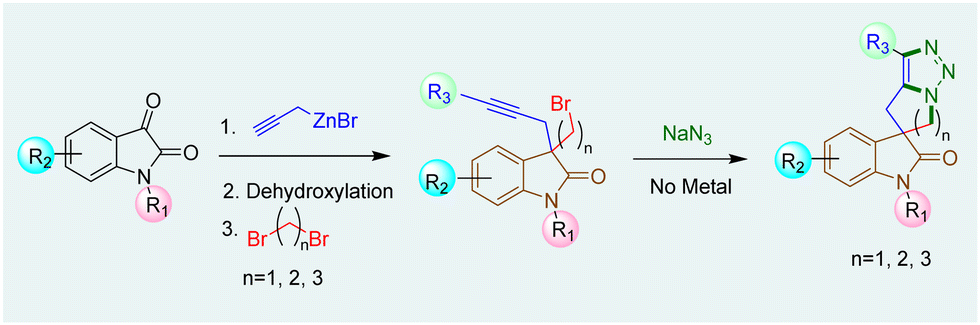 | ||
| Scheme 1 Synthesis of 1,2,3-triazole fused spirooxindoles. | ||
The synthesis commenced with the zinc-mediated propargylation on N-protected isatin derivatives (1a–k) in tetrahydofuran, leading to the synthesis of 3-hydroxy-3-propargyl substituted 2-oxindole derivatives 2a–k (Scheme 2). A catalytic amount of HfCl4 was employed to enhance the yield. For N-methyl isatin 1a, we obtained 91% yield of 2a, comparable to the report by Alcaide and co-workers.16 We then explored the scope and generality of this approach on several N-protected isatins (1b–d), as well as isatins having different types of electron withdrawing (1i–k) and electron donating (1e–h) groups on different positions of the aromatic ring (Scheme 2, see also ESI†). High yields of products (2a–k) were obtained overall. However, isatins with electron-donating groups on the aromatic ring gave slightly lower yields than those with electron-withdrawing substituents, as expected due to increased electrophilicity of the carbonyl group when electron withdrawing groups are present. To carry out dehydroxylation of 2, we first adopted a procedure similar to our previously reported SnCl2 in HCl/glacial AcOH method for N-protected 3-allyl-3-hydroxy-2-oxindole derivatives.17 However, a low yield of N-methyl protected 3-propargyl-2-oxindole 3a was obtained (Scheme 2). To overcome the problem, we converted the hydroxy group to chloro using thionyl chloride and DIPEA in dichloromethane. The reaction mixture was immediately utilized for the next step after workup without any purification. Upon treatment with Zn/AcOH in anhydrous tetrahydofuran, dechlorination occurred with moderate to good yield of the produced 3-propargyl-2-oxindole derivatives (3a–m) (Scheme 2).18 The yield of 3 was quite a bit higher for N-phenyl substituted 2-oxindole 3c compared to the yields obtained with Me, benzyl or allyl protections 3a–b, 3d. The substrate 2h containing OCF3 produced a relatively low yield of 3h, probably due to the decomposition of the starting material to some extent. Subsequently, 3-propargyl substituted 2-oxindoles 3a–m were treated with dibromides of different chain lengths in the presence of solid KOH in DMF. The reaction took place at the active hydrogen site resulting in the formation of 3,3-disubstituted 2-oxindoles 4–6 (Scheme 3). When dibromomethane was employed as the electrophile for 3a, the corresponding 3-bromomethyl-3-propargyl-2-oxindole 4a was obtained in 85% yield. However, upon using 1,2-dibromoethane and 1,3-dibromopropane, the yields of the corresponding oxindoles, 5a with a 2-bromoethyl substituent and 6a with a 3-bromopropyl group, decreased to 75% and 70%, respectively. A comparable decrease in yield was observed for substrates having different nitrogen protecting groups (3b–d) and functional groups in the aromatic ring (3e–k). This may be attributed to side reactions like SN2/E2 reactions becoming more prominent with longer alkyl chains, leading to undesired by-products or the partial reactant decomposition, reducing the overall yield of the desired product.
 | ||
Scheme 2 Synthesis of 3-propargyl substituted oxindoles from isatins. Reaction conditions: (i) 1 (2 mmol), propargyl bromide (6 mmol), Zn dust (12 mmol), HfCl4 (0.4 mmol), THF/aq. NH4Cl (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, 20 mL) at 0 °C. (ii) 2 (1 mmol), SOCl2 (1.2 mmol), DIPEA (3 mmol) in DCM (10 mL) at 0 °C-rt; (iii) Zn dust (6 mmol), AcOH (3 mL) in THF (10 mL) at 0 °C. :5, 20 mL) at 0 °C. (ii) 2 (1 mmol), SOCl2 (1.2 mmol), DIPEA (3 mmol) in DCM (10 mL) at 0 °C-rt; (iii) Zn dust (6 mmol), AcOH (3 mL) in THF (10 mL) at 0 °C. | ||
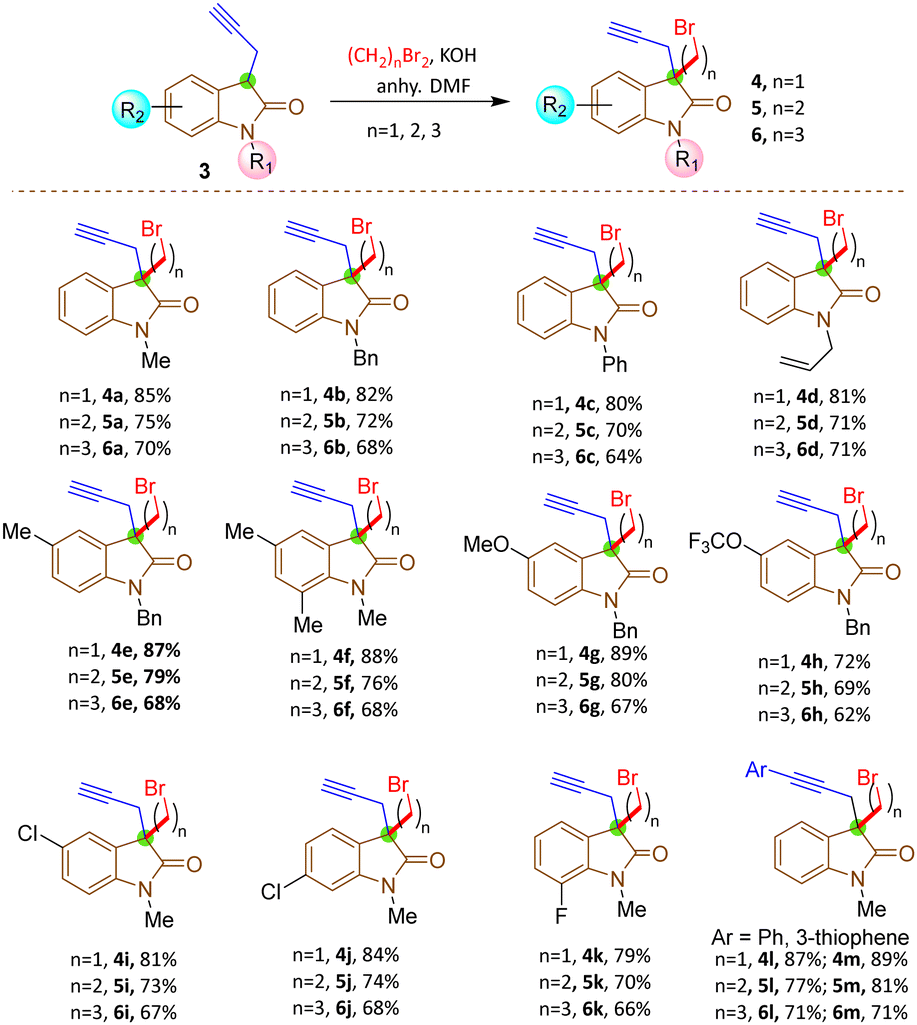 | ||
| Scheme 3 Synthesis of 3,3-disubstituted oxindoles from 3-propargyl substituted oxindole. Reaction conditions: 3 (0.25 mmol), dibromoalkane (0.50 mmol), powdered KOH (0.50 mmol) in DMF (0.5 mL) at 0 °C-rt. Isolated yields. | ||
Next, 3-bromomethyl-3-propargyl 2-oxindole 4a was treated with NaN3 in DMF at 80 °C for two hours initially, then increased to 100 °C for another 22 hours (Scheme 4). The reaction led to the formation of a triazole fused five-membered spirooxindole derivative 7a in 86% yield. The reaction involved a nucleophilic substitution, replacing the bromine atom with an azide group, followed by a metal-free intramolecular Huisgen cycloaddition,19 resulting in the formation of a triazole-fused spirooxindole derivative 7a (Scheme 4). Under identical conditions, the reaction was conducted on different N-protected 3-bromomethyl substituted 2-oxindoles (4b–d), as well as substrates having different functional groups on the aromatic rings (4e–k). Impressively, each reaction yielded good to excellent results. The structure of compound 7g was confirmed by single crystal X-ray analysis.‡ However, substrates 4h, 4k containing F and OCF3 groups afforded comparatively lower yields of the corresponding products 7h and 7k, possibly due to some extent of decomposition of the starting materials.
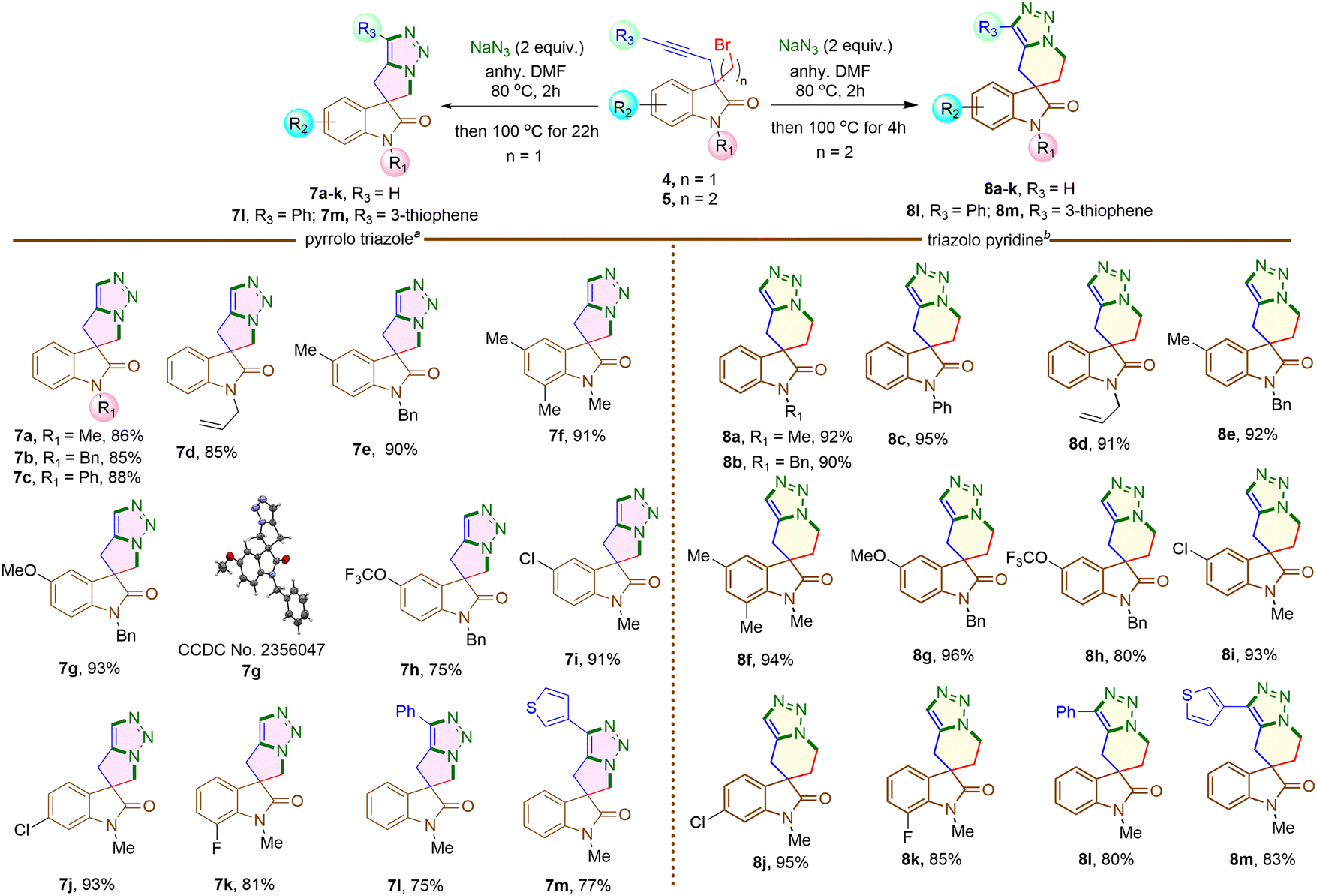 | ||
Scheme 4 Synthesis of pyrrolo triazoles and triazolo pyridine containing spirooxindole from 3,3-disubstituted 2-oxindoles. Reaction conditions: a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) 4 (0.15 mmol), NaN3 (0.30 mmol) in anhy. DMF (1 mL) at 80 °C for the 1st 2 hours then temperature raised to 100 °C for another 22 hours; b5 (0.15 mmol), NaN3 (0.30 mmol) in anhy. DMF (1 mL) at 80 °C for 2 hours then temperature raised to 100 °C for another 4 hours. Isolated yields. 4 (0.15 mmol), NaN3 (0.30 mmol) in anhy. DMF (1 mL) at 80 °C for the 1st 2 hours then temperature raised to 100 °C for another 22 hours; b5 (0.15 mmol), NaN3 (0.30 mmol) in anhy. DMF (1 mL) at 80 °C for 2 hours then temperature raised to 100 °C for another 4 hours. Isolated yields. | ||
When 3-bromomethyl-2-oxindoles containing internal alkynes such as 4I–m were treated with NaN3, a slightly lower yield of the cycloaddition products 7I–m was obtained. We next performed the one-pot azide formation and intramolecular cycloaddition of 3-(2-bromoethyl) oxindole derivatives 5a–l with two equivalents of NaN3 (Scheme 4). The reaction mixture was heated at 80 °C in DMF for two hours, following the same procedure as with 3-(bromomethyl) derivatives 4a–m. However, upon increasing the temperature to 100 °C, the cycloaddition of 5a–m completed after four hours, resulting in triazole-containing piperidine-fused spirooxindole derivatives 8a–m in 80–96% yields. The generality of the reaction was explored with substrates containing different N-protecting groups 5a–d as well as electron-withdrawing 5i–k and electron-donating substituents 5e–h on different positions of the aromatic ring. In each case, good to excellent yields of cycloaddition products 8a–k were obtained. The oxindoles containing internal alkynes 5l–m also furnished the desired products 8l–m in 80% and 83% yields, respectively, slightly lower than the terminal alkyne 5a–k. Thus, the substituents had minimal effect on the overall yield of the cycloaddition reaction. The yields and reaction rates were significantly influenced by the ring size of the heterocyclic compounds formed during the cycloaddition reaction; 6-membered heterocyclic ring-fused spirooxindoles 8a–m were formed more readily compared to the synthesis of five-membered rings 7a–m, which required longer reaction times. After successfully synthesizing triazole-containing five and six-membered ring-fused spirooxindole derivatives (7a–m, 8a–m), we explored our methodology for synthesizing seven-membered ring fused spirooxindole derivatives due to the limited number of reports available in the literature for this class of compounds.1c Compound 3-bromopropyl 3-propargyl 2-oxindole 6a was treated with 2 equivalents of NaN3 at 80 °C for two hours and then at 120 °C for another 46 hours (Scheme 5) for complete conversion. The desired 2-spirooxindole fused to triazoloazepine 9a was obtained in 72% yield. We then explored the scope of the reaction on various N-protected (6b–d) as well as different substituents at the different positions of the benzene ring of 3-bromopropyl 2-oxindoles (6e–k). In each case, triazole-fused seven-membered rings spiro-linked to 2-oxindoles (9a–k) were obtained in moderate to good yields. However, these yields were lower compared to the corresponding five and six-membered counterparts. The structure of compound 9d was established through single crystal X-ray analysis.‡ Following the successful synthesis of five, six and seven membered ring-fused spirooxindoles, we attempted to apply our methodology for the synthesis of eight and nine membered ring fused spirooxindoles. Unfortunately, we were unable to isolate any eight or nine membered ring-fused spirooxindoles from the reaction mixture under the optimized conditions (see ESI† for the synthesis of the substrates).
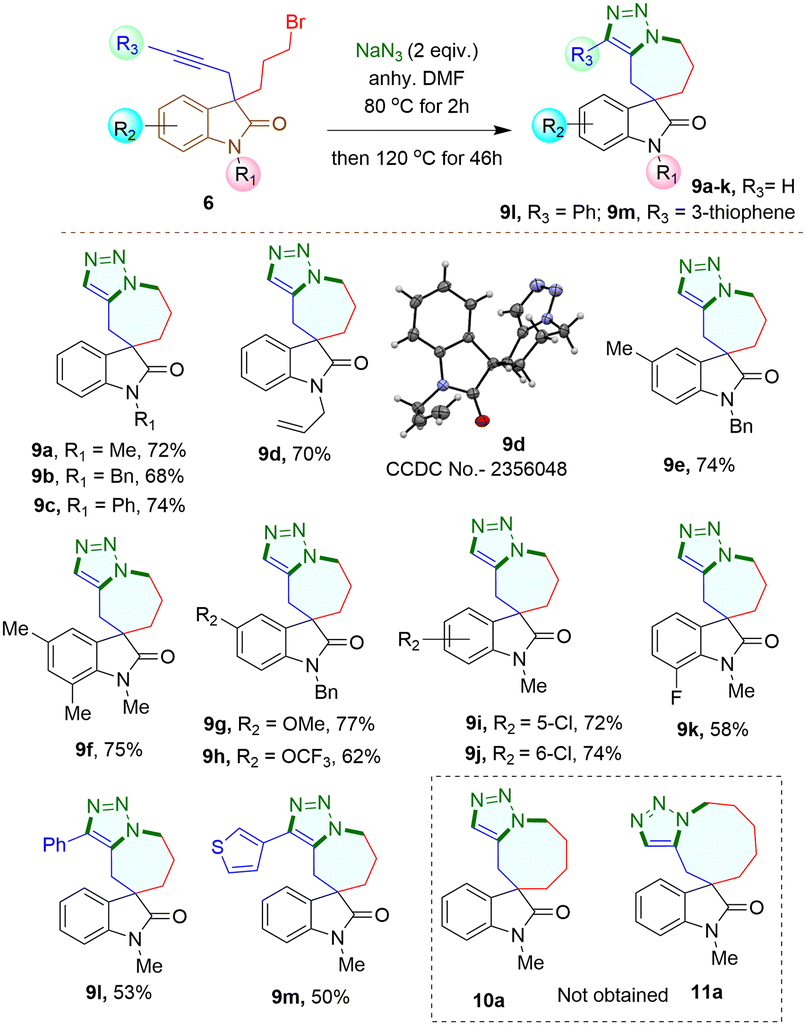 | ||
| Scheme 5 Synthesis of triazolo azepine-containing spirooxindoles from 3,3-disubstituted oxindoles. Reaction conditions: 6 (0.15 mmol), NaN3 (0.30 mmol) in anhy. DMF (1 mL) at 80 °C for the 1st 2 hours, then another 46 hours at 120 °C. Isolated yields. | ||
In summary, we have devised a novel and efficient approach for synthesizing spirooxindole-derived pyrrolidine, piperidine, and azepine-fused-1,2,3-triazole derivatives. This method involves propargylation at C-3 of isatin, dehydroxylation, one-pot substitution at C-3 with dibromoalkane and metal-free Huisgen cycloaddition. We found that spirooxindoles fused with 6-membered rings are formed more readily, while those with 7-membered rings result in lower yields. While existing reports mainly focus on 5 and 6-membered rings, our method effectively produces 5, 6, and 7-membered spirooxindole derivatives. Spirooxindoles fused to 6-membered rings are formed more readily than those with 5 or 7 membered rings. Our ongoing research will further explore these structures and evaluate their bioactivity.
JD thanks Wellcome Trust-DBT India Alliance (Grant Number IA/S/18/2/503986) and SERB-CRG (CRG/2021/04525) for funding. SM and NP thank CSIR, India for the research fellowship. JH thanks ICMR for the research fellowship.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected examples, see: (a) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104 CrossRef CAS PubMed; (b) S. Nasri, et al. , Top. Curr. Chem., 2021, 25, 379 Search PubMed; (c) A. J. Boddy and J. A. Bull, Org. Chem. Front., 2021, 8, 1026–1084 RSC; (d) M. Ganesh and S. Suraj, Org. Biomol. Chem., 2022, 20, 5651 RSC.
- S. L. Degorce, et al. , ACS Med. Chem. Lett., 2019, 10, 1198 CrossRef CAS PubMed.
- (a) S. T. Al-Rashood, et al. , J.Enzym. Inhib. Med. Chem., 2020, 35, 831–839 CrossRef CAS PubMed; (b) L. Chen and J. He, J. Org. Chem., 2020, 85, 5203–5219 CAS; (c) R. Murugan, et al. , Eur. J. Med. Chem., 2009, 44, 3272–3279 CrossRef CAS PubMed; (d) N. H. de Silva, et al. , Eur. J. Med. Chem., 2022, 240, 1–8 CrossRef PubMed; (e) M. Costa, et al. , Eur. J. Med. Chem., 2016, 123, 487–507 CrossRef CAS PubMed; (f) V. V. Vintonyak, et al. , Angew. Chem., Int. Ed., 2010, 49, 5902–5905 CrossRef CAS PubMed; (g) L. L. Zhang, et al. , Tetrahedron, 2019, 75, 1689–1696 CrossRef CAS; (h) L. Chen, et al. , J. Agric. Food Chem., 2016, 64, 6508 CrossRef CAS PubMed; (i) G. Zhou, et al. , Chin. J. Org. Chem., 2016, 36, 143–150 CrossRef CAS.
- (a) H. Yoon, et al. , Angew. Chem., Int. Ed., 2017, 56, 10920–10923 CrossRef CAS PubMed; (b) C. Shao, et al. , Chem. Commun., 2017, 53, 10429–10432 RSC; (c) C.-H. Chen, et al. , J. Org. Chem., 2017, 82, 6417–6425 CrossRef CAS PubMed; (d) J.-R. Song, et al. , Adv. Synth. Catal., 2020, 362, 500–505 CrossRef CAS; (e) D.-Y. Zhang, et al. , Chem. – Eur. J., 2015, 21, 10314–10317 CrossRef CAS PubMed; (f) S. K. Arupula, et al. , J. Org. Chem., 2018, 83(5), 2660–2675 CrossRef CAS PubMed; (g) M. Zhang, et al. , ACS Sustainable Chem. Eng., 2017, 5, 6175–6182 CrossRef CAS.
- (a) J. D. White, et al. , J. Org. Chem., 2010, 75, 3569–3577 CrossRef CAS PubMed; (b) H. Kamisaki, et al. , Chem. Eur. J., 2011, 17, 626–633 CrossRef CAS PubMed.
- I. V. Efremov, et al. , J. Med. Chem., 2012, 55, 9069–9088 CrossRef CAS PubMed.
- C. B. Cui, et al. , J. Antibiot., 1996, 49, 832–835 CrossRef CAS PubMed.
- P. Vachal, et al. , J. Med. Chem., 2012, 55, 2945–2959 CrossRef CAS PubMed.
- K. R. Senwar, et al. , Eur. J. Med. Chem., 2015, 102, 413–424 CrossRef CAS PubMed.
- J. J. Liu, et al., Hillsborough, US Pat., 2008/0287421 A1, 2008 Search PubMed.
- S. Pellegrino, et al. , Bioorg. Med. Chem., 2013, 21, 5470–5479 CrossRef CAS PubMed.
- (a) F. Yu, et al. , Green Chem., 2013, 15, 453 RSC; (b) H. Xu, et al. , Chem. Commun., 2016, 52, 8002–8005 RSC; (c) J. L. Meloche and B. L. Ashfeld, Angew. Chem. Int. Ed., 2017, 56, 6604–6608 Search PubMed; (d) G.-J. Mei and F. Shi, Chem. Commun., 2018, 54, 6607–6621 RSC; (e) S. Xing, et al. , Adv. Synth. Catal., 2021, 363, 1044–1049 CrossRef CAS.
- S. R. Kandimallaa and G. Sabitha, Adv. Synth. Catal., 2017, 359, 3444–3453 CrossRef.
- (a) K. Dhara, et al. , Org. Biomol. Chem., 2011, 9, 3801–3807 RSC; (b) K. Dhara, et al. , J. Org. Chem., 2012, 77(18), 8071–8082 Search PubMed; (c) K. Dhara, et al. , Angew. Chem. Int. Ed., 2015, 54, 15831–15835 CrossRef CAS PubMed.
- (a) P. Saha, et al. , Chem. Commun., 2019, 55, 731–750 RSC; (b) P. Saha, et al. , Annual Rep. Med. Chem., 2020, 54, 287–324 Search PubMed; (c) P. Saha, et al. , Chem. Soc. Rev., 2023, 52, 4248–4291 RSC.
- B. Alcaide, et al. , J. Org. Chem., 2005, 70(8), 3198–3204 CrossRef CAS PubMed.
- T. Mandal, et al. , Org. Lett., 2018, 20(16), 4759–4763 CrossRef CAS PubMed.
- J. E. Thomson, et al. , Tetrahedron, 2010, 66, 3801–3813 CrossRef CAS.
- M. Breugst and H. U. Reissig, Angew. Chem., Int. Ed., 2020, 59, 12293–12307 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2356047 and 2356048. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02534b |
| ‡ CCDC 2356047 and 2356048† contain the supplementary crystallo-graphic data for compounds 7g and 9d (see also ESI†) |
| This journal is © The Royal Society of Chemistry 2024 |